
Abstract
Background
The homologous recombination (HR) repair pathway for DNA damage, particularly the BRCA1 and BRCA2 genes, has become a target for cancer therapy, with poly ADP-ribose polymerase (PARP) inhibitors showing significant outcomes in treating germline BRCA1/2 (gBRCA1/2) mutated breast cancer. Recent studies suggest that some patients with somatic BRCA1/2 (sBRCA1/2) mutation or mutations in HR-related genes other than BRCA1/2 may benefit from PARP inhibitors as well, particularly those with PALB2 mutations. The current analysis aims to evaluate the prevalence of genetic alterations specific to BRCA1, BRCA2, and PALB2 in a large cohort of Taiwanese breast cancer patients through tumor-targeted sequencing.
Methods
A total of 924 consecutive assays from 879 Taiwanese breast cancer patients underwent tumor-targeted sequencing (Thermo Fisher Oncomine Comprehensive Assay v3). We evaluated BRCA1, BRCA2, and PALB2 mutational profiles, with variants annotated and curated by the ClinVAR, the Oncomine™ Knowledgebase Reporter, and the OncoKB™. We also conducted reflex germline testing using either whole exome sequencing (WES) or whole genome sequencing (WGS), which is ongoing.
Results
Among the 879 patients analyzed (924 assays), 130 had positive mutations in BRCA1 (3.1%), BRCA2 (8.6%), and PALB2 (5.2%), with a total of 14.8% having genetic alterations. Co-occurrence was noted between BRCA1/BRCA2, BRCA1/PALB2, and BRCA2/PALB2 mutations. In BRCA1-mutated samples, only p.K654fs was observed in three patients, while other variants were observed no more than twice. For BRCA2, p.N372H was the most common (26 patients), followed by p.S2186fs, p.V2466A, and p.X159_splice (5 times each). For PALB2, p.I887fs was the most common mutation (30 patients). This study identified 176 amino acid changes; 60.2% (106) were not documented in either ClinVAR or the Oncomine™ Knowledgebase Reporter. Using the OncoKB™ for annotation, 171 (97.2%) were found to have clinical implications. For the result of reflex germline testing, three variants (BRCA1 c.1969_1970del, BRCA1 c.3629_3630del, BRCA2 c.8755-1G > C) were annotated as Pathogenic/Likely pathogenic (P/LP) variants by ClinVar and as likely loss-of-function or likely oncogenic by OncoKB; while one variant (PALB2 c.448C > T) was not found in ClinVar but was annotated as likely loss-of-function or likely oncogenic by OncoKB.
Conclusion
Our study depicted the mutational patterns of BRCA1, BRCA2, and PALB2 in Taiwanese breast cancer patients through tumor-only sequencing. This highlights the growing importance of BRCA1/2 and PALB2 alterations in breast cancer susceptibility risk and the treatment of index patients. We also emphasized the need to meticulously annotate variants in cancer-driver genes as well as actionable mutations across multiple databases.
Similar content being viewed by others

Background
Since the discovery of the BRCA1 and BRCA2 genes in 1994 and 1995 [1, 2], the homologous recombination (HR) repair pathway for DNA damage has become a focus area for tumorigenesis and cancer therapy. Preclinical studies in 2005 demonstrated that poly ADP-ribose polymerase inhibitors (PARP) inhibitors selectively target BRCA-deficient cells, a decade after the discovery of the BRCA genes [3]. Nowadays, numerous phase III clinical trials have shown that various PARP inhibitors can improve treatment outcomes in both early and advanced germline BRCA1/2 (gBRCA1/2)-mutant breast cancer, as well as achieve a better quality of life [4,5,6,7], opposed to cytotoxic chemotherapy.
Although PARP inhibitors have shown tremendous treatment outcomes in prolonging progression-free survival for gBRCA1/2-mutant breast cancer and extending overall survival for high-risk early-stage cancer, the prevalence of these mutations is estimated to be only 2–5% in unselected general population [8,9,10,11]. Therefore, it is important to identify patients beyond gBRCA1/2 carriers whose cancers may be sensitive to PARP inhibition, given the limited population of gBRCA1/2 mutations in breast cancer patients.
Studies in prostate and ovarian cancer have suggested that some patients with somatic BRCA1/2 (sBRCA1/2) mutation or mutations in HR-related genes other than BRCA1/2 may benefit from PARP inhibitors [12,13,14,15]. According to the results of the TBCRC 048, an investigator-initiated phase 2 trial to assess Olaparib response in metastatic breast cancer patients with sBRCA1/2 mutations or germline/somatic mutations in HR-related genes other than BRCA1/2, responses were observed only in patients with sBRCA1/2 mutations (objective response rate [ORR] 50%) or gPALB2 mutations (ORR 82%) [16]. The Talazoparib Beyond BRCA (TBB) trial included any solid tumor with germline or somatic mutations in HR-related genes other than BRCA1 and BRCA2 in cohort B. They reported a 31% overall response rate in breast cancer patients. Furthermore, all five breast cancer patients with gPALB2 mutations had treatment-associated tumor regression [17]. Among all HR-related genes, PALB2 is responsible for loading RAD51 onto ssDNA, stimulating RAD51-mediated strand exchange and D-loop formation via the BRCA complex (BRCA1-PALB2-BRCA2-RAD51) [18, 19]. Germline PALB2 mutation is estimated to be present in about 1% of breast cancer patient populations [19,20,21,22] and is known for its remarkable increased risk of breast cancer and pancreatic cancer [22]. Better understanding of the patterns regarding both somatic and germline mutations in BRCA1/2 and PALB2 can lead to improved treatment outcomes for these specific populations.
The epidemiology for breast cancer is quite different between Taiwanese (ethnically Han Chinese origin) and Caucasian populations [23]. The median age of disease onset is younger in Taiwanese breast cancer patients, and young breast cancer patients in Taiwan carried a greater risk for disease progression and a shorter interval to secondary contralateral breast cancer than in Western women [23,24,25]. As the early onset and bilaterality of breast cancer are more likely to be related to genetic predisposing factors [26, 27], it is important to identify potential genetic alterations underpinning Taiwanese patients.
The clinical characteristics and outcomes of gBRCA1/2-mutant Taiwanese breast cancer patients had been studied in various studies, which revealed a prevalence rate of 3.8% for BRCA pathogenic variants in the Taiwanese breast cancer cohort, with a higher proportion among triple-negative breast cancer (TNBC), and an increased risk in contralateral breast cancer [28,29,30,31]. However, limited tumor-targeted sequencing analysis had been reported.
Our previous study evaluated mutational profiles in Taiwanese breast cancers by tumor-targeted sequencing. The latest results from the study of 621 enrolled breast cancer patients showed that HR-related genes were altered in 122 (19%) of the population. Other than BRCA1/2, the most prevalent HR-related mutant genes were ARID1A (7%), PALB2 (7%), and PTEN (6%). In total, 164 (25%) of the 648 Taiwanese breast cancer samples had at least one mutation among the HR-related genes. BRCA1 and BRCA2 had affected 3% and 5% of the study population, respectively, and were collectively altered in 6%, with co-occurrence of BRCA1/2 in 7 breast cancers [32]. In current analysis, we further extended the number of enrolled subjects and evaluated the prevalence of genetic alterations specific in BRCA1, BRCA2, and PALB2, with additional annotation from well-established database.
