
Abstract
Identification of causal mutations in barley and wheat is hampered by their large genomes and suppressed recombination. To overcome these obstacles, we have developed MutChromSeq, a complexity reduction approach based on flow sorting and sequencing of mutant chromosomes, to identify induced mutations by comparison to parental chromosomes. We apply MutChromSeq to six mutants each of the barley Eceriferum-q gene and the wheat Pm2 genes. This approach unambiguously identified single candidate genes that were verified by Sanger sequencing of additional mutants. MutChromSeq enables reference-free forward genetics in barley and wheat, thus opening up their pan-genomes to functional genomics.
Similar content being viewed by others


Background
The extraordinary success of wheat and barley as major worldwide crops is underpinned by their adaptability to diverse environments, high yield and nutritional content. Identification and manipulation of the genes controlling these traits will help to sustainably increase yields and ensure global food security. However, the large genomes of barley (5.5 Gb) and wheat (17 Gb) coupled with extensive regions of suppressed recombination [1, 2] make traditional map-based gene isolation in these crops both time-consuming and costly. In plants with small genomes, such as Arabidopsis and rice, whole genome sequencing on mapping populations has proven a powerful approach to aid gene isolation [3, 4]. Furthermore, whole genome sequencing and comparison of multiple independently derived mutants belonging to the same complementation group permits direct gene identification with little or no requirement for recombination [5, 6]. Because of the genome size of barley and wheat, sequencing whole genomes in multiple mutants is not practical due to the cost and the difficulty of comparison and interpretation of very large datasets. In allopolyploid wheat, this analysis is further complicated by the presence of homoeologues and multiple gene duplications [2, 7].
Various approaches can be used to reduce the sequence complexity of the barley and wheat genomes (Table 1). Exome capture sequencing (ExomeSeq) has been used in barley [8] and wheat [9, 10] to define induced mutations, for forward genetics to define a candidate for the barley HvPHYTOCHROME C gene [11] and to clone the barley many noded dwarf gene [12] and the wheat stem rust resistance genes Sr22 and Sr45 [13]. An exome capture design, however, is biased by only incorporating the known gene space annotated in reference genomes and therefore risks missing out on genes present in the species’ pan genome. A case in hand concerns the pleiotropic resistance gene Yr36, a START kinase, which is absent in post Green Revolution wheat [14]. Transcriptome sequencing (RNAseq) can overcome some of these limitations but is biased by the tissue sampled, the time of sampling and the sequencing depth. Also, assembly of RNAseq data from polyploid wheat is problematic due to the co-expression of near-sequence identical homoeologues and RNAseq will only directly reveal mutations in transcribed sequences (while regulatory sequences will be overlooked). Therefore, ExomeSeq and RNAseq have important limitations for isolating genes underpinning adaptive diversity in non-reference accessions.
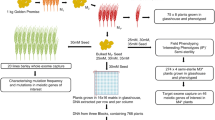
Chromosome flow sorting and sequencing (ChromSeq) represents a powerful, lossless and sequence-unbiased approach to genome complexity reduction [7]. Recent advances in labelling repetitive DNA on chromosomes prior to flow cytometric chromosome analysis allow purification of the seven barley and 21 bread wheat chromosomes independent of the cultivar [15, 16]. We reasoned that the sequence comparison of multiple independently derived mutant flow sorted chromosomes (MutChromSeq) would allow the identification of induced, causal mutations without the need for positional fine mapping (Fig. 1).

