
Abstract
Background
Although hybridization is thought to be relatively rare in animals, the raw genetic material introduced via introgression may play an important role in fueling adaptation and adaptive radiation. The butterfly genus Heliconius is an excellent system to study hybridization and introgression but most studies have focused on closely related species such as H. cydno and H. melpomene. Here we characterize genome-wide patterns of introgression between H. besckei, the only species with a red and yellow banded ‘postman’ wing pattern in the tiger-striped silvaniform clade, and co-mimetic H. melpomene nanna.
Results
We find a pronounced signature of putative introgression from H. melpomene into H. besckei in the genomic region upstream of the gene optix, known to control red wing patterning, suggesting adaptive introgression of wing pattern mimicry between these two distantly related species. At least 39 additional genomic regions show signals of introgression as strong or stronger than this mimicry locus. Gene flow has been on-going, with evidence of gene exchange at multiple time points, and bidirectional, moving from the melpomene to the silvaniform clade and vice versa. The history of gene exchange has also been complex, with contributions from multiple silvaniform species in addition to H. besckei. We also detect a signature of ancient introgression of the entire Z chromosome between the silvaniform and melpomene/cydno clades.
Conclusions
Our study provides a genome-wide portrait of introgression between distantly related butterfly species. We further propose a comprehensive and efficient workflow for gene flow identification in genomic data sets.
Similar content being viewed by others

Background
Hybridization, or interbreeding between species, has the potential to influence adaptation and speciation in a variety of ways. For instance, occasional hybridization between incompletely isolated species may contribute to either accelerating speciation or breaking down species barriers [1]. In addition, hybridization can also lead to adaptive introgression by transmitting beneficial alleles between species via backcross hybrids [2–4], which can happen during either sympatric speciation or the secondary contact phase of allopatric speciation. There are a number of striking examples of adaptive introgression in plants, both between hybridizing wild species as well as between crops and their wild relatives [5–7]. Furthermore, there is a growing list of examples of adaptive introgression in animals. For instance, an allele associated with rodenticide warfarin resistance at the gene Vkorc1 has been shared by mouse species [8], and an insecticide resistance mutation has been transferred between Anopheles sibling species [9, 10]. Similarly, haplotypes at the ALX1 gene, which is strongly associated with beak shape in Darwin’s finches, appear to be shared among species due to hybridization [11]. In addition, recent examples suggest modern humans have benefited by adaptive introgression from different populations and extinct species: introgressed Neanderthal alleles may have helped modern humans adapt to non-African environments [12, 13], and high-altitude adaptive traits have been contributed to Tibetans from Denisovans and Nepalese Sherpa [14, 15].
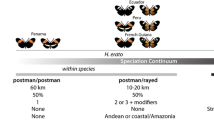
Another striking example of adaptive introgression in animals involves wing pattern mimicry in Heliconius butterflies [16, 17]. The genus Heliconius is a group of neotropical butterflies that display diverse Müllerian mimetic wing patterns to warn predators of their toxicity [18, 19]. Given a long history of research and rapidly developing genomic resources, Heliconius is also an excellent system to address a variety of evolutionary questions related to adaptation and hybridization [20–25]. The evolution of mimicry in Heliconius has resulted from introgression between closely related species as well as convergent evolution between more distantly related species. For instance, Heliconius species belonging to the melpomene-cydno-timareta clade, which hybridize regularly but are subject to Haldane’s rule, share wing color patterns due to introgression between species [16, 17, 26–28]. On the other hand, between distantly related co-mimetic pairs, such as between Heliconius melpomene and Heliconius erato, nearly identical wing patterns have evolved independently [29–31]. Between these two extremes are rare instances of mimicry between the melpomene-cydno-timareta and silvaniform clades; these clades are ecologically and morphologically distinct and hybridization between them is rare [32, 33]. Previously, gene flow has been documented from the melpomene-cydno-timareta clade into the silvaniform clade [34], and Heliconius elevatus, a silvaniform species that displays a melpomene-like rayed color pattern was shown to have acquired its mimicry phenotype via introgression from H. melpomene [16]. One of the most widespread color patterns across the broad distribution of co-mimics H. melpomene and H. erato is the red and yellow banded ‘postman’ phenotype seen throughout Central America, much of Brazil and isolated patches in Peru and Colombia. In coastal Brazil, H. melpomene nanna and H. erato phyllis co-occur with the only silvaniform species that shares this postman color pattern, Heliconius besckei (Fig. 1). The origin of mimicry in H. besckei remains elusive, however, and given rare ongoing hybridization among melpomene and silvaniform group species, could have been the product of introgression or convergent evolution.

The geographical distribution of Heliconius butterfly species. The distributions of Heliconius melpomene-cydno-timareta and silvaniform clades are shown in different colors, along with wing pattern images of focal taxa
Recently, next generation sequencing technologies have enabled genome-wide studies to depict global introgression patterns in a variety of systems [11, 16, 35–38]. As a result, a diverse set of statistical tests have been developed that utilize genome-wide data for the detection of introgression between closely related species. For instance, the four taxon ABBA-BABA test was originally designed to detect interbreeding between modern humans and Neanderthals [35, 39] and then was widely applied to test for ancient admixture among other systems such as Heliconius and swallowtail butterflies, spiny lizards, Zimmerius flycatchers and crows [16, 40–43]. Here, we examine genome-wide patterns of divergence and introgression in Heliconius butterflies using full genome resequencing data, focusing on potential adaptive introgression between H. melpomene and H. besckei. We integrate a series of population genetic and phylogenetic approaches to infer introgression and propose a comprehensive and efficient workflow for gene flow identification in large population genomic data sets. Our results provide a genome-wide portrait of polarized introgression between distantly related species, yielding multiple discrete instances of putative adaptive introgression.
