
Abstract
We have witnessed an explosion in our understanding of the evolution and structure of plant genomes in recent years. Here, we highlight three important emergent realizations: (1) that the evolutionary history of all plant genomes contains multiple, cyclical episodes of whole-genome doubling that were followed by myriad fractionation processes; (2) that the vast majority of the variation in genome size reflects the dynamics of proliferation and loss of lineage-specific transposable elements; and (3) that various classes of small RNAs help shape genomic architecture and function. We illustrate ways in which understanding these organism-level and molecular genetic processes can be used for crop plant improvement.
Similar content being viewed by others

Background
The readers of Genome Biology are likely to agree that we are living in a tremendously exciting time to be a biologist, perhaps one that in the future will be thought of as a ‘golden era’, replete with technological and conceptual breakthroughs. These breakthroughs are synergistic twins, of course, as novel analytical methods lead to applications that generate biological discoveries and hypotheses that are conceptually transformative. This synergy is particularly evident in the study of plant genome evolution, in which massively parallel sequencing approaches have revealed genomic diversity in exquisite detail, which has led to many insights into genome function and evolution. Our purpose in this short review is to highlight progress made in the understanding of plant genome evolution, with a focus on crop plants and on recent key insights. We highlight that modern plant genomes derive from processes set in motion by a history of repeated, episodic whole-genome doubling events, and that the extraordinary variation in genome size across plant species largely reflects differences in proliferation and survival of various classes and families of transposable elements (TEs), often in a lineage-specific manner. Furthermore, we discuss the connections between genomic architecture and small RNA function. As our review is mainly focused on crop plant genomes, we also discuss how plant genomics is relevant to crop improvement and food security.
Whole-genome doubling: wash, rinse, repeat
One of the important realizations of the genomics era is that whole-genome doubling (WGD), or polyploidy, is far more prevalent in the evolutionary history of plants than previously recognized. Classic estimates based on comparative cytogenetic studies [1–3] and stomatal guard cell sizes [4] have indicated that chromosome doubling is common in many genera and families, with estimates of the frequency of polyploid ancestry ranging from 35 % to 70 %. Thus, polyploidy has long been appreciated as important in angiosperm diversification and as an active mode of speciation in many groups. Polyploidy leading to speciation can arise by several means [5], either within single individuals or following hybridization between closely related populations (autopolyploidy), or from interspecific or, more rarely, intergeneric hybridization events (allopolyploidy) [6].
The ubiquity and cyclical nature of polyploidy
Genomic analyses over the past 15 years have demonstrated that all flowering plants are polyploid, and multiply so [7–9]. That is, the phylogenetic history of angiosperms abounds with WGD events, the most recent of which are superimposed on earlier duplications that took place early in angiosperm evolution, and before that on duplications that occurred at the root of the seed plants [7]. Our understanding of the cyclical nature of polyploidy was first suggested by analyses of expressed sequence tags (ESTs) in many different plant species (or genera). These analyses revealed ‘peaks’ of sequence similarity among genes within genomes representing multiple gene duplicates, whose collective existence and features suggest they traced to a common origin [10]. In many cases, several such peaks existed within individual genomes, which ostensibly reflects progressively more ancient WGD events. This emerging view of the canonical angiosperm genome as one that has experienced multiple episodic polyploidy events has been confirmed by recent genome sequencing efforts (Table 1). These studies revealed a widespread pattern of nested, intragenomic syntenies, often shared among close relatives but varying widely and in a lineage-specific fashion among different angiosperm groups. Therefore, we can rightfully replace the obsolete question ‘is this species polyploid?’ with the more appropriate ‘when did genome duplication occur and how many rounds of genome doubling have occurred in the history of this particular species?’
Genomic responses to polyploidy
This enhanced appreciation of the history of plant genomes might make one ask why this history of repeated, episodic polyploidy was not recognized earlier. The answer to this question lies in the surprisingly varied spectrum of genomic responses to polyploidy [11–19], which range in timing from those accompanying the initial genome merging and doubling, to others operating over millions of years. As modeled in Fig. 1, the immediate responses to the formation of a polyploid (mostly allopolyploid) genome include DNA-level and expression-level responses. Examples of the DNA-level responses include reciprocal or non-reciprocal homoeologous exchange, mutational loss of duplicated genes, intersubgenomic spread of TEs (which can be activated by genome merging and polyploidization), and divergence in molecular evolutionary rates. Expression-level alterations accompanying or set in motion by polyploidy encompass a variety of forms of duplicate gene expression bias, and subfunctionalization and neofunctionalization of expression patterns. Long-term responses include genome-wide subfunctionalization and neofunctionalization [20–23] and massive genome structural rearrangements (Fig. 2). These structural rearrangements include reductions in chromosome numbers and the large-scale loss of repetitive sequences and duplicate genes [24–26]. Thus, new polyploid species, most of which have experienced multiple cycles of polyploidization, eventually experience massive loss of ‘redundant’ DNA and chromosome restructuring, and recurrent genome downsizing [26]. Thus, neopolyploid species ultimately become diploidized by mechanistically diverse processes, such that contemporary descendants increasingly behave cytogenetically as normal diploid species while harboring in their genomes the vestigial evidence of past WGD events.

Processes and patterns of polyploidy in plants. Flowering plant genomes have an evolutionary history that includes multiple, lineage-specific, whole-genome doubling events. A model of a hypothetical allopolyploid genome derived from two progenitor diploid genomes (A and B) is shown in the figure. Hybridization and genome doubling set in motion short-term and long-term genomic processes, at the level of DNA sequence (top) and at the expression level (bottom). Young allopolyploids, which initially contain two genomes (A T and B T ) inherited from the parental genome donors, often display homoeologous recombination (‘gene conversion’), mutational loss and/or silencing of duplicated genes, intergenomic spread of TEs and differential rates of molecular evolution, as well as many different forms of biased or altered gene expression, including biased homoeolog expression and expression level dominance (not shown), and expression subfunctionalization (shown as partitioning of ancestral blue and green expression domains, bottom left) and neofunctionalization (novel red expression domain, bottom right)

Episodic, cyclical episodes of polyploidy in plants. All plant lineages are paleopolyploid, differing only in the number of rounds of whole-genome doubling in their ancestry, and in the timing of these events. Long-term processes following whole-genome doubling include differential retention of duplicated genes (here, more circles than squares are lost), biased genome fractionation (shown as greater loss of the B genome), massive genome downsizing and reduction in the number of chromosomes
The fate of duplicated genes
An intriguing facet of this cyclical process of genome downsizing is that it may be non-random with respect to the fate of duplicate genes. Genes restored to single copy status often have broader expression domains and higher expression levels than those retained in duplicate; they are also enriched for essential housekeeping functions, chloroplast-related functions, and functions in DNA replication and repair [27]. Although much remains to be learned in this active area of investigation, the evolutionary forces underlying the fate of duplicated genes include those emerging from the selective demands of stoichiometry during protein complex assembly, or the necessity of maintaining balanced protein interactions, and other possibilities involving higher-order interactions of protein function within biological networks [27–30]. For example, genes encoding proteins that function as monomers with few interacting protein partners or that function in downstream parts of biological pathways are expected to experience fewer functional constraints than those encoding proteins that have numerous protein–protein interactions, function as parts of protein complexes, are highly connected in biological networks, or function in upstream parts of pathways with multiple downstream epistatic effects.
