
Abstract
Promoters are genomic regions where the transcription machinery binds to initiate the transcription of specific genes. Computational tools for identifying bacterial promoters have been around for decades. However, most of these tools were designed to recognize promoters in one or few bacterial species. Here, we present Promotech, a machine-learning-based method for promoter recognition in a wide range of bacterial species. We compare Promotech’s performance with the performance of five other promoter prediction methods. Promotech outperforms these other programs in terms of area under the precision-recall curve (AUPRC) or precision at the same level of recall. Promotech is available at https://github.com/BioinformaticsLabAtMUN/PromoTech.
Similar content being viewed by others
Background
Promoters are DNA segments essential for the initiation of transcription at a defined location in the genome, which are recognized by a specific RNA polymerase (RNAP) holoenzyme (E σ) [1]. E σ is formed by RNAP and a σ factor. σ factors are bacterial DNA-binding regulatory proteins of transcription initiation that enable specific binding of RNAP to promoters [1]. Recognizing promoters is critical for understanding bacterial gene expression regulation. There have been numerous bioinformatics tools developed to recognize bacterial promoter sequences [2–17] (summarized in Supplementary Table S1). However, most of these tools were designed to recognize promoters in Escherichia coli or in few (2 or 3) bacterial species, and their applicability to a wider range of bacterial species is unproven. Additionally, the performance of current tools rapidly decreases when applied to whole genomes, and thus, it is common practice to restrict the size of the input sequence to a few hundred nucleotides.
Shahmuradov et al. [11] evaluated the performance of their method (bTSSfinder) and other three methods on ten bacterial species belonging to five different phyla. The best average sensitivity (recall) values obtained were 59% and 49% by bTSSfinder and BPROM [17], respectively, while bTSSfinder achieved higher accuracy than the other three assessed tools. These results are promising as they showed that it is possible to recognize promoters of several bacterial species even when the methods were designed for specific bacterial species. BPROM uses five relatively conserved motifs from E. coli to identify promoters, and bTSSfinder focuses on E. coli and three species of Cyanobacteria. Based on this, we hypothesized that predictive performance can be improved if a method is trained on data from a diverse group of bacterial species.
Another bacterial promoter detection method evaluated on a multi-species data set is G4PromFinder[10]. G4PromFinder utilizes conserved motifs and focuses on Streptomyces coelicolor A3(2) and Pseudomonas aeruginosa PA14. Di Salvo et al. comparatively assessed G4PromFinder’s performance in terms of F1-score with that of bTSSfinder, PePPER [14], and PromPredict [16, 18] and found that G4PromFinder outperformed the other three tools in GC-rich bacterial genomes. There are several recently published E. coli promoter prediction methods such as MULTiPly [4], SELECTOR [2], iPromoter-BnCNN [3], IBPP [7], and iPromoter-2L [19], among others (Supplementary Table S1). Cassiano and Silva-Rocha [20] carried out a comparative assessment of bacterial promoter prediction tools for identifying E. coli σ70 promoters. In their benchmark, they found that iPro70-FMWin [6] achieved the best results in terms of accuracy and MCC.
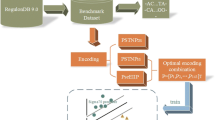
Here, we developed a general (species independent) bacterial promoter recognition method, Promotech, trained on a large data set of promoter sequences of nine distinct bacterial species belonging to five different phyla (namely, Actinobacteria, Chlamydiae, Firmicutes, Proteobacteria, and Spirochaetes). As promoters are typically located directly upstream of the transcription start site (TSS), we used published TSS global maps obtained using sequencing technology such as dRNA-seq[21] and Cappable-seq[22] to define promoter sequences. We trained and evaluated twelve random forests and recurnrent neural network models using these data to select Promotech’s classification model (Fig. 1). Finally, we compared the performance of Promotech with that of five other bacterial promoter prediction methods on independent data from four bacterial species. Promotech outperformed all these five methods in terms of the area under the ROC curve (AUROC), the area under the precision-recall curve (AUPRC), and precision at a specific recall level.

Flowchart illustrating our methodology
Results and discussion
Variety of training and validation data
We obtained a large amount of promoter sequences from published global TSS maps (listed in Table 10). On both the training and the validation data, we had bacterial species belonging to distinct phyla and having a wide range (from 30 to 72%) of GC content (Tables 1 and 2). In total, our training data contained 27,766 promoter sequences, and our validation data contained 11,615 promoter sequences (Supplementary Tables S2 and S3).