Results
Whole genome phylogenetic analyses
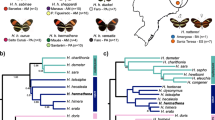
We first characterized the evolutionary history of Heliconius butterflies using genome-wide single nucleotide polymorphism (SNP) data (approximately 23 million SNPs) from 73 butterflies representing 29 species (Fig. 2). This analysis was used to define a sister taxon for H. besckei for introgression scans between H. besckei and H. melpomene nanna using the D-statistic. In Brazil, H. besckei co-occurs with silvaniform species Heliconius numata robigus and Heliconius ethilla narcaea but our results strongly supported H. numata + Heliconius ismenius as the sister clade to H. besckei so we selected H. besckei, H. numata and H. m. nanna as the three ingroup taxa for Patterson’s D-statistic tests. Moreover, the time-calibrated maximum-likelihood tree of 30 samples showed that the split between H. besckei and H. numata was estimated at 1.93 million years ago (Mya), which was earlier than the split between the Heliconius cydno and H. melpomene lineages (1.34 Mya) (Figure S1 in Additional file 1). This result suggests that if introgression occurred between H. besckei and H. m. nanna, it could have happened at one or more distinct time points: between H. besckei and the melpomene-cydno-timareta ancestor, between H. besckei and the melpomene ancestor, and/or between H. besckei and H. m. nanna.

Genome-wide phylogeny of Heliconius butterflies. The maximum-likelihood phylogenetic tree is based on the genome-wide SNP data (23 Mb alignment) with the clades indicated as color-coded bars. The scale bar represents the percentage of substitutions per site
Searching for large-scale introgression between H. m. nanna and H. besckei
Since H. m. nanna and H. besckei are sympatric species that are potentially interfertile and display similar wing patterns, our a priori hypothesis was that wing pattern mimicry was shared via introgression, either originating in H. melpomene and then being passed to the ancestor of H. besckei or vice versa. To test this hypothesis, we calculated Patterson’s D-statistic incorporating H. numata as the third ingroup taxon and Heliconius wallacei as the outgroup taxon. We used H. wallacei as the outgroup because this sample had the most genotype calls among the three potential outgroup samples, H. wallacei, Heliconius burneyi and Heliconius doris (Table S1 in Additional file 2). For each fixed 50 kb window across 21 chromosomes, we estimated derived SNP allele frequencies supporting either ‘ABBA’ or ‘BABA’ patterns among the ingroup taxa and then calculated the mean D-statistic value for each chromosome (Table S2 in Additional file 2). The results suggested no large-scale gene flow between H. besckei and H. m. nanna, but rather, greater gene flow between H. numata and H. m. nanna, although the D-statistic values were close to zero (Table S2 in Additional file 2). This marginal but significant D-statistic between H. numata and H. m. nanna was unexpected. Given the large overlap in the distributions of H. numata and H. melpomene, it is possible that these two species do exchange more gene flow than between H. besckei and H. melpomene.
Evidence of gene flow at the B/D mimicry locus
Heliconius wing patterning is controlled by a small number of Mendelian switch loci with the red forewing band and yellow hind wing band of the H. melpomene postman phenotype specifically controlled by the B/D and Yb loci, respectively [44, 45]. Previous analysis showed evidence of introgression around the B/D and Yb mimicry loci between co-mimetic postman as well as rayed H. melpomene and Heliconius timareta [16]. We applied a 5 kb window size to plot D-statistic values along B/D and Yb genomic intervals to infer potential gene flow between H. besckei and H. m. nanna. For comparison, we also plotted D-statistic values along B/D and Yb genomic intervals in co-mimetic Heliconius melpomene amaryllis and H. timareta (see also [16]). The results revealed two sharp peaks of elevated D in the B/D interval indicating genetic similarity between H. besckei and H. m. nanna, and the physical position of this region coincided perfectly with the genomic region showing evidence of introgression between H. m. amaryllis and H. timareta (Fig. 3). Statistical tests using a fixed 5 kb window through the B/D interval were significantly different from zero (D numata, besckei, m. nanna, wallacei = 0.385 ± 0.109, P = 0.0004), indicative of a strong introgression signal at this mimicry locus between H. besckei and H. m. nanna. The Yb locus, in contrast, showed no clear evidence of elevated D, and this region also showed a more diffuse signature of allele sharing in the H. m. amaryllis and H. timareta comparison (Fig. 3). Statistical tests through the region of the Yb locus that showed the strongest evidence of introgression in the H. m. amaryllis and H. timareta comparison (500 kb to 1 Mb) revealed a pattern similar to the genomic background, with greater allele sharing between H. numata and H. m. nanna in comparison with H. besckei and H. m. nanna (D numata, besckei, m. nanna, wallacei = −0.084 ± 0.034, P = 0.01318). These results suggest wing pattern mimicry, but perhaps only variation at the B/D mimicry locus, was introgressed between H. melpomene and H. besckei.

Patterson’s D-statistic scans along the B/D and Yb mimicry loci scaffolds. For the tree topology above (a), clear introgression patterns (labeled with green bars) are observed along B/D between H. m. amaryllis and H. timareta and between H. besckei and H. m. nanna, using 5 kb adjacent windows (b)
Patterns of genome-wide introgression
To further examine genome-wide patterns of introgression between H. m. nanna and H. besckei, we calculated the D-statistic for every 5 kb, 10 kb and 50 kb window across the genome separately and used the maximum signature of putative H. besckei–H. m. nanna introgression at B/D as a minimum threshold (D numata, besckei, m. nanna, wallacei ≥ 0.74 for 5 kb window, D numata, besckei, m. nanna, wallacei ≥ 0.56 for 10 kb window, D numata, besckei, nanna, wallacei ≥ 0.34 for 50 kb window). We kept candidate loci only if supported by more than five valid SNPs per 5 kb, which yielded 97 unique candidate introgression loci: 60 5-kb segments, 32 10-kb segments and 5 50-kb segments (Table S3 in Additional file 2). Next, we calculated the f-statistic for the 97 focal intervals and extracted those with D and f d significantly different from zero (P < 0.01; Table S3 in Additional file 2). The f-statistic was originally designed to estimate the proportion of the genome exchanged as a result of introgression [35], but it was recently modified to complement the D-statistic in identifying introgressed genomic segments [46]. A total of 50 candidate introgression loci were significant with both the D and f d tests (Table S3 in Additional file 2).