A second, fascinating aspect of this ‘duplicate gene diploidization’ phenomenon is that the origin of the retained genes, when compared with the origin of the genes that are lost, may be strikingly non-random with respect to the two donor diploid genomes. This ‘biased fractionation’, which has now been detected in both monocots and eudicots [24, 31, 32], is an utterly unexpected process that has even been reported to have occurred after allopolyploid events that trace to the start of the Tertiary [33]. In this example, differential retention of ancestral genomes involved in a 60-million-year-old polyploidization event in the ancestry of cotton remains evident in modern cotton diploid species. The evolutionary drivers of biased fractionation are incompletely understood and might be different in different taxa, but are likely to involve, among other factors, the interplay between selection and adjacency of genes to TEs that might have a repressive effect on gene expression (and thereby render these genes more ‘expendable’ than their homoeologs) [25, 33].
Transposable elements and genome size variation
“The history of the earth is recorded in the layers of its crust; the history of all organisms is inscribed in the chromosomes” (H. Kihara [34]).
On completion of the first plant genome, that of Arabidopsis thaliana, it was already clear that even the ‘simplest’ of plant genomes is a mosaic derived from multiple rounds of polyploidy events [35]. Since then, dozens of additional genomes have been sequenced, including those of most major crop plants (Table 1) [36]. Much like ancient palimpsests, sequenced genomes metaphorically reveal, at the sequence level, the reused manuscript pages from previous authors, or, as described above and in Fig. 2, the nested remnants of previous WGD events. Many of these surviving duplicated regions regulate gene activity and function, so genomic archaeology and even paleontology are essential to reveal the scriptio inferior, the history and hidden messages contained in genome sequences.
One revelation emanating from these studies concerns the genesis of the extraordinary variation in plant genome sizes [37–39]. WGD events are an obvious route to genome expansion, but many ‘diploid’ species have enormous genomes. For example, the barley genome is ~11.5 times larger than that of another cereal, rice (5.1 Gbp and 0.43 Gbp, respectively). In addition to polyploidy, genome size can saltationally increase owing to rapid proliferation of TEs [40], notwithstanding mechanisms for removal of these elements, such as unequal and illegitimate recombination [41]. Lineage-specific amplification, and potentially deletion, of TEs is common in plants, even among closely related species, such as between subspecies of domesticated rice, Oryza sativa subsp. indica and subsp. japonica [42]. Within the same genus, O. australiensis has a genome that is more than twice the size of that of O. sativa, mostly as a result of the addition of ~400 Mbp of DNA in the past few million years by three individual retrotransposable element families [43]. A clade of Australian cotton (Gossypium) diploid species have a nearly three-fold larger genome than those of the American diploid clade, owing to lineage-specific proliferation and deletion of different families of TEs [44, 45]. These examples highlight that the majority of variation in plant genome size reflects the dynamics of TE proliferation and clearance, superimposed on a history of WGD [38, 39]. Although this pattern is now known, the underlying causes of TE proliferation are far less well understood. Why are some TEs amplified in some genomes but not in others, even when they are present? For instance, the elements that resulted in doubling of the O. australiensis genome are present in all other Oryza lineages but have remained largely inactive, except for the TE Gran3 of O. granulata, which caused a ~200 Mbp retroelement burst of activity approximately 2 million years ago in this species. Gran3 is related to the Wallabi TE of O. australiensis [43, 46]. Are there certain ecological conditions that govern or trigger these TE proliferation events?
Constancy of genic content yet enormous variation in genome size
Despite their extraordinary range in size, from the tiny 60 Mbp genome of Genlisea aurea to the enormous >150 Gbp genome of Paris japonica, plant genomes have comparatively little variation in gene content [47]. This fact reflects the combined effects of TE proliferation, which dwarfs the effects of tandem or dispersed gene duplication in increasing genomic DNA content, and the process of long-term genomic fractionation, which is associated with loss of most gene duplications following WGD (Fig. 2). TEs have been implicated as important factors in gene regulation and adaptation, particularly with gene content being fairly consistent across plants and the rapid accumulation and removal of TEs [48–50].
Implications for genome assembly and interpretation
Most of the insights about plant genomes were enabled by cytogenetics, molecular genetics and, now, high-throughput sequencing technology. In fact, the majority of our food and fiber crops have at least one genome assembly in the public domain (Table 1). However, the quality of these genome assemblies varies considerably, reflecting a transition from map-based Sanger sequencing (e.g., [35, 51]) to second-generation, low-cost, short-read, whole-genome shotgun sequencing that generally yields ‘gene space’ assemblies. The complexities of genome sequencing in plants with large genomes or in those that have experienced recent polyploidy have often been quite vexing because of the high sequence similarity among recently merged or doubled genomes. This challenge has been particularly true for large allopolyploid genomes, such as that of wheat (~15 Gbp), Triticum aestivum, for which a high-quality reference genome has yet to be released. The preponderance of highly similar repetitive elements in these genomes means that these are often excluded from whole-genome assemblies. This exclusion is an important consideration not just for the sake of genome completeness per se, but also because many of these repeats are the primary targets of epigenetic/chromatin remodeling pathways that often affect the expression or structure of genes [39, 52]. Third-generation, long-read (5 to >40 kbp read length) sequencing technologies from platforms such as Pacific BioSciences [53] and Oxford Nanopore [54] are bringing us to a future of high-quality, gap-free genome sequences, which are necessary to more fully understand genome structure and function. Within the next two to three years we anticipate that most of the assemblies listed in Table 1 will be upgraded, or even replaced, using these new technologies.
Resequencing and pangenomes
Reference genome sequences are but snapshots of single genomes frozen in time. However, plants continue to evolve, adapt and diversify, so the genetic variation revealed in a single genome sequence fails to adequately represent the variation present within a species. Reference genomes have become highly useful as templates for ‘mapping’ resequencing data from additional accessions, which has led to insights into the structure and history of genetic variation within a crop plant or other species [55]. Resequencing, however, is limited by the inefficiency of mapping short reads in variable genomes, particularly in species with abundant genomic variation and TE activity. Accordingly, variants larger than single nucleotides or small insertions or deletions (indels) are often not captured in resequencing datasets, so many intergenic sequences that might be important in gene regulation are missed [56]. Moreover, the effect of TEs on presence–absence variation and on the evolution of new genes (with Pack-MULE [57] or TRIM [58] TEs being examples of the latter effect) within a genus or species might not be captured in a single genome sequence. Pantranscriptomes [59] and pangenomes have emerged as tools to effectively capture this additional layer of variation. This strategy entails sequencing multiple genomes within a species, as in soybean (Glycine soja) [60] or maize [61, 62], or even an entire genus (e.g., Oryza [63]), such that diverged and rearranged sequences can be mapped and analyzed. High-quality pangenome references capture natural variation and rare variants that are important for the identification of genes or regions associated with adaptation to environmental conditions and for crop improvement. DivSeek [64] and the Global Crop Diversity Trust [65] are examples of organizations seeking to coordinate resequencing efforts of entire germplasm collections. The International Rice Research Institute (IRRI), the Chinese Academy of Sciences (CAS) and the Beijing Genomics Institute (BGI) also recently coordinated the resequencing of ~3000 diverse rice accessions [64]. Notably, only a single high-quality reference genome exists for Asian cultivated rice, and so a large proportion of the resequencing data are unmappable. This observation demonstrates the need for high-quality pangenome reference sets, not just for rice but for the majority of crop plants.