Germplasm structuring and gene isolation by MutChromSeq. a Mutagenesis of wheat seed with ethyl methanesulfonate (EMS), screening for mutant plants in the M2 generation and progeny testing in the M3 generation. Additional genetics may be required for some traits to determine complementation groups. b Preparation of liquid suspensions of mitotic chromosomes from M3 roots and labelling of chromosomes with fluorescently labelled DNA probes followed by flow sorting based on DNA content and fluorochrome signal. Pure chromosome preparations are amplified by phi DNA polymerase. c Sequencing of wild type and mutant chromosomes, sequence comparison, and candidate gene identification
Results
As a proof of concept we initially tested MutChromSeq on six ethyl methanesulphonate (EMS)-derived mutants of the recently cloned barley Eceriferum-q gene required for epicuticular aliphatic wax accumulation [17, 18] (Fig. 2a; Additional file 1: Figure S1 and Table S1). Flow sorting of chromosome 2H, to which Eceriferum-q has been assigned [18–20], followed by multiple displacement amplification (MDA) [21] yielded chromosome DNA preps of high purity (88 to 98 %), quantity (6.6 to 8.2 μg) and molecular weight (3 to 20 kb) (Additional file 1: Table S1 and Figure S2a). We sequenced the amplified chromosomal DNA using Illumina short read sequencing-by-synthesis technology (Additional file 1: Table S2) and performed a de novo assembly of the wild-type cultivar Foma. This resulted in 405,419 contiguous genomic sequences (contigs) of more than 500 bp, totalling 598 Mb with an N50 of 1.4 kb (Additional file 1: Table S3). We then compared the sequence reads from the six mutants to the wild-type assembly and looked at the mutation overlap. We found 57 contigs with single nucleotide variants (SNVs) in three of the mutants, five contigs with SNVs in four mutants and a single 12-kb contig with SNVs in five of the six mutants (Table 2). Closer inspection of the mutant line eceriferum-q.334 that did not have a mutation in our candidate contig revealed a SNV density of 1 in 1580 bp, whereas the frequency in the other mutants ranged from 1 per 380 kb to 1 per 741 kb (Additional file 1: Table S4). On this basis we concluded that the line eceriferum-q.334 was a cultivar contaminant and it was excluded from further analysis.

Cloning of the barley Eceriferum-q and the wheat Pm2 genes by MutChromSeq. a Wax covered leaf sheath of wild-type barley cultivar Foma and six eceriferum (not bearing wax) mutants. Line 334 was deemed to be a cultivar contaminant following sequence analysis. b Powdery mildew infected wheat leaves of line CI12632/8*Cc (Pm2), six EMS-derived susceptible mutants (pm2) and the susceptible control cultivar Chancellor. c, d The Eceriferum (c) and Pm2 (d) loci showing intron–exon boundaries, protein domains and 5′ and 3′ untranslated regions (UTR). Mutations identified by MutChromSeq are indicated by red vertical lines, while mutations identified by Sanger sequencing of additional mutants are indicated by black vertical lines. A number above the line indicates identical mutations occurring in independent lines. Non-sense mutations are indicated by asterisks. Two mutations identified in the pm2 mutant 80_355 are joined by an arched line. CC, coiled-coil; NB-ARC, nucleotide-binding adaptor shared by APAF-1, R proteins, and CED-4; LRR, leucine-rich repeat
Sequence homology (BLAST) search of the 12-kb candidate contig against publicly available barley resources [22] and analysis of whole transcriptome reads obtained from wild-type leaf sheath tissue identified a single transcriptional unit supported by one full-length barley cDNA and RNAseq reads (Additional file 1: Figure S2b) with 100 % identity to the recently cloned Eceriferum-q gene [18]. The intronless gene spans 1229 bp from the start to the stop codons and contains 5′ and 3′ UTRs of 94 and 331 bp, respectively (Fig. 2c). We confirmed the five mutations identified by MutChromSeq by PCR amplification and Sanger sequencing and sequenced an additional six EMS-derived Eceriferum-q mutants and identified another six mutations. All the EMS-derived mutations are G/C-to-A/T transitions that cause nonsense (1) or missense (10) mutations (Additional file 1: Table S5). Based on the EMS canonical mutation frequency and the GC distribution observed in the five EMS-treated and sequenced Foma 2H chromosome contigs, we calculated the probability for a mutation to occur in the same contig in 11 EMS-treated individuals by chance alone to be 1 in 4 × 1014 (see “Methods”). In conclusion, these data provide compelling evidence for successful gene identification by MutChromSeq in barley.