Model selection
To select Promotech’s classification model, we built two and ten models using random forest (RF) [23, 24] and recurrent neural networks (RNN) [25] (Fig. 1), respectively. The RF models consisted of one trained with hot-encoded features (RF-HOT) and another trained with tetra-nucleotide frequencies (RF-TETRA). To calculate the tetra-nucleotide frequency vector of a given sequence, the number of occurrences of each possible 4-nucleotide DNA sequence (4-mer) in that sequence is divided by the total number of 4-mers in it. The optimal parameters of the RF models were selected using 10-fold grid search cross-validation on 75% of the training data. The RNN models consisted of five long short-term memory (LSTM) [26] and five Gated recurrent unit (GRU) [27] models having zero to four hidden layers and a word embedding [28] layer to obtain a numerical representation of the promoter sequences (Table S4 in Additional file 1). From now on, these RNN models are denoted as GRU-X or LSTM-X, where X indicates the number of hidden layers. All the models were trained using an unbalanced dataset with a 1:10 ratio of positive to negative instances to simulate the small number of promoters in a whole bacterial genome. Due to time constraints, we were unable to run grid search cross-validation for the RNNs and assessed these models by randomly splitting the training data into 75% for training and 25% for testing. The best performing models per machine learning method in terms of AUPRC and AUROC were RF-HOT, GRU-1, and LSTM-4, as shown in Table 3. RF-HOT was the model with the highest AUPRC overall.
Model interpretation
To interpret the models created, we performed feature importance analysis to find out motifs recognized by the models. To do this, we obtained the feature importance ranking from the RF models. First, the importance scores were calculated using permutation-based importance (also called mean decrease in accuracy) [29] and impurity-based importance [24] on RF-TETRA. The most important tetra-mers based on the impurity-based importance score from RF-TETRA were TATA, ATAA, TAAT, TTAT, AAAA, and TTTT. The test was repeated using the permutation-based importance score and permuting each feature five times. Both tests produced similar results having the same tetra-nucleotide sequences appearing at the top of the ranking, only varying their relative ranking position and score (Tables 4 and 5).
The feature importance analysis was repeated on the RF-HOT model. In RF-HOT, each feature represents the presence of one of the four possible nucleotides: adenine (A), thymine (T), guanine (G), and cytosine (C) for the current position in the range of − 39 to 0 relative to the TSS. Each nucleotide was represented as a 4-digit binary number, i.e., A (1000), G (0100), C (0010), and T (0001). The permutation and impurity-based feature importance ranking generated by RF-HOT provided the most important positions in the range of − 39 to 0 relative to the TSS and the nucleotide with the most relevance for each position. To have visual representations of these results, each nucleotide’s importance score was plotted on a bar graph (Figs. 2 and 3). These results suggest that having adenine (A) and thymine (T) in the range of − 8 to − 12 relative to the TSS is highly important for promoter recognition. Additionally, these results suggest that the RF models learn to identify the Pribnow-Schaller box [30, 31], which is a six-nucleotide consensus sequence (TATAAT), commonly located around 10 bp upstream from the TSS.

Impurity-based feature importance scores per nucleotide per position relative to the TSS as calculated from the RF-HOT model

Permutation-based feature importance scores per nucleotide per position relative to the TSS as calculated from the RF-HOT model
Genome-wide promoter prediction assessment
We designed our first two assessments to demonstrate that Promotech was able to make predictions for a whole bacterial genome. Five models were selected for this assessment: the best models per machine learning method (RF-HOT, GRU-1, and LSTM-4), GRU-0, and LSTM-3. GRU-0 and LSTM-3 were selected to evaluate the effect in performance if the model had one less hidden layer. As the models were trained on 40-nt-long sequences, to do whole-genome predictions, we needed to cut the genome in 40-nt-long sequences. Thus, we traversed each genome with a sliding window with a one nt step and a 40-nt window size. The sliced sequences were then pre-processed and fed to the model twice, first, using a forward strand configuration and then using a backwards strand configuration. These steps were repeated for each bacterium on the validation set, namely, M. smegmatis, L. phytofermentans, B. amyloliquefaciens, and R. capsulatus. This was a computationally demanding assessment, as, for example, the sliding window created 6,988,167 sequences of 40 nt when used on the M. smegmatis genome. Each sequence was given a second time with a backward strand configuration ending up with 13,976,336 sequences. Thus, each of the four selected models was executed roughly 14 million times for the M. smegmatis genome. The process took around 4 h to run per bacterium per model, including the sliding window, data pre-processing, and model’s execution. Increasing the step size decreased the execution time but also decreased the model performance.
In the first assessment, predicted promoters were considered true positives if they have at least 10% sequence overlap with an actual promoter. Table 6 shows the average AUPRC and AUROC obtained by each model. The PRC and ROC obtained per bacterium are shown in Supplementary Figs. 1-4 in Additional file 1.