Methods
The objective of this study was to assess the prevalence of BRCA1, BRCA2, and PALB2 mutations in Taiwanese breast cancer patients using targeted sequencing using tumor-only samples. The Institutional Review Board of Taipei Veterans General Hospital approved the study (protocol number: 2018-09-007A). Written informed consent was obtained from all participants prior to enrollment.
Study population and patient recruitment
The full protocol of the VGH-TAYLOR study (Veterans General Hospital Taipei—Yung-Ling foundation sinO-canceR study, ClinicalTrials.gov: NCT04626440), which focuses on the heterogeneity of Taiwanese breast cancer patients and included initial targeted sequencing of 380 and 648 assays, has been described elsewhere [32,33,34].
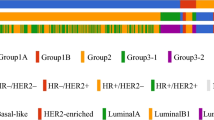
All patients had been evaluated by their clinicians upon diagnosis. The treatment options were determined by physicians according to patients’ characteristics, molecular subtypes, and clinical stages. The concept of share-decision making (SDM) was fully informed and the process of SDM was carried out before the initiation of treatment. Enrolled subjects were subsequently assigned into Group 1 [planned to receive surgery as the first-line treatment and followed by adjuvant therapy, Group 2 [planned to receive neoadjuvant therapy as the first-line treatment and followed by surgery], and Group 3 [diagnosed with de novo and treatment naïve stage IV breast cancer, or stage IV breast cancer with recurrence beyond three years after surgery] (Fig. 1). Three years of enrollment and 4 years of follow-up after enrollment were planned.

Study protocol of the VGH-TAYLOR study
Collection of breast cancer samples and clinical information
Informed consent was obtained from all participants after a thorough explanation by investigators (CCH and LMT). Clinical parameters were assessed by immunohistochemistry (IHC) with estrogen receptor (ER) and progesterone receptor (PR) positivity defined as at least 1% of tumor cells exhibiting nuclear staining. Hormone receptor positivity was defined as either ER-positive or PR-positive. Patients with human epidermal growth factor receptor II (HER2) testing scored as IHC 3+ (positive) or 2+ (equivocal) and with fluorescence in situ hybridization (FISH)-confirmed amplification were considered HER2-positive. All patients received treatment according to the contemporary practice guidelines of the Comprehensive Breast Health Center at Taipei Veterans General Hospital, which were based on the NCCN and St. Gallen guidelines [35, 36].
Tumor-only targeted sequencing
The details of tumor only targeted sequencing have been described elsewhere [32, 33]. For next-generation sequencing (NGS) library preparation, the Ion Torre Oncomine™ Comprehensive Assay v3 (Thermo Fisher Scientific, Waltham, MA) was used, enabling the detection of 161 cancer-related genes and identification of single nucleotide variants (SNVs), copy number variations (CNVs), gene fusions, and indels.
A total of 879 consecutive breast cancer patients, representing 924 assays, were enrolled in the study. Formalin-fixed paraffin-embedded specimens were assayed with sequencing data analyzed using the Torrent Suite software. The data were further aligned and annotated by the Ion Reporter with the default Oncomine BRCA (5.12) filter applied. Software versions were Torrent Suite (v5.10.0), Ion Reporter (v5.10), Coverage Analysis (v5.10.0.3), SampleID (v5.10.0.1), and VariantCaller (v5.10.0.18). In current study, we focused on BRCA1, BRCA2, and PALB2 mutations.
Variant calling and annotation
Variants were further filtered with the Oncomine™ Knowledgebase Reporter (Thermo Fisher Scientific). To further correct spurious findings due to transethnic discrepancies, the online VariED tool was consulted to filter out Taiwan Biobank polymorphisms [37]. The mutational consequences of filtered variants were determined with the ClinVAR database, a freely accessible public archive of reports on the relationships between human variations and phenotypes [38].
We also used the OncoKB™ to assess the clinical implications of variants that were not identified or confirmed by the Oncomine™ Knowledgebase Reporter or the ClinVAR. The OncoKB™ is a knowledge base for precision oncology that offers information on the therapeutic implications of particular genetic alterations in cancer. The database is curated by a group of oncologists, researchers, and bioinformaticians who meticulously assess the available evidence related to each variant, including clinical trial results, FDA approvals, and guidelines from professional organizations. The OncoKB™ also provides annotations for the functional and structural impact of each variant, as well as the level of evidence supporting the annotation. By using the OncoKB™ to evaluate variants that other databases have not identified or confirmed, we can gain a more thorough understanding of the potential clinical implications of these variants [39]. Tumor sequencing variant annotations were functionally annotated using the SNPnexus [40].
To ensure quality control, over 90% of amplicons with > 100× coverage were used as a parameter. Additionally, a minimum coverage of 250 × was deemed acceptable for detecting SNVs and indels with allele fractions of 10% and 20%, respectively [41, 42]. Actionability was determined based on the joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists [43].
Reflex germline testing
When patients' tumor samples tested positive for pathogenic or likely pathogenic variants as identified by ClinVar, reflex germline testing was conducted using whole exome sequencing (WES) through blood sample collection. In some cases, whole genome sequencing (WGS) may have been utilized prior to WES. Both WGS and WES are essential in detecting germline mutations linked to a variety of genetic disorders. WGS offers an exhaustive analysis by sequencing the entire genome, allowing for the identification of mutations across both coding and non-coding regions, as well as the detection of structural variations and copy number variations for a detailed examination. In comparison, WES, while more cost-effective and quicker, concentrates on coding regions or exons, potentially overlooking certain mutations that would be detected by WGS.
Alternative methods for determining germline and somatic mutations
To distinguish germline from somatic mutations with tumor-only sequencing, we employed algorithms including the LOH-germline inference calculator (LOHGIC) [44] and the somatic-germline-zygosity (SGZ) method as alternatives for patients not ready for germline testing [45]. We integrated both methods, considering tumor purity, allele frequency, ploidy, and copy number variations for mutation classification. Only tumor purity and allele frequency were considered as our cohort showed no copy number variations for BRCA1/2 and all samples were diploid. We simplified both the LOHGIC and the SGZ by directly comparing observed allele frequencies against those expected in germline and somatic mutations.
For germline mutations, expected in both tumor and normal tissues, the allele frequency should be around 50% in pure normal samples and vary in tumor samples due to loss of heterozygosity. In contrast, somatic mutations, present only in tumor cells, will have an allele frequency dependent on tumor purity, typically near 50% in pure tumor samples but lower in samples with less tumor purity intertwined with adjacent normal tissues.
Result
Enrolled Taiwanese breast cancers
In current study, we presented the updated results of 924 Thermo Fisher (TMO) OCP v3 assays obtained from 879 breast cancer patients, with 43 patients being assayed twice, and 1 patient being assayed thrice from the VGH-TAYLOR study. The patient distributions of clinical scenarios were as follows: Group 1A (surgery first, n = 578, 65.8%); Group 1B (recurrence within 3 years, n = 22, 2.5%); Group 2 (neoadjuvant therapy, n = 117, 13.3%); Group 3–1 (de novo stage IV, n = 40, 4.6%); and Group 3–2 (recurrence beyond 3 years, n = 67, 7.6%). In addition, there were samples of 55 patients (6.3%) from the retrospective biobank cohort. Figure 2 displays the distributions of IHC results and molecular subtypes.