Small RNAs, mediators of interactions in duplicated genomes
Small RNAs are important modulators and mitigating factors of the effects of genome duplications and TE-driven genomic expansion on genome architecture. Several recent reviews have highlighted the diversity of small RNAs and their ability to function in trans to direct or communicate their silencing effects across members of gene or TE families [66, 67]. These molecules are classified into three major classes that have distinct roles in gene or TE regulation: (1) microRNAs (miRNAs) that are derived from mRNA precursors produced by the processing activity of Dicer-like 1 (DCL1) and that function in post-transcriptional control of target mRNAs; (2) secondary small interfering RNAs (siRNAs) that are also processed from mRNAs by a Dicer enzyme (DCL4 or DCL5) and typically have a phased configuration (phasiRNAs), which can function against other mRNAs as trans-acting siRNAs (tasiRNAs); and (3) heterochromatic siRNAs (hc-siRNAs), which derive from precursors transcribed by plant-specific Pol IV and Pol V enzymes and are processed by yet another Dicer enzyme (DCL3) (Fig. 3). Each of these three classes of small RNA has a suppressive activity: 21-nucleotide or 22-nucleotide mature miRNAs reduce protein levels, typically by reducing the expression of their target transcripts, in diverse pathways often related to development or stress responses; 21-nucleotide or 22-nucleotide tasiRNAs or phasiRNAs have roles that are similar to those of miRNAs or have yet uncharacterized roles; and 24-nucleotide hc-siRNAs function as ‘guardians of the genome’, providing stable, multigenerational protection against invasive transposons. Extensive analysis of sequenced plant genomes has shown each of these classes of small RNAs has distinct evolutionary paths and influences on genome structure that reflect their functional roles [66, 67].

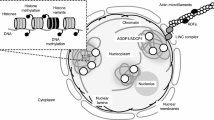
The role of small RNAs in plant genome evolution. a Plant genomes are rich sources of small RNAs, which are predominantly the products of three major pathways. (1) Heterochromatic siRNAs (hc-siRNAs) are 24-nucleotide products of the activity of the plant-specific Pol IV and Pol V pathways; hc-siRNAs derive from heterochromatic regions and target those regions for reinforcement of silencing chromatin marks. (2) MicroRNAs (miRNAs) are ~21-nucleotide or 22-nucleotide products of processed long noncoding mRNAs that function to suppress target mRNAs either by (2a) blocking translation, or (2b) directing Argonaute-mediated slicing of targets. Plant miRNAs typically function by directing slicing. (3) Some miRNAs, typically 22 nucleotides long, trigger the production of secondary siRNAs, which are products of DCL4 and are 21 nucleotide long, from their target mRNAs. This pathway requires RDR6, and some resulting siRNAs can act in trans to slice additional targets; hence their name of trans-acting siRNAs (tasiRNAs). b hc-siRNAs are typically derived from TEs, the predominant component of inactive chromatin in plant genomes. Transposons (colored parallelograms in 1–3) can be silenced (blue triangles) as a result of RNA-directed DNA methylation mediated by hc-siRNAs derived from those elements (thin blue arrows). Some transposons can escape DNA methylation and silencing (white dots), to later remobilize. Transposons can be additionally silenced by hc-siRNAs functioning in trans from related TEs (dashed lines). After allopolyploid individuals form (2), the genomic context changes for TEs from the progenitor genomes, and these elements can be silenced by hc-sRNAs derived from sequence-similar TEs residing in the added genome (dashed, curvy lines). After this period of adaptation (3), some TEs can be newly silenced (gold triangles), whereas a few might remain unsilenced and active, and might amplify into the added genome (thin black arrows). UTR untranslated region
Small RNA responses to WGD events and TE proliferation
Immediately following WGD events, duplicated genes and TEs are expected to experience a relaxation of selection that is due to functional redundancy at the time of duplication. The implications of this relaxed selection vary among genes and TEs, and among the types of small RNAs that have regulatory roles. Mature miRNAs are processed from transcripts of precursor genes (MIRNAs) that are influenced by the same events occurring at the whole-genome scale that shape gene and genome evolution, including polyploidy and other mechanisms underlying gene duplication [68]. Like for protein-coding genes, the emergence of lineage-specific miRNAs is fairly common, although a core set of well-conserved miRNAs also exists [69]. In soybean, duplicated (i.e., multi-copy) MIRNAs were retained at a higher rate than single-copy MIRNAs, with this higher rate resulting from functional constraints and genomic duplication events local to the MIRNAs [68]. The evolutionary basis for this finding is unclear, but this observation could reflect the functional importance of miRNA dosage when target genes are duplicated. As a consequence of genomic duplications, some well-conserved miRNAs are found at increased copy numbers in species that underwent recent polyploidy. For example, there are 22 copies of the miR165/166 family found in the recently duplicated soybean genome, whereas nine copies are observed in the Arabidopsis genome. This high dosage is not yet known to have functional relevance, but the evolutionary decay of duplicated MIRNA genes might be slowed because the most functionally relevant portion of the mRNA precursor of a miRNA is presumably the hairpin structure, which is just a few hundred nucleotides in length. The importance of precursor components 5’ and 3’ of this stem-loop are, however, still poorly characterized in plants. Strong selection for sequence conservation in miRNAs in regions other than their promoter elements is likely to be largely limited to nucleotides within the hairpin that are needed for processing, plus those in the mature miRNA that are required for successful targeting. The net effect of this limited selection for conservation is that miRNAs might have fewer positions at which mutations would be functionally equivalent to nonsense or missense mutations than protein-coding genes; hence, miRNAs might have a longer half-life than protein-coding genes following WGD events.
Like miRNAs, phasiRNAs are generated from mRNAs, and thus their precursors (PHAS loci) are duplicated or lost through the many processes that also affect deletion and replication of other genomic regions. As far as we know, the important functionally constrained nucleotides in phasiRNA mRNA precursors include promoter elements, the miRNA target site and the typically few phasiRNAs that have important targets. To date, the targets of phasiRNA derived from long, non-coding RNAs are largely unknown, although in a few cases they have been well-described, such as the 21-nucleotide “tasiARF” in TAS3 [70]; in the case of TAS3, it is just one or two of many 21-nucleotide phasiRNAs from the locus that is believed to be functional. Thus, as MIRNA genes, PHAS genes might be slower to pseudogenize than protein-coding genes, and, therefore, their retention time after polyploidization might be longer than that of protein-coding duplicates. The appearance of novel PHAS loci has also been recorded. In the Medicago and soybean genomes, for example, non-conserved, flower-enriched or anther-enriched PHAS loci exist. Some of these loci seem to target transposons, perhaps as a mechanism to suppress TE activity during reproduction [71, 72]. However, most changes in PHAS loci are likely to be spontaneous events, independent of polyploidy events.