To further put MutChromSeq to the test we decided to target a plant resistance (R) gene in hexaploid wheat. R genes typically encode nucleotide binding and leucine-rich repeat containing proteins (NLRs), belong to large multigene families, reside in complex gene clusters with multiple sequence-related paralogues and display extreme sequence and copy number variation between accessions [23]. This often obscures orthogonal relationships and makes functional dissection of R gene clusters by recombination alone impractical. We obtained 12 EMS-derived susceptible mutants of the dominant powdery mildew resistance gene 2 (Pm2; Additional file 1: Table S5), which has been mapped to chromosome 5D and originally derives from the Ulka donor variety [24]. We chose six of the Pm2 mutants and the wild-type parent for MutChromSeq (Fig. 2b) and obtained highly pure chromosome samples, which were sequenced on the Illumina platform (Additional file 1: Tables S1 and S2).
Applying the same analysis pipeline we used before, we identified two candidate contigs >1 kb. One could be discarded after manual inspection due to the very large number of SNVs observed between this contig and the mapped wild-type reads indicative of an assembly artefact (Additional file 1: Figure S3), while the other contig contained a full-length NLR type gene encoding domains with homology to a coiled coil, a nucleotide binding site and leucine-rich repeats (Fig. 2d). We confirmed the nature of this short read-assembled 10-kb contig by long-range PCR amplification and sequencing of the complete coding region (Additional file 1: Figure S2d). We converted the contig into a PCR molecular marker and showed that it genetically co-localises with Pm2-mediated resistance to a 1.3-cM interval on chromosome 5D (Additional file 1: Figure S2e). Further pivotal confirmation that this gene is Pm2 was obtained by Sanger sequencing of the remaining six mutants (Additional file 1: Table S5). All mutants were found to contain G/C-to-A/T transitions typical of EMS, which gave rise to predicted nonsense (5) and missense (7) mutations. No other mutations were found in the Pm2 10-kb short read-assembled contig in the six chromosome sequenced mutants. Based on the EMS mutation frequency and GC distribution in the six sequenced Pm2 chromosome contigs, we calculated that the probability of getting 12 mutations in the same gene in 12 pm2 mutants by chance alone to be 1 in 3 × 1011 (see “Methods”).
Discussion
We have used MutChromSeq to successfully reclone the barley Eceriferum-q gene and clone de novo the wheat Pm2 gene. Prior to chromosome flow sorting the target gene must be assigned to a chromosome. This usually requires the phenotypic and genotypic analysis of progeny derived from a sexual cross to allow for the independent assortment of chromosomes. However, recombination per se is not required. This makes the method particularly attractive for cloning genes in recombination-sparse regions. Indeed, the method does not require fine mapping or the construction of a physical reference sequence across a map interval. The approach is fast and robust and in the Eceriferum-q example allowed the identification of the correct gene despite the inclusion of a false positive. Our ensuing mapping data and the sequencing of multiple additional independent mutants support the validity and accuracy of MutChromSeq as a powerful approach for gene isolation.
Chromosome flow sorting is a technically demanding procedure requiring expertise in cytogenetics, flow cytometry and molecular biology and the availability of a flow sorter [15, 16]. A group aiming to clone a small number of genes is unlikely to justify the effort required to establish a chromosome sorting pipeline. However, the successful projects on draft genome sequencing in barley [25, 26], rye [27] and wheat [7] demonstrate the viability of collaborative projects.
In the MutChromSeq approach, amplified chromosomal DNA is sequenced. Only tens of thousands of copies of sorted chromosomes are required. These quantities can be purified in less than one day. Thus, we believe that only a small number of specialised laboratories can provide sufficient capacity to satisfy the community demand for chromosome sorting from agronomically important species with large genomes for which protocols for chromosome sorting are available.