In order to help distinguish between introgression and ancestral variation, we calculated DNA sequence divergence (dxy) for each candidate introgression interval and compared this with chromosomal mean dxy (Table S4 in Additional file 2) because introgressed regions generally show lower absolute genetic divergence [47]. A total of 91 loci showed lower H. besckei–H. m. nanna dxy compared with neighboring regions and their chromosome as a whole (Table S5 in Additional file 2). Martin et al. [46] showed that dxy can be correlated with D due to factors other than gene flow so we further tested for evidence of allele sharing between H. besckei and H. m. nanna by comparing H. besckei–H. m. nanna dxy to that between other species comparisons, H. besckei–H. numata, and H. m. nanna–H. numata. We found that at 39 of 97 intervals, H. besckei–H. m. nanna dxy was lower than both the silvaniform comparison, H. besckei–H. numata, and the H. m. nanna–H. numata comparison (Table S5 in Additional file 2), again pointing to allele sharing between H. besckei and H. melpomene.
We also checked sequencing read depths for all candidate introgression loci among the four D-statistic taxa to rule out false positives caused by potential read mapping artifacts (Table S6 in Additional file 2). For instance, a signature of allele sharing between taxa could result from a subset of reads most similar to the reference genome, H. melpomene in this case, mapping back to the reference better than more divergent reads. This read mapping artifact could produce an apparent signature of introgression between species (specifically H. melpomene introgression into other species) but it should also yield reduced read coverage in the affected area. On the other hand, unusually high read depth might be due to read misalignment and could influence allele frequency estimates. Based on the sample statistics shown in Tables S1 and S6 in Additional file 2, loci with high (above 40) or low read depth (below 5) were filtered out. In total, we found that 85 candidate introgression loci were within an acceptable sequencing coverage range. By overlaying the results from all statistics, we identified a subset of 41 candidate introgression loci, including two adjacent 50 kb regions from the B/D mimicry locus, that were supported by all measures (Table 1). These genomic regions were distributed among 16 of the 21 chromosomes in H. melpomene. Many of the 5 kb segments were filtered out but most of the 10 kb and 50 kb segments were retained, which indicates a relatively robust introgression pattern of these larger genomic regions.
Time and direction of gene flow among Heliconius species
According to our phylogenetic analysis, H. besckei originated prior to diversification within the melpomene-cydno-timareta clade so introgression could have occurred before or after the origin of H. m. nanna. Furthermore, because evidence of introgression emerged using relatively small (5 kb, 10 kb and 50 kb) window sizes, it is likely that gene flow between H. besckei and H. melpomene was relatively ancient, so that signals of longer introgressed haplotypes were lost over time due to recombination and subsequent substitutions. To trace the history of introgression and infer the direction of gene flow, we reconstructed genealogies for each 5 kb and 10 kb candidate introgression locus using both the original window and a larger window size (original + 5 kb on either side for 5 kb candidate loci, original + 10 kb on either side for 10 kb candidate loci). This approach was motivated by the need to both maximize phylogenetic signal, potentially lacking in small genomic segments, and determine the physical extent of introgressed haplotypes. For each 50 kb candidate introgression loci, we reconstructed genealogies for the original 50 kb as well as 10 kb subsections (Fig. 4; Figures S2–S9 in Additional file 1).

Signatures of introgression between H. besckei and H. m. nanna along four candidate introgression loci. For each candidate locus, the Patterson’s D-statistic, f d and dxy scans are performed and the maximum likelihood phylogenetic tree using 10 kb window size is constructed to infer the timing and direction of gene flow
We used the relationships of taxa on gene genealogies of candidate introgression loci, and discordance from the genome-wide tree, to infer the history of gene flow at each locus. For instance, visual inspection of the genealogy for candidate introgression locus 77 places all H. besckei haplotypes as a sister-clade to all melpomene-cydno-timareta haplotypes (Fig. 4). From this relationship we inferred that there was ancient gene flow between H. besckei and the common ancestor of the melpomene-cydno-timareta clade (Table 2), but because this is the oldest time point and we lack another clade between these taxa and the outgroup, we could not infer the direction of gene flow. In contrast, for candidate introgression loci 96 and 97, which are from the B/D mimicry locus, H. besckei haplotypes clustered not with other silvaniform species, but with a subset of melpomene-cydno-timareta clade species (Fig. 4). From this we inferred that introgression occurred from H. melpomene into H. besckei and that it occurred historically (Table 2), prior to some diversification among H. melpomene subspecies, since this haplotype is shared among a number of H. melpomene subspecies (Fig. 4). Inferring the history of introgression in this way requires that the inferred genealogy is discordant from the genome-wide tree, with respect to the focal taxa, but that discordance across the tree as a whole is not so pervasive as to make the historical pattern uninterpretable. Therefore, we restricted these analyses to candidate introgression loci for which gene genealogies showed discordance specific to the focal taxa but otherwise limited discordance over the rest of the tree. Gene trees for 25 of 44 candidate introgression loci met these criteria (Fig. 4; Figures S2–S9 in Additional file 1) and we were able to infer the timing and direction of gene flow for 23 of them (Table 2).
We were surprised to find that inspection of many trees suggested potential gene flow between H. melpomene and silvaniform species other than H. besckei. For instance, trees for loci 61, 73, 75, and 94 clustered H. m. nanna with Heliconius ethilla as opposed to H. besckei (Figures S3, S4 and S7 in Additional file 1). All of our genome-wide analyses used H. besckei’s closest relative, Heliconius numata, for comparison but other silvaniform species also co-occur in this Brazilian community and could be exchanging genes with the focal taxa as well. It is possible that some of the strong signatures of putative H. besckei–H. m. nanna introgression could actually be a result of introgression from another silvaniform species. Therefore, we calculated D-statistics to test potential gene flow between H. m. nanna and silvaniform species such as H. ethilla, Heliconius pardalinus, Heliconius hecale and H. ismenius for 23 candidate introgression loci (Table S7 in Additional file 2). These results suggest that other silvaniform species have indeed been involved in as many as two-thirds of the inferred introgression events (Table 2). Furthermore, the dynamics appear to differ between introgression events involving H. besckei versus those involving other silvaniform species (Table 2). Specifically, all introgression involving H. besckei appears to have been in the direction of gene flow into H. besckei. These events also appear to be older, often involving the H. melpomene ancestor or the melpomene-cydno-timareta ancestor. In contrast, introgression involving all other silvaniform species appears to move in the opposite direction, into H. melpomene, and is often inferred to have occurred at more recent time points, frequently involving H. m. nanna specifically (Table 2).