Distributions of clinical variables across study groups
Distribution and patterns of BRCA1, BRCA2, and PALB2 mutations
Of the 924 assays, 281 were positive for mutant BRCA1, BRCA2 and PLAB2 in 130 patients. These mutations impacted 3.1% (27 patients), 8.6% (76 patients), and 5.2% (46 patients), respectively. In total, genetic alterations were noted in 14.8% (130 patients). Details of distribution and patterns of BRCA1, BRCA2, and PALB2 mutations were portrayed in Table 1, Figs. 3, 4, and 5.

Mutation map of BRCA1 gene

Mutation map of BRCA2 gene

Mutation map of PLAB2 gene
The BRCA1 mutation cohort is associated with a higher proportion of advanced stages compared to those without. Additionally, the BRCA2-mutant patients show a higher incidence of family history of ovarian cancer, resulting in a significant difference in the number of mutant patients with a family history of ovarian cancer (Table 1).
In terms of IHC phenotypes, 13 (2.3%) of the BRCA1 mutant breast cancers were HR+/HER2−, 3 (3.3%) were HR+/HER2+, 2 (2.6%) were HR−/HER2+, and 9 (6.9%) were HR−/HER2−. For BRCA2 mutated cases, 51 (9.0%) were HR+/HER2−, 9 (9.9%) were HR+/HER2+, 6 (7.9%) were HR−/HER2+, and 9 (6.9%) were HR-/HER2-. Among the PALB2 mutated patients, 30 (5.3%) were HR+/HER2−, 4 (4.4%) were HR+/HER2+, 3 (3.9%) were HR−/HER2+, and 9 (6.9%) were HR−/HER2− (Table 1).
The study revealed the co-occurrence of BRCA1/2 in 13 breast cancer samples (log2 odds ratio: > 3, p-value < 0.001, and q-value < 0.001). Additionally, the co-occurrence of BRCA1 and PALB2 was found in 8 samples (log2 odds ratio: > 3, p-value < 0.001, and q-value < 0.001), and the co-occurrence of BRCA2 and PALB2 was found in 8 samples (log2 odds ratio: 2.401, p-value < 0.001, and q-value < 0.001). These findings are presented in Table 2.
Among these patients, 5 had both BRCA1/2 mutations, 1 had both BRCA2 and PALB2 mutations, none had both BRCA1 and PALB2 mutations, and 7 had all three mutations. The list of these patients and the variants that they harbored are shown in Table 3. Twenty-four patients had two or more variants. Of those, 10 patients had dual variants, two had triple variants, one had four variants, three had five variants, one had seven variants, one had eight variants, one had nine variants, one had 15 variants, one had 16 variants, one had 19 variants, one had 24 variants, and one had 25 variants. (Table 3).
Among all the variants, p.I887fs was observed 30 times in PALB2-mutant assays. For BRCA2-mutant assays, p.S2186fs, p.V2466A, and p.X159_splice were observed 5 times, while p.T912fs and p.E33* were observed 3 times each. In BRCA1-mutated assays, only p.K654fs was observed three times, while other variants were observed no more than twice. It should be noted that although p.N372H was observed 26 times in BRCA2-mutated assays, it has been confirmed to be a benign variant. The list of those recurrent variants with clinical implications is displayed in Fig. 6.

Amino acid changes, repeatedly mutated, with evidence of clinical implication, validated by the OncoKB™
Functional annotations and clinical implications
The study analyzed various genetic variants and identified 176 amino acid (AA) changes, and the characteristics stratified by genes and the clinical implications are listed in Table 2. There were four variant that did not notice any AA change, and three novel splice site variants (BRCA1 c.5256+1G > A, BRCA1 c.5215+1G > A, and BRCA2 c.-38-3CAG > C) were identified, all listed in Table 4. Although these novel variants had no AA change, they were classified as Pathogenic/Likely pathogenic (P/LP) by the ClinVAR database. Notably, 60.2% (106) of the discovered AA changes were not documented in either ClinVAR or the Oncomine™ Knowledge database. Using the OncoKB™ for annotation, 171 (97.2%) AA changes were found to have clinical implications. Half of all missense mutations without clinical implications (4 out of 8) were deemed insignificant, and 23.1% (3 out of 13) of AA changes not recorded in the ClinVAR or the Oncomine™ Knowledge database was not considered clinically relevant. The BRCA2 mutation cohort exhibited the highest proportion of P/LP variants. This cohort also contained the only benign variant identified and had the highest number of AA changes without clinical implications, as per the OncoKB™ (Table 5).
Reflex germline testing
In our study, reflex germline testing was conducted for patients until November 20, 2023. These procedures are summarized in Table 6, which concentrates on WGS and WES analyses for individuals who had positive results from tumor-only sequencing. Specifically, 48 cases, constituting 36.9%, were identified with pathogenic or likely pathogenic variants via tumor-targeted sequencing, as classified by the ClinVar database, and were subsequently recalled for further investigation through WGS or WES.Among 130 cases examined, 7 cases (5.4%) completed WGS uncovering crucial genetic variations. None harbored germline mutations in BRCA1/2 and PALB2. Another 20 cases (15.4%) were reached and 9 had completed WES (6.9%). The study group encountered enormous challenges including loss of follow-up and 14 were deceased.
The combined results of whole genome sequencing (WGS) and whole exome sequencing (WES) have provided in-depth insights into the origins of identified variants. Notably, 4 cases (3.1%) exhibited pathogenic germline mutations. Reflex germline testing results showed that three variants (BRCA1 c.1969_1970del, BRCA1 c.3629_3630del, BRCA2 c.8755-1G > C) were classified as Pathogenic/Likely pathogenic (P/LP) by ClinVar and as likely loss-of-function or likely oncogenic by OncoKB. Meanwhile, one variant (PALB2 c.448C > T) was not listed in ClinVar, but OncoKB annotated it as likely loss-of-function or likely oncogenic. Additionally, there were 2 cases (1.5%) of germline mutations with uncertain significance, and 5 cases (3.8%) with benign germline alterations, which emphasize the genetic intricacy involved in the development of breast cancer. The result for all the variants from reflex germline testing is presented in Additional file 1: Table S1.
Alternative methods for determining germline and somatic mutations
Table 7 illustrates the results of LOHGIC and SGZ analyses. Among the 281 samples, 40 were identified as germline mutations and 169 samples (60.1%) were of somatic origin. Borderline cases comprised 26 samples (9.3%). Lastly, there were 46 samples, (16.4%) unclassifiable due to missing data in tumor purity.
Discussion
Our study presents the one of the largest cohorts of breast cancer patients with BRCA1, BRCA2, and PALB2 mutations detected through tumor-only sequencing in Taiwan. We analyzed 924 Thermo Fisher OCP v3 assays from 879 breast cancer patients, dividing them into different groups based on their clinical scenarios. Out of the 924 assays conducted, 281 were positive for mutant genes in 130 patients, with BRCA1, BRCA2, and PALB2 mutations identified in 27 patients (3.1%), 76 patients (8.6%), and 46 patients (5.2%), respectively. Overall, genetic alterations were observed in 14.8% of the assays. The high detection rate of breast cancer susceptibility genes could lead to more patients undergoing germline testing and receiving appropriate treatments.