Unlike methylation filtration [28], high-C0 t fractionation [29] or duplex-specific nuclease digestion [30], complexity reduction by chromosome sorting is lossless and all sequences from a particular chromosome are sequenced. Apart from reducing the complexity of the DNA sample to be sequenced, an important advantage of the targeted approach is that it simplifies DNA sequence analysis by avoiding homoeologues in polyploids (wheat) and paralogues and pseudogenes present on other chromosomes (all species).
We recently used an NLR exome capture to clone two resistance genes in wheat by sequence comparison of multiple independently derived EMS mutants [13]. However, not all R genes are NLRs. For example, pleiotropic adult plant resistance (APR) genes in wheat encode proteins belonging to disparate structural classes, including an ABC transporter (Lr34), a START kinase (Yr36) and a hexose transporter (Lr67) [14, 31, 32]. Furthermore, genes conferring resistance to necrotrophs, hemibiotrophs and pathogens with an apoplastic lifestyle are less likely to be encoded by NLRs. MutChromSeq avoids the potential problems in gene cloning due to the bias introduced by exome-based capture approaches, including the absence of some genes in reference genome assemblies. Furthermore, compared to ExomeSeq and RNAseq, MutChromSeq is more likely to directly identify mutations in regulatory regions and assemble into the same contig exons separated by large introns. The bridging of exons separated by large introns and identification of native regulatory elements may also be achieved by combining exome capture with single molecule long read sequencing technology [33]. However, long-read technology would at present be expensive to apply to multiple, complete wheat exomes.
Conclusions
We propose MutChromSeq, a gene cloning method which does not rely on recombination or fine-mapping. Our approach combines mutagenesis, genome complexity reduction by chromosome flow sorting (which is established for a number of plants, including members of the Triticeae) and high-throughput sequencing. Thus, the requirements for our method to work are that the plant species is amenable to mutagenesis, that the target gene can be associated with a clear phenotype and knowledge of which chromosome the gene is on. For genes fulfilling these prerequisites, we offer a fast and inexpensive method that opens up the possibility to clone a large number of previously intractable genes.
Methods
The aim of this study was to develop a gene cloning method in wheat and barley that would overcome the challenges imposed by their large genomes and extensive regions of suppressed recombination. To this end we combined mutagenesis, which is independent of recombination, with chromosome flow sorting and sequencing. By only sequencing the chromosome a gene has been rough-mapped to, we obtained 21-fold and sevenfold reductions in complexity in wheat and barley, respectively. Comparing the chromosome sequence of multiple mutants allowed the rapid identification of a single candidate gene.
Chromosome flow sorting and preparation of amplified chromosomal DNA
Liquid suspensions of mitotic metaphase chromosomes were prepared from synchronised root tips of barley [34] and wheat seedlings [35]. GAA microsatellites on chromosomes in suspension were labelled with a fluorescein isothiocyanate (FITC) conjugate [16] and chromosomal DNA was stained by 4′,6-diamidino-2-phenylindole (DAPI) at 2 μg/ml. The chromosome samples were analysed at a rate of 2000 chromosomes/s by a FACSAria SORP (BD Biosciences, San Jose, CA, USA) and sort windows were set up on FITC versus DAPI dot plots to sort chromosome 2H in barley and 5D in wheat. The chromosomes were sorted at rates of 25/s into 0.5-ml PCR tubes containing 40 μl deionised water. Three independent samples of 50,000 chromosomes were sorted from each line and their DNA amplified by multiple displacement amplification [21] using the Illustra GenomiPhi V2 DNA amplification kit (GE Healthcare Life Sciences, Pittsburgh, PA, USA). Amplified DNA samples derived from each line were pooled to achieve higher sequence representation. In order to determine the purity in flow-sorted fractions, during each sort run, 2000 chromosomes were sorted into a 5-μl drop of P5 buffer on a microscope slide and sorted chromosomes were identified by FISH using probes for GAA microsatellites in barley and GAA microsatellites and Afa family repeats in wheat [36].