Annotation of introgression loci
We found that protein coding genes were located in 32 of 41 putatively introgressed loci (Table S8 in Additional file 2), supporting the idea that introgression may have functional consequences. The list of annotated genes contained in the candidate introgression loci suggests a number of interesting biological functions potentially associated with introgression, including collagen and cuticle matrix formation, metabolism, embryonic patterning, synapse function, and heat stress, to name a few. Furthermore, the candidate introgression loci that do not contain protein coding sequences are likely to have functional consequences as well. A clear example of this is the strong signature of introgression we detected between H. melpomene and H. besckei at the B/D mimicry locus. This region on chromosome 18 does not contain genes but it is immediately adjacent to the gene optix, which is known to control all red color pattern variation across Heliconius butterflies [44]. This is the same genomic region that has been introgressed among co-mimetic H. melpomene, H. timareta, and H. elevatus in Peru and Colombia and contains the regulatory elements governing differential expression of optix [16, 17].
Analysis of the Z chromosome
Previous work comparing H. cydno and H. melpomene found pronounced genetic differentiation and greatly reduced interspecific gene flow on the Z chromosome, relative to autosomes [48, 49], a population genetic pattern consistent with the large Z effect on hybrid female sterility in crosses between these two species [50], as well as in other Heliconius butterflies [51], and the disproportionately large role of the Z chromosome in mediating reproductive isolation in female heterogametic taxa in general [52]. We compared H. m. nanna with H. besckei and H. numata and also found reduced introgression on the Z chromosome relative to autosomes (Table S2 in Additional file 2). However, comparisons between H. m. nanna and other silvaniform species, such as H. ethilla, revealed a distinct and unexpected signature of introgression on the Z chromosome, apparent as highly elevated D (Table S9 in Additional file 2). In addition to elevated D on the Z chromosome, this signature also appeared as reduced dxy and FST on the Z chromosome relative to comparisons between other species (Table S9 in Additional file 2), although the FST results were not as striking as the other two statistics. Also, Z chromosome FST values were still generally higher than autosomes, consistent with the idea that this Z chromosomal introgression was relatively old. By swapping taxa in the four-taxon D-statistic test, we found that the signature of shared variation on the Z chromosome was not exclusive to H. m. nanna and H. ethilla but could be traced back to sharing between the entire melpomene-cydno-timareta clade and the silvaniform subclade that includes H. ethilla, H. hecale, and H. pardalinus (Table S9 in Additional file 2). Furthermore, a genealogy based on all Z chromosome SNP variation revealed a deep discordance with the genome-wide tree with the H. ethilla, H. hecale, and H. pardalinus clade sister to the melpomene-cydno-timareta clade, as opposed to the other silvaniform clade (Figure S10 in Additional file 1). As a whole, these results suggest ancient introgression of the entire Z chromosome between just one of these two silvaniform subclades and the ancestor of the entire melpomene-cydno-timareta clade.
Discussion
The fate of introgressed genetic variation
Gene flow between species can play diverse and even opposing roles in the evolutionary process, from homogenizing genetic variation and eroding the species barrier to providing novel alleles that facilitate adaptation. Phylogenetic discordance we see today is the result of interactions among multiple processes including sorting of ancestral variation, introgression, and recombination over the combined history of two related species. Hybridization and introgression offer the opportunity for exchange of genetic material and ultimately selection plays a central role in the integration of introduced alleles into the recipient genome. For instance, introgression of a deleterious allele is liable to be prevented, while a neutral allele should undergo random recombination and accumulate further neutral substitutions over time and has a low probability of fixation, and a beneficial allele should spread in the new population with a higher probability of fixation. The length of introgressed haplotypes also provides information about the timing of gene flow and the pace of selective sweeps because recombination and subsequent substitutions will erode them over time. Overall, therefore, we expect that small, introgressed genomic regions originating from the oldest gene flow events are likely to be adaptive.
In this study, we found various instances consistent with historical adaptive introgression between H. melpomene and H. besckei in Brazil. Our analyses suggest little recent gene flow between these taxa and point to gene exchange at time points older than when H. melpomene aglaope donated alleles at the B/D mimicry locus to H. timareta florencia, which could be detected at a chromosome-wide level [16]. We also found that protein coding genes are located in a majority of putatively introgressed loci (Table S8 in Additional file 2). Furthermore, a number of the inferred introgression events occurred between other silvaniform species and H. melpomene, which suggests a more widespread pattern of gene flow among these lineages, not only limited to co-mimics that share a similar wing pattern.
Potential functional consequences of introgression
The persistence of small, apparently introgressed genomic regions over time is suggestive of a functional role, although it is often difficult to determine the exact nature of those roles given our limited knowledge of butterfly functional genetics. In the case of mimicry, our data suggest that H. besckei acquired its mimetic wing pattern from H. melpomene. Our analyses reveal one other genomic region that appears to share the same history as the B/D mimicry locus on chromosome 18, this being a 50 kb interval on chromosome 1 (locus 93) that we infer to have been transferred along with wing pattern mimicry from the H. melpomene ancestor to H. besckei (Table 2). This candidate introgression locus contains collagen type IV subunit α-2 and collagen type IV subunit α-1 lies right outside this candidate locus. Collagen IV is the main constituent of basement membranes and plays an active morphogenetic role in determining organ shape and animal form [53]. Furthermore, collagen type IV subunits α-1 and α-2 play important roles in muscle function across animals and they have recently been implicated in flight muscle function in the migratory monarch butterfly, Danaus plexippus [54]. Mimicry in Heliconius butterflies is well known to involve flight behavior in addition to wing pattern. Srygley and colleagues have shown that aspects of flight kinematics, primarily wing-beat frequency [55, 56], converge among co-mimetic Heliconius species, as do wing and body morphology associated with biomechanics [57, 58]. While highly speculative, our genomic data offer the intriguing possibility that H. besckei may have acquired a behavioral component of mimicry, locomotor mimicry, via introgression from H. melpomene in addition to wing pattern mimicry. This hypothesis will require further functional tests in the future.
A unique evolutionary signature on the Z chromosome
Sex-linked genes often play an important role in generating reproductive isolation between closely related species [52, 59] and therefore sex chromosomes might generally be expected to experience reduced interspecific gene flow in comparison with autosomes. Consistent with this, previous studies on nightingales and Heliconius species in the cydno-melopmene clade found a pattern of reduced introgression on the Z chromosome [49, 60]. Similarly, reduced gene flow on the X chromosome was recently documented in Anopheles mosquitoes where autosomal introgression is pervasive [38]. Our analysis of Z chromosome variation among more distantly related Heliconius species revealed a surprising result. Comparison of H. melpomene and H. besckei showed the expected pattern of reduced introgression on the Z chromosome relative to autosomes. However, comparisons with other silvaniforms suggest ancient introgression of the entire Z chromosome between the silvaniform subclade that contains H. hecale, H. pardalinus, and H. ethilla, and the ancestor of the melpomene-cydno-timareta clade.
Efficiency of detecting introgressed loci
The methods for generating high-density SNP data in virtually any organism are now relatively straightforward, and with this new technology come statistical tools to examine genome-wide patterns of introgression. Established analytical pipelines for examining gene flow between species include both divergence-based genome scans (linkage disequilibrium, FST and D-statistic) and likelihood/model-based methods [61, 62]. Given that our sampling fit the requirements of the D-statistic (two sister species, a third species potentially involved in introgression, and an outgroup species), our goal was to integrate D-statistic, f-statistic, and DNA sequence divergence approaches to infer introgression and disentangle incomplete lineage sorting from gene flow. In order to maximize the power of our approach, we further defined a baseline of expected signatures of introgression using the observed patterns around the B/D mimicry locus. This is a genomic region for which we had an a priori hypothesis and for which the data yielded a strong signature of introgression. Finally, we used phylogenetics, comparing trees inferred using genome-wide SNP data with those from candidate introgression loci, to infer the timing and direction of introgression between species. Here we applied a progressive phylogenetic analysis with multiple window sizes which enabled us to characterize the introgression histories for 23 of 41 loci. Importantly, our pipeline offers an efficient solution to identify and polarize genome-wide introgression events that can be applied widely to emerging genome-wide polymorphism data.
Conclusions
Previous work has shown that closely related Heliconius species that mimic one another evolved that similarity by exchanging wing patterning alleles through hybridization. However, the potential for adaptive introgression across the rest of the genome has not been explored, nor has the extent of gene flow among more distantly related species. Here we focused on H. besckei, an enigmatic species from Brazil that has a color pattern that matches sympatric co-mimics but differs completely from its closest relatives, making it a good candidate for introgression of wing pattern mimicry. We tested this and found a strong signature of introgression at the gene optix, which controls red wing patterning in Heliconius, and then we used these population genetic signatures as a minimum threshold for genome-wide comparisons. This approach yielded 39 additional genomic regions that also showed strong evidence of introgression. Analysis of these putatively introgressed genomic regions revealed that gene flow has been on-going, bi-directional between clades, and complex, involving multiple extant species as well as their ancestors. We also found evidence of ancient introgression of the entire Z chromosome between lineages, which is unexpected because this chromosome has previously been shown to be resistant to introgression between closely related Heliconius species. As a whole, our results substantially expand the potential impact of introgressive hybridization throughout the evolutionary history of Heliconius butterflies.
Methods
Sample preparation and sequencing
Twenty-five adult butterflies were collected in the field in Brazil, Ecuador and Costa Rica. For each individual, wings were carefully separated and genomic DNA was extracted from thoracic tissue using a DNeasy Blood & Tissue Kit (Qiagen). Illumina paired-end libraries were constructed using the Illumina Truseq protocol and then were pooled and sequenced using an Illumina HiSeq2000. Raw reads were demultiplexed according to their barcodes.
Data collection and genotyping calling
We downloaded 48 available whole genome resequencing datasets from NCBI (PRJNA226620) [48, 63] and ENA (ERP002440) [49]. Low quality reads with fewer than 90 % bases that had a minimum quality score above 10 were removed after quality filtering from 73 genome resequencing datasets and the rest were aligned to the H. melpomene v1.1 [16] using Bowtie2 v2.0.0-beta7 [64] with parameter -very-sensitive-local and then re-ordered and sorted by Picard v1.84 (http://broadinstitute.github.io/picard/). PCR duplicates were removed using Picard. RealignerTargetCreator and IndelRealigner [65] in GATK v2.1 were used to realign indels and UnifiedGenotyper [66] was used to call genotypes across 73 individuals using the following parameters: heterozygosity 0.01, stand_call_conf 50, stand_emit_conf 10, dcov 250. SNPs with good quality (Qual > 30) were finally used in the subsequent analyses (Table S1 in Additional file 2).
Genome-wide phylogeny and divergence time estimation
Polymorphism genotype calls existing in all 73 individuals with good quality (around 23.14 Mb) were aligned and converted into PHYLIP format and a genome-wide maximum-likelihood phylogenetic tree was constructed using RAxML [67] with the GTRGAMMA model and 100 bootstrap replicates. The tree image was created using iTOL [68]. A genome-wide tree topology including 32 taxa was extracted as input for the software PhyTime [69], which was used to estimate divergence times, calibrated using the mean split time estimates between H. cydno and H. melpomene (1.4 Mya) and between H. cydno and H. pachinus (0.43 Mya) from a previous population genomics study [48]. The PhyTime output tree was processed using TreeAnnotator [70]. For candidate introgression loci, maximum-likelihood trees were generated using PhyML3.0 [71] with the GTR model and 100 bootstrap replicates to infer the direction of gene flow.
Detecting gene flow among H. besckei, H. numata and H. m. nanna
We integrated both Patterson’s D-statistic [35, 39] and a modified f-statistic (f d) [46] to better identify potential introgressed loci across the whole genome among three ingroup taxa (H. m. nanna, H. besckei and H. numata) using H. wallacei as an outgroup. The D-statistic was used to examine the phylogenetic distribution of derived alleles at loci that display either an ABBA or BABA allelic configuration (Fig. 2). Since we had four individuals per taxon, the frequency of the derived allele at each site in each population was used instead of binary counts of fixed ABBA and BABA sites with [39]:
where P 1 , P 2 , P 3 and P 4 are the four taxa of the comparison and \( {\hat{P}}_{ij} \) is the observed frequency of SNP i in population j. For whole genome estimation, we assigned scaffolds to 21 chromosomes according to H. melpomene linkage mapping and selected a block size of 50 kb, greater than the estimated linkage disequilibrium in Heliconius, to calculate the standard errors on D-statistics across 21 chromosomes [16]. An R package, bootstrap 2012-04, was used to perform the leave-one-out jackknife approach [72]. To identify candidate introgression loci, a small fixed window size (5 kb, 10 kb and 50 kb) was chosen for both D-statistic and the modified f-statistic (f d). f d was calculated as [46]:
where P 1 , P 2 , P 3 and O are the four taxa of the comparison and P D can be either P 2 or P 3 , which has the higher frequency of the derived allele. For each 5 kb, 10 kb or 50 kb window, the standard error was calculated using a moving block bootstrap approach with the optimal block size being equal to n 1/3, where n is the total number of values. Then all the tests were followed by two tailed z-tests to determine if the standard error for each D or f value was significantly different from zero, which indicates potential gene flow. For some of the candidate introgression loci, we also used the D-statistic to examine the potential of introgression between H. m. nanna and other silvaniform species, including H. ethilla, H. ismenius, H. hecale and H. pardalinus (Table S7 in Additional file 2). Because we tested all 97 candidate introgression loci with the D and f d statistics (and dxy, see below), we controlled for multiple testing using the Benjamini-Hochberg false discovery rate (FDR) method [73]. We used an FDR of 0.01 for all three tests, yielding adjusted significance thresholds of P < 0.006289 for D, P <0.007216 for f d, and P < 0.009381 for dxy.
Population genetic analyses and sequencing depth estimation
To rule out false positive introgression loci due to incomplete lineage sorting, mean pairwise sequence divergence (dxy) was calculated among H. m. nanna, H. besckei and H. numata as a complementary analysis to the D-statistic and modified f-statistic [47]. Four individuals of the same species were used to calculate mean dxy for each chromosome using a block size of 50 kb using the following equation [47]:
where p x and p y refer to reference allele frequency in taxon x and y. Then, standard error was calculated for each chromosome using a jackknife approach. For each candidate introgression locus, dxy was calculated using a smaller block size of 100 bp to allow moving block bootstrapping. If the mean dxy value of a putatively introgressed locus was lower than the mean value of its chromosome, the two values were compared statistically using a Mann-Whitney U-test. Sequencing depth and FST values of 50 kb adjacent windows were also calculated for the 21 chromosomes and candidate loci using VCFtools package [74].
Annotating candidate introgressed loci
We extracted coding sequence regions for each candidate locus based on the genome annotation of H. melpomene [16] and performed NCBI-BLASTX against the nr database to characterize them.
Ethical approval
No ethical approval was required.
Availability of supporting data
Illumina paired-end whole genome resequencing data are available from NCBI Sequence Read Archive (SRA); all accession numbers are in Table S1 in Additional file 2. The H. melpomene reference genome, including annotation of version 1.1, is available at the Heliconius Genome Project website, http://www.butterflygenome.org/, as well as at Lepbase, http://lepbase.org/.
Abbreviations
- dxy :
-
mean pairwise sequence divergence
- FDR:
-
False discovery rate
- Mya:
-
Million years ago
- SNP:
-
Single nucleotide polymorphism
References
Mallet J. Hybridization as an invasion of the genome. Trends Ecol Evol. 2005;20:229–37.
Arnold ML. Transfer and origin of adaptations through natural hybridization: were Anderson and Stebbins right? Plant Cell. 2004;16:562–70.
Hedrick PW. Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol Ecol. 2013;22:4606–18.
Racimo F, Sankararaman S, Nielsen R, Huerta-Sánchez E. Evidence for archaic adaptive introgression in humans. Nat Rev Genet. 2015;16:359–71.
Stewart CN, Halfhill MD, Warwick SI. Transgene introgression from genetically modified crops to their wild relatives. Nat Rev Genet. 2003;4:806–17.
Martin NH, Bouck AC, Arnold ML. Detecting adaptive trait introgression between Iris fulva and I. brevicaulis in highly selective field conditions. Genetics. 2006;172:2481–9.
Whitney KD, Randell RA, Rieseberg LH. Adaptive introgression of herbivore resistance traits in the weedy sunflower Helianthus annuus. Am Nat. 2006;167:794–807.
Song Y, Endepols S, Klemann N, Richter D, Matuschka FR, Shih CH, et al. Adaptive introgression of anticoagulant rodent poison resistance by hybridization between old world mice. Curr Biol. 2011;21:1296–301.
Clarkson CS, Weetman D, Essandoh J, Yawson AE, Maslen G, Manske M, et al. Adaptive introgression between Anopheles sibling species eliminates a major genomic island but not reproductive isolation. Nat Commun. 2014;5:4248.
Norris LC, Main BJ, Lee Y, Collier TC, Fofana A, Cornel AJ, et al. Adaptive introgression in an African malaria mosquito coincident with the increased usage of insecticide-treated bed nets. Proc Natl Acad Sci U S A. 2015;112:815–20.
Lamichhaney S, Berglund J, Almén MS, Maqbool K, Grabherr M, Martinez-Barrio A, et al. Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature. 2015;518:371–5.
Sankararaman S, Mallick S, Dannemann M, Prüfer K, Kelso J, Pääbo S, et al. The genomic landscape of Neanderthal ancestry in present-day humans. Nature. 2014;507:354–7.
Vernot B, Akey JM. Resurrecting surviving Neandertal lineages from modern human genomes. Science. 2014;343:1017–21.
Huerta-Sánchez E, Jin X, Asan, Bianba Z, Peter BM, Vinckenbosch N, et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature. 2014;512:194–7.
Jeong C, Alkorta-Aranburu G, Basnyat B, Neupane M, Witonsky DB, Pritchard JK, et al. Admixture facilitates genetic adaptations to high altitude in Tibet. Nat Commun. 2014;5:3281.
Heliconius Genome Consortium. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature. 2012;487:94–8.
Pardo-Diaz C, Salazar C, Baxter SW, Merot C, Figueiredo-Ready W, Joron M, et al. Adaptive introgression across species boundaries in Heliconius butterflies. PLoS Genet. 2012;8:e1002752.
Sheppard PM, Turner J, Brown K, Benson W, Singer M. Genetics and the evolution of Muellerian mimicry in Heliconius butterflies. Phil Trans R Soc B. 1985;308:433–610.
Joron M, Jiggins CD, Papanicolaou A, McMillan WO. Heliconius wing patterns: an evo-devo model for understanding phenotypic diversity. Heredity. 2006;97:157–67.
Bates HW. XXXII. Contributions to an insect fauna of the Amazon Valley. Lepidoptera: Heliconidæ. Trans Linn Soc Lond. 1862;23:495–566.
Brown Jr KS. The biology of Heliconius and related genera. Annu Rev Entomol. 1981;26:427–57.
Turner JG. Adaptation and evolution in Heliconius: a defense of neo-Darwinism. Ann Rev Ecol Syst. 1981;12:99–121.
Mallet J. Rapid speciation, hybridization and adaptive radiation in the Heliconius melpomene group. In: Butlin R, Bridle J, Schluter D, editors. Speciation and patterns of diversity. Cambridge: Cambridge University Press; 2009. p. 177–94.
Kronforst MR, Papa R. The functional basis of wing patterning in Heliconius butterflies: the molecules behind mimicry. Genetics. 2015;200:1–19.
Merrill RM, Dasmahapatra KK, Davey JW, Dell’Aglio DD, Hanly JJ, Huber B, et al. The diversification of Heliconius butterflies: what have we learned in 150 years? J Evol Biol. 2015;28:1417–38.
Bull V, Beltrán M, Jiggins CD, McMillan WO, Bermingham E, Mallet J. Polyphyly and gene flow between non-sibling Heliconius species. BMC Biol. 2006;4:11.
Kronforst MR, Young LG, Blume LM, Gilbert LE. Multilocus analyses of admixture and introgression among hybridizing Heliconius butterflies. Evolution. 2006;60:1254–68.
Nadeau NJ, Martin SH, Kozak KM, Salazar C, Dasmahapatra KK, Davey JW, et al. Genome-wide patterns of divergence and gene flow across a butterfly radiation. Mol Ecol. 2013;22:814–26.
Baxter SW, Papa R, Chamberlain N, Humphray SJ, Joron M, Morrison C, et al. Convergent evolution in the genetic basis of Müllerian mimicry in Heliconius butterflies. Genetics. 2008;180:1567–77.
Hines HM, Counterman BA, Papa R, Albuquerque de Moura P, Cardoso MZ, Linares M, et al. Wing patterning gene redefines the mimetic history of Heliconius butterflies. Proc Natl Acad Sci U S A. 2011;108:19666–71.
Supple MA, Hines HM, Dasmahapatra KK, Lewis JJ, Nielsen DM, Lavoie C, et al. Genomic architecture of adaptive color pattern divergence and convergence in Heliconius butterflies. Genome Res. 2013;23:1248–57.
Dasmahapatra KK, Silva-Vásquez A, Chung JW, Mallet J. Genetic analysis of a wild-caught hybrid between non-sister Heliconius butterfly species. Biol Lett. 2007;3:660–3.
Mallet J, Beltrán M, Neukirchen W, Linares M. Natural hybridization in heliconiine butterflies: the species boundary as a continuum. BMC Evol Biol. 2007;7:28.
Kronforst MR. Gene flow persists millions of years after speciation in Heliconius butterflies. BMC Evol Biol. 2008;8:98.
Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, et al. A draft sequence of the Neandertal genome. Science. 2010;328:710–22.
Ellegren H, Smeds L, Burri R, Olason PI, Backström N, Kawakami T, et al. The genomic landscape of species divergence in Ficedula flycatchers. Nature. 2012;491:756–60.
Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J, et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature. 2012;484:55–61.
Fontaine MC, Pease JB, Steele A, Waterhouse RM, Neafsey DE, Sharakhov IV, et al. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science. 2014;347:1258524.
Durand EY, Patterson N, Reich D, Slatkin M. Testing for ancient admixture between closely related populations. Mol Biol Evol. 2011;28:2239–52.
Leaché AD, Harris RB, Maliska ME, Linkem CW. Comparative species divergence across eight triplets of spiny lizards (Sceloporus) using genomic sequence data. Genome Biol Evol. 2013;5:2410–9.
Zhang W, Kunte K, Kronforst MR. Genome-wide characterization of adaptation and speciation in tiger swallowtail butterflies using de novo transcriptome assemblies. Genome Biol Evol. 2013;5:1233–45.
Poelstra JW, Vijay N, Bossu CM, Lantz H, Ryll B, Müller I, et al. The genomic landscape underlying phenotypic integrity in the face of gene flow in crows. Science. 2014;344:1410–4.
Rheindt FE, Fujita MK, Wilton PR, Edwards SV. Introgression and phenotypic assimilation in Zimmerius flycatchers (Tyrannidae): population genetic and phylogenetic inferences from genome-wide SNPs. Syst Biol. 2014;63:134–52.
Reed RD, Papa R, Martin A, Hines HM, Counterman BA, Pardo-Diaz C, et al. optix drives the repeated convergent evolution of butterfly wing pattern mimicry. Science. 2011;333:1137–41.
Nadeau NJ, Whibley A, Jones RT, Davey JW, Dasmahapatra KK, Baxter SW, et al. Genomic islands of divergence in hybridizing Heliconius butterflies identified by large-scale targeted sequencing. Phil Trans R Soc Lond B. 2012;367:343–53.