Genetic mutations and variants
Recent research has revealed a significant occurrence of harmful variants in three key genes associated with breast cancer risk, as analyzed through tumor genomic profiling. In a significant study by the Breast Cancer Association Consortium in 2021 involving over 60,000 breast cancer cases and 53,000 controls, sequencing was conducted on 34 potential risk genes. The standout genes were BRCA1, BRCA2, and PALB2. Variants leading to truncating/incomplete proteins in these genes correlated with an increased breast cancer risk, which were statistically significant (all P-values less than 0.0001), underscoring their crucial role in the genetic landscape of breast cancer. These genes are also recognized markers for determining the eligibility for PARP inhibitor therapies, warranting further investigation into their contribution to breast cancer risk [46]. The frequency of mutations in these genes varies by the method of testing and the population studied. Research focusing on Asian patients with BRCA1/2 mutations found a wide prevalence range, with BRCA1 mutations present in 2.3–42% and BRCA2 mutations in 2.3–11.4% of the group [47]. In Taiwan, a study showed a 3.8% prevalence of these mutations in an unselected patient population [30]. Reports on PALB2 mutations in Asian populations are scarce; however, a thorough global review indicated that PALB2 pathogenic variants occur in 0.9% to 3.2% [19].
While germline mutations in breast cancer susceptibility genes have been extensively studied, reports of somatic mutations are rare. However, with the advent of tumor-targeted sequencing, we can explore both germline and somatic mutations simultaneously. In 2022, the Dana-Farber/Harvard Cancer Center conducted a tumor-targeted sequencing study across a broad range of malignancies on 7575 patients, which included 1514 breast cancer patients. If a patient was identified with any P/LP variants of BRCA1, BRCA2, or PALB2 within the tumor, clinical germline testing (CGT) would be performed. The study found that BRCA1 and BRCA2 mutations were present in 2.5% and 3.7% of breast cancer patients, respectively, while PALB2 mutations were present in 0.6% of then. Out of all P/LP variants from tumor-sequencing, 70.5% were confirmed as germline mutations [48].
The study analyzed various genetic variants and identified 176 amino acid (AA) changes. All BRCA1, BRCA2, and PLAB2 mutation information was curated and confirmed by the ClinVAR, the Oncomine™ Knowledgebase Reporter, and the OncoKB™. There were 4 variants that did not have AA changes reported, and 3 of them were novel variants (BRCA1 c.5256+1G > A, BRCA1 c.5215+1G > A, and BRCA2 c.-38-3CAG > C), which were reported the first time in this Taiwanese cohort. The most common BRCA1 mutation was p.K654fs (c.1960_1961insG), which is a frameshift insertion and deleterious mutation (three cases). Although not recorded by the ClinVAR or the ACMG 73 genes the OncoKB™ has confirmed its clinical implication. The most common BRCA2 mutations were p.N372H (c.1114A > C, 26 cases), p.S2186fs (c.6556_6557insA; 5 cases), p.V2466A (c.7397T > C; 5 cases), and p.X159_splice (c.476−2A > G/c or 476−3C > T; 5 cases). The variant p.N372H was initially reported in 2000 and is one of the common non-synonymous polymorphisms [49, 50]. It has been a research focus in the scientific community and has drawn increasing attention [50,51,52,53,54,55,56,57,58]. A meta-analysis of 22 studies, involving 22,515 cases and 22,388 controls, found no significant association between the BRCA2 p.N372H polymorphism and breast cancer risk. This suggests that the BRCA2 p.N372H allele may be non-pathogenic. Although p.S2186fs is not annotated by the ClinVAR or listed in the ACMG 73 genes, it is a frameshift insertion and deleterious mutation with clinical implications. This has been confirmed by the OncoKB™. For p.V2466A, the ClinVAR provides conflicting interpretations, with some considering it a benign entity. Regarding p.X159_splice, it is interesting to note that there are two different coding variants being recorded. c.476-3C > T has conflicting implications and has been seen in patients from Central/Eastern Europe. In contrast, c.476-2A > G has been ascertained as pathogenic and has been mentioned in Italian and Chinese population. The most common PALB2 mutation observed was p.I887fs (c.2659_2660delAT), a deleterious frameshift mutation that was observed in 30 patients. Although this mutation is not listed in the ClinVAR or ACMG 73 genes list, its clinical implications have been approved by the OncoKB™.
Gene–gene interactions
The study revealed a significant tendency of co-occurrence between BRCA1/2, BRCA1-PALB2, and BRCA2-PALB2 mutations. It should be noted that 17 samples were discarded due to missing values in at least one of the interrogated genes, preventing analysis of mutual exclusivity. Constructing a gene–gene interaction (GGI) network is important for understanding breast carcinogenesis, as single gene or protein alterations are not sufficient to induce cancer. Rather, the interactions with other genes or microenvironment play a key role. A large-scale study on the interaction between genes was conducted on European Non-Small Cell Lung Cancer (NSCLC) risk, using a total of 445,221 participants from various projects [59]. The study found important gene–gene interactions in the 5p15.33 and 6p21.32 regions, which can be used to improve lung cancer screening models.
It was found that BRCA1 interacts with RAD51 to play a role in DNA repair, while BRCA2 co-localizes with RAD51 and BRCA1, indicating a similar function [60]. PALB2, first reported by Xia et al. in 2006 [61], plays a fundamental role in HR. It acts as a bridging molecule that connects the BRCA complex (BRCA1-PALB2-BRCA2-RAD51) and facilitates the function of RAD51, a protein that is vital for strand invasion during HR [18, 19].
A large-scale mutational analysis in 7,325 individuals identified four interactions between mutations in the breast cancer susceptibility genes [62]. These interactions include ATM and CHEK2 with BRCA1 and BRCA2, ATM and BRCA, CHEK2 and BRCA1/BRCA2 combined, and CHEK2 and BRCA1 or BRCA2. The results show a lower risk of breast cancer than that predicted by the multiplicative product of the constituent risks. These findings likely reflect functional relationships between the encoded proteins in DNA repair and have important implications for models of disease predisposition and clinical translation.
Currently, limited studies have addressed the gene–gene interaction among BRCA1, BRCA, and PALB2 in real-world settings of large-scale breast cancer gene analysis. Our results may shed light on exploring the association between breast cancer susceptibility genes and the possibility of creating a genetic panel for predicting and prognosing hereditary breast cancer.
Clinical features
The BRCA1 mutation cohort has a higher proportion of advanced-stage disease compared to others, which may be due to various reasons. One possible explanation is that patients with BRCA1 mutations are more likely to have triple-negative breast cancer [63], which is associated with a higher risk of distant recurrence [64, 65]. Another possible reason is that BRCA1 mutations may be associated with a higher likelihood of developing bilateral breast cancer, which could increase the risk of disease progression [66]. Additionally, patients with BRCA1 mutations are more likely to develop breast cancer at a younger age, when the disease may be more aggressive and therefore more likely to be diagnosed at an advanced stage [67, 68]. Despite having more advanced disease, evidence is mixed as to whether BRCA-associated breast cancer has poorer outcomes. Some studies have shown that carriers of a BRCA1 mutation have worse overall survival [69], while others have found no significant difference in outcomes [70].