lllumina library construction and sequencing
The barley Illumina libraries were constructed following the Broad Institute’s DISCOVAR protocol (http://www.broadinstitute.org/software/discovar/blog/?page_id=375) for PCR free libraries, except New England Biolab reagents were used instead of Kapa. Each mutant library was sequenced on one lane of a HiSeq 2500 with 125 bp paired-end (PE) reads (for each of the mutants) and 250 bp PE reads for wild-type Foma. The wheat libraries were constructed using the Illumina TruSeq protocol with a 250-bp insert size and subjected to 125-bp PE sequencing on an Illumina HiSeq2000 platform. Libraries corresponding to Federation*4/Ulka, Federation and Chancellor were sequenced using independent lanes. In the case of CI12362/8*Cc and the mutants, three libraries were pooled per lane. The average on target chromosome coverage, after correcting for PCR duplicates, ranged from 27 (for the Eceriferum-q.295 line) to 35 (for the Pm2 wild-type line). The details of raw sequence data generated are shown in Additional file 1: Table S2.
ChromSeq mutant hunter
All raw data were quality trimmed using sickle v.1.2 (https://github.com/najoshi/sickle). We created draft de novo assemblies of chromosome 2H of the wild-type barley cultivar Foma and chromosome 5D of the wild-type wheat line CI12632/8*Cc (Additional file 1: Table S3) using CLC Assembly Cell (https://www.qiagenbioinformatics.com/; version 4.3.0) and standard parameters. The assemblies were masked for repeats using RepeatMasker (http://repeatmasker.org) and the Triticeae Repeat Database (TREP; http://wheat.pw.usda.gov/ITMI/Repeats/) as external library. We then mapped the raw data of the chromosomes from the EMS mutants to the repeat-masked assemblies using BWA [37] v0.7.12. Mappings were filtered for reads not mapping as a pair (samtools view –f2) and PCR duplicates (samtools rmdup) using SAMtools [38] v0.1.19. Mappings were subsequently converted to the mpileup format (samtools mpileup –BQ0). The individual data sets in mpileup format were then integrated to count the number of mutants that had a SNV per contig. Potential SNVs were discarded if the allele frequency was below 99.99 %, the coverage was below 15 and the position was not mutated in more than two mutants. Note, the values above are input parameters to a custom script (http://github.com/steuernb/MutChromSeq). The stringency is dependent on the quality and depth of input data.
Calculating mutation probabilities
Taking into account that (i) the majority of EMS mutations are G/C-to-A/T transitions [39], (ii) that the majority of our repeat-masked barley and wheat contigs had a similar GC content (Additional file 1: Figure S4) and (iii) that EMS mutations tend to be randomly distributed [40–42], we calculated the probability of a false positive as a function of the length of the contig (Additional file 1: “Supplementary materials and methods” and Table S8). Thus, in barley, the largest probability of a 12-kb contig being mutated in a single mutant was calculated to be 0.05 based on the canonical mutation density observed in Eceriferum.q-327 (Additional file 1: Tables S4 and S9). Therefore, the probability of the contig being mutated across all 11 mutants by chance alone is (0.05)11 or 1 in 4 × 1014. Similarly, in wheat, we calculated the largest probability of a 10-kb contig being mutated as 0.11 based on the canonical mutation density observed in mutant pm2_78.56 (Additional file 1: Tables S4 and S9). Therefore, the probability of the contig being mutated across all 12 Pm2 mutants is (0.11)12 or 1 in 3 × 1011.
Further details of materials and methods are shown in Additional file 1: “Supplementary materials and methods”.
References
Kunzel G, Korzun L, Meister A. Cytologically integrated physical restriction fragment length polymorphism maps for the barley genome based on translocation breakpoints. Genetics. 2000;154:397–412.