In comparison to miRNAs and phasiRNAs, hc-siRNAs, as derivatives of TEs, are subject to numerous stochastic and selective evolutionary forces that shape genomes, and are likely to be critical in the modulation or mitigation of the effects of WGD events. hc-siRNAs function as primary defenses against ‘invasive’ TEs acting as a sort of “vaccine” against deleterious elements. Even so, they are derived directly from TEs through the specialized pathway of RNA-directed DNA methylation (RdDM), produced via TE precursors transcribed as RNAs long enough to generate a hc-siRNA, but too short to encode a functional TE [73]. Their derivation from TEs allows them to provide direct genomic defenses against TEs, but their transcription by Pol IV and Pol V and their retention in the nucleus prevents their translation into invasive TEs, thereby avoiding any potential adverse effects. Evolutionary analysis indicates that the RdDM pathway is fairly ancient in plants, with components dating to before the divergence of mosses, in which 23-nucleotide siRNAs direct RdDM [74]; later, in gymnosperms, hc-siRNAs achieved their ‘modern’ size of 24 nucleotides [75], with substantial diversification in the components of the pathway occurring throughout the evolution of gymnosperms and later in angiosperms [76]. Perhaps this elaboration of the machinery for silencing facilitated polyploidization events by providing an effective mechanism for suppressing TE activity, which otherwise might have been more rampant, and hence disruptive, following genomic mergers.
hc-siRNAs are believed to function in trans to direct silencing at related elements via sequence homology [77], although this hypothesis has not been thoroughly tested and we do not have a good idea of the degree of homology that is required for such trans activity. Nevertheless, we can speculate that novel and important interactions occur between the two suites of distinct hc-siRNAs and TEs that become suddenly merged within the same genome during allopolyploidization events (Fig. 3b). One possible outcome of this form of biological reunion is that hc-siRNAs function to suppress TEs both in cis and in trans, and hence that TEs are no more likely to mobilize than in the originally separate genomes. Alternatively, interspecific hybridization and WGD events might be accompanied by a burst of TE proliferation, perhaps as a direct consequence of a destabilized or altered population of hc-siRNAs and their influence on DNA methylation or chromatin states (Fig. 3b) [78–80]. Perhaps TEs escape silencing by flying under the genomic surveillance radar [81, 82], and thereby proliferate and invade new genomic space, which would have multiple effects on genomic structure and gene evolution [39]. These effects would be particularly important in reproductive tissues, in which TE silencing is less effective; this hypothesis is supported by growing evidence [83]. The TE complement of plant genomes usually consists of various TE families that massively amplified through ancient bursts of proliferation (as in O. australiensis [43]), and many of these genomic explosions are likely to represent a ‘failure of the vaccine’ — an escape from detection and suppression of TEs. The proximal trigger of bursts of TE proliferation is not understood, but could involve mechanisms that disable defenses via suppression of silencing or ephemeral developmental periods during which RdDM is less active in germline cells, or perhaps during the formation of zygotes. For example, asymmetric contributions of the maternal and paternal gametes, including siRNAs or modifiers of silencing processes [84–87], could differentially influence the TEs in the resulting zygote, potentially allowing some TEs to proliferate.
Conclusions and future perspectives
The genomes of the approximately 300,000 species of flowering plants exhibit extraordinary variation in size and their complement of genomic elements. This variation is the outcome of temporally dynamic and phylogenetically variable, even idiosyncratic, interplay among processes set in motion by episodes of polyploidy, TE proliferation and regulatory events mediated by small RNAs. These events are all molded by even more complex biotic and abiotic interactions between the organisms and their environments. What are the broad implications of this new and improved view of the origin of the modern angiosperm genome architecture? This perspective might be fundamental to much of plant biology, as many different processes, be they metabolic, physiological or ecological, are specified by the size and functional diversification of contemporary multigene family structures, gene expression patterns and the systems biology context of various genomic elements. These processes all operate within a genomic milieu of TEs and small RNAs that partly originate from the survivors of past ‘wash–rinse–repeat’ cycles of polyploidization followed by non-random and incomplete diploidization. These endpoints, having been shaped by diverse selective and, presumably, neutral forces, have generated the genic and genomic architecture that underlies all plant phenotypes, be they physiological, ecological or morphological [8, 27, 88, 89]. An exciting area for future research is the exploration of the connections between the short-term and long-term responses to WGD and the interconnections of these responses with TE proliferation and small RNA evolution, both in terms of molecular mechanisms and implications for natural selection. This challenge will necessitate a multidisciplinary, integrative approach and biological investigation of multiple model allopolyploid systems and natural ecological settings. The use of experimentally tractable systems, including synthetic polyploids and their natural relatives, to explore the interconnections between the phenomena we have highlighted and the evolutionary ecology of specific lineages is an exciting prospect. Now that large-scale ‘omics’ datasets of genomes, transcriptomes, epigenomes, etc. are increasingly becoming available within or across species, trans-disciplinary teams will be more able to understand plant responses to varying environments and long-term adaptation. These studies will contribute to understanding basic biological processes and are a prelude to engineering these process for the betterment of humankind.
This fundamental genomic understanding is likely to be valuable for crop improvement. Oliver et al. [50] tabulated 65 examples of TE insertions in regulatory or coding sequences that affect a wide range of phenotypic traits, such as skin color in grape [90] and anthocyanin accumulation in blood orange [91]. The most famous example involving a TE insertion and crop productivity is perhaps the insertion of the Hopskotch TE in the far-upstream regulatory region of tb1 in maize, which enhanced tb1 expression and promoted the typical architecture of the maize plant relative to that of its progenitor, teosinte [92]. Gene and genome doubling have also been shown to be important in agriculture, as summarized by Olsen and Wendel [93]. Examples of this importance are seen in major grains such as wheat and rice, as well as in other crop plants such as tomato and sunflower. In addition to cases in which known TE insertions or duplicated genes have been shown to affect crop plant traits, the more general importance of these events has been appreciated, even when the specific lesions are not understood. For example, in the most important species of cotton (G. hirsutum), which is allopolyploid, the two co-resident genomes have intermingled and contribute unequally to fiber quality and yield [94–98]. In maize, large genotype–phenotype association studies have shown that modern paralogs descended from the most recent WGD are ~50 % more likely to be associated with functional and phenotypic variation than singleton genes, which highlights the importance of genome-wide neofunctionalization in generating new variation [99]. As is the case for TEs and WGD events, diversification, evolution and selection of small RNAs are potentially important processes in crop plants, including rice [49, 64] and cotton [99]. In cotton, only one of two homoeologs of an mRNA that encodes a MYB transcription factor underwent preferential degradation during cotton fiber development, which makes this case particularly illustrative of a direct link between a recent WGD event and miRNA behavior. Further work is needed to understand the interplay between TE proliferation, insertion/retention bias in polyploid plants and small RNA biology, and how to harness this biology to enhance traits of agronomic importance.
Genome sequences also provide many insights into the paleogenomic record of plant life, but, as with paleontology, not all features fossilize equally well and the record is incomplete.