Clinical implications and practice of tumor-targeted sequencing
Nowadays, tumor-only targeted sequencing has gained increasing attention. One advantage of tumor-only testing is that it can reveal P/LP variants in genes associated with cancer predisposition and potential therapeutics with a higher level of coverage. Identification of mutations through tumor-only sequencing may lead to reflex germline testing, which can identify individuals at an increased risk for cancer and allow for early detection and intervention.
Recent studies have indicated that direct tumor sequencing could be more advantageous than solely testing for inherited mutations. The comprehensive tumor sequencing effort followed by clinical genetic testing at Dana-Farber/Harvard Cancer Center has not only highlighted the commonness of pathogenic/likely pathogenic mutations in genes such as BRCA1, BRCA2, and PALB2, but has also underscored the significance of this targeted sequencing approach [48]. They found that patients with BRCA mutations were more likely to undergo CGT than those without. Interestingly, over half (52.9%) of the tumor-identified P/LP patients did not meet any personal or family history criteria for CGT. Additionally, 32.7% of patients with BRCA1/2 or PALB2 P/LP variants did not have any other clinical indication for germline testing. Nonetheless, 70.5% of P/LP variants identified through CGT were germline origin. These results show the potentiality of tumor-only sequencing in detecting P/LP mutations in cancer predisposition genes across malignancies. Furthermore, they highlighted the necessity of expanding the indications for CGT beyond traditional criteria. A significant proportion of patients with these mutations may not have any personal or family history of cancer, which may have a significant impact on cancer risk assessment, surveillance, and treatment decisions in the future. Before universal germline and tumor sequencing becomes feasibility, tumor-targeted sequencing seems to be a reasonable choice for personalized therapy.
Both germline and somatic alterations can affect treatment decisions and outcomes. For breast cancer patients, the results of the TBCRC-048 and TBB trials suggested further exploration of PARP inhibitors in metastatic or advanced breast cancers with HR-associated mutations beyond BRCA1 and BRCA2 [16, 17]. Identifying additional biomarkers to expand this treatment in somatic BRCA1/2-mutant or HR-related-gene-mutant advanced breast or ovarian cancers could significantly benefit patients who would otherwise receive chemotherapies as the only regimen. These efforts may reveal a patient population that would benefit from targeted therapy, improving patient outcomes and reducing the complications associated with cytotoxic chemotherapy.
Reflex germline testing and other alternative methods for identifying the origin of variants
The significance and clinical applicability of tumor-targeted sequencing is well acknowledged, yet the unique importance of reflex germline testing cannot be overemphasized. We are continuously expanding cases for such testing. However, the extent of reflex testing conducted to date is still limited, with only 7 cases underwent WGS and 9 with WES (Table 6). This has led us to explore alternative methods to determine whether the reported mutation is germline or somatic origin.
The refined LOHGIC and SGZ methodologies are designed to assess three crucial factors: tumor purity (the proportion of cancer cells in a sample), allele frequency (the incidence of mutations in DNA sequencing reads), and confirmation of a diploid genome (maintaining two copies of each gene). These methods are crucial for differentiating between somatic mutations, which occur in somatic (non-reproductive) cells, and germline mutations, which are inherited. Although these algorithms are useful, they have their limitations; in an analysis of 130 cases, 10 could not be conclusively classified as either germline or somatic due to varying origins of the mutations. Nonetheless, there was a noticeable pattern, with 60% of the mutations being identified as somatic. This observation highlights the imperative for more refined techniques to accurately determine the origins of mutations.
Annotation and curation
In the study, 60.2% (106) of the reported AA changes were not documented in either the ClinVAR or the commercial the Oncomine™ Knowledge base Reporter. However, when using the OncoKB™ for annotation, 171 AA changes were found to have clinical implications. Nearly half (N = 80, 45.5%) of these variants with AA changes were due to frameshift deletions or insertions, which were all clinically significant according to the OncoKB™. Interestingly, half of missense mutations without clinical implications (4 out of 8) were deemed insignificant, and 23.1% (3 out of 13) of AA changes not recorded in the ClinVAR or the Oncomine™ Knowledge base Reporter were not considered clinically relevant. Regarding the 40 splice site mutations, 62.5% (25) were not reported as deleterious mutations by the Oncomine™ Knowledgebase Reporter, but the ClinVAR identified 60% (15 out of 25) of these mutations as P/LP variants. Furthermore, the OncoKB™ identified that 85% (34 out of 40) of all splice site mutations to be clinically significant.
Accurate interpretation of genetic variants is critical in both clinical and research settings. Before reporting detected variants, appropriately trained and certified molecular diagnostic procedures must be carefully carried out in the context of clinical scenarios, including histologic features [43]. However, the classification criteria can vary between submitters, and the evidence for a particular variant may be conflicting, leading to difficulties in an unbiased interpretation. Numerous studies have explored potential indicators for reinterpreting pathogenic variants within specific databases, as well as across distinct platforms. For example, in a recent comparison of the RefSeq and Gencode human gene databases, only 27.5% of transcripts annotated in the Gencode are shared by the RefSeq [71]. Whiffin et al. selected 43 variants from the ClinVAR classified as P/LP, which were not rare enough in at least one of the Exome Aggregation Consortium populations [72]. Their analysis showed that 42 of these variants should be considered variants of uncertain significance (VUS) instead of P/LP. Xiang et al. analyzed common P/LP variants in the ClinVAR database and identified indicators associated with reclassification, indicating missing opportunities due to misinterpretation [73]. The study selected 217 variants in 173 genes for manual interpretation according to guidelines, with 40% of which downgraded to benign or VUS, while 2% identified as more likely risk alleles. Inappropriate classification was associated with low-rank, older annotation, higher allele frequency, and collection through methods other than clinical testing. It is important to note that the reinterpretation of cancer predisposition genes requires a multidisciplinary effort involving clinicians, genetic counselors, bioinformaticians, and researchers [74].
In summary, the reinterpretation of cancer predisposition genes due to annotation inconsistencies among distinct database is an important issue that requires careful consideration and multidisciplinary collaboration. The use of updated annotation and rigid guideline follow-up and the integration of multiple types of genomic data can help improve the accuracy of cancer risk assessment and inform personalized prevention and treatment strategies.
Strength and limitations
This study analyzing BRCA1, BRCA2, and PALB2 mutations through tumor-targeted sequencing boosts several notable strengths. Firstly, it is the first large-scale analysis of its kind in Taiwan and Asia, involving a substantial number of breast cancer cases. Additionally, the study benefits from the availability of data on family history, molecular subtypes, and early or advanced breast cancer status. Moreover, by utilizing updated annotation databases and guidelines during interpretation and annotation, the study achieves enhanced comprehensiveness and accuracy in its results.
There are several limitations to consider. First, additional germline sequencing has been conducted with compromised recalled rates. Second, the sample size varied among different clinical groups, which may pose a challenge. Third, current study confined to only three genes, potentially narrowing the assessment of the genetic landscape. Lastly, a more in-depth evaluation of clinical outcomes and subgroup analyses, incorporating clinical-pathological factors and treatment regimens, is essential for a deeper understanding of Taiwanese breast cancer and should be conducted in future studies.
Conclusion
In conclusion, our study reported a cohort of Taiwanese breast cancers harboring mutations in BRCA1, BRCA2, and PALB2 through tumor-only sequencing, which underscores the impact of BRCA1/2 and PALB2 on breast cancer risk and potential therapeutic opportunities. Tumor-only sequencing has enabled a greater number of patients to uncover their genomic alterations, which offers additional insights for management strategies. These include recommendations for germline testing and the prospective utilization of PARP inhibitors to augment treatment efficacy. Nonetheless, supplementary germline testing remains critical, and investigating alternative methods for distinguishing whether variants are germline or somatic origin are invaluable.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- HR:
-
Homologous recombination
- PARP inhibitors:
-
Poly ADP-ribose polymerase inhibitors
- gBRCA1/2 :
-
Germline BRCA1/2
- sBRCA1/2 :
-
Somatic BRCA1/2
- ORR:
-
Objective response rate
- TNBC:
-
Triple-negative breast cancer
- SDM:
-
Share-decision making
- IHC:
-
Immunohistochemistry
- ER:
-
Estrogen receptor
- PR:
-
Progesterone receptor
- HER2:
-
Human epidermal growth factor receptor II
- NGS:
-
Next-generation sequencing
- SNVs:
-
Single nucleotide variants
- CNVs:
-
Copy number variations
- TMO:
-
Thermo Fisher
- AA:
-
Amino acid
- P/LP:
-
Pathogenic/Likely pathogenic
- GGI:
-
Gene–gene interaction
- NSCLC:
-
Non-Small Cell Lung Cancer
- CGT:
-
Clinical germline testing
- VUS:
-
Variants of uncertain significance
- WES:
-
Whole exome sequencing
- WGS:
-
Whole genome sequencing
- LOHGIC:
-
LOH-germline inference calculator
- SGZ:
-
Somatic-germline-zygosity
References
Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71. https://doi.org/10.1126/science.7545954.
Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378(6559):789–92. https://doi.org/10.1038/378789a0.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21. https://doi.org/10.1038/nature03445.
Geyer CE Jr, Garber JE, Gelber RD, Yothers G, Taboada M, Ross L, et al. Overall survival in the OlympiA phase III trial of adjuvant Olaparib in patients with germline pathogenic variants in BRCA1/2 and high-risk, early breast cancer. Ann Oncol. 2022;33(12):1250–68. https://doi.org/10.1016/j.annonc.2022.09.159.
Robson ME, Tung N, Conte P, Im SA, Senkus E, Xu B, et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann Oncol. 2019;30(4):558–66. https://doi.org/10.1093/annonc/mdz012.
Litton JK, Hurvitz SA, Mina LA, Rugo HS, Lee KH, Gonçalves A, et al. Talazoparib versus chemotherapy in patients with germline BRCA1/2-mutated HER2-negative advanced breast cancer: final overall survival results from the EMBRACA trial. Ann Oncol. 2020;31(11):1526–35. https://doi.org/10.1016/j.annonc.2020.08.2098.
Diéras V, Han HS, Kaufman B, Wildiers H, Friedlander M, Ayoub JP, et al. Veliparib with carboplatin and paclitaxel in BRCA-mutated advanced breast cancer (BROCADE3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21(10):1269–82. https://doi.org/10.1016/S1470-2045(20)30447-2.
Anglian Breast Cancer Study Group. Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Anglian Breast Cancer Study Group. Br J Cancer. 2000;83(10):1301–8. https://doi.org/10.1054/bjoc.2000.1407.
Malone KE, Daling JR, Doody DR, Hsu L, Bernstein L, Coates RJ, et al. Prevalence and predictors of BRCA1 and BRCA2 mutations in a population-based study of breast cancer in white and black American women ages 35 to 64 years. Cancer Res. 2006;66(16):8297–308. https://doi.org/10.1158/0008-5472.CAN-06-0503.
John EM, Miron A, Gong G, Phipps AI, Felberg A, Li FP, West DW, et al. Prevalence of pathogenic BRCA1 mutation carriers in 5 US racial/ethnic groups. JAMA. 2007;298(24):2869–76. https://doi.org/10.1001/jama.298.24.2869.
Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23(4):517–25. https://doi.org/10.1038/nm.4292.
Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697–708. https://doi.org/10.1056/NEJMoa1506859.
Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18(1):75–87. https://doi.org/10.1016/S1470-2045(16)30559-9.
Abida W, Campbell D, Patnaik A, Shapiro JD, Sautois B, Vogelzang NJ, et al. Non-BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: analysis from the Phase II TRITON2 study. Clin Cancer Res. 2020;26(11):2487–96. https://doi.org/10.1158/1078-0432.CCR-20-0394.
Mateo J, Porta N, Bianchini D, McGovern U, Elliott T, Jones R, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020;21(1):162–74. https://doi.org/10.1016/S1470-2045(19)30684-9.
Tung NM, Robson ME, Ventz S, Santa-Maria CA, Nanda R, Marcom PK, et al. TBCRC 048: phase II study of Olaparib for metastatic breast cancer and mutations in homologous recombination-related genes. J Clin Oncol. 2020;38(36):4274–82. https://doi.org/10.1200/JCO.20.02151.
Gruber JJ, Afghahi A, Timms K, DeWees A, Gross W, Aushev VN, et al. A phase II study of talazoparib monotherapy in patients with wild-type BRCA1 and BRCA2 with a mutation in other homologous recombination genes. Nat Cancer. 2022;3(10):1181–91. https://doi.org/10.1038/s43018-022-00439-1.
Wu S, Zhou J, Zhang K, Chen H, Luo M, Lu Y, et al. Molecular mechanisms of PALB2 function and its role in breast cancer management. Front Oncol. 2020;10:301. https://doi.org/10.3389/fonc.2020.00301.
Toh M, Ngeow J. Homologous recombination deficiency: cancer predispositions and treatment implications. Oncologist. 2021;26(9):e1526–37.
Erkko H, Xia B, Nikkilä J, Schleutker J, Syrjäkoski K, Mannermaa A, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007;446(7133):316–9. https://doi.org/10.1038/nature05609.
Foulkes WD, Ghadirian P, Akbari MR, Hamel N, Giroux S, Sabbaghian N, et al. Identification of a novel truncating PALB2 mutation and analysis of its contribution to early-onset breast cancer in French-Canadian women. Breast Cancer Res. 2007;9(6):R83. https://doi.org/10.1186/bcr1828.
Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkäs K, Roberts J, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371(6):497–506. https://doi.org/10.1056/NEJMoa1400382.
Shen YC, Chang CJ, Hsu C, Cheng CC, Chiu CF, Cheng AL. Significant difference in the trends of female breast cancer incidence between Taiwanese and Caucasian Americans: implications from age-period-cohort analysis. Cancer Epidemiol Biomarkers Prev. 2005;14(8):1986–90. https://doi.org/10.1158/1055-9965.EPI-04-0932.
Kuo WH, Yen AM, Lee PH, Hou MF, Chen SC, Chen KM, et al. Incidence and risk factors associated with bilateral breast cancer in area with early age diagnosis but low incidence of primary breast cancer: analysis of 10-year longitudinal cohort in Taiwan. Breast Cancer Res Treat. 2006;99(2):221–8. https://doi.org/10.1007/s10549-006-9194-z.
Kuo WH, Yen AM, Lee PH, Chen KM, Wang J, Chang KJ, et al. Cumulative survival in early-onset unilateral and bilateral breast cancer: an analysis of 1907 Taiwanese women. Br J Cancer. 2009;100(4):563–70. https://doi.org/10.1038/sj.bjc.6604898.
Malone KE, Begg CB, Haile RW, Borg A, Concannon P, Tellhed L, et al. Population-based study of the risk of second primary contralateral breast cancer associated with carrying a mutation in BRCA1 or BRCA2. J Clin Oncol. 2010;28(14):2404–10. https://doi.org/10.1200/JCO.2009.24.2495.
Lynch HT, Silva E, Snyder C, Lynch JF. Hereditary breast cancer: part I. Diagnosing hereditary breast cancer syndromes. Breast J. 2008;14(1):3–13. https://doi.org/10.1111/j.1524-4741.2007.00515.x.
Kuo WH, Lin PH, Huang AC, Chien YH, Liu TP, Lu YS, et al. Multimodel assessment of BRCA1 mutations in Taiwanese (ethnic Chinese) women with early-onset, bilateral or familial breast cancer. J Hum Genet. 2012;57(2):130–8. https://doi.org/10.1038/jhg.2011.142.
Wang YA, Jian JW, Hung CF, Peng HP, Yang CF, Cheng HS, et al. Germline breast cancer susceptibility gene mutations and breast cancer outcomes. BMC Cancer. 2018;18(1):315. https://doi.org/10.1186/s12885-018-4229-5.
Chian J, Sinha S, Qin Z, Wang SM. BRCA1 and BRCA2 Variation in Taiwanese General Population and the Cancer Cohort. Front Mol Biosci. 2021;21(8):685174. https://doi.org/10.3389/fmolb.2021.685174.
Lin PH, Chen SC, Tseng LM, Chang KJ, Huang AC, Cheng KC, et al. Impact of BRCA mutation on the survival and risk of contralateral breast cancer in Asian breast cancer patients. Breast Cancer Res Treat. 2022;192(3):629–37. https://doi.org/10.1007/s10549-021-06446-7.
Huang CC, Tsai YF, Liu CY, Lien PJ, Lin YS, Chao TC, Feng CJ, Chen YJ, Lai JI, Phan NN, Hsu CY, Chiu JH, Tseng LM. Prevalence of tumor genomic alterations in homologous recombination repair genes among taiwanese breast cancers. Ann Surg Oncol. 2022;29(6):3578–90. https://doi.org/10.1245/s10434-022-11347-0.
Huang CC, Tsai YF, Liu CY, et al. Comprehensive molecular profiling of Taiwanese breast cancers revealed potential therapeutic targets: prevalence of actionable mutations among 380 targeted sequencing analyses. BMC Cancer. 2021;21:199. https://doi.org/10.1186/s12885-021-07931-4.
Liu CY, Huang CC, Tsai YF, Chao TC, Lien PJ, Lin YS, et al. VGH-TAYLOR: comprehensive precision medicine study protocol on the heterogeneity of Taiwanese breast cancer patients. Future Oncol. 2021. https://doi.org/10.2217/fon-2021-0131.
NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®), Version 3.2023
Burstein HJ, Curigliano G, Thürlimann B, Weber WP, Poortmans P, Regan MM, et al. Panelists of the St Gallen Consensus Conference. Customizing local and systemic therapies for women with early breast cancer: the St. Gallen International Consensus Guidelines for treatment of early breast cancer 2021. Ann Oncol. 2021;32(10):1216–1235. https://doi.org/10.1016/j.annonc.2021.06.023.
Lee CY, Chattopadhyay A, Chiang LM, Juang JJ, Lai LC, Tsai MH, et al. VariED: the first integrated database of gene annotation and expression profiles for variants related to human diseases. Database (Oxford). 2019;2019:baz075. https://doi.org/10.1093/database/baz075.
Landrum MJ, Chitipiralla S, Brown GR, Chen C, Gu B, Hart J, et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 2020;48(D1):D835–44. https://doi.org/10.1093/nar/gkz972.
Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017. https://doi.org/10.1200/PO.17.00011.
Oscanoa J, Sivapalan L, Gadaleta E, DayemUllah AZ, Lemoine NR, et al. SNPnexus: a web server for functional annotation of human genome sequence variation (2020 update). Nucleic Acids Res. 2020;48(W1):W185–92. https://doi.org/10.1093/nar/gkaa420.
Jennings LJ, Arcila ME, Corless C, Kamel-Reid S, Lubin IM, Pfeifer J, et al. Guidelines for validation of next-generation sequencing-based oncology panels: a joint consensus recommendation of the association for molecular pathology and college of American Pathologists. J Mol Diagn. 2017;19(3):341–65.
Dehghani M, Rosenblatt KP, Li L, Rakhade M, Amato RJ. Validation and clinical applications of a comprehensive next generation sequencing system for molecular characterization of solid cancer tissues. Front Mol Biosci. 2019;25(6):82.
Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19(1):4–23.
Khiabanian H, Hirshfield KM, Goldfinger M, Bird S, Stein M, Aisner J, Toppmeyer D, Wong S, Chan N, Dhar K, Gheeya J, Vig H, Hadigol M, Pavlick D, Ansari S, Ali S, Xia B, Rodriguez-Rodriguez L, Ganesan S. Inference of germline mutational status and evaluation of loss of heterozygosity in high-depth, Tumor-Only Sequencing Data. JCO Precis Oncol. 2018. https://doi.org/10.1200/PO.17.00148.
Sun JX, He Y, Sanford E, Montesion M, Frampton GM, Vignot S, et al. A computational approach to distinguish somatic versus germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLoS Comput Biol. 2018;14(2):e1005965.
Breast Cancer Association Consortium. Breast cancer risk genes—association analysis in more than 113,000 women. N Engl J Med. 2021;384(5):428–39.
Kwong A, et al. Comprehensive spectrum of BRCA1 and BRCA2 deleterious mutations in breast cancer in Asian countries. J Med Genet. 2016;53:15–23. https://doi.org/10.1136/jmedgenet-2015-103132.
Bychkovsky BL, Li T, Sotelo J, Tayob N, Mercado J, Gomy I, et al. Identification and management of pathogenic variants in BRCA1, BRCA2, and PALB2 in a tumor-only genomic testing program. Clin Cancer Res. 2022;28(11):2349–60. https://doi.org/10.1158/1078-0432.CCR-21-2861.
Healey CS, Dunning AM, Teare MD, Chase D, Parker L, Burn J, et al. A common variant in BRCA2 is associated with both breast cancer risk and prenatal viability. Nat Genet. 2000;26(3):362–4. https://doi.org/10.1038/81691.
Qiu LX, Yao L, Xue K, Zhang J, Mao C, Chen B, et al. BRCA2 N372H polymorphism and breast cancer susceptibility: a meta-analysis involving 44,903 subjects. Breast Cancer Res Treat. 2010;123(2):487–90. https://doi.org/10.1007/s10549-010-0767-5.
Spurdle AB, Hopper JL, Chen X, Dite GS, Cui J, McCredie MR, et al. The BRCA2 372 HH genotype is associated with risk of breast cancer in Australian women under age 60 years. Cancer Epidemiol Biomarkers Prev. 2002;11(4):413–6.