Choulet F, Alberti A, Theil S, Glover N, Barbe V, Daron J, Pingault L, Sourdille P, Couloux A, Paux E, et al. Structural and functional partitioning of bread wheat chromosome 3B. Science. 2014;345:1249721.
Schneeberger K, Ossowski S, Lanz C, Juul T, Petersen AH, Nielsen KL, Jorgensen JE, Weigel D, Andersen SU. SHOREmap: simultaneous mapping and mutation identification by deep sequencing. Nat Methods. 2009;6:550–1.
Takagi H, Tamiru M, Abe A, Yoshida K, Uemura A, Yaegashi H, Obara T, Oikawa K, Utsushi H, Kanzaki E, et al. MutMap accelerates breeding of a salt-tolerant rice cultivar. Nat Biotechnol. 2015;33:445–9.
Nordstrom KJ, Albani MC, James GV, Gutjahr C, Hartwig B, Turck F, Paszkowski U, Coupland G, Schneeberger K. Mutation identification by direct comparison of whole-genome sequencing data from mutant and wild-type individuals using k-mers. Nat Biotechnol. 2013;31:325–30.
Schneeberger K. Using next-generation sequencing to isolate mutant genes from forward genetic screens. Nat Rev Genet. 2014;15:662–76.
International Wheat Genome Sequencing C. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 2014;345:1251788.
Mascher M, Richmond TA, Gerhardt DJ, Himmelbach A, Clissold L, Sampath D, Ayling S, Steuernagel B, Pfeifer M, D'Ascenzo M, et al. Barley whole exome capture: a tool for genomic research in the genus Hordeum and beyond. Plant J. 2013;76:494–505.
Henry IM, Nagalakshmi U, Lieberman MC, Ngo KJ, Krasileva KV, Vasquez-Gross H, Akhunova A, Akhunov E, Dubcovsky J, Tai TH, Comai L. Efficient genome-wide detection and cataloging of EMS-induced mutations using wxome capture and next-generation sequencing. Plant Cell. 2014;26:1382–97.
King R, Bird N, Ramirez-Gonzalez R, Coghill JA, Patil A, Hassani-Pak K, Uauy C, Phillips AL. Mutation scanning in wheat by exon capture and Next-Generation Sequencing. PLoS One. 2015;10:e0137549.
Pankin A, Campoli C, Dong X, Kilian B, Sharma R, Himmelbach A, Saini R, Davis SJ, Stein N, Schneeberger K, von Korff M. Mapping-by-sequencing identifies HvPHYTOCHROME C as a candidate gene for the early maturity 5 locus modulating the circadian clock and photoperiodic flowering in barley. Genetics. 2014;198:383–96.
Mascher M, Jost M, Kuon JE, Himmelbach A, Assfalg A, Beier S, Scholz U, Graner A, Stein N. Mapping-by-sequencing accelerates forward genetics in barley. Genome Biol. 2014;15:R78.
Steuernagel B, Periyannan SK, Hernández-Pinzón I, Witek K, Rouse MN, Yu G, Hatta A, Ayliffe M, Bariana H, Jones JDG, et al. Rapid cloning of disease-resistance genes in plants using mutagenesis and sequence capture. Nat Biotechnol. 2016. In press.
Fu D, Uauy C, Distelfeld A, Blechl A, Epstein L, Chen X, Sela H, Fahima T, Dubcovsky J. A kinase-START gene confers temperature-dependent resistance to wheat stripe rust. Science. 2009;323:1357–60.
Dolezel J, Vrana J, Capal P, Kubalakova M, Buresova V, Simkova H. Advances in plant chromosome genomics. Biotechnol Adv. 2014;32:122–36.
Giorgi D, Farina A, Grosso V, Gennaro A, Ceoloni C, Lucretti S. FISHIS: Fluorescence in situ hybridization in suspension and chromosome flow sorting made easy. PLoS One. 2013;8:e57994.
Lundqvist U, Lundqvist A. Mutagen specificity in barley for 1580 Eceriferum mutants localized to 79 loci. Hereditas. 1988;108:1–12.
Schneider LM, Adamski NM, Christensen CE, Stuart DB, Vautrin S, Hansson M, Uauy C, von Wettstein-Knowles P. The Cer-cqu gene cluster determines three key players in a beta-diketone synthase polyketide pathway synthesizing aliphatics in epicuticular waxes. J Exp Bot. 2016. doi:10.1093/jxb/erw105.
Takahashi R, Yamamoto J, Yasuda S, Itano Y. Inheritance and linkage studies in barley. Ber Ohara Inst landw Forsch. 1953;10:29–52.
Tsuchiya T. Cytogenetics of telotrisomics in barley. Barley Genet Newsl. 1972;2:93–8.
Simkova H, Svensson JT, Condamine P, Hribova E, Suchankova P, Bhat PR, Bartos J, Safar J, Close TJ, Dolezel J. Coupling amplified DNA from flow-sorted chromosomes to high-density SNP mapping in barley. BMC Genomics. 2008;9:294.
Colmsee C, Beier S, Himmelbach A, Schmutzer T, Stein N, Scholz U, Mascher M. BARLEX--the Barley Draft Genome Explorer. Mol Plant. 2015;8:964–6.
Kuang H, Woo SS, Meyers BC, Nevo E, Michelmore RW. Multiple genetic processes result in heterogeneous rates of evolution within the major cluster of disease resistance genes in lettuce. Plant Cell. 2004;16:2870–94.
Pugsley AT, Carter MV. The resistance of twelve varieties of Triticum vulgare to Erysiphe graminis tritici. Aust J Biol Sci. 1953;6:335–46.
Mayer KF, Taudien S, Martis M, Simkova H, Suchankova P, Gundlach H, Wicker T, Petzold A, Felder M, Steuernagel B, et al. Gene content and virtual gene order of barley chromosome 1H. Plant Physiol. 2009;151:496–505.
Mayer KFX, Martis M, Hedley PE, Simková H, Liu H, Morris JA, Steuernagel B, Taudien S, Roessner S, Gundlach H, et al. Unlocking the barley genome by chromosomal and comparative genomics. Plant Cell. 2011;23:1249–63.
Martis MM, Zhou R, Haseneyer G, Schmutzer T, Vrána J, Kubaláková M, König S, Kugler KG, Scholz U, Hackauf B, et al. Reticulate evolution of the rye genome. Plant Cell. 2013;25:3685–98.
Rabinowicz PD, Schutz K, Dedhia N, Yordan C, Parnell LD, Stein L, McCombie WR, Martienssen RA. Differential methylation of genes and retrotransposons facilitates shotgun sequencing of the maize genome. Nat Genet. 1999;23:305–8.
Barbazuk WB, Bedell JA, Rabinowicz PD. Reduced representation sequencing: a success in maize and a promise for other plant genomes. Bioessays. 2005;27:839–48.
Matvienko M, Kozik A, Froenicke L, Lavelle D, Martineau B, Perroud B, Michelmore R. Consequences of normalizing transcriptomic and genomic libraries of plant genomes using a duplex-specific nuclease and tetramethylammonium chloride. PLoS One. 2013;8:e55913.
Lagudah ES, Krattinger SG, Herrera-Foessel S, Singh RP, Huerta-Espino J, Spielmeyer W, Brown-Guedira G, Selter LL, Keller B. Gene-specific markers for the wheat gene Lr34/Yr18/Pm38 which confers resistance to multiple fungal pathogens. Theor Appl Genet. 2009;119:889–98.
Moore JW, Herrera-Foessel S, Lan C, Schnippenkoetter W, Ayliffe M, Huerta-Espino J, Lillemo M, Viccars L, Milne R, Periyannan S, et al. A recently evolved hexose transporter variant confers resistance to multiple pathogens in wheat. Nat Genet. 2015;47:1494–8.