The majority of plant genome sequences are from crop plants. Crop genome sequences anchor large commodity-based communities around a single resource that can be leveraged in numerous directions for crop improvement and basic discoveries. Reference genomes can now be used by germplasm banks worldwide. These banks contain domesticated crop relatives that are adapted to grow under varied environmental conditions and that harbor untapped reservoirs of traits that can be used for crop improvement. How can one exploit the knowledge of genomic evolutionary processes to tap into these resources and thereby create new traits that will empower the next green revolution? An initial step would be to genotype gene bank collections [100]. A landmark example of this approach was the recent resequencing of 3000 cultivated rice accessions representative of two large rice gene banks, from which more than 18.9 million new single nucleotide polymorphisms were discovered [64, 101]. Another example is the Seeds of Discovery project at International Maize and Wheat Improvement Center (CYMMIT) in Mexico, where 27,500 and 30,000 maize and wheat accessions, respectively, have been genotyped and are being phenotyped [102]. As discussed above, pangenomic resources will be needed to more efficiently capture the variation from these resequencing and genotyping projects. Such data can then be integrated into genomic selection breeding programs to drive the generation of tomorrow’s crops.
The importance of this agenda is difficult to overstate. The United Nations projects that the world population will exceed 9.7 billion by 2050, with the majority of growth coming from Africa and Asia [103]. One of the biggest challenges we face is how to feed an additional ~2.4 billion people in less than 35 years in a sustainable and environmentally responsible way. By unraveling the history of plant genomes and their genomic ecosystems we can begin to understand how natural selection shaped genomes in time and space to adapt to different environmental conditions. Genomic information will allow us to develop high yielding and sustainable genotypic combinations that are more efficient in the use of nutrients and water, resistant to insects and pathogens, and more nutritious.
Abbreviations
- EST:
-
Expressed sequence tag
- hc-siRNA:
-
Heterochromatic siRNA
- phasiRNA:
-
Phased, secondary siRNA
- RdDM:
-
RNA-directed DNA methylation
- siRNA:
-
Small interfering RNA
- tasiRNA:
-
Trans-acting siRNA
- TE:
-
Transposable element
- WGD:
-
Whole-genome doubling
References
Grant V. Plant speciation. New York: Columbia; 1981.
Stebbins GL. Types of polyploids: their classification and significance. Adv Genet. 1947;1:403–29.
Stebbins GL. Chromosomal evolution in higher plants. London: Edward Arnold; 1971.
Masterson J. Stomatal size in fossil plants: evidence for polyploidy in majority of angiosperms. Science. 1994;264:421–4.
Ramsey J, Schemske DW. Pathways, mechanisms, and rates of polyploid formation in flowering plants. Annu Rev Ecol Syst. 1998;29:467–501.
Wendel JF, Doyle JJ. Polyploidy and evolution in plants. In: Henry RJ, editor. Plant diversity and evolution. Wallingford, UK: CABI Publishing; 2005. p. 97–117.
Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, et al. Ancestral polyploidy in seed plants and angiosperms. Nature. 2011;473:97–100.
Soltis DE, Albert VA, Leebens-Mack J, Bell CD, Paterson AH, Zheng C, et al. Polyploidy and angiosperm diversification. Am J Bot. 2009;96:336–48.
Paterson AH, Wang X, Li J, Tang H. Ancient and recent polyploidy in monocots. In Soltis P, Soltis DE, editors. Polyploidy and genome evolution. Berlin: Springer; 2012. p. 93–108.
Vanneste K, Van de Peer Y, Maere S. Inference of genome duplications from age distributions revisited. Mol Biol Evol. 2013;30:177–90.
Soltis PS, Soltis DE. Polyploidy and genome evolution. Berlin: Springer; 2012.
Doyle JJ, Flagel LE, Paterson AH, Rapp RA, Soltis DE, Soltis PS, et al. Evolutionary genetics of genome merger and doubling in plants. Annu Rev Genet. 2008;42:443–61.
Wendel JF. Genome evolution in polyploids. Plant Mol Biol. 2000;42:225–49.
Chen ZJ. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu Rev Plant Biol. 2007;58:377–406.
Chen ZJ, Ni Z. Mechanisms of genomic rearrangements and gene expression changes in plant polyploids. Bioessays. 2006;28:240–52.
Grover C, Gallagher J, Szadkowski E, Yoo M, Flagel L, Wendel J. Homoeolog expression bias and expression level dominance in allopolyploids. New Phytol. 2012;196:966–71.
Hu G, Houston NL, Pathak D, Schmidt L, Thelen JJ, Wendel JF. Genomically biased accumulation of seed storage proteins in allopolyploid cotton. Genetics. 2011;189:1103–15.
Jackson S, Chen ZJ. Genomic and expression plasticity of polyploidy. Curr Opin Plant Biol. 2010;13:153–9.
Koh J, Chen S, Zhu N, Yu F, Soltis PS, Soltis DE. Comparative proteomics of the recently and recurrently formed natural allopolyploid Tragopogon mirus (Asteraceae) and its parents. New Phytol. 2012;196:292–305.
Liu S-L, Adams KL. Dramatic change in function and expression pattern of a gene duplicated by polyploidy created a paternal effect gene in the Brassicaceae. Mol Biol Evol. 2010;27:2817–28.
Liu Z, Xin M, Qin J, Peng H, Ni Z, Yao Y, et al. Temporal transcriptome profiling reveals expression partitioning of homeologous genes contributing to heat and drought acclimation in wheat (Triticum aestivum L.). BMC Plant Biol. 2015;15:152.
Hughes TE, Langdale JA, Kelly S. The impact of widespread regulatory neofunctionalization on homeolog gene evolution following whole-genome duplication in maize. Genome Res. 2014;24:1348–55.
Renny-Byfield S, Gallagher JP, Grover CE, Szadkowski E, Page JT, Udall JA, et al. Ancient gene duplicates in Gossypium (Cotton) exhibit near-complete expression divergence. Genome Biol Evol. 2014;6:559–71.
Freeling M, Woodhouse MR, Subramaniam S, Turco G, Lisch D, Schnable JC. Fractionation mutagenesis and similar consequences of mechanisms removing dispensable or less-expressed DNA in plants. Curr Opin Plant Biol. 2012;15:131–9.
Woodhouse MR, Cheng F, Pires JC, Lisch D, Freeling M, Wang X. Origin, inheritance, and gene regulatory consequences of genome dominance in polyploids. Proc Natl Acad Sci U S A. 2014;111:5283–8.
Leitch A, Leitch I. Genomic plasticity and the diversity of polyploid plants. Science. 2008;320:481–3.
De Smet R, Adams KL, Vandepoele K, Van Montagu MC, Maere S, Van de Peer Y. Convergent gene loss following gene and genome duplications creates single-copy families in flowering plants. Proc Natl Acad Sci U S A. 2013;110:2898–903.
Conant GC, Wolfe KH. Turning a hobby into a job: How duplicated genes find new functions. Nat Rev Genet. 2008;9:938–50.
Birchler JA, Veitia RA. Gene balance hypothesis: connecting issues of dosage sensitivity across biological disciplines. Proc Natl Acad Sci U S A. 2012;109:14746–53.
Conant GC, Birchler JA, Pires JC. Dosage, duplication, and diploidization: clarifying the interplay of multiple models for duplicate gene evolution over time. Curr Opin Plant Biol. 2014;19:91–8.
Schnable JC, Springer NM, Freeling M. Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc Natl Acad Sci U S A. 2011;108:4069–74.
Cheng F, Wu J, Fang L, Sun S, Liu B, Lin K, et al. Biased gene fractionation and dominant gene expression among the subgenomes of Brassica rapa. PloS One. 2012;7:e36442.
Renny-Byfield S, Gong L, Gallagher JP, Wendel JF. Persistence of sub-genomes in paleopolyploid cotton after 60 million years of evolution. Mol Biol Evol. 2015;32:1063–71.
Crow JF. Hitoshi Kihara, Japan's pioneer geneticist. Genetics. 1994;137:891–4.
Vision TJ, Brown DG, Tanksley SD. The origins of genomic duplications in Arabidopsis. Science. 2000;290:2114–7.
Michael TP, VanBuren R. Progress, challenges and the future of crop genomes. Curr Opin Plant Biol. 2015;24:71–81.
Michael TP. Plant genome size variation: bloating and purging DNA. Brief Funct Genomics. 2014;13:308–17.
Leitch IJ, Leitch AR. Genome size diversity and evolution in land plants. Berlin: Springer; 2013.
Bennetzen JL, Wang H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu Rev Plant Biol. 2014;65:505–30.
Verde I, Abbott AG, Scalabrin S, Jung S, Shu S, Marroni F, et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet. 2013;45:487–94.
Ma J, Devos KM, Bennetzen JL. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 2004;14:860–9.
Ma J, Bennetzen JL. Rapid recent growth and divergence of rice nuclear genomes. Proc Natl Acad Sci U S A. 2004;101:12404–10.
Piegu B, Guyot R, Picault N, Roulin A, Sanyal A, Kim H, et al. Doubling genome size without polyploidization: dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 2006;16:1262–9.
Hawkins JS, Kim H, Nason JD, Wing RA, Wendel JF. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006;16:1252–61.
Hawkins JS, Proulx SR, Rapp RA, Wendel JF. Rapid DNA loss as a counterbalance to genome expansion through retrotransposon proliferation in plants. Proc Natl Acad Sci U S A. 2009;106:17811–6.
Ammiraju JS, Zuccolo A, Yu Y, Song X, Piegu B, Chevalier F, et al. Evolutionary dynamics of an ancient retrotransposon family provides insights into evolution of genome size in the genus Oryza. Plant J. 2007;52:342–51.
Albert VA, Barbazuk WB, Der JP, Leebens-Mack J, Ma H, Palmer JD, et al. The Amborella genome and the evolution of flowering plants. Science. 2013;342:1241089.
Stapley J, Santure AW, Dennis SR. Transposable elements as agents of rapid adaptation may explain the genetic paradox of invasive species. Mol Ecol. 2015;24:2241–52.
Casacuberta E, González J. The impact of transposable elements in environmental adaptation. Mol Ecol. 2013;22:1503–17.
Oliver KR, McComb JA, Greene WK. Transposable elements: powerful contributors to angiosperm evolution and diversity. Genome Biol Evol. 2013;5:1886–901.
International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature. 2005;436:793–800.
Hollister JD, Smith LM, Guo Y-L, Ott F, Weigel D, Gaut BS. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc Natl Acad Sci U S A. 2011;108:2322–7.
Levene MJ, Korlach J, Turner SW, Foquet M, Craighead HG, Webb WW. Zero-mode waveguides for single-molecule analysis at high concentrations. Science. 2003;299:682–6.
Howorka S, Cheley S, Bayley H. Sequence-specific detection of individual DNA strands using engineered nanopores. Nat Biotechnol. 2001;19:636–9.
Lai J, Li R, Xu X, Jin W, Xu M, Zhao H, et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nat Genet. 2010;42:1027–30.
Marroni F, Pinosio S, Morgante M. Structural variation and genome complexity: is dispensable really dispensable? Curr Opin Plant Biol. 2014;18:31–6.
Jiang N, Bao Z, Zhang X, Eddy SR, Wessler SR. Pack-MULE transposable elements mediate gene evolution in plants. Nature. 2004;431:569–73.
Gao D, Li Y, Kim KD, Abernathy B, Jackson SA. Landscape and evolutionary dynamics of terminal repeat retrotransposons in miniature in plant genomes. Genome Biol. 2016;17:1–17.
Hirsch CN, Foerster JM, Johnson JM, Sekhon RS, Muttoni G, Vaillancourt B, et al. Insights into the maize pan-genome and pan-transcriptome. Plant Cell. 2014;26:121–35.
Li YH, Zhou G, Ma J, Jiang W, Jin LG, Zhang Z, et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat Biotechnol. 2014;32:1045–52.
Hansey CN, Vaillancourt B, Sekhon RS, De Leon N, Kaeppler SM, Buell CR. Maize (Zea mays L.) genome diversity as revealed by RNA-sequencing. PloS One. 2012;7:e33071.
Lu F, Romay MC, Glaubitz JC, Bradbury PJ, Elshire RJ, Wang T, et al. High-resolution genetic mapping of maize pan-genome sequence anchors. Nat Commun. 2015;6:6914.
Jacquemin J, Bhatia D, Singh K, Wing RA. The international Oryza map alignment project: development of a genus-wide comparative genomics platform to help solve the 9 billion-people question. Curr Opin Plant Biol. 2013;16:147–56.
Li J-Y, Wang J, Zeigler RS. The 3000 rice genomes project: new opportunities and challenges for future rice research. GigaScience. 2014;3:1–3.
The Crop Trust. https://www.croptrust.org. 2016. Accessed 17 Feb 2016.
Axtell MJ. Classification and comparison of small RNAs from plants. Annu Rev Plant Biol. 2013;64:137–59.
Fei Q, Xia R, Meyers BC. Phased, secondary, small interfering RNAs in posttranscriptional regulatory networks. Plant Cell. 2013;25:2400–15.
Zhao M, Meyers BC, Cai C, Xu W, Ma J. Evolutionary patterns and coevolutionary consequences of miRNA genes and microRNA targets triggered by multiple mechanisms of genomic duplications in soybean. Plant Cell. 2015;27:546–62.
Montes RAC, De Paoli E, Accerbi M, Rymarquis LA, Mahalingam G, Marsch-Martínez N, et al. Sample sequencing of vascular plants demonstrates widespread conservation and divergence of microRNAs. Nat Commun. 2014;5:3722.
Axtell MJ, Jan C, Rajagopalan R, Bartel DP. A two-hit trigger for siRNA biogenesis in plants. Cell. 2006;127:565–77.
Zhai J, Jeong D-H, De Paoli E, Park S, Rosen BD, Li Y, et al. MicroRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Genes Dev. 2011;25:2540–53.
Arikit S, Xia R, Kakrana A, Huang K, Zhai J, Yan Z, et al. An atlas of soybean small RNAs identifies phased siRNAs from hundreds of coding genes. Plant Cell. 2014;26:4584–601.
Matzke MA, Mosher RA. RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat Rev Genet. 2014;15:394–408.
Coruh C, Cho SH, Shahid S, Liu Q, Wierzbicki A, Axtell MJ. Comprehensive annotation of Physcomitrella patens small RNA loci reveals that the heterochromatic short interfering RNA pathway is largely conserved in land plants. Plant Cell. 2015;27:2148–62.
Chávez Montes RA, Rosas-Cárdenas FF, De Paoli E, Accerbi M, Rymarquis LA, Mahalingam G, et al. Sample sequencing of vascular plants demonstrates widespread conservation and divergence of microRNAs. Nat Commun. 2014;5:3722.
Huang Y, Kendall T, Forsythe ES, Dorantes-Acosta A, Li S, Caballero-Pérez J, et al. Ancient origin and recent innovations of RNA polymerase IV and V. Mol Biol Evol. 2015;32:1788–99.