Wenham RM, Schildkraut JM, McLean K, Calingaert B, Bentley RC, Marks J, et al. Polymorphisms in BRCA1 and BRCA2 and risk of epithelial ovarian cancer. Clin Cancer Res. 2003;9(12):4396–403.
Teare MD, Cox A, Shorto J, Anderson C, Bishop DT, Cannings C. Heterozygote excess is repeatedly observed in females at the BRCA2 locus N372H. J Med Genet. 2004;41(7):523–8. https://doi.org/10.1136/jmg.2003.017293.
Palli D, Falchetti M, Masala G, Lupi R, Sera F, Saieva C, et al. Association between the BRCA2 N372H variant and male breast cancer risk: a population-based case-control study in Tuscany, Central Italy. BMC Cancer. 2007;3(7):170. https://doi.org/10.1186/1471-2407-7-170.
Guidugli L, Carreira A, Caputo SM, Ehlen A, Galli A, Monteiro AN, et al; ENIGMA consortium. Functional assays for analysis of variants of uncertain significance in BRCA2. Hum Mutat. 2014;35(2):151–64. https://doi.org/10.1002/humu.22478.
Johnston JJ, Rubinstein WS, Facio FM, Ng D, Singh LN, Teer JK, et al. Secondary variants in individuals undergoing exome sequencing: screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibility genes. Am J Hum Genet. 2012;91(1):97–108. https://doi.org/10.1016/j.ajhg.2012.05.021.
Bodian DL, McCutcheon JN, Kothiyal P, Huddleston KC, Iyer RK, Vockley JG, et al. Germline variation in cancer-susceptibility genes in a healthy, ancestrally diverse cohort: implications for individual genome sequencing. PLoS ONE. 2014;9(4):e94554. https://doi.org/10.1371/journal.pone.0094554.
Van der Merwe NC, Combrink HM, Ntaita KS, Oosthuizen J. Prevalence of clinically relevant germline BRCA variants in a large unselected south African breast and ovarian cancer cohort: a public sector experience. Front Genet. 2022;8(13):834265. https://doi.org/10.3389/fgene.2022.834265.
Zhang R, Shen S, Wei Y, Zhu Y, Li Y, Chen J, et al. A large-scale genome-wide gene-gene interaction study of lung cancer susceptibility in Europeans with a trans-ethnic validation in Asians. J Thorac Oncol. 2022;17(8):974–90. https://doi.org/10.1016/j.jtho.2022.04.011.
Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature. 2000;408(6811):429–32. https://doi.org/10.1038/35044000.
Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22(6):719–29. https://doi.org/10.1016/j.molcel.2006.05.0222.
Turnbull C, Seal S, Renwick A, Warren-Perry M, Hughes D, Elliott A, et al. Gene–gene interactions in breast cancer susceptibility. Hum Mol Genet. 2012;21(4):958–62. https://doi.org/10.1093/hmg/ddr525.
Hu C, Polley EC, Yadav S, Lilyquist J, Shimelis H, Na J, et al. The contribution of germline predisposition gene mutations to clinical subtypes of invasive breast cancer from a clinical genetic testing cohort. J Natl Cancer Inst. 2020;112(12):1231–41. https://doi.org/10.1093/jnci/djaa023.
Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 Pt 1):4429–34. https://doi.org/10.1158/1078-0432.CCR-06-3045.
Lin NU, Claus E, Sohl J, Razzak AR, Arnaout A, Winer EP. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: high incidence of central nervous system metastases. Cancer. 2008;113(10):2638–45. https://doi.org/10.1002/cncr.23930.
Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. 2017;317(23):2402–16. https://doi.org/10.1001/jama.2017.7112.
Kast K, Rhiem K, Wappenschmidt B, Hahnen E, Hauke J, Bluemcke B, et al.; German Consortium for Hereditary Breast and Ovarian Cancer (GC-HBOC). Prevalence of BRCA1/2 germline mutations in 21 401 families with breast and ovarian cancer. J Med Genet. 2016;53(7):465–71. https://doi.org/10.1136/jmedgenet-2015-103672.
Schmidt MK, van den Broek AJ, Tollenaar RA, Smit VT, Westenend PJ, Brinkhuis M, et al. Breast cancer survival of BRCA1/BRCA2 mutation carriers in a hospital-based cohort of young women. J Natl Cancer Inst. 2017. https://doi.org/10.1093/jnci/djw329.
Baretta Z, Mocellin S, Goldin E, Olopade OI, Huo D. Effect of BRCA germline mutations on breast cancer prognosis: a systematic review and meta-analysis. Medicine (Baltimore). 2016;95(40): e4975. https://doi.org/10.1097/MD.0000000000004975.
Zhong Q, Peng HL, Zhao X, Zhang L, Hwang WT. Effects of BRCA1- and BRCA2-related mutations on ovarian and breast cancer survival: a meta-analysis. Clin Cancer Res. 2015;21(1):211–20. https://doi.org/10.1158/1078-0432.CCR-14-1816.
Pertea M, Shumate A, Pertea G, Varabyou A, Breitwieser FP, Chang YC, et al. CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol. 2018;19(1):208. https://doi.org/10.1186/s13059-018-1590-2.
Whiffin N, Minikel E, Walsh R, O’Donnell-Luria AH, Karczewski K, Ing AY, Barton PJR, Funke B, Cook SA, MacArthur D, Ware JS. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med. 2017;19(10):1151–8. https://doi.org/10.1038/gim.2017.26.
Xiang J, Yang J, Chen L, Chen Q, Yang H, Sun C, et al. Reinterpretation of common pathogenic variants in ClinVar revealed a high proportion of downgrades. Sci Rep. 2020;10(1):331. https://doi.org/10.1038/s41598-019-57335-5.
Rahman N. Realizing the promise of cancer predisposition genes. Nature. 2014;505(7483):302–8. https://doi.org/10.1038/nature12981.
Acknowledgements
Partial materials were presented during the Taiwan Surgical Association 2023 Meeting on March 19, 2023. We would like to express our gratitude to Dr. Morris Chang, Taiwan Clinical Oncology Research Foundation and Melissa Lee Cancer Foundation for their kind help during the study period. The authors would also like to thank National Science and Technology Council for their support (NSTC 111-2314-B-075-063-MY3).
Funding
The study was exclusively sponsored by YongLin Healthcare Foundation under the clinical study protocol No. QCR18002. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
HFC and CCH drafted the manuscript and conducted the analysis. CYL joined in part of the study design. TCC, PJL, YSL, CJF, and JHC recruited subjects and attended panel discussion. CYH processed specimens and ascertained diagnostic accuracy of pathology. LMT initiated the whole study, took full responsibility of execution and finalized the submitted manuscript. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The whole study protocol was reviewed and approved by Institutional Review Board of Taipei Veterans General Hospital and written consent for participation was obtained (protocol number: 2018-09-007A).
Consent for publication
All included patients provided written informed consent for publication.
Competing interests
All authors declare that there are no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Table S1
. The result of all the variants from reflex germline testing.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cheng, HF., Tsai, YF., Liu, CY. et al. Prevalence of BRCA1, BRCA2, and PALB2 genomic alterations among 924 Taiwanese breast cancer assays with tumor-only targeted sequencing: extended data analysis from the VGH-TAYLOR study. Breast Cancer Res 25, 152 (2023). https://doi.org/10.1186/s13058-023-01751-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13058-023-01751-z