Martin SH, Davey JW, Jiggins CD. Evaluating the use of ABBA-BABA statistics to locate introgressed loci. Mol Biol Evol. 2014;32:244–57.
Smith J, Kronforst MR. Do Heliconius butterfly species exchange mimicry alleles? Biol Lett. 2013;9:20130503.
Kronforst MR, Hansen ME, Crawford NG, Gallant JR, Zhang W, Kulathinal RJ, et al. Hybridization reveals the evolving genomic architecture of speciation. Cell Rep. 2013;5:666–77.
Martin SH, Dasmahapatra KK, Nadeau NJ, Salazar C, Walters JR, Simpson F, et al. Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Res. 2013;23:1817–28.
Naisbit RE, Jiggins CD, Linares M, Salazar C, Mallet J. Hybrid sterility, Haldane’s rule and speciation in Heliconius cydno and H. melpomene. Genetics. 2002;161:1517–26.
Jiggins CD, Linares M, Naisbit RE, Salazar C, Yang ZH, Mallet J. Sex-linked hybrid sterility in a butterfly. Evolution. 2001;55:1631–8.
Qvarnström A, Bailey RI. Speciation through evolution of sex-linked genes. Heredity. 2009;102:4–15.
Pastor-Pareja JC, Xu T. Shaping cells and organs in Drosophila by opposing roles of fat body-secreted Collagen IV and perlecan. Dev Cell. 2011;21:245–56.
Zhan S, Zhang W, Niitepõld K, Hsu J, Haeger JF, Zalucki MP, et al. The genetics of monarch butterfly migration and warning colouration. Nature. 2014;514:317–21.
Srygley RB. Locomotor mimicry in Heliconius butterflies: contrast analyses of flight morphology and kinematics. Phil Trans R Soc Lond B. 1999;354:203–14.
Srygley RB. Evolution of the wave: aerodynamic and aposematic functions of butterfly wing motion. Proc R Soc B. 2007;274:913–7.
Srygley RB, Dudley R. Correlations of the position of center of body mass with butterfly escape tactics. J Exp Biol. 1993;174:155–66.
Srygley RB. Locomotor mimicry in butterflies? The associations of positions of centres of mass among groups of mimetic, unprofitable prey. Phil Trans R Soc Lond B. 1994;343:145–55.
Presgraves DC. Sex chromosomes and speciation in Drosophila. Trends Genet. 2008;24:336–43.
Storchová R, Reif J, Nachman MW. Female heterogamety and speciation: reduced introgression of the Z chromosome between two species of nightingales. Evolution. 2010;64:456–71.
Sousa V, Hey J. Understanding the origin of species with genome-scale data: modelling gene flow. Nat Rev Genet. 2013;14:404–14.
Lohse K, Frantz LAF. Neandertal admixture in Eurasia confirmed by maximum-likelihood analysis of three genomes. Genetics. 2014;196:1241–51.
Gallant JR, Imhoff VE, Martin A, Savage WK, Chamberlain NL, Pote BL, et al. Ancient homology underlies adaptive mimetic diversity across butterflies. Nat Commun. 2014;5:4817.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90.
Letunic I, Bork P. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 2011;39:W475–8.
Guindon S. Bayesian estimation of divergence times from large sequence alignments. Mol Biol Evol. 2010;27:1768–81.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–73.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21.
Tibshirani R, Leisch F. Functions for the book “An introduction to the bootstrap”. In: An Introduction to the Bootstrap. 1st ed. Chapman & Hall/CRC; 2012. https://cran.r-project.org/web/packages/bootstrap/bootstrap.pdf
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;289–300.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8.
Acknowledgments
We thank D. Massardo for assistance with sample preparation. We also thank reviewers for their insightful comments that improved the manuscript.
Funding
The project was funded by NIH grant GM108626 and NSF grant IOS-1452648 to M.R.K. GRPM was supported by CNPq grant 201806/2009-6.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WZ, KKD, JM, GRPM and MRK conceived and designed the study; WZ and KKD analyzed the data and performed statistical analyses; KKD, JM, GRPM and MRK contributed samples; WZ and MRK wrote the paper with input from KKD, JM, and GRPM; all authors read and approved the final manuscript.
Additional files
Additional file 1: Figure S1.
A maximum-likelihood phylogeny of 32 samples combined with dating of the nodes. Divergence times were calibrated using a fast Bayesian approach based on the separation time of melpomene and cydno-timareta clades (1.3 ± 0.1 Mya) and the separation time of H. pachinus from the cydno clade (0.4 ± 0.1 Mya). Samples from H. besckei and H. m. nanna are highlighted in red. Numbers are in million years and blue bars stand for 95 % confidence intervals. Figures S2-S9. Maximum-likelihood phylogenetic trees were constructed for all candidate introgression loci using original windows (5 kb, 10 kb and 50 kb), expanded windows (original 5 kb + 5 kb on either side, original 10 kb + 10 kb on either side) and multiple 10 kb windows within original 50 kb windows. The window size (as shown in each figure) may vary due to actual scaffold length and position. Figure S10. A maximum-likelihood phylogeny of Heliconius butterflies based on the SNP data from the Z chromosome. The cydno-timareta-melpomene clade is grouped with a subset of silvaniform species (highlighted in blue). (PDF 715 kb)
Additional file 2: Table S1.
Sample information and sequencing statistics. Table S2. Results of chromosomal D-statistics. Table S3. D-statistics and f d -statistics for all the predicted candidate introgression loci. Table S4. Results of chromosomal dxy. Table S5. Results of dxy for all the candidate introgression loci. Table S6. Read depth analyses for all the predicted candidate introgression loci among four focal species. Table S7. Results of D-statistics to examine gene flow between other species and H. m. nanna. Table S8. Gene annotations of 41 candidate introgression loci. Table S9. Autosome versus Z chromosome population genetic statistics. (DOCX 123 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, W., Dasmahapatra, K.K., Mallet, J. et al. Genome-wide introgression among distantly related Heliconius butterfly species. Genome Biol 17, 25 (2016). https://doi.org/10.1186/s13059-016-0889-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13059-016-0889-0