Schrader L, Kim JW, Ence D, Zimin A, Klein A, Wyschetzki K, et al. Transposable element islands facilitate adaptation to novel environments in an invasive species. Nat Commun. 2014;5:5495.
Kawakami T, Strakosh SC, Zhen Y, Ungerer MC. Different scales of Ty1/copia-like retrotransposon proliferation in the genomes of three diploid hybrid sunflower species. Heredity. 2010;104:341–50.
Kenan-Eichler M, Leshkowitz D, Tal L, Noor E, Melamed-Bessudo C, Feldman M, et al. Wheat hybridization and polyploidization results in deregulation of small RNAs. Genetics. 2011;188:263–72.
Madlung A, Masuelli RW, Watson B, Reynolds SH, Davison J, Comai L. Remodeling of DNA methylation and phenotypic and transcriptional changes in synthetic Arabidopsis allotetraploids. Plant Physiol. 2002;129:733–46.
Mirouze M, Reinders J, Bucher E, Nishimura T, Schneeberger K, Ossowski S, et al. Selective epigenetic control of retrotransposition in Arabidopsis. Nature. 2009;461:427–30.
Ito H, Gaubert H, Bucher E, Mirouze M, Vaillant I, Paszkowski J. An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature. 2011;472:115–9.
Kawashima T, Berger F. Epigenetic reprogramming in plant sexual reproduction. Nat Rev Genet. 2014;15:613–24.
Mosher RA, Melnyk CW, Kelly KA, Dunn RM, Studholme DJ, Baulcombe DC. Uniparental expression of PolIV-dependent siRNAs in developing endosperm of Arabidopsis. Nature. 2009;460:283–6.
Rodrigues JA, Ruan R, Nishimura T, Sharma MK, Sharma R, Ronald PC, et al. Imprinted expression of genes and small RNA is associated with localized hypomethylation of the maternal genome in rice endosperm. Proc Natl Acad Sci U S A. 2013;110:7934–9.
García-Aguilar M, Gillmor CS. Zygotic genome activation and imprinting: parent-of-origin gene regulation in plant embryogenesis. Curr Opin Plant Biol. 2015;27:29–35.
Boavida LC, Hernandez-Coronado M, Becker JD. Setting the stage for the next generation: epigenetic reprogramming during sexual plant reproduction. In: Pontes O, Jin H, editors. Nuclear functions in plant transcription, signaling and development. New York: Springer; 2015. p. 93–118.
Rensing SA. Gene duplication as a driver of plant morphogenetic evolution. Curr Opin Plant Biol. 2014;17:43–8.
Ebel ER, DaCosta JM, Sorenson MD, Hill RI, Briscoe AD, Willmott KR, et al. Rapid diversification associated with ecological specialization in neotropical Adelpha butterflies. Mol Ecol. 2015;24:2392–405.
Kobayashi S, Goto-Yamamoto N, Hirochika H. Retrotransposon-induced mutations in grape skin color. Science. 2004;304:982.
Butelli E, Licciardello C, Zhang Y, Liu J, Mackay S, Bailey P, et al. Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell. 2012;24:1242–55.
Studer A, Zhao Q, Ross-Ibarra J, Doebley J. Identification of a functional transposon insertion in the maize domestication gene tb1. Nat Genet. 2011;43:1160–3.
Olsen KM, Wendel JF. A bountiful harvest: genomic insights into crop domestication phenotypes. Annu Rev Plant Biol. 2013;64:47–70.
Jiang C-X, Wright RJ, El-Zik KM, Paterson AH. Polyploid formation created unique avenues for response to selection in Gossypium (cotton). Proc Natl Acad Sci U S A. 1998;95:4419–24.
Paterson AH, Wendel JF, Gundlach H, Guo H, Jenkins J, Jin D, et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature. 2012;492:423–7.
Li F, Fan G, Lu C, Xiao G, Zou C, Kohel RJ, et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat Biotechnol. 2015;33:524–30.
Zhao X-P, Si Y, Hanson RE, Crane CF, Price HJ, Stelly DM, et al. Dispersed repetitive DNA has spread to new genomes since polyploid formation in cotton. Genome Res. 1998;8:479–92.
Wendel JF, Schnabel A, Seelanan T. Bidirectional interlocus concerted evolution following allopolyploid speciation in cotton (Gossypium). Proc Natl Acad Sci U S A. 1995;92:280–4.
Wallace JG, Bradbury PJ, Zhang N, Gibon Y, Stitt M, Buckler ES. Association mapping across numerous traits reveals patterns of functional variation in maize. PLoS Genet. 2014;10:e1004845.
McCouch S, Baute GJ, Bradeen J, Bramel P, Bretting PK, Buckler E, et al. Agriculture: feeding the future. Nature. 2013;499:23–4.
3K RGP. The 3000 rice genomes project. GigaScience. 2014;3:7.
Seeds of Discovery. http://seedsofdiscovery.org/en/. 2012. Accessed 17 Feb 2016.
United Nations, Department of Economic and Social Affairs, Population Division. 2015. World Population Prospects: The 2015 Revision, Key Findings and Advance Tables. ESA/P/WP.241. http://esa.un.org/unpd/wpp/publications/. Accessed 15 Apr 2016.
Tilman D, Balzer C, Hill J, Befort BL. Global food demand and the sustainable intensification of agriculture. Proc Natl Acad Sci U S A. 2011;108:20260–4.
Ming R, VanBuren R, Liu Y, Yang M, Han Y, Li LT, et al. Genome of the long-living sacred lotus (Nelumbo nucifera Gaertn.). Genome Biol. 2013;14:R41.
Dohm JC, Minoche AE, Holtgrawe D, Capella-Gutierrez S, Zakrzewski F, Tafer H, et al. The genome of the recently domesticated crop plant sugar beet (Beta vulgaris). Nature. 2014;505:546–9.
Bombarely A, Rosli HG, Vrebalov J, Moffett P, Mueller LA, Martin GB. A draft genome sequence of Nicotiana benthamiana to enhance molecular plant-microbe biology research. Mol Plant Microbe Interact. 2012;25:1523–30.
Potato Genome Sequencing Consortium, Xu X, Pan S, Cheng S, Zhang B, Mu D, et al. Genome sequence and analysis of the tuber crop potato. Nature. 2011;475:189–95.
Hirakawa H, Shirasawa K, Miyatake K, Nunome T, Negoro S, Ohyama A, et al. Draft genome sequence of eggplant (Solanum melongena L.): the representative solanum species indigenous to the old world. DNA Res. 2014;21:649–60.
Kim S, Park M, Yeom SI, Kim YM, Lee JM, Lee HA, et al. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat Genet. 2014;46:270–8.
Polashock J, Zelzion E, Fajardo D, Zalapa J, Georgi L, Bhattacharya D, et al. The American cranberry: first insights into the whole genome of a species adapted to bog habitat. BMC Plant Biol. 2014;14:165.
Huang S, Ding J, Deng D, Tang W, Sun H, Liu D, et al. Draft genome of the kiwifruit Actinidia chinensis. Nat Commun. 2013;4:2640.
Denoeud F, Carretero-Paulet L, Dereeper A, Droc G, Guyot R, Pietrella M, et al. The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science. 2014;345:1181–4.
