
Abstract
Objective
To evaluate the safety, feasibility, and efficacy of combined adrenergic blockade with propranolol and clonidine in patients with severe traumatic brain injury (TBI).
Background
Administration of adrenergic blockade after severe TBI is common. To date, no prospective trial has rigorously evaluated this common therapