
Abstract
Background
The shoot apical meristem (SAM), from which all above-ground tissues of plants are derived, is critical to plant morphology and development. In maize (Zea mays), loss-of-function mutant studies have identified several SAM-related genes, most encoding homeobox transcription factors (TFs), located upstream of hierarchical networks of hundreds of genes.
Results
Here, we collect 46 transcriptome and 16 translatome datasets across 62 different tissues or stages from the maize inbred line B73. We construct a dynamic regulome for 27 members of three SAM-related homeobox subfamilies (KNOX, WOX, and ZF-HD) through machine-learning models for the detection of TF targets across different tissues and stages by combining tsCUT&Tag, ATAC-seq, and expression profiling. This dynamic regulome demonstrates the distinct binding specificity and co-factors for these homeobox subfamilies, indicative of functional divergence between and within them. Furthermore, we assemble a SAM dynamic regulome, illustrating potential functional mechanisms associated with plant architecture. Lastly, we generate a wox13a mutant that provides evidence that WOX13A directly regulates Gn1 expression to modulate plant height, validating the regulome of SAM-related homeobox genes.
Conclusions
The SAM-related homeobox transcription-factor regulome presents an unprecedented opportunity to dissect the molecular mechanisms governing SAM maintenance and development, thereby advancing our understanding of maize growth and shoot architecture.
Similar content being viewed by others

Background
In plants, the shoot apical meristem (SAM) is a domed structure at the aerial growth tip that contains stem cells that generate daughter cells that will differentiate to become above-ground organs. The balance of organ initiation and stem-cell maintenance leads to constraints in SAM size and shape [1, 2]. SAM architecture is determined in part by the genetic background, and several genes contributing to the gross morphology of the SAM have been identified through mutant screens [3]. In the world’s most-produced crop maize (Zea mays L.), several SAM-related genes have been identified in this manner; these genes show distinct SAM-specific expression patterns and act at the top of hierarchical networks comprising hundreds of genes [4,5,6,7]. Some cloned SAM-related genes are homeobox genes encoding a protein containing a typical DNA-binding domain of 60 amino acids, known as a homeodomain, which is present in dozens of transcription factors (TFs).
Homeobox proteins can be classified into 14 distinct classes in plants, encompassing well-established subfamilies such as KNOTTED1-like homeobox (KNOX), zinc-finger homeodomain (ZF-HD), WUSCHEL-related homeobox (WOX), and homeodomain leucine zipper (HD-ZIP), as well as emerging evolutionary categories such as plant homeodomain (PHD) and DNA-binding homeobox (DDT) [8]. The fundamental role of homeobox proteins lies in the control they exert over the progression of growth and development. Typically, members of the KNOX subfamily have major roles in the proper regulation of orchestrating plant growth and development, including the regulation of meristem formation and the maintenance of organ morphogenesis [9,10,11]. The HD-ZIP subfamily exhibits substantial potential in enhancing plant growth behind plant responses to environmental stressors [12, 13]. Similarly, the WOX subfamily contributes to early embryogenesis, the sustained activity of meristematic stem cells, and the development of lateral organs [14,15,16]. Research on HD-containing proteins in plants began in maize, wherein Knotted1 was first identified in controlling leaf differentiation [17, 18]. Subsequently, the functional importance of numerous genes encoding proteins with HD domains has been corroborated across diverse plant species, including Rough sheath1 (Rs1), Gnarley1 (Gn1), Narrow sheath1 (Ns1), and Ns2, among others [19, 5, 20]. Collectively, these findings underscore the immense regulatory potential of the homeobox TF family in shaping plant growth, development, and responses to environmental stimulus.
Homeobox TFs interact with the promoter regions of specific target genes. A single TF can potentially target hundreds to thousands of genes, and the collective interactions among multiple TFs and their respective targets establish a central regulatory network that governs nearly all aspects of organismal biology [21, 22]. Despite the biological significance of TF regulatory networks, there has been a persistent bottleneck in the identification of such networks. This bottleneck necessitates the collection of vast amounts of information through individual experiments, each targeting a single TF [23, 24]. In the case of plants, additional challenges arise due to the relative lack of specific antibodies that can be used for chromatin immunoprecipitation followed by sequencing (ChIP-seq) assays, as well as the recalcitrance or difficulty of crop transformation for the expression of a construct encoding an epitope-tagged version of a TF of interest [21, 25]. Recent attempts to address this issue have employed genome-wide analyses that combine single ChIP experiments with high-throughput sequencing. For example, a regulatory network consisting of 112 TFs was identified in human colorectal-cancer cells and a network comprising 104 leaf-related TFs was reconstructed in maize [26, 27]. However, it is worth noting that such studies remain relatively scarce, and elucidation of the landscape underlying regulatory networks within plant genomes remains challenging.
We previously devised a transient and simplified cleavage under targets and tagmentation method known as tsCUT&Tag, which can deduce the targets of TFs across different plant tissues and stages based on a comprehensive machine-learning model [28]. Here, we employed tsCUT&Tag, co-expression networks, and gene-regulatory networks to dissect the regulome of homeobox TFs that are strongly associated with SAM maintenance and development in maize. We constructed regulatory networks, offering a valuable resource for dissecting the intricate regulatory mechanisms conferring plant architecture in maize via homeobox TFs. We provide evidence for the pivotal role of WOX13A in regulating Gn1, ultimately influencing plant height. Our findings shed light on the regulatory patterns of SAM-related homeobox TFs and well-known SAM functional genes.
Results
SAM-related homeobox genes are enriched in KNOX, WOX, and ZF-HD subfamilies
Homeobox genes are TFs that control tissue differentiation and development [29]. Several key homeobox genes have been cloned and shown to function in the SAM in maize. To ascertain the functional relevance of these genes in SAM development, we first assembled 26 well-known SAM functional genes and described their mutant phenotypes [30,31,32,33,34,35,36,37,38,39,40] (Fig. 1A, Additional file 2: Table S1). This includes nine homeobox TFs. Of these, Kn1, Gn1, ZmBLH12, and ZmBLH14 displayed elevated expression levels within the SAM and related meristems (Additional file 1: Fig. S1) [4, 5, 17]. We leveraged an existing maize multi-omics integrative network [41] to define the direct interconnected network for these 26 genes at both the co-expression and co-translation levels (Fig. 1B, Additional file 3: Table S2). Focusing on the homeobox family within the co-translation and co-expression integrated network (co-network) of SAM functional genes, three homeobox subfamilies are either co-expressed or co-translated alongside SAM functional genes (Fig. 1C). The KNOX, WOX, and ZF-HD subfamilies have a statistically significantly higher co-expression ratio with SAM functional genes compared to randomly selected control genes (Fig. 1C), suggesting that these subfamilies are more likely to function in the SAM. Next, we established a gene-regulatory network (GRN) centered on the three-most-significantly enriched subfamilies in the co-network (Fig. 1C), namely KNOX, WOX, and ZF-HD, to scrutinize in more detail the regulatory mechanisms of SAM functional genes.

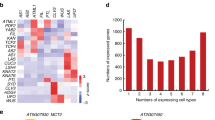
Genes co-expressed and co-translated with known SAM functional genes are statistically significantly enriched in the KNOX, ZF-HD, and WOX subfamilies. A Heatmap representation of the expression levels of known SAM functional genes in the SAM, differentiated reproductive tissues, and other mature tissues. B Co-expression and co-translation genome networks of known SAM functional genes. MaxS: max scaling. C Proportion of known SAM functional genes in co-expression and co-translation networks for different homeobox subfamilies compared to random control genes (an equal random selection of genes in all co-networks). ** represents significance, ns represents no-significance
Genome-wide binding sites of maize SAM-related TFs
Toward dissecting the molecular mechanisms by which maize SAM-related TFs shape tissue differentiation and development, we cloned full-length coding sequences of SAM-related TF genes from the KNOX, WOX, and ZF-HD homeobox subfamilies from a maize TFome (Additional file 4: Table S3) [42]. To check sequence homology between maize V3 and V5 genome-annotation releases, we aligned protein sequences for each TF. Twenty-three of the 27 studied TFs have identical V3 and V5 protein sequences. Four genes, Zm00001eb354880, Zm00001eb058930, Zm00001eb295920, and Zm00001eb217470, have minor differences between the two versions (Additional file 1: Fig. S2). We conducted tsCUT&Tag experiments by integrating CUT&Tag in B73 protoplasts, and ATAC-seq and RNA-seq across various tissues and stages [28] to catalog genomic binding sites for each TF. We successfully performed nearly 100 tsCUT&Tag experiments on three TF subfamilies. Subsequently, a standardized pipeline was used for data processing with stringent criteria for normalized strand cross-correlation coefficient (NSC) and relative strand cross-correlation coefficient (RSC), both set at > 1.05 and 1.0, respectively (Additional file 5: Table S4). The Pearson correlation coefficient for reproducibility between biological replicates was set at ≥ 0.8 (Additional file 1: Fig. S3). A dataset comprising 27 TFs, each with at least two biological replicates, was compiled. Among them, seven members belong to the KNOX subfamily and 10 each to the WOX and ZF-HD subfamilies. We delineated the genomic relationships among these homeobox TFs through a phylogenetic analysis, finding higher phylogenetic affinity within subfamilies except for the KNOX subfamily member Ns1, which is more closely related to the WOX subfamily members (Additional file 1: Fig. S4).
Following a series of stringent bioinformatics analyses and filters (see the “Methods” section), we identified a total of 30,915 reproducible TF-bound sites for these SAM-related TFs (Fig. 2A). The number of binding peaks showed considerable variability among TFs, with a median value of approximately 5002 binding sites per TF (Inter-quantile range [IQR] 1016 to 8677) (Additional file 1: Fig. S5A, Additional file 6: Table S5). The number of regulated target genes detected by tsCUT&Tag experiments ranged from 962 to 7227 per TF (Additional file 1: Fig. S5B, Additional file 6: Table S5). The genomic distribution of binding peaks showed a bias toward the promoter regions of target genes, as previously described [43] (Fig. 2B and C, P < 7.2E − 10). Most tested TFs preferentially bound sequences containing core ATTA and CCAA motifs, consistent with Arabidopsis homeobox TFs (Additional file 7: Fig. S6) [44]. With these target-gene datasets for each SAM-related homeobox TF, we then constructed a comprehensive regulome.

The regulome of maize SAM-related homeobox subfamilies. A Total number and percentage of binding peaks identified by tsCUT&Tag for KNOX, ZF-HD, and WOX subfamily members. B Metaplot showing the distribution of TF binding sites for KNOX, ZF-HD, and WOX family members tested. C Heatmap representation of tsCUT&Tag and ATAC-seq signals over ~ 3 kb of sequence upstream and downstream of the transcription start site (TSS) for the SAM-related homeobox TFs Rs1 and Ns1. D Zm00001eb007470 (ZmRPH1) in the genome browser. ATAC-seq and TF binding results are shown. SAM, shoot apical meristem
We performed gene-ontology (GO) enrichment analysis on the putative target genes of using -Log10(FDR) as the enrichment standard. We saw statistically significant enrichment in functions related to transport processes, growth and development, and responses to hormones and stress (-Log10(FDR) > 2) (Additional file 7: Table S6). For example, several target genes are related to plant-hormone stimuli, including auxin, abscisic acid, gibberellin, cytokinin, and brassinosteroids. Other enriched target genes are involved in organ development and morphogenesis. Taken together, genes from the regulome of SAM-related homeobox TFs have physiological relevance and constitute a network resource for further understanding maize growth and development.
Target hubs and HOT regions are enriched for regulatory genes
Genomic regions with high TF occupancy are frequently associated with important functions [45, 46]. Here, we aggregated all TF-target-gene interactions for the 27 tested TFs. 77.2% of potential target genes are bound simultaneously by multiple TFs. As the number of binding TFs increases, the number of co-regulated target genes decreases, indicating that most TFs co-regulate a limited number of target genes (Additional file 1: Fig. S7A). Notably, the proportion of overlapping target genes regulated by TFs within subfamilies is statistically significantly higher than between subfamilies (P = 0.004, KS-test) (Additional file 1: Fig. S7B).
Hub target genes engage in crosstalk between different signaling pathways and represent important TF targets [47]. We built a random TF-target-gene distribution to delineate the hub target genes by randomizing the relationship between TFs and their potential target genes, while preserving the number of potential target genes for each TF, as previously described (Additional file 1: Fig. S8A) [48]. Based on the 99th percentile values of the randomized distributions, we defined 3,168 potential target genes without enhancer enrichment, which are bound by 13 or more TFs, as hub targets (Additional file 8: Table S7). Complementary to hub target genes, we characterized “hot” regions in the genome as regions bound by many TFs. Hot regions are distinct from hub genes in that they can be bound by many TFs at different locations. Their occupancy follows a negative exponential curve [48] (Additional file 1: Fig. S8B). A total of 3053 hot regions without enhancer enrichment were identified, defined as regions bound by 12 or more TFs (Additional file 8: Table S7). These hot regions are linked with 2773 target genes. We observed that 2,608 (~ 82.3%) of 3168 hub targets overlap with hot regions, with the remaining 17.7% of hub genes not being associated with hot regions because of the many TFs that bind to their loci target at different regions (Additional file 1: Fig. S8C).
Next, we surveyed regulatory complexity and identified a specific set of low-complexity genes, i.e. those bound by a single TF (Additional file 8: Table S7). GO-term enrichment analysis on these genes revealed their association with protein-complex biogenesis and assembly, and growth (Additional file 1: Fig. S6D). However, hub genes exhibited a pronounced enrichment within critical pathways, including signal transduction, photoprotection, defense response, and hormone-signaling pathways, underscoring their important roles in regulatory networks (Additional file 1: Fig. S8E).
We also identified several potential target genes bound by more than 20 TFs. For example, Zm00001eb007470 (ZmRPH1, encoding a microtubule-associated protein) showed a high-TF-occupancy region bound by 24 TFs (Fig. 2D). ZmRPH1 contributes to cytoskeletal architecture and its over-expression induces a dwarf phenotype [49].
KNOX, WOX, and ZF-HD subfamilies partially overlap in binding-site specificity
Some TFs function collaboratively in organisms. To further delineate TF combinations, we visualized genome-wide mapping and quantified the frequency of co-localization of all possible pairs of TFs to target genes, based on the presence of their respective binding peaks within each TF sample (Additional file 1: Fig. S9A and Additional file 1: Fig. S9B). Specifically, we considered all TF pairs that bind to the same target genes via non-overlapping motifs. Most pairs of TFs showed a low degree of co-localization for their binding sites within target genes and co-localizing TFs predominantly belong to the same subfamily.
To evaluate binding-site specificity within the three homeobox subfamilies, we performed a comparative analysis and saw that ~ 51% of all detected peaks are present for at least two subfamilies (Fig. 2A). This finding indicates that although these three subfamilies have distinct binding profiles, they also show substantial overlap in their binding sites among individual members within each subfamily. We performed motif analysis using the upstream and downstream regulatory regions of peak summits bound by KNOX-only, WOX-only, ZF-HD-only members, or bound by all subfamilies referred to as shared peaks. Shared-peak subsets were enriched for the core motifs ATTA and CCAA. However, WOX-only and KNOX-only peaks only retuned one over-represented motif, CCAA. Notably, we detected ATTA-enriched motifs in ZF-HD-only peaks (Fig. 3A).

Binding specificity of KNOX, ZF-HD and WOX subfamily TFs to unique and shared peaks in their target genes. A Sequence logos for the top motifs identified for sites bound by WOX-only, ZF-HD-only, KNOX-only TFs, or peaks shared among them. Values represent the E-value of each motif. B Venn diagram of target genes overlapping among KNOX, ZF-HD, and WOX subfamilies. C Functional enrichment of WOX-only, ZF-HD-only, KNOX-only, or shared target genes determined by GO analysis. SN, mean SumNormalizer
Although different TFs within the same subfamily shared more target genes (53% average) than different TFs across different subfamilies (49% average; P = 0.004, Additional file 1: Fig. S7B), we observed that at least 74–89% of all target genes from one of the three TF subfamilies were also among the targets of at least one of the other two subfamilies (Fig. 3B). This indicates that the target genes specifically regulated by each subfamily accounted for only 10.7–26.4% of the total regulated by that subfamily. We then tested whether the four categories of target genes (Fig. 3C) were associated with unique functions. GO-term enrichment analysis revealed that genes co-occupied by all members of all three TF homeobox families are statistically significantly enriched in terms related to “plant-organ development,” “flower development,” “leaf development,” “response to hormone,” “fruit development,” and “signal transduction” (Fig. 3C). However, genes exclusively targeted by KNOX-type TFs (KNOX-only) were enriched for terms associated with “plant-type cell wall,” and WOX-only targets were enriched for “chloroplast” and “meristem maintenance.” We detected no statistically significant enrichment for GO terms among high-confidence genes exclusively targeted by ZF-HD-specific TFs (Fig. 3C). Collectively, these results underscore the propensity of KNOX, WOX, and ZF-HD subfamily TFs to frequently co-regulate the same locus.
Functional differentiation of SAM-related TFs
Gene duplication is a fundamental mechanism underpinning the functional diversification of paralogous genes resulting from whole-genome duplication (WGD), tandem duplication (TD), duplication mediated by transposition (TRD), proximal duplication (PD), and dispersed duplication (DSD) [50, 51]. To enhance the construction of the regulome, we additionally gathered published ChIP-seq data for three subfamilies, namely Kn1 of KNOX, for analysis [51]. We identified 14 duplicated gene pairs, which we classified as having arisen from WGD (three pairs), PD (one pair), TRD (five pairs), and DSD (five pairs) (Additional file 9: Table S8) [51]. These duplications predominantly arose during the divergence of the Magnoliophyta, Petrosaviidae, Commelinids, and Poaceae and ultimately the speciation of Z. mays (Additional file 1: Fig. S10A and Additional file 1: Fig. S10B, Additional file 9: Table S8).
To probe the functional fates that genes may assume after duplication events, we examined the temporal and spatial expression patterns of duplicated genes across maize development by RNA-seq. We identified two duplicate pairs showing the same overall pattern of expression (i.e., conserved duplications), with the remaining two pairs showing distinct expression patterns (i.e., divergent duplications), with ~ 70% of the duplications showing partial expression divergence (Additional file 9: Table S8). Protein-sequence divergence might also lead to functional differentiation. To explore regulome divergence between duplicated gene pairs, we introduce an index—divergence score (DS) [52] to assess differentiation between two regulatory networks. Notably, as protein-sequence similarity increased, new regulatory networks emerged (Fig. 4A, R = 0.56, P = 0.01). The randomized DS displays a normal distribution (Fig. S11). Consequently, we categorized duplications as conserved, partially diverged, or diverged, with a 95% threshold (Fig. 4B, Additional file 9: Table S8). The duplicated pair Zhd1 and Zhd21 is conserved at the regulome level, indicating that these TFs regulate the expression of the same genes. By contrast, four duplicate pairs, such as Kn1 and Lg3, showed divergence in the cohort of genes that each encoded TF regulates, and the remaining pairs (11/14) fell in between, leading us to define them as partially diverged (Fig. 4B).

Conservation and divergence in networks among duplicated genes. A Increased protein-sequence similarity is associated with greater similarity in gene-regulatory networks (GRN) generated by tsCUT&Tag. B Network rewiring between duplicate pairs of TFs may remain unchanged after duplication (conserved), involve both common and unique connections (partly diverged), or exhibit few common connections (diverged). Examples from each scenario in the classes of duplicates are shown at right. Nodes are denoted as 1:1000. C Pattern of conservation or divergence between transcriptome and regulome data for the same duplicate pairs of TFs. D Differential expression analysis based on RNA-seq between wild type and the kn1 mutant. E GO terms of target genes regulated by Kn1 and Lg3
By integrating the differentiation pattern observed at the transcriptome and regulome levels, we noticed one divergent duplication with the same behavior across these two regulatory levels (Fig. 4C). Kn1 and Liguleless3 (Lg3) (a TRD), showed functional differentiation and bear distinct cis-elements within their promoter regions and have different gene structures, resulting in a diversification of combinatorial regulation (Additional file 1: Fig. S12, A and B). The duplicate genes also differed in their expression patterns, with Kn1 more highly expressed than others in differentiated tissues, but similar in mature tissues (Fig. 12C). Additionally, analysis of Kn1 target genes and differentially expressed genes (DEGs) in the kn1 mutant revealed that Kn1 targets Lg3 and up-regulates its expression (Fig. 4D) [17]. Functional enrichment analysis of target genes indicated that both Kn1 and Lg3 exhibit a broad spectrum of hormone-related regulatory functions (Fig. 4E). However, Kn1 targets specifically participate in regulation of circadian rhythm, vegetative phase change, and growth and development of flowers and leaves (Fig. 4E). Meanwhile, Lg3 targets are involved in regulation of leaf senescence (Fig. 4E). These findings align with previous findings that Kn1 primarily functions in regulating the differentiation of SAM into cell groups with distinct functions, and in growth and development of lateral organs. Moreover, Kn1 often interacts with Lg3 and Lg4 to jointly regulate leaf development [51]. We conclude that Kn1 duplication that gave rise to Lg3 led to the subfunctionalization of fate differentiation. This example revealed that divergence in the connectivity of the regulome between duplicated genes is accompanied by divergence in expression, hinting at functional divergence.
Tissue- or stage-dynamic networks of SAM-related TFs
Plant development and phenotypic variation are governed by precise and multifaceted GRNs [41, 53, 54]. Here, we constructed a GRN using information integrated from all target genes identified by tsCUT&Tag, co-expression, and co-translation [41]. This network contained 235,043 edges and 18,463 nodes, providing a substantial resource to facilitate the functional dissection of maize SAM-related homeobox TFs.
In a typical network, the in-degree of nodes represents the number of genes that can be regulated or interact with a given node. We evaluated the distribution of in-degree values, which showed the characteristics of a scale-free network for the SAM-related homeobox TFs (Additional file 1: Fig. S13A). Moreover, converting in-degree values to their log2 values resulted in a pattern consistent with a power-law distribution (R2 = 0.87) (Additional file 1: Fig. S13B). Hot nodes within the network had more connectivity than others, signifying their importance in shaping information-flow pathways. We selected the top 10% of nodes with the highest degree of connectivity, corresponding to 1844 genes; of these, ~ 45% (822 genes) were also hub targets bound by 13 TFs or more, as determined by our tsCUT&Tag experiments (Additional file 1: Fig. S13C, Additional file 10: Table S9). GO enrichment analysis of these genes underscored their involvement in physiology, including “signal transduction,” “response to hormone,” “organ senescence,” and “defense response,” indicative of the important roles these nodes play within the network (Additional file 1: Fig. S13D).
To further dissect the functional roles of SAM-related homeobox TFs, we gathered comprehensive RNA-seq, ribosome profiling (Ribo-seq), and ATAC-seq datasets collected from various B73 tissues: leaves, SAM, internodes, stems, tassels and endosperm (Additional file 11: Table S10). We defined 18,242–26,493 genes across different tissues or stages as being expressed, with a minimum expression level of transcripts (TPM > 1) (Additional file 1: Fig. S14A, Additional file 11: Table S10). We also detected between ~ 50% and ~ 80% of the target genes identified by tsCUT&Tag as being expressed in a specific tissue or stage (Additional file 1: Fig. S14B). ATAC-seq unveiled 4187–27,145 accessible-chromatin regions spanning tissue stages. Between these two datasets, 10–74% of the target genes identified by tsCUT&Tag overlap with ATAC-seq open regions in specific tissues or stages. Within accessible-chromatin regions across different tissues, each TF regulates 521–5353 target genes (Additional file 1: Fig. S14C and Additional file 1: S14D). The transcriptome GRN and translatome GRN also showed a substantial overlap with the target genes for each TF, ranging from 0.3–27% and 0–30% of target genes as detected by tsCUT&Tag (Additional file 1: Fig. S14E and Additional file 1: Fig. S14F).
To construct tissue-specific dynamic regulatory networks, we first designed a machine-learning model using ATAC-seq and tsCUT&Tag data from etiolated seedlings. The accuracy of the multi-layer perceptron models for the 27 TFs ranged from 0.75 to 0.96 (Additional file 12: Table S11). The constructed models were then used to predict TF binding sites in the SAM based on ATAC-seq data. We predicted hundreds of TF binding sites in the SAM (Additional file 13: Table S12). These binding sites were used to build the SAM dynamic regulatory network.
We constructed SAM dynamic networks by integrating multiple datasets. To acquire high-confidence TF-DNA regulatory associations, we required the target genes predicted by tsCUT&Tag and ATAC-seq to coexist in both the transcriptome and translatome GRNs. Simultaneously, we required the target genes expressed in the SAM (TPM > 1). Ultimately, we constructed a dynamic SAM network (Fig. 5A, Additional file 14: Table S13). It contained several known functional genes that influence plant architecture, flowering, and grain development, such as BEL1-like homeodomain12 (ZmBLH12), BEL1-like homeodomain12 (ZmBLH14), and Fasciated Ear4 (Fea4) (Fig. 5A). GO enrichment analysis demonstrated that the SAM dynamic network is predominantly enriched for genes related to “meristem development,” “meristem initiation,” “meristem maintenance,” “shoot system development,” “hormone-mediated signaling pathway” (gibberellin, ethylene, and auxin), “tissue development” (flower, fruit, seed, leaves), and cell differentiation (Fig. 5B). Together, these findings highlight the biological significance inherent in SAM dynamic networks, providing insights into the functional characteristics of hitherto unclassified genes.

SAM dynamic regulatory landscape of maize TFs. A–B The dynamic regulatory network in the SAM (A) and their functional annotation by GO enrichment (B). RichFactor represents the ratio of the input gene number in the pathway to the background gene number in the pathway
Networks of SAM-related TFs associated with plant height
Plant height is an important agronomic trait with a complex regulatory mechanism [55]. We collated a list of published functional genes related to maize plant height and divided them into more than 12 distinct modules encompassing hormone-signaling pathways (gibberellin, auxin, brassinosteroid, cytokinin, ethylene, abscisic acid, and strigolactone), phytochrome signaling, as well as pathways involved in vegetative and reproductive development, SAM maintenance and determinacy, microtubule cellulose, sugar metabolism, and nutrition. We then constructed a plant-height functional network selected by an integrated network based on this list of ~ 78 known functional genes and TF genes (Fig. 6A, Additional file 15: Table S14). An example of the resulting network, namely for Knox6, illustrates how homeobox TFs exert their influence on plant height through a multitude of distinct pathways (Fig. 6B). Each functional gene appeared to be affected by multiple homeobox TFs, as shown for Vanishing tassel2 (Vt2), demonstrating the complexity of the molecular network governing plant height (Fig. 6C). As an example, Vt2 is involved in auxin biosynthesis, this gene is simultaneously regulated by KNOX members Knox6 and Hb20 (Fig. 7D). Moreover, Knox6 also bound to the Hb20 locus, indicating that one KNOX TF can regulate another KNOX gene (Fig. 7D). To validate these regulatory relationships, we conducted firefly luciferase (LUC) reporter assays in maize protoplasts. We cloned 2-kb Vt2 and Hb20 promoter regions upstream of the LUC reporter gene and used Knox6 and Hb20 as effectors. Knox6 and Hb20 repressed the transcription of the Vt2, Knox6 activated the Hb20 (Fig. 6E and F), indicating that Knox6 and Hb20 have regulatory roles in auxin biosynthesis, thereby influencing plant height [56].

Network of known plant-height functional genes and TFs in maize. A Regulatory network showing the connection between SAM-related TFs and known plant-height functional genes, with different colors indicating the various pathways depicted to the right. B Knox6 regulates diverse biological pathways. Color-coded dots correspond to categories shown in A. CVt2, a functional target gene, is regulated by multiple TFs. Color-coded dots are the same as A. D Genome browser view of the association of Knox6 and Hb20 with the Vt2 and Hb20 promoters. E, F Dual-luciferase reporter assay (E) and diagram of the proposed module (F) indicating that transcription from the Vt2 promoter is repressed by Knox6 and Hb20, whereas transcription from the Hb20 promoter is induced by Knox6. “Treatment” refers to the co-transformation of the pM999 vector containing the TF effector and the p0800 vector containing the 2-kb promoter region of the target gene. “Control” refers to the co-transformation of the empty pM999 vector with the p0800 vector containing the 2-kb promoter

WOX13A regulates the expression of Gn1 and affects maize architecture. AWOX13A expression across different tissues and stages. B Strategy for the CRISPR–Cas9-based isolation of a WOX13A knockout mutant. C Differential expression analysis based on RNA-seq of V6-stage internodes from wild type and wox13a mutant. The position of Gn1 is annotated. D Venn diagram showing the overlap between target genes identified by tsCUT&Tag and DEGs obtained by RNA-seq. E Dual-luciferase reporter assay showing Gn1 transcription is induced by WOX13A. “Treatment” refers to the co-transformation of the pM999 vector containing the TF effector and the p0800 vector containing the 2-kb promoter region of the target gene. “Control” refers to the co-transformation of the empty pM999 vector with the p0800 vector containing the 2-kb promoter. F Representative phenotypes wild-type KN5585 and the wox13a mutant. Scale bar: 20 cm. G Plant height and ear height of KN5585 and wox13a plants grown in Shandong and Hubei, China. Statistical significance was determined by Student’s t test. **, P < 0.01; ***, P < 0.001
Loss of ZmWOX13A function contributes to plant-height variation in maize
To test the functional hypothesis of genes inferred from the network, we selected WOX13A, which was most highly expressed in internodes and was highly connected in the plant-height transcriptional GRN, suggesting that it might play a role in determining plant height (Fig. 7A). We used CRISPR-Cas9-mediated genome editing to generate a loss-of-function wox13a mutant carrying a 1-bp deletion from the second exon, causing a frameshift and leading to premature termination of translation (Fig. 7B). We performed RNA-seq on internode samples at the V6 stage on the wild type (KN5585) and wox13a mutant, finding 490 DEGs (P < 0.05 and absolute Log2(FC) > 1) (Fig. 7C, Additional file 16: Table S15). We also defined 5839 genes as direct WOX13A targets by tsCUT&Tag; the overlap between the two gene lists identified 74 genes as WOX13A-regulated genes (Fig. 7C and D). Gn1 was among these — a well-known SAM-related and plant-height gene (Fig. 7B and D). By transient luciferase-reporter assay using a Gn1pro:LUC reporter and a WOX13A effector, we confirmed WOX13A activates the Gn1 expression (Fig. 7E). To rigorously examine wox13a developmental defects, we grew wild-type KN558 and wox13a mutants in the field at two locations in China over two locations. Plant and ear height in wox13a mutants were consistently statistically significantly shorter than KN5585 controls (Fig. 7F and G). These findings strongly suggest that WOX13A induces Gn1 expression, consequently influencing the overall plant architecture of maize.
Discussion
Attaining optimal plant architecture in maize represents a critical determinant for achieving high yields. The establishment of plant architecture depends largely on the development of the SAM [57, 58]. In maize, some of the cloned SAM-related functional genes belong to the homeobox-protein family, underscoring their role in plant architecture. Toward identifying the molecular networks governing maize architecture, we generated a comprehensive molecular regulatory network and dynamic regulatory networks for key plant-architecture tissues by focusing on 27 SAM homeobox TFs. We compared the generated homeobox integrated network resource with the genes influenced by known SAM functional genes in maize. Our study found that 84.3% (4758/5647) of target genes identified by FEA4 ChIP-seq were present in our homeobox integrated network (Additional file 1: Fig. S15A) [59]. Additionally, 41.7% of the nodes in our generated SAM dynamic network overlapped with FEA4 target genes (Additional file 1: Fig. S15B). Approximately 62.6% of the differentially expressed genes between the grx triple mutant and wild type were also found in the homeobox integrated network (Additional file 1: Fig. S15C) [38]. Similarly, about 58% of DEGs between the ub2/ub3 double mutant tassel and wild-type tassel at the V5 stage were present in the homeobox integrated network (Additional file 1: Fig. S15D) [60]. These findings demonstrate that the targets affected by known SAM functional genes are largely present in the generated homeobox integrated network, validating its biological significance. Furthermore, the homeobox integrated network includes a larger number of network nodes and potential functions, providing an invaluable resource and foundation for in-depth exploration into the multifaceted functions of homeobox TFs within the context of maize growth and development.
tsCUT&Tag offers advantages such as a straightforward experimental procedure, time and cost efficiency, and high throughput. However, it still has some limitations in plant epigenetic studies. Enzymatic digestion of the plant cell wall is a stress treatment that may affect the regulatory networks of certain TFs, especially those related to chemical stimuli [49]. Additionally, tsCUT&Tag is reliant on the availability of high-quality protoplasts. Obtaining such protoplasts can be challenging due to the spatiotemporal specificity of TFs [61]. Integrating data from techniques like ATAC-seq and RNA-seq from specific tissues can aid in constructing the tissue regulatory landscape of TFs and enhance accuracy in analyzing tissue dynamic networks [28]. Although tsCUT&Tag uses a machine-learning model based on the ATAC-seq and RNA-seq data and could predict the targets of TFs across different tissues and stages with an accuracy of ~ 70% [28], we still need to be cautious since there are some false positives.
Gene duplications constitute a fundamental mechanism underlying the expansion of gene families and the diversification of gene functions, thereby serving as a significant catalyst for genome evolution. A deep understanding of DNA-binding specificity among distinct TF subfamily members is crucial for characterizing genetic redundancy and diversity, functional differentiation of duplications, and more. In this study, we established that members originating from the same subfamily exhibit only relatively minor differences in binding site specificity and target genes, indicating a substantial degree of functional redundancy within individual homeobox TF families. Furthermore, we saw contrasting fates of duplications to SAM-related homeobox genes at the transcriptome and regulome levels. Kn1 and Lg3 are an example of a divergent duplication, with Kn1 regulating Lg3 expression to ensure proper inflorescence growth, indicative of subfunctionalization.
Dissecting regulatory networks is an efficient method for exploring highly dynamic and complex principles of functional genomics [62]. Here, we curated an expansive regulatory network encompassing 27 homeobox TFs, comprising 235,043 edges and 18,463 nodes, reflecting a complexity and redundancy of regulation and supervision that is consistent with performance in animals [63,64,65]. Compared to other investigations around homeobox functional genes [17, 19, 20], the GRNs we identified encapsulate a comprehensive spectrum of genetic-information flow across diverse pathways. Notably, all TFs investigated here regulate at least three distinct biological pathways, and at least two or more TFs converge to control genes within the same pathway, collectively revealing a multifaceted mode of transcriptional coordination.
Rational deployment of favorable gene combinations may potentially contribute to functional enhancements. Our understanding of TFs remains somewhat limited in the context of integrated networks, prompting us to integrate additional datasets to create dynamic regulatory networks across various maize growth stages and tissues. This network within distinct tissues harbors distinct functional pathways, comprised both known functional genes and those with unknown functions. Such networks aid in the rapid discovery of functional genes and their regulatory relationships. Exploring the functions of target genes is a valuable tool for the prediction of the biological roles associated with individual TFs. Through empirical validation, we demonstrated that Knox6 and Hb20 repress the Vt2 transcription, a key functional gene involved in auxin biosynthesis. This empirical confirmation reinforces a model wherein Knox6 and Hb20 participate in the regulation of auxin biosynthesis. We generated a wox13a mutant via gene editing, which demonstrated that WOX13A influences plant height through its regulation of the functional gene Gn1. These findings collectively underscore the powerful predictive capabilities of GRNs in delineating the functions of TFs and scoping the complex regulatory interplay among TFs themselves, as well as between TFs and their target genes, within diverse biological pathways.
Conclusions
In summary, we generated a comprehensive dynamic regulome of SAM-related homeobox genes with tsCUT&Tag data of maize homeobox genes. By reproducing the regulatory associations between plant-height functional genes and SAM-related homeobox genes, our findings contribute to the identification and characterization of regulatory mechanisms governing plant architecture. We identify a novel role for Wox13A in control of plant height, further assisting rational improvement of agronomic traits.
Methods
Plant materials and vector construction
Seeds of maize (Zea mays inbred line B73) were planted in a growth chamber in controlled conditions (25 °C, in continuous darkness) for approximately 10 days. The secondary yellow leaves were used for protoplast isolation and tsCUT&Tag. B73 seeds were also planted in a greenhouse under local environmental conditions in Wuhan. Samples from the shoot apical meristem of V4-stage seedlings, as well as the internode and stem of V5-stage seedlings, were collected for ATAC-seq. KN5585 and the wox13a mutant were planted in a greenhouse, also under local environmental conditions. Internode samples from V6-stage plants were harvested for RNA-seq. KN5585 and wox13a were cultivated for phenotypic observation in Zibo, Shandong during the spring of 2022, and in both Zibo, Shandong, and Wuhan, Hubei (China) during the spring of 2023.
Thirty-two maize vectors containing genes listed in Additional file 4: Table S3 were amplified from the maize TFome using primers TF-F and TF-R, as previously described [42]. Full-length coding sequences were cloned into linearized pM999-GFP at the XbaI restriction site through homologous recombination. Transformation-grade plasmids were then prepared according to previously described methods [54].
Protoplast isolation, transformation, and tsCUT&Tag assay
Isolate and transformation of maize protoplasts was done as previously described [28]. For CUT&Tag assays, the tsCUT&Tag procedure [28] was followed, based on CUT&Tag procedure with adjustments [66]. The CUT&Tag assays were conducted using a commercial kit (Vazyme, In-Situ ChIP Library Prep Kit for Illumina, TD901-TD902). The TF–target DNA libraries were constructed with distinct indexes (Vazyme, TruePrep™ Index Kit V4 for Illumina, TD204) and sequenced on an Illumina NovaSeq platform at Annoroad (Beijing, China).
ATAC-seq
The construction of ATAC-seq libraries for the SAM, stem and internode samples was performed following a previously established protocol with some modifications [67, 68]. In brief, native nuclei were extracted and pelleted by centrifugation and then resuspended in 20 μL 1 × TTBL buffer (VAHTS, TD501). For each library, ~ 10,000 nuclei were treated with Tn5 (VAHTS, TD501) in the presence of 0.3% v/v Triton X-100 at 37 °C for 30 min. Samples were purified using a Qiagen MinElute kit and then amplified to construct a library with a TruePrep DNA Library Prep Kit V2 for Illumina sequencing (TD501, Vazyme). The resulting amplicons underwent size selection using DNA clean beads (KAPA) and were prepared for 150-bp paired-end sequencing.
Analysis of TF peaks and open chromatin
Clean reads were aligned to the B73 maize reference genome version 5.0 using Bowtie2 software [69,70,71]. Aligned reads were filtered to retain those with mapping quality (MAPQ) > 30 and exclude reads mapping to multiple locations in the genome by retaining AS:I: < N > tag and removing the XS:i: < N > tag. We used BEDTools to eliminate reads at the TF site to mitigate vector contamination [72]. SAMtools rmdup was applied to discard duplicated sequences, yielding the BAM file for subsequent analysis [73]. TF peak calling was performed using MACS2 (version 2.1.1) with control background subtraction and specifying the genome size (-g 2.1e + 9) [74]. Macs2 peaks are re-sized to ~ 150 bp around the summit and aggregate signal values. The merged-peak calling criteria employed a 1% (0.01) irreproducible discovery rate (IDR) threshold. Open-chromatin regions were identified with a cutoff P value of 0.00001 by MACS2-call-summit [74]. Target genes were defined as the closest gene containing a peak summit within 2-kb upstream and downstream of the TSS and annotated via ChIPseeker [75]. Cross-correlation analysis was conducted using phantompeakqualtools, yielding two metrics, normalized strand cross-correlation coefficent (NSC) and relative strand cross-correlation coefficient (RSC) [76]. Conversion of.bam files to.bigwig files was carried out using deepTools bamCoverage, applying reads-per-genome coverage (RPGC) normalization [77]. Heatmaps were generated using deepTools computeMatrix, plotHeatmap, and plotProfile [77]. Motif analysis was conducted using MEME-chip with the following parameters: -meme-mod anr -meme-minw 4 -meme-maxw 15 -meme-nmotifs 10 -meme-p 8. Input fasta sequence files for MEME-chip were generated by extracting 50 bp of sequence upstream and downstream of the peak summit using BEDtools and getfasta [72, 78]. To evaluate the reproducibility between biological replicates, Pearson’s correlation coefficients were calculated using deepTools (version 3.2.0) multiBamSummary. Biological replicates with Pearson’s correlation coefficient ≥ 0.8 were retained for further analyses.
Identification of hub genes and hot regions
Hub targets were defined as described in previous studies [46, 79]. The identification of hub-target genes followed the protocol described in their respective procedures. Hub genes were defined as genes identified by more TFs than the 99th percentile of the maximal value in 100 randomizations of the columns in the TF-to-gene matrix, while preserving the total number of TF targets in each randomization. Similarly, the identification of hot regions followed the same strategy as that used for hub genes. Specifically, hub genes were defined as genes targeted by more than 13 TFs, while hot regions were characterized as those being bound by more than 12 TFs. Hotspot regions were screened using candidate enhancers identified previously [80].
RNA-seq and differential expression analysis
To generate an expression map throughout the growth period, RNA-seq data were compiled from multiple sources [41] [81]. RNA-seq data obtained from the SAM 10 functional domains were included [82]. Ribo-seq data from 16 tissues or stages were used as reported by [41]. Internode samples were collected in triplicate from KN5585 and the wox13a mutant at the V6 stage. Total RNA was extracted using a Direct-zol RNA Microprep kit (Zymo Research) following the manufacturer’s instructions. RNA-seq libraries were prepared and sequenced as 150-bp paired-end reads at BGI (Shenzhen, China), through BGISEQ-500RS.
For all RNA-seq analyses, RSEM [83] was used to align the data to the B73 reference genome version 5.0 and estimate gene-expression levels. Genes differentially expressed between WT and mutant were identified with DESeq2 [84] with the following parameters: P value < 0.05, absolute Log2(Fold-Change) > 1. GO-term enrichment analysis was conducted using singular-enrichment analysis with agriGO (version 2.0, http://systemsbiology.cau.edu.cn/agriGOv2/), with the threshold set at FDR < 0.01 [85].
TF co-regulation
The degree of colocalization between TFs was quantified within each sample. The step in deciphering the combinatorial regulatory code is to identify the location of TFs. Initially, a heatmap was employed to illustrate the enrichment level of genome-wide peak regions for the 27 TFs using R. For the coregulatory matrix, the TFs were clustered based on the Jaccard distance (1—Jaccard Index) between their peak sets using average-linkage hierarchical clustering based on R.
Evaluation of gene divergence
Duplications were selected from three subfamilies derived from the download genome duplications reported by [51]. To predict cis-acting elements within the promoter regions, 1-kb regions upstream of the transcription start site were extacted for each gene using BEDtools, followed by analysis with PlantCARE [86]. To investigate the variation in spatiotemporal expression of duplicated genes, the ‘Scipy’ module in Python was employed to compute the Pearson’s correlation coefficient (R) between pairs of duplicated genes across different tissues. For a more in-depth exploration of regulatory divergence in duplicated genes, the Divergence score (DS) index was introduced, defined by the formula reported in \(1-\frac{\text{Number of shared genes between the two TFs targets}}{\text{Total number of genes in the two TFs}}\) [52].
Additionally, 100 gene pairs were randomly generated in TFs, from which the DS values were calculated, the DS value perform normal distribution based on MASS and lattice R packages. The 95th percentile of the DS-value distribution was selected as the threshold for statistically significant regulatory differentiation among duplicated gene pairs, with DS values indicating conserved (> 0.863), divergent (< 0.515), or intermediate levels of genetic differentiation (0.515 to 0.863) between duplicated genes.
Inference of an integrated transcription-regulatory network and tissue-dynamic networks
The integrated transcription regulatory network combines TF–target genes identified through tsCUT&Tag and published Kn1 ChIP and gene pairs identified, and co-expression and co-translation gene pairs from omics databases [17, 41]. Regulatory weight is obtained by normalizing the signal value on Min–Max Scaling. Conformance to a power-law distribution in the network was verified using Python.
To predict the dynamic binding sites of each TF in different tissues, we used PyTorch to construct a simple Multi-layer Perceptron (MLP) model to perform supervised learning on the tsCUT&Tag and ATAC-seq data. We trimmed the peaks of tsCUT&Tag and ATAC-seq to 100-bp upstream and downstream of the peak summit. The training data were generated by overlapping peak sequences between tsCUT&Tag and ATAC-seq. Peaks in the yellow leaves’ open chromatin were labeled as 1, and open-chromatin sequences without TF binding were labeled as 0, allowing the model to learn the features of sequence binding. These labeled sequences were then randomly divided into training and test sets. The trained model was then used to predict TF binding sites by analyzing the ATAC-seq peaks from the SAM.
To construct tissue-specific dynamic regulatory networks, expression levels were integrated from various tissue samples. For the SAM, expression levels were integrated from the 10 SAM domains, as well as the SAMs of V1- and V3-stage seedlings. Using a machine-learning model to predict each TF binding site in SAM, we required that the predicted target genes also show an overall expression level above the threshold TPM > 1. Additionally, these genes must overlap with the GRN generated by GENIE3 using gene-expression and translation levels as previously described [42, 73]. The regulatory weight is the sum of the weights identified by tsCUT&Tag and the weights predicted by the transcriptome and translatome GRNs.
Transient luciferase-reporter assays
Transient-expression assays were conducted using maize leaf protoplasts [87]. The promoters of Hb20, Vt2, and Gn1 were cloned upstream of firefly luciferase (LUC) via the KpnI and PstI sites in pGreenII 0800-LUC through homologous recombination. The full-length coding sequences of Knox6, Hb20, and WOX13A were cloned into the pGreenII PM999 vector using a homologous combination. Plasmids were mixed in appropriate combinations and transformed into protoplasts. After 14 h of incubation in darkness, protoplasts were collected and processed using a Dual-Luciferase Reporter Gene Assay Kit (DL101, Vazyme) following the manufacturer’s recommended protocol. LUC and REN activity were measured using a microplate reader. “Treatment” refers to the co-transformation of the pM999 vector containing the TF effector and the p0800 vector containing the 2-kb promoter region of the target gene. “Control” refers to the co-transformation of the empty pM999 vector with the p0800 vector containing the 2-kb promoter.
....
Availability of data and materials
The sequencing data generated in this paper and BAM files are available at the Genome Sequence Archive (GSA) at the Big Data Center (National Genomics Data Center, NGDC) [88]. The data and machine learning models in this study were deposited under MIT license in GitHub (https://github.com/LuoZiya/ML_in_regulatome_of_HB_TFs/) [89] and in Zenodo (https://zenodo.org/records/13479358) [90]. The peak and open-chromatin-region files and the integrated regulome files have been deposited online at http://zeasystemsbio.hzau.edu.cn/dataset.html. The published Ribo-seq and RNA-seq datasets of samples are available at Sequence Read Archive (SRA) at the National Center for Biotechnology Information (NCBI), the detailed accession numbers for each sample are in Additional file 17: Table S16 [41].
References
Laux T, Mayer KFX, Berger J, Jurgens G. The WUSCHEL gene is required for shoot and floral meristem integrity in Arabidopsis. Development. 1996;122:87–96.
Pautler M, Tanaka W, Hirano HY, Jackson D. Grass meristems I: shoot apical meristem maintenance, axillary meristem determinacy and the floral transition. Plant Cell Physiol. 2013;54:302–12.
Long JA, Moan EI, Medford JI, Barton MK. A member of the KNOTTED class of homeodomain proteins encoded by the STM gene of Arabidopsis. Nature. 1996;379:66–9.
Smith LG, Greene B, Veit B, Hake S. A dominant mutation in the maize homeobox gene, knotted-1, causes its ectopic expression in leaf-cells with altered fates. Development. 1992;116:21–000.
Becraft PW, Freeling M. Genetic-analysis of rough-sheath1 developmental mutants of maize. Genetics. 1994;136:295–311.
Conklin PA, Johnston R, Conlon BR, Shimizu R, Scanlon MJ. Plant homeodomain proteins provide a mechanism for how leaves grow wide. Development. 2020;147(20):dev193623.
Muehlbauer GJ, Fowler JE, Girard L, Tyers R, Harper L, Freeling M. Ectopic expression of the maize homeobox gene liguleless3 alters cell fates in the leaf. Plant Physiol. 1999;119:651–62.
Mukherjee K, Brocchieri L, Burglin TR. A comprehensive classification and evolutionary analysis of plant homeobox genes. Mol Biol Evol. 2009;26:2775–94.
Shani E, Yanai O, Ori N. The role of hormones in shoot apical meristem function. Curr Opin Plant Biol. 2006;9:484–9.
Jasinski S, Piazza P, Craft J, Hay A, Woolley L, Rieu I, Phillips A, Hedden P, Tsiantis M. KNOX action in Arabidopsis is mediated by coordinate regulation of cytokinin and gibberellin activities. Curr Biol. 2005;15:1560–5.
Sakamoto T, Kamiya N, Ueguchi-Tanaka M, Iwahori S, Matsuoka M. KNOX homeodomain protein directly suppresses the expression of a gibberellin biosynthetic gene in the tobacco shoot apical meristem. Genes Dev. 2001;15:581–90.
Sharif R, Raza A, Chen P, Li Y, El-Ballat EM, Rauf A, Hano C, El-Esawi MA. HD-ZIP Gene family: potential roles in improving plant growth and regulating stress-responsive mechanisms in plants. Genes (Basel). 2021;12(8):1256.
Elhiti M, Stasolla C. Structure and function of homodomain-leucine zipper (HD-Zip) proteins. Plant Signal Behav. 2009;4:86–8.
Schoof H, Lenhard M, Haecker A, Mayer KF, Jurgens G, Laux T. The stem cell population of Arabidopsis shoot meristems in maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell. 2000;100:635–44.
Wu X, Dabi T, Weigel D. Requirement of homeobox gene STIMPY/WOX9 for Arabidopsis meristem growth and maintenance. Curr Biol. 2005;15:436–40.
Ueda M, Zhang Z, Laux T. Transcriptional activation of Arabidopsis axis patterning genes WOX8/9 links zygote polarity to embryo development. Dev Cell. 2011;20:264–70.
Bolduc N, Yilmaz A, Mejia-Guerra MK, Morohashi K, O’Connor D, Grotewold E, Hake S. Unraveling the KNOTTED1 regulatory network in maize meristems. Genes Dev. 2012;26:1685–90.
Vollbrecht E, Veit B, Sinha N, Hake S. The developmental gene Knotted-1 is a member of a maize homeobox gene family. Nature. 1991;350:241–3.
Nardmann J, Ji J, Werr W, Scanlon MJ. The maize duplicate genes narrow sheath1 and narrow sheath2 encode a conserved homeobox gene function in a lateral domain of shoot apical meristems. Development. 2004;131:2827–39.
Foster T, Yamaguchi J, Wong BC, Veit B, Hake S. Gnarley1 is a dominant mutation in the knox4 homeobox gene affecting cell shape and identity. Plant Cell. 1999;11:1239–52.
Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–502.
Neph S, Stergachis AB, Reynolds A, Sandstrom R, Borenstein E, Stamatoyannopoulos JA. Circuitry and dynamics of human transcription factor regulatory networks. Cell. 2012;150:1274–86.
Davidson EH, Rast JP, Oliveri P, Ransick A, Calestani C, Yuh CH, Minokawa T, Amore G, Hinman V, Arenas-Mena C, et al. A genomic regulatory network for development. Science. 2002;295:1669–78.
Kim J, Chu J, Shen X, Wang J, Orkin SH. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–61.
Franco-Zorrilla JM, Lopez-Vidriero I, Carrasco JL, Godoy M, Vera P, Solano R. DNA-binding specificities of plant transcription factors and their potential to define target genes. Proc Natl Acad Sci U S A. 2014;111:2367–72.
Yan J, Enge M, Whitington T, Dave K, Liu J, Sur I, Schmierer B, Jolma A, Kivioja T, Taipale M, Taipale J. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell. 2013;154:801–13.
Tu X, Mejia-Guerra MK, Valdes Franco JA, Tzeng D, Chu PY, Shen W, Wei Y, Dai X, Li P, Buckler ES, Zhong S. Reconstructing the maize leaf regulatory network using ChIP-seq data of 104 transcription factors. Nat Commun. 2020;11:5089.
Wu L, Luo Z, Shi Y, Jiang Y, Li R, Miao X, Yang F, Li Q, Zhao H, Xue J, et al. A cost-effective tsCUT&Tag method for profiling transcription factor binding landscape. J Integr Plant Biol. 2022;64:2033–8.
Shepherd JCW, Mcginnis W, Carrasco AE, Derobertis EM, Gehring WJ. Fly and frog homoeo domains show homologies with yeast mating type regulatory proteins. Nature. 1984;310:70–1.
Bommert P, Lunde C, Nardmann J, Vollbrecht E, Running M, Jackson D, Hake S, Werr W. thick tassel dwarf1 encodes a putative maize ortholog of the Arabidopsis CLAVATA1 leucine-rich repeat receptor-like kinase. Development. 2005;132:1235–45.
Bommert P, Je BI, Goldshmidt A, Jackson D. The maize Galpha gene COMPACT PLANT2 functions in CLAVATA signalling to control shoot meristem size. Nature. 2013;502:555–8.
Wu Q, Xu F, Liu L, Char SN, Ding Y, Je BI, Schmelz E, Yang B, Jackson D. The maize heterotrimeric G protein beta subunit controls shoot meristem development and immune responses. Proc Natl Acad Sci U S A. 2020;117:1799–805.
Wang F, Yu Z, Zhang M, Wang M, Lu X, Liu X, Li Y, Zhang X, Tan BC, Li C, Ding Z. ZmTE1 promotes plant height by regulating intercalary meristem formation and internode cell elongation in maize. Plant Biotechnol J. 2022;20:526–37.
Guo M, Rupe MA, Dieter JA, Zou J, Spielbauer D, Duncan KE, Howard RJ, Hou Z, Simmons CR. Cell Number Regulator1 affects plant and organ size in maize: implications for crop yield enhancement and heterosis. Plant Cell. 2010;22:1057–73.
Chuck GS, Brown PJ, Meeley R, Hake S. Maize SBP-box transcription factors unbranched2 and unbranched3 affect yield traits by regulating the rate of lateral primordia initiation. Proc Natl Acad Sci U S A. 2014;111:18775–80.
Moon J, Candela H, Hake S. The Liguleless narrow mutation affects proximal-distal signaling and leaf growth. Development. 2013;140:405–12.
Brooks L 3rd, Strable J, Zhang X, Ohtsu K, Zhou R, Sarkar A, Hargreaves S, Elshire RJ, Eudy D, Pawlowska T, et al. Microdissection of shoot meristem functional domains. PLoS Genet. 2009;5:e1000476.
Yang RS, Xu F, Wang YM, Zhong WS, Dong L, Shi YN, Tang TJ, Sheng HJ, Jackson D, Yang F. Glutaredoxins regulate maize inflorescence meristem development via redox control of TGA transcriptional activity. Nat Plants. 2021;7:1589–601.
Liu X, Galli M, Camehl I, Gallavotti A. RAMOSA1 ENHANCER LOCUS2-mediated transcriptional repression regulates vegetative and reproductive architecture. Plant Physiol. 2019;179:348–63.
Becraft PW, Stinard PS, McCarty DR. CRINKLY4: a TNFR-like receptor kinase involved in maize epidermal differentiation. Science. 1996;273:1406–9.
Han LQ, Zhong WS, Qian J, Jin ML, Tian P, Zhu WC, Zhang HW, Sun YH, Feng JW, Liu XG, et al. A multi-omics integrative network map of maize. Nat Genet. 2023;55:144.
Burdo B, Gray J, Goetting-Minesky MP, Wittler B, Hunt M, Li T, Velliquette D, Thomas J, Gentzel I, Brito MD, et al. The Maize TFome - development of a transcription factor open reading frame collection for functional genomics. Plant J. 2014;80:356–66.
Lambert SA, Jolma A, Campitelli LF, Das PK, Yin YM, Albu M, Chen XT, Taipale J, Hughes TR, Weirauch MT. The Human Transcription Factors. Cell. 2018;172:650–65.
O’Malley RC, Huang SSC, Song L, Lewsey MG, Bartlett A, Nery JR, Galli M, Gallavotti A, Ecker JR. Cistrome and epicistrome features shape the regulatory DNA landscape (vol 165, pg 1280, 2016). Cell. 2016;166:1598–1598.
Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JR, Guenther MG, Kumar RM, Murray HL, Jenner RG, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–56.
Heyndrickx KS, Van de Velde J, Wang CM, Weigei D, Vandepoele K. A functional and evolutionary perspective on transcription factor binding in arabidopsis thaliana. Plant Cell. 2014;26:3894–910.
Barabasi AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5:101-U115.
Marbach D, Roy S, Ay F, Meyer PE, Candeias R, Kahveci T, Bristow CA, Kellis M. Predictive regulatory models in Drosophila melanogaster by integrative inference of transcriptional networks. Genome Res. 2012;22:1334–49.
Li W, Ge FH, Qiang ZQ, Zhu L, Zhang SS, Chen LM, Wang XQ, Li JS, Fu Y. Maize ZmRPH1 encodes a microtubule-associated protein that controls plant and ear height. Plant Biotechnol J. 2020;18:1345–7.
Freeling M. Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu Rev Plant Biol. 2009;60:433–53.
Qiao X, Li QH, Yin H, Qi KJ, Li LT, Wang RZ, Zhang SL, Paterson AH. Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 2019;20(1):38.
De Smet R, Sabaghian E, Li Z, Saeys Y, Van de Peer Y. Coordinated functional divergence of genes after genome duplication in arabidopsis thaliana. Plant Cell. 2017;29:2786–800.
Zhu WC, Miao XX, Qian J, Chen SJ, Jin QX, Li MZ, Han LQ, Zhong WS, Xie D, Shang XY, Li L. A translatome-transcriptome multi-omics gene regulatory network reveals the complicated functional landscape of maize. Genome Biol. 2023;24(1):60.
Tu XY, Mejia-Guerra MK, Franco JAV, Tzeng D, Chu PY, Shen W, Wei YY, Dai XR, Li PH, Buckler ES, Zhong SL. Reconstructing the maize leaf regulatory network using ChIP-seq data of 104 transcription factors. Nat Commun. 2020;11(1):5089.
Wang YH, Li JY. Molecular basis of plant architecture. Annu Rev Plant Biol. 2008;59:253–79.
Phillips KA, Skirpan AL, Liu X, Christensen A, Slewinski TL, Hudson C, Barazesh S, Cohen JD, Malcomber S, McSteen P. vanishing tassel2 Encodes a grass-specific tryptophan aminotransferase required for vegetative and reproductive development in maize. Plant Cell. 2011;23:550–66.
Wang B, Smith SM, Li JY. Genetic Regulation of Shoot Architecture. Annu Rev Plant Biol. 2018;69(69):437–68.
Sanchez P, Nehlin L, Greb T. From thin to thick: major transitions during stem development. Trends Plant Sci. 2012;17:113–21.
Pautler M, Eveland AL, LaRue T, Yang F, Weeks R, Lunde C, Il Je B, Meeley R, Komatsu M, Vollbrecht E, et al. FASCIATED EAR4 encodes a bZIP transcription factor that regulates shoot meristem size in maize. Plant Cell. 2015;27:104–20.
Kong D, Li C, Xue W, Wei H, Ding H, Hu G, Zhang X, Zhang G, Zou T, Xian Y, et al. UB2/UB3/TSH4-anchored transcriptional networks regulate early maize inflorescence development in response to simulated shade. Plant Cell. 2023;35:717–37.
Suter DM. Transcription factors and DNA play hide and seek. Trends Cell Biol. 2020;30:491–500.
Trewavas A. A brief history of systems biology. “Every object that biology studies is a system of systems.” Francois Jacob (1974). Plant Cell. 2006;18:2420–30.
Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104.
Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804.
Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489:91–100.
Kaya-Okur HS, Wu SJ, Codomo CA, Pledgers ES, Bryson TD, Henikoff JG, Ahmad K, Henikoff S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun. 2019;10(1):1930.
Sun YH, Dong L, Zhang Y, Lin D, Xu WZ, Ke CX, Han LQ, Deng LL, Li GL, Jackson D, et al. 3D genome architecture coordinates trans and cis regulation of differentially expressed ear and tassel genes in maize. Genome Biol. 2020;21(1):143.
Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357-U354.
Jiao YP, Peluso P, Shi JH, Liang T, Stitzer MC, Wang B, Campbell MS, Stein JC, Wei XH, Chin CS, et al. Improved maize reference genome with single-molecule technologies. Nature. 2017;546:524.
Hufford MB, Seetharam AS, Woodhouse MR, Chougule KM, Ou SJ, Liu JN, Ricci WA, Guo TT, Olson A, Qiu YJ, et al. De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes. Science. 2021;373:655-+.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, Li H. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10(2):giab008.
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based Analysis of ChIP-Seq (MACS). Genome Biology. 2008;9(9):R137.
Yu GC, Wang LG, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–3.
Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–31.
Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–5.
Machanick P, Bailey TL. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011;27:1696–7.
Shalgi R, Lieber D, Oren M, Pilpel Y. Global and local architecture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput Biol. 2007;3:1291–304.
Oka R, Zicola J, Weber B, Anderson SN, Hodgman C, Gent JI, Wesselink JJ, Springer NM, Hoefsloot HCJ, Turck F, Stam M. Genome-wide mapping of transcriptional enhancer candidates using DNA and chromatin features in maize. Genome Biol. 2017;18:137.
Zhou P, Hirsch CN, Briggs SP, Springer NM. Dynamic patterns of gene expression additivity and regulatory variation throughout maize development. Mol Plant. 2019;12:410–25.
Knauer S, Javelle M, Li L, Li XR, Ma XL, Wimalanathan K, Kumari S, Johnston R, Leiboff S, Meeley R, et al. A high-resolution gene expression atlas links dedicated meristem genes to key architectural traits. Genome Res. 2019;29:1962–73.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. Bmc Bioinformatics. 2011;12:323.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Tian T, Liu Y, Yan HY, You Q, Yi X, Du Z, Xu WY, Su Z. agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017;2017(45):W122–9.
Lescot M, Dehais P, Thijs G, Marchal K, Moreau Y, Van de Peer Y, Rouze P, Rombauts S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002;30:325–7.
Song SS, Huang H, Gao H, Wang JJ, Wu DW, Liu XL, Yang SH, Zhai QZ, Li CY, Qi TC, Xie DX. Interaction between MYC2 and ETHYLENE INSENSITIVE3 Modulates Antagonism between Jasmonate and Ethylene Signaling in Arabidopsis. Plant Cell. 2014;26:263–79.
Luo Z, Wu LM, Miao XX, Zhang S, Wei NN, Zhao S, Shang XY, Hu HY, Xue J, Zhang TF, et al. A dynamic regulome of shoot-apical-meristem-related homeobox transcription factors modulates plant architecture in maize. Genome Sequence Archive. 2024. https://ngdc.cncb.ac.cn/search/all?q=CRA012938.
Luo Z, Wu LM, Miao XX, Zhang S, Wei NN, Zhao S, Shang XY, Hu HY, Xue J, Zhang TF, et al. A dynamic regulome of shoot-apical-meristem-related homeobox transcription factors modulates plant architecture in maize. Github. 2024. https://github.com/LuoZiya/ML_in_regulatome_of_HB_TFs.
Luo Z, Wu LM, Miao XX, Zhang S, Wei NN, Zhao S, Shang XY, Hu HY, Xue J, Zhang TF, et al. A dynamic regulome of shoot-apical-meristem-related homeobox transcription factors modulates plant architecture in maize. Zenodo. 2024. https://zenodo.org/records/13479358.
Acknowledgements
We thank the high-performance computing platform at the National Key Laboratory of Crop Genetic Improvement in Huazhong Agricultural University. We really appreciate Professor Gary J. Muehlbauer from University of Minnesota for the discussion and supervision at the early stage of this project. We also thank Dr. Weifu Li and Mr. Hao Wu from the college of Informatics of Huazhong Agricultural University for the code debugging of machine learning.
Peer review information
Wenjing She was the primary editor of this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Review history
The review history is available as Additional file 18.
Funding
This research was supported by the National Key Research and Development Program of China (2023YFF1000100, 2023ZD04076), the National Natural Science Foundation of China (32321005, 32261143463), and the Outstanding Youth Team Cultivation Project of Center Universities (2662023PY007).
Author information
Authors and Affiliations
Contributions
L.L. and S.X. designed and supervised the study. Z.L., L.W., and X.M. performed the experiments with assistance from S.Z., S. Z., H.H., X.S., and N.W. Z.L. performed the bioinformatic analyses. J.X. and T.Z. provided ideas on the design and useful discussion on the study. Z.L. and L.L. prepared the manuscript. F.Y. helped provide mutant materials.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
13059_2024_3391_MOESM1_ESM.pdf
Additional file 1: Supplementary Figures S1–S15: Figure S1. Heatmap representation of the expression levels of known SAM functional genes in the SAM, differentiated tissues and other mature tissues. Figure S2. Alignment of TF V3 and V5 protein sequences for the 27 SAM TFs examined in this study."Query" represents the V5 version, and "Sbjct" represents the V3 version. Figure S3. Scatter plots of 27 TFome tsCUT&Tag data from SAM-related homeobox genes. Pearson correlations were calculated in deepTools. Figure S4. Gene structures and phylogenetic tree based on amino-acid sequence for SAM-related homeobox proteins. Blue represents the ZF-HD subfamily, green represents the KNOX subfamily, pink represents the WOX subfamily. Figure S5. Distribution of bound peaks (A), target-gene number (B) and FRiP (C) for 27 TFs examined in this study. Figure S6. Top motifs identified for upstream and downstream 50 bp of each TF peaks by MEME-ChIP software. Figure S7. Characteristics of bound TFs per potential target gene. (A) Histogram showing the percentage of bound TFs per potential target gene. (B) Overlap for between-subfamily and within-subfamily TFs’ target genes. P value was calculated by KS test. Figure S8. Organization of hub genes and hot regions. Characteristics of bound TFs per potential target gene. (A) Bound TFs per potential target gene. (A-B) Bound TFs per potential target gene (A) and per peak region (B). (C) Number of overlapping genes between hub genes and genes regulated by hot regions. (D, E) Functional enrichment of target genes regulated by only 1 TF (D) and hub genes (E). Figure S9. Colocalization of binding sites for KNOX, ZFHD and WOX subfamily TFs. (A) Distribution of genome-wide peak regions. (B) TF co-binding matrix based on common potential-peak regions and average-linkage hierarchical clustering based on Jaccard Index. Figure S10. Identification of duplication pairs in 27 SAMrelated homeobox genes. Percentage of duplicate genes at different evolutionary groups of plant taxa (A) and in different subfamilies (B). Figure S11. Distribution of regulatory divergence score (DS). Figure S12. The duplication of KN1 andLg3. (A–C) cis-elements in promoter regions (A), Gene structure (B) and expression levels of KN1 and Lg3 (C). Figure S13. Features of the integrated network. (A, B) Distribution of node in-degree values (A) fit a power-law (red line, B). (C, D) Overlapping genes between high-occupied genes identified by tsCUT&Tag and nodes of the top-10% indegree in network (C), and their functional annotation (D). Figure S14. Summary of ATAC-seq and RNA-seq datasets and gene pairs characteristic of the network. (A) Number of expressed genes identified by RNA-seq in six different tissues. (B) Percentage of expressed target genes in a specific tissue. (C) Number of peaks identified by ATAC-seq in six different tissues. (D) Overlap of tsCUT&Tag target genes with ATAC-seq data. (E, F) Number and rate of overlapping tsCUT&Tag target genes with transcriptome GRN (E) and translational GRN (F). Figure S15. Overlapping genes between homeobox network and known SAM functional TFs’ targets. (A) Overlapping genes between homeobox merge network and FEA4 target genes by ChIP-seq of ∼2-5mm ear. (B) Overlapping genes between homeobox SAM dynamic network and FEA4 target genes by ChIP-seq of ∼2-5mm ear. (C) Overlapping genes between homeobox merge network and differentially expressed genes between grx triple mutant and wild type. (D) Overlapping genes between homeobox merge network and differentially expressed genes between ub2/3 double mutant and wild type.
13059_2024_3391_MOESM15_ESM.xlsx
Additional file 15: Table S14. Sources of well-known functional genes and SAM-related TF genes associated with plant height.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Luo, Z., Wu, L., Miao, X. et al. A dynamic regulome of shoot-apical-meristem-related homeobox transcription factors modulates plant architecture in maize. Genome Biol 25, 245 (2024). https://doi.org/10.1186/s13059-024-03391-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13059-024-03391-8