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–7.
Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science. 2006;313:1596–604.
Wang Z, Hobson N, Galindo L, Zhu S, Shi D, McDill J, et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 2012;72:461–73.
Chan AP, Crabtree J, Zhao Q, Lorenzi H, Orvis J, Puiu D, et al. Draft genome sequence of the oilseed species Ricinus communis. Nat Biotechnol. 2010;28:951–6.
Prochnik S, Marri PR, Desany B, Rabinowicz PD, Kodira C, Mohiuddin M, et al. The cassava genome: current progress, future directions. Trop Plant Biol. 2012;5:88–94.
Rahman AY, Usharraj AO, Misra BB, Thottathil GP, Jayasekaran K, Feng Y, et al. Draft genome sequence of the rubber tree Hevea brasiliensis. BMC Genomics. 2013;14:75.
Huang S, Li R, Zhang Z, Li L, Gu X, Fan W, et al. The genome of the cucumber, Cucumis sativus L. Nat Genet. 2009;41:1275–81.
Garcia-Mas J, Benjak A, Sanseverino W, Bourgeois M, Mir G, Gonzalez VM, et al. The genome of melon (Cucumis melo L.). Proc Natl Acad Sci U S A. 2012;109:11872–7.
Guo S, Zhang J, Sun H, Salse J, Lucas WJ, Zhang H, et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat Genet. 2013;45:51–8.
Shulaev V, Sargent DJ, Crowhurst RN, Mockler TC, Folkerts O, Delcher AL, et al. The genome of woodland strawberry (Fragaria vesca). Nat Genet. 2011;43:109–16.
Velasco R, Zharkikh A, Affourtit J, Dhingra A, Cestaro A, Kalyanaraman A, et al. The genome of the domesticated apple (Malus x domestica Borkh.). Nat Genet. 2010;42:833–9.
Wu J, Wang Z, Shi Z, Zhang S, Ming R, Zhu S, et al. The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res. 2013;23:396–408.
van Bakel H, Stout JM, Cote AG, Tallon CM, Sharpe AG, Hughes TR, et al. The draft genome and transcriptome of Cannabis sativa. Genome Biol. 2011;12:R102.
Natsume S, Takagi H, Shiraishi A, Murata J, Toyonaga H, Patzak J, et al. The draft genome of hop (Humulus lupulus), an essence for brewing. Plant Cell Physiol. 2015;56:428–41.
Liu S, Liu Y, Yang X, Tong C, Edwards D, Parkin IA, et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat Commun. 2014;5:3930.
International Peach Genome Initiative, Verde I, Abbott AG, Scalabrin S, Jung S, Shu S, et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet. 2013;45:487–94.
Young ND, Debelle F, Oldroyd GE, Geurts R, Cannon SB, Udvardi MK, et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature. 2011;480:520–4.
Varshney RK, Song C, Saxena RK, Azam S, Yu S, Sharpe AG, et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol. 2013;31:240–6.
Sato S, Nakamura Y, Kaneko T, Asamizu E, Kato T, Nakao M, et al. Genome structure of the legume, Lotus japonicus. DNA Res. 2008;15:227–39.
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, et al. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463:178–83.
Varshney RK, Chen W, Li Y, Bharti AK, Saxena RK, Schlueter JA, et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat Biotechnol. 2012;30:83–9.
Schmutz J, McClean PE, Mamidi S, Wu GA, Cannon SB, Grimwood J, et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat Genet. 2014;46:707–13.
Kang YJ, Kim SK, Kim MY, Lestari P, Kim KH, Ha BK, et al. Genome sequence of mungbean and insights into evolution within Vigna species. Nat Commun. 2014;5:5443.
Yang H, Tao Y, Zheng Z, Zhang Q, Zhou G, Sweetingham MW, et al. Draft genome sequence, and a sequence-defined genetic linkage map of the legume crop species Lupinus angustifolius L. PloS One. 2013;8:e64799.
Zhang T, Hu Y, Jiang W, Fang L, Guan X, Chen J, et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat Biotechnol. 2015;33:531–7.
Argout X, Salse J, Aury JM, Guiltinan MJ, Droc G, Gouzy J, et al. The genome of Theobroma cacao. Nat Genet. 2011;43:101–8.
Wu GA, Prochnik S, Jenkins J, Salse J, Hellsten U, Murat F, et al. Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nat Biotechnol. 2014;32:656–62.
Ming R, Hou S, Feng Y, Yu Q, Dionne-Laporte A, Saw JH, et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature. 2008;452:991–6.
Wang X, Wang H, Wang J, Sun R, Wu J, Liu S, et al. The genome of the mesopolyploid crop species Brassica rapa. Nat Genet. 2011;43:1035–9.
Chalhoub B, Denoeud F, Liu S, Parkin IA, Tang H, Wang X, et al. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science. 2014;345:950–3.
Moghe GD, Hufnagel DE, Tang H, Xiao Y, Dworkin I, Town CD, et al. Consequences of whole-genome triplication as revealed by comparative genomic qnalyses of the wild radish Raphanus raphanistrum and three other Brassicaceae species. Plant Cell. 2014;26:1925–37.
Al-Dous EK, George B, Al-Mahmoud ME, Al-Jaber MY, Wang H, Salameh YM, et al. De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nat Biotechnol. 2011;29:521–7.
Singh R, Ong-Abdullah M, Low ET, Manaf MA, Rosli R, Nookiah R, et al. Oil palm genome sequence reveals divergence of interfertile species in Old and New worlds. Nature. 2013;500:335–9.
D'Hont A, Denoeud F, Aury JM, Baurens FC, Carreel F, Garsmeur O, et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature. 2012;488:213–7.
Wang M, Yu Y, Haberer G, Marri PR, Fan C, Goicoechea JL, et al. The genome sequence of African rice (Oryza glaberrima) and evidence for independent domestication. Nat Genet. 2014;46:982–8.
International Barley Genome Sequencing Consortium, Mayer KF, Waugh R, Brown JW, Schulman A, Langridge P, et al. A physical, genetic and functional sequence assembly of the barley genome. Nature. 2012;491:711–6.
Mayer KF, Rogers J, Doležel J, Pozniak C, Eversole K, Feuillet C, et al. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 2014;345:1251788.
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–5.
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457:551–6.
Zhang G, Liu X, Quan Z, Cheng S, Xu X, Pan S, et al. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat Biotechnol. 2012;30:549–54.
Cannarozzi G, Plaza-Wuthrich S, Esfeld K, Larti S, Wilson YS, Girma D, et al. Genome and transcriptome sequencing identifies breeding targets in the orphan crop tef (Eragrostis tef). BMC Genomics. 2014;15:581.
Renny-Byfield S, Wendel JF. Doubling down on genomes: polyploidy and crop plants. Am J Bot. 2014;101:1711–25.
Acknowledgements
Research on plant genomes in all four authors’ laboratories has been largely supported by the National Science Foundation, whose support we gratefully acknowledge.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors wrote, read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wendel, J.F., Jackson, S.A., Meyers, B.C. et al. Evolution of plant genome architecture. Genome Biol 17, 37 (2016). https://doi.org/10.1186/s13059-016-0908-1
Published:
DOI: https://doi.org/10.1186/s13059-016-0908-1