RF-HOT achieved the best overall AUPRC (0.14) and AUROC (0.82) (Table 6). An AUPRC of 0.14 might seem low, but if one considers that there are millions of 40-nt-long sequences in a bacterial genome and only a few thousand of these sequences are actual promoters, then this performance is much better than random guessing. For example, M. smegmatis has four thousand actual promoters (Table 10) and 14 million 40-nt-long genomic sequences; thus, a random classifier has an average AUPRC of 0.0003. RF-HOT achieved an AUPRC of 0.27 in M. smegmatis genome which is roughly a thousand-fold improvement over random performance.
To gain insight into the behavior of the models, we visually inspected the location of the predicted promoters and observed that many predicted promoters were located nearby the actual promoters (Fig. 4). To account for this, we re-evaluated the models’ performance to count as correct predictions those within 100 nt of an actual promoter. We called this task “the cluster promoter prediction.” Assessing the performance of the models using the cluster promoter prediction method increased AUPRC 2 to 6 times and AUROC by 1.5 times the values obtained in the first assessment (Figs. 5, 6, 7, and 8 and Table 7). This suggests that our models predict promoters in the proximity of actual promoters but are unable to recognize the exact genomic location of the actual promoters.

Predicted promoters observed in actual promoters’ proximity but not overlapping. Blue squares on the first row indicate the location of actual promoters while blue squares on the second and third rows indicate the location of predicted promoters with a predicted probability of 0.6 and 0.5, respectively. Within each circle a predicted promoter cluster is shown

PR curves (a) and ROC curves (b) per model obtained when counting predicted promoters nearby actual promoters as true positives on M. smegmatis str. MC2 155 bacterium. The numbers between brackets beside the model ID indicate AUPRC (a) and AUROC (b) of that model

PR curves (a) and ROC curves (b) per model obtained when counting predicted promoters nearby actual promoters as true positives on L. phytofermentans ISDg bacterium. Numbers between brackets beside the model ID indicate AUPRC (a) and AUROC (b) of that model

PR curves (a) and ROC curves (b) per model obtained when counting predicted promoters nearby actual promoters as true positives on R. capsulatus SB 1003 bacterium. The numbers between brackets beside the model ID indicate AUPRC (a) and AUROC (b) of that model

PR curves (a) and ROC curves (b) per model obtained when counting predicted promoters nearby actual promoters as true positives on B. amyloliquefaciens XH7 bacterium. The numbers between brackets beside the model ID indicate AUPRC (a) and AUROC (b) of that model
Performance comparison with existing methods
To compare Promotech’s performance with that of other existing methods, we used four independent test datasets containing promoters found by global TSS mapping using sequencing technologies. As in the previous assessments, the independent datasets included B. amyloliquefaciens, L. phytofermentans, M. smegmatis, and R. capsulatus. On this assessment, the datasets have a 1:1 ratio of positive to negative instances (Supplementary Table S3). As these datasets contained thousands instead of millions of sequences, we were able to include the RF-TETRA model that failed to run on a whole genome (due to memory issues).
As Promotech’s goal is to be applicable to a wide range of bacterial species, we compared Promotech models with other multi-species methods such as bTSSFinder [11] and G4PromFinder [10]. Additionally, we included BPROM [17] in the comparative assessment, as it is the most commonly used promoter prediction program (as per Google Scholar, BPROM’s manuscript has been cited 547 times). Finally, we also compared Promotech’s performance with two recent E. coli-specific methods: MULTiPly [4], designed for various sigma factors, and iPro70-FMWin [6], designed for sigma 70. In total, this benchmark included Promotech’s six models and five other bacterial promoter prediction tools.
Promotech’s random forests models (RF-TETRA and RF-HOT) consistently achieved the highest AUPRC and AUROC across the four bacterial species (Tables 8 and 9). RF-TETRA achieved the highest average AUPRC and AUROC among all the methods. Among the five other bacterial promoter prediction tools, iPro70-FMWin showed the best predictive performance but still substantially lower than Promotech’s. Based on these results, we selected RF-HOT as Promotech’s predictive model for genome-wide promoter prediction. For recognizing promoters on 40-nt-long genomic sequences in data sets containing up to thousands of sequences, we recommend both RF-HOT and RF-TETRA models.
Additionally, we compared the performance of RF-TETRA and RF-HOT for identifying E. coli promoters against that of E. coli-specific tools. To do this, we obtained Promotech’s predictions on a balanced data set with 2860 experimentally validated E. coli promoters collected from RegulonDB [32]. This data set has been used to evaluate the performance of several E. coli promoter prediction tools [3, 19, 33]. The average 5-fold cross-validation MCC and accuracy reported on this data set [3, 19, 33] are in the range of [0.498,0.763] and [0.748,0.882], respectively. RF-HOT achieved on this data set a MCC of 0.54, accuracy of 0.77, AUPRC of 0.845, and AUROC of 0.84, while RF-TETRA achieved a MCC of 0.47, accuracy of 0.734, AUPRC of 0.830, and AUROC of 0.808. Thus, RF-HOT is in the range of performance-level observed in programs specifically developed to identify E. coli promoters. These results demonstrate that Promotech is indeed suitable for predicting promoters on various bacterial species.
Conclusions
Based on our results, we recommend (1) to use E. coli-specific tools to predict E. coli promoters as they can identify E. coli promoters more accurately than a general bacterial promoter identification method such as Promotech and (2) to use Promotech to identify promoters in bacterial species other than E. coli, as we have shown Promotech outperforms other promoter prediction tools including iPro70-FMWin, one of the most accurate E. coli-specific tools [20], for identifying promoters on a variety of bacterial species (Tables 8 and 9).
In sum, Promotech is a promoter recognition tool suited for general (species independent) bacterial promoter detection that is able to perform promoter recognition on a whole bacterial genome. Promotech is available under the GNU General Public License v3.0 at [34, 35].
Methods
The goal of this study was to develop a general tool to recognize bacterial promoters. To do this, we assembled a large data set of promoter sequences from various bacterial species, generated twelve machine learning models, and selected the best models based on AUROC and AUPRC. Our best models were compared with five existing tools (BPROM [17], bTSSFinder [11], MULTIPly [4], iPro70-FMWin [6], and G4PromFinder [10]) using a validation data set, not used for training, of four bacterial species.
Materials
Collecting data
Bacterial TSS detected by next-generation sequencing (NGS) approaches, namely, dRNA-seq[21] and Cappable-seq[22], were collected from the literature (Table 10). We obtained promoter genomic coordinates and the corresponding sequences using BEDTools [36]. E σ covers DNA from roughly 55 bp upstream to 15 bp downstream of the TSS [1]. As the promoter region is not located downstream of the TSS and E σ covers 15 bp downstream of the TSS, we excluded 15 bp from both sides of the region covered by E σ and took as the promoter sequence the 40 bp upstream of the TSS. The bacterial species included in this study are listed in Table 10.
Generating positive and negative instance sets
A Nextflow [37] pipeline was designed to obtain the promoter (positive) sequences from the TSS coordinates by taking the genome FASTA file and the TSS coordinates as input and obtaining the promoter coordinates as 40 bp upstream from the TSS to the TSS using BEDTools’ slop command. The BEDTools’ subtract command was used to delete duplicates, and the getfasta command was used to obtain the FASTA sequences from the promoter coordinates.
To obtain non-promoter (negative) sequences, we used BEDTools’ random command to obtain random genomic coordinates and the getfasta command to obtain the corresponding genomic sequences. Negative sequences overlapping positive sequences were excluded from the training data. Note that some of these negative instances might in fact be actual promoters, and thus, predictive performance is conservatively assessed.
The training data sets created have a 1:10 ratio of positive to negative instances (unbalanced). For the validation data set, we created a data set with a 1:1 ratio of positive to negative instances (balanced). The total number of positive and negative instances per bacterium is shown in Supplementary Tables S2 and S3 in Additional file 1.
Machine learning models
We used two machine learning methods: recurrent neural networks (RNNs) [25] and random forest (RF) [23, 24]. Both methods have been successfully used before to classify genomic sequences. Random forest is a popular machine learning method for its ability to identify feature importance and handles many data types (continuous, categorical, and binary). It is well-suited for high-dimensional data and avoids over-fitting by its voting scheme among the ensemble of trees within it [38]. RNNs are also well-suited for genomic sequence analysis due to their ability to handle variable-length inputs, detecting sequential patterns and retaining information through time.
Model selection
Due to the lengthy training time of the RNNs, we were unable to run CV for the RNN models. Thus, to select the best model, we split our training data in 75% for training and 25% for testing. Models were trained with 75% of the data and then compared to each other on their performance in the 25% left-out data. After selecting the best models, these models were retrained using all of the training data and the resulting models used for whole-genome promoter prediction and comparative assessment with the other tools.
Random forest
Two RF models were generated; the first was trained using hot-encoded features; this meant that the nucleotides (A, G, C, T) were transformed into binary vector representations [1000], [0100], [0010], and [0001], respectively. This model is henceforth referred to as RF-HOT. The second model was trained using tetra-nucleotide frequencies calculated using the scikit-bio library [39] and denoted as RF-TETRA. The models were created using the Sklearn’s RandomForestClassifier [40] combined with a 10-fold grid search CV to handle the hyper-parameter optimization. The hyper-parameter search space was max_features (m): [None, “sqrt”, “log2”] and n_estimators (n): [1000, 2000, 3000]; both models were trained using an unbalanced data set with a 1:10 ratio of positive to negative instances. The best hyper-parameters found by grid search CV for both RF models were m = “log2” and n = 2000 with class weights values of {0 : 0.53, 1 : 10.28}.
Recurrent neural networks
Two types of RNNs were trained, long short-term memory unit (LSTM) [26] and gated recurrent unit (GRU) [27], using word embeddings representation of the tetra-nucleotide sequences calculated using the Keras’ Tokenizer class [41]. The models were designated as GRU-X or LSTM-X where X indicates the number of hidden layers. All models were manually tuned with an architecture consisting of one embedding layer, one GRU or LSTM layer, zero to four dense layers with dropout to reduce overfitting, one binary output, Adam optimizer function, and binary cross-entropy loss function (Table S4 in Additional file 1).
Computer infrastructure
All RF and RNN models were trained on the Compute Canada’s Beluga Cluster [42] configured with four NVidia V100SXM2 16GB GPUs, eight Intel Gold 6148 Skylake @ 2.4 GHz CPUs, and managed using SLURM commands.
Model assessment
Three assessments were performed to evaluate the models’ performance. The first consisted in scanning each bacterial genome using a 40-nt sliding window. In total, the number of generated sequences ranged from 4 to 7 million depending on the genome size. Models were given as input each of the sliding window sequences. Models then outputted per 40-nt sequence the probability of having a promoter within that sequence. To be counted as a true positive, the predicted promoter sequence had to have at least 10% sequence overlap with an actual promoter sequence. All other predicted promoters were considered false positives. To determine whether a predicted promoter overlapped with an actual promoter, we used the BEDTools intersect command with the parameters -s and -f 0.1.
In the second assessment, we also scanned each bacterial genome using a 40-nt sliding window. However, in this assessment, we considered a predicted promoter a true positive if it was within 100 nt of an actual promoter. In this setting, we use BEDtools’ closest command to find the five predicted promoters closest to an actual promoter. Then, those closest predicted promoters less than 100 nt away, upstream or downstream, from an actual promoter were counted as true positives. All other predicted promoters were considered false positives.
In the third assessment, we used the validation balanced data set obtained as described above. In this assessment, we included BPROM, bTSSfinder, G4Promfinder, iPro70-FMWin, and MULTiPLy. MULTiPLy and G4PromFinder accept 40-nt-long sequences, but BPROM and bTSSFinder require sequences 250-nt-long and iPro70-FMWin requires 81-nt-long sequences. We used BEDTools’ slopBed [38] command to extend the sequences in the data set from 40 to the required length. As G4Promfinder is written in Python, we integrated into our own pipeline and fed the sequences directly to G4Promfinder. To run, BPROM and bTSSfinder, we wrote each sequence to a file and then ran the programs through a shell script called from our own pipeline. MULTiPLy was tested separately, as it was developed in Matlab, so a short script was written to feed it each bacterium’s data set. We ran BPROM and MULTiPLy with their default values. bTSSFinder was run with the parameters -c 1 -t e and -h 2 indicating to search for the highest ranking promoter regardless of promoter class, use E. coli mode, and search on both strands. All other bTSSFinder parameters were left at their default values. G4Promfinder only requires as input sequences in FASTA format. iPro70-FMWin’s results were obtained from its website inputting the sequences in FASTA format.
Performance metrics used were (1) the area under the precision-recall curve (AUPRC), where precision is the number of true positives divided by the total number of predicted positives and recall (also called sensitivity or true-positive rate) is the number of true positives divided by the total number of actual positives and (2) the area under the ROC curve (AUROC), where true-positive rate is the same as recall and false-positive rate is the number of false positives divided by the total number of predicted positives.
References
Mejía-Almonte C, Busby SJW, Wade JT, van Helden J, Arkin AP, Stormo GD, Eilbeck K, Palsson BO, Galagan JE, Collado-Vides J. Redefining fundamental concepts of transcription initiation in bacteria. Nat Rev Genet. 2020; 21(11):699–714. https://doi.org/10.1038/s41576-020-0254-8.
Li F, Chen J, Ge Z, Wen Y, Yue Y, Hayashida M, Baggag A, Bensmail H, Song J. Computational prediction and interpretation of both general and specific types of promoters in Escherichia coli by exploiting a stacked ensemble-learning framework. Brief Bioinform. 2021; 22(2):2126–40. https://doi.org/10.1093/bib/bbaa049.
Amin R, Rahman CR, Ahmed S, Sifat MHR, Liton MNK, Rahman MM, Khan MZH, Shatabda S. iPromoter-BnCNN: a novel branched CNN-based predictor for identifying and classifying sigma promoters. Bioinformatics. 2020; 36(19):4869–75. https://doi.org/10.1093/bioinformatics/btaa609.
Zhang M, Li F, Marquez-Lago TT, Leier A, Fan C, Kwoh CK, Chou K-C, Song J, Jia C. MULTiPly: a novel multi-layer predictor for discovering general and specific types of promoters. Bioinformatics. 2019; 35(17):2957–65.
Lai H-Y, Zhang Z-Y, Su Z-D, Su W, Ding H, Chen W, Lin H. iProEP: a computational predictor for predicting promoter. Mol Therapy-Nucleic Acids. 2019; 17:337–46.
Rahman MS, Aktar U, Jani MR, et al.iPro70-FMWin: identifying Sigma70 promoters using multiple windowing and minimal features. Mol Gen Genomics. 2019; 294(1):69–84. https://doi.org/10.1007/s00438-018-1487-5.
Wang S, Cheng X, Li Y, Wu M, Zhao Y. Image-based promoter prediction: a promoter prediction method based on evolutionarily generated patterns. Sci Rep. 2018; 8(1):1–9.
Liu B, Yang F, Huang D-S, Chou K-C. iPromoter-2L: a two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformatics. 2018; 34(1):33–40. https://doi.org/10.1093/bioinformatics/btx579.
He W, Jia C, Duan Y, Zou Q. 70propred: a predictor for discovering sigma70 promoters based on combining multiple features. BMC Syst Biol. 2018; 12(4):44. https://doi.org/10.1186/s12918-018-0570-1.
Salvo MD, Pinatel E, Talà A, Fondi M, Peano C, Alifano P. G4PromFinder: an algorithm for predicting transcription promoters in GC-rich bacterial genomes based on AT-rich elements and G-quadruplex motifs. BMC Bioinformatics. 2018; 19(1):36.
Shahmuradov IA, Razali RM, Bougouffa S, Radovanovic A, Bajic VB. bTSSfinder: a novel tool for the prediction of promoters in Cyanobacteria and Escherichia coli. Bioinformatics. 2017; 33(3):334–40.
Umarov RK, Solovyev VV. Recognition of prokaryotic and eukaryotic promoters using convolutional deep learning neural networks. PloS ONE. 2017; 12(2):e0171410. https://doi.org/10.1371/journal.pone.0171410.
Song K. Recognition of prokaryotic promoters based on a novel variable-window z-curve method. Nucleic Acids Res. 2012; 40(3):963–71. https://doi.org/10.1093/nar/gkr795.
de Jong A, Pietersma H, Cordes M, Kuipers OP, Kok J. PePPER: a webserver for prediction of prokaryote promoter elements and regulons. BMC Genomics. 2012; 13(1):299.
de Avila e Silva S, Echeverrigaray S, Gerhardt GJL. BacPP: bacterial promoter prediction—a tool for accurate sigma-factor specific assignment in enterobacteria. J Theor Biol. 2011; 287:92–99.
Rangannan V, Bansal M. High-quality annotation of promoter regions for 913 bacterial genomes. Bioinformatics. 2010; 26(24):3043–50.
Salamov VSA, Solovyevand A. Automatic annotation of microbial genomes and metagenomic sequences. In: Metagenomics and its applications in agriculture. Hauppauge: Nova Science Publishers: 2011. p. 61–78.
Rangannan V, Bansal M. Relative stability of DNA as a generic criterion for promoter prediction: whole genome annotation of microbial genomes with varying nucleotide base composition. Mol BioSyst. 2009; 5(12):1758–69.
Liu B, Yang F, Huang D-S, Chou K-C. iPromoter-2L: a two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformatics. 2018; 34(1):33–40.
Cassiano MHA, Silva-Rocha R. Benchmarking bacterial promoter prediction tools: potentialities and limitations. mSystems. 2020; 5(4). https://doi.org/10.1128/mSystems.00439-20.
Sharma CM, Vogel J. Differential RNA-seq: the approach behind and the biological insight gained. Curr Opin Microbiol. 2014; 19:97–105. https://doi.org/10.1016/j.mib.2014.06.010.