Witek K, Jupe F, Witek A, Baker D, Clark MD, Jones JDG. Accelerated cloning of a potato late blight-resistance gene using RenSeq and SMRT sequencing. Nat Biotechnol. 2016. In press.
Lysak MA, Cihalikova J, Kubalakova M, Simkova H, Kunzel G, Dolezel J. Flow karyotyping and sorting of mitotic chromosomes of barley (Hordeum vulgare L.). Chromosome Res. 1999;7:431–44.
Vrana J, Kubalakova M, Simkova H, Cihalikova J, Lysak MA, Dolezel J. Flow sorting of mitotic chromosomes in common wheat (Triticum aestivum L.). Genetics. 2000;156:2033–41.
Kubalakova M, Valarik M, Bartos J, Vrana J, Cihalikova J, Molnar-Lang M, Dolezel J. Analysis and sorting of rye (Secale cereale L.) chromosomes using flow cytometry. Genome. 2003;46:893–905.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, GPDPS. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Uauy C, Paraiso F, Colasuonno P, Tran RK, Tsai H, Berardi S, Comai L, Dubcovsky J. A modified TILLING approach to detect induced mutations in tetraploid and hexaploid wheat. BMC Plant Biol. 2009;9:115.
Farrell A, Coleman BI, Benenati B, Brown KM, Blader IJ, Marth GT, Gubbels MJ. Whole genome profiling of spontaneous and chemically induced mutations in Toxoplasma gondii. BMC Genomics. 2014;15:354.
Shirasawa K, Hirakawa H, Nunome T, Tabata S, Isobe S. Genome-wide survey of artificial mutations induced by ethyl methanesulfonate and gamma rays in tomato. Plant Biotechnol J. 2016;14:51–60.
Lindner H, Kessler SA, Muller LM, Shimosato-Asano H, Boisson-Dernier A, Grossniklaus U. TURAN and EVAN mediate pollen tube reception in Arabidopsis Synergids through protein glycosylation. PLoS Biol. 2015;13:e1002139.
Acknowledgements
We thank Jarmila Číhalíková, Romana Šperková and Zdenka Dubská (IEB) for help with chromosome sorting and preparation of amplified chromosomal DNA, and Darren Heavens (Earlham Institute, Norwich) for sequencing advice.
Funding
This research was supported with funding from the JIC Innovation Fund IF2015BW22, the Biotechnology and Biological Sciences Research Council, the Swiss National Science Foundation grants 310030B_144081, 310030_163260 and PZ00P3_154694, the Czech Science Foundation (award number P501/12/G090), the Czech Republic National Program of Sustainability (award number LO1204), and the Plant Fellows Program, co-funded by the COFUND FP7 EU (grant agreement number 267423) to JSM.
Availability of data and material
DNA and RNA raw sequencing data are available from the European Bioinformatics Institute (EBI) Short Read Archive (SRA) under accession numbers PRJEB12101 (barley cultivar Foma RNA sequence), PRJEB12151 (MutChromSeq on Eceriferum-q), PRJEB12098 (MutChromSeq on Pm2) and PRJEB12158 (flow-sorted chromosomes 5D of Chancellor, Federation and Federation*4/Ulka).
The MutChromSeq custom scripts are available from GitHub (http://github.com/steuernb/MutChromSeq) and have been assigned an MIT open source license with the DOI 10.5281/zenodo.60879.
Authors’ contributions
JSM, BS, SG, GH, SH, NA, TW, SGK, JV and MK performed experiments. JSM, BS, JD, BK and BBHW wrote the manuscript. JSM, BS, JD, BK and BBHW contributed to the design of the study. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Additional file
Additional file 1:
Supplementary material and methods, Figures S1–S4 and Tables S1–S9. (DOCX 2154 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sánchez-Martín, J., Steuernagel, B., Ghosh, S. et al. Rapid gene isolation in barley and wheat by mutant chromosome sequencing. Genome Biol 17, 221 (2016). https://doi.org/10.1186/s13059-016-1082-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13059-016-1082-1