Ettwiller L, Buswell J, Yigit E, Schildkraut I. A novel enrichment strategy reveals unprecedented number of novel transcription start sites at single base resolution in a model prokaryote and the gut microbiome. BMC Genomics. 2016; 17(1):199.
Ho TK. Random decision forests. In: Proceedings of 3rd International Conference on Document Analysis and Recognition, vol. 1. IEEE: 1995. p. 278–82. https://doi.org/10.1109/ICDAR.1995.598994.
Breiman L. Random forests. Mach Learn. 2001; 45(1):5–32.
Rumelhart DE, Hinton GE, Williams RJ. Learning representations by back-propagating errors. Nature. 1986; 323(6088):533–36.
Hochreiter S, Schmidhuber J. Long short-term memory. Neural Comput. 1997; 9(8):1735–80.
Cho K, van Merriënboer B, Gulcehre C, Bahdanau D, Bougares F, Schwenk H, Bengio Y. Learning phrase representations using RNN encoder–decoder for statistical machine translation. In: Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP). Doha: Association for Computational Linguistics: 2014. p. 1724–34. https://doi.org/10.3115/v1/D14-1179.
Bengio Y, Ducharme R, Vincent P, Jauvin C. A neural probabilistic language model. J Mach Learn Res. 2003; 3(Feb):1137–55.
Altmann A, Toloşi L, Sander O, Lengauer T. Permutation importance: a corrected feature importance measure. Bioinformatics. 2010; 26(10):1340–47. https://doi.org/10.1093/bioinformatics/btq134.
Pribnow D. Nucleotide sequence of an RNA polymerase binding site at an early T7 promoter. Proc Natl Acad Sci. 1975; 72(3):784–88.
Schaller H, Gray C, Herrmann K. Nucleotide sequence of an RNA polymerase binding site from the DNA of bacteriophage fd. Proc Natl Acad Sci. 1975; 72(2):737–41.
Santos-Zavaleta A, Salgado H, Gama-Castro S, Sánchez-Pérez M, Gómez-Romero L, Ledezma-Tejeida D, García-Sotelo JS, Alquicira-Hernández K, Muñiz-Rascado LJ, Peña-Loredo P, Ishida-Gutiérrez C, Velázquez-Ramírez DA, Del Moral-Chávez V, Bonavides-Martínez C, Méndez-Cruz C-F, Galagan J, Collado-Vides J. Regulondb v 10.5: tackling challenges to unify classic and high throughput knowledge of gene regulation in E. coli k-12. Nucleic Acids Res. 2019; 47(D1):212–20. https://doi.org/10.1093/nar/gky1077.
Lin H, Deng E-Z, Ding H, Chen W, Chou K-C. iPro54-PseKNC: a sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res. 2014; 42(21):12961–72. https://doi.org/10.1093/nar/gku1019.
Chevez-Guardado R, Peña-Castillo L. BioinformaticsLabAtMUN/Promotech: Promotech v1.0. Zenodo. 2021. https://doi.org/10.5281/zenodo.4737459. https://doi.org/10.5281/zenodo.4737459.
Chevez-Guardado R, Peña-Castillo L. BioinformaticsLabAtMUN/PromoTech. GitHub. 2020. https://github.com/BioinformaticsLabAtMUN/PromoTech. Accessed 29 Oct 2021.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26(6):841–42.
Di Tommaso P, Chatzou M, Floden EW, Barja PP, Palumbo E, Notredame C. Nextflow enables reproducible computational workflows. Nat Biotechnol. 2017; 35(4):316–19.
Zhang C, Ma Y. Ensemble machine learning: methods and applications. Redmond: Springer; 2012.
Knight R, Huttley G, McDonald D. scikit-bio. 2014. http://scikit-bio.org/. Accessed 29 Oct 2021.
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. Scikit-learn: machine learning in python. J Mach Learn Res. 2011; 12:2825–30.
Chollet F. keras. GitHub. 2015. https://github.com/fchollet/keras. Accessed 29 Oct 2021.
Baldwin S. Compute Canada: advancing computational research. In: Journal of Physics: Conference Series, vol. 341. IOP Publishing: 2012. p. 012001. https://doi.org/10.1088/1742-6596/341/1/012001.
Berger P, Knödler M, Förstner K, Berger M, Bertling C, Sharma CM, Vogel J, Karch H, Dobrindt U, Mellmann A. The primary transcriptome of the Escherichia coli O104: H4 pAA plasmid and novel insights into its virulence gene expression and regulation. Sci Rep. 2016; 6:35307.
Thomason MK, Bischler T, Eisenbart SK, Förstner K, Zhang A, Herbig A, Nieselt K, Sharma CM, Storz G. Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J Bacteriol. 2015; 197(1):18–28.
Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Sittka SFA, Chabas S, Reiche K, Hackermüller J, Reinhardt R. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010; 464(7286):250–55.
Yu S-H, Vogel J, U Förstner K. ANNOgesic: a Swiss army knife for the RNA-seq based annotation of bacterial/archaeal genomes. GigaScience. 2018; 7(9):096.
Dugar G, Herbig A, Förstner KU, Heidrich N, Reinhardt R, Nieselt K, et al.High-Resolution Transcriptome Maps Reveal Strain-Specific Regulatory Features of Multiple Campylobacter jejuni Isolates. PLoS Genet. 2013; 9(5):e1003495. https://doi.org/10.1371/journal.pgen.1003495.
Rosinski-Chupin I, Sauvage E, Fouet A, Poyart C, Glaser P. Conserved and specific features of Streptococcus pyogenes and Streptococcus agalactiae transcriptional landscapes. BMC Genomics. 2019; 20(1):236.
Kröger C, Dillon SC, Cameron ADS, Papenfort K, Sivasankaran SK, Hokamp K, Chao Y, Sittka A, Hébrard M, Händler K. The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc Natl Acad Sci. 2012; 109(20):1277–86.
Albrecht M, Sharma CM, Dittrich MT, Müller T, Reinhardt R, Vogel J, Rudel T. The transcriptional landscape of Chlamydia pneumoniae. Genome Biol. 2011; 12(10):98.
Shao W, Price MN, Deutschbauer AM, Romine MF, Arkin AP. Conservation of transcription start sites within genes across a bacterial genus. MBio. 2014; 5(4):01398–14.
Zhukova A, Fernandes LG, Hugon P, Pappas CJ, Sismeiro O, Coppée J-Y, Becavin C, Malabat C, Eshghi A, Zhang J-J. Genome-wide transcriptional start site mapping and sRNA identification in the pathogen Leptospira interrogans. Front Cell Infect Microbiol. 2017; 7:10.
Jeong Y, Kim J-N, Kim MW, Bucca G, Cho S, Yoon YJ, Kim B-G, Roe J-H, Kim SC, Smith CP. The dynamic transcriptional and translational landscape of the model antibiotic producer Streptomyces coelicolor A3 (2). Nat Commun. 2016; 7:11605.
Martini MC, Zhou Y, Sun H, Shell SS. Defining the transcriptional and post-transcriptional landscapes of Mycobacterium smegmatis in aerobic growth and hypoxia. Front Microbiol. 2019; 10:591.
Boutard M, Ettwiller L, Cerisy T, Alberti A, Labadie K, Salanoubat M, Schildkraut I, Tolonen AC. Global repositioning of transcription start sites in a plant-fermenting bacterium. Nat Commun. 2016; 7(1):1–9.
Grüll M. Transcriptomic studies of the bacterium rhodobacter capsulatus. PhD thesis: Memorial University of Newfoundland; 2019.
Liao Y, Huang L, Wang B, Zhou F, Pan L. The global transcriptional landscape of Bacillus amyloliquefaciens XH7 and high-throughput screening of strong promoters based on RNA-seq data. Gene. 2015; 571(2):252–62.
Acknowledgments
This research was enabled in part by support provided by ACENET (www.ace-net.ca/) and Compute Canada (www.computecanada.ca).
Peer review information
Tim Sands was the primary editor of this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Review history
The review history is available as Additional file 2.
Funding
This research was partially supported by a grant from the Natural Sciences and Engineering Research Council of Canada (NSERC) to L.P.-C. (Grant number RGPIN: 2019-05247). R.C. was partially supported by funding from Memorial University (MUN)’s School of Graduate Studies. Neither NSERC or MUN have a role in the study.
Author information
Authors and Affiliations
Contributions
L.P.-C. conceived the Promotech. R.C. implemented the Promotech and performed all the experiments under the supervision of L.P.-C. All authors discussed the results and contributed to the final manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
The Additional file 1 is a PDF file that includes the following tables: Table S1 — A summary of promoter prediction approaches in the last twelve years. Table S2 — The number of TSS per each bacterium in the training data set. Table S3 — The number of TSS per each bacterium in the validation data set. Table S4 — RNN hyper-parameters. Supplementary Figs. 1 - 4 — PRC and ROC per model and per bacterium when requiring 10% sequence overlap between predicted promoters and actual promoters to count them as true positives.
Additional file 2
Review history.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chevez-Guardado, R., Peña-Castillo, L. Promotech: a general tool for bacterial promoter recognition. Genome Biol 22, 318 (2021). https://doi.org/10.1186/s13059-021-02514-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13059-021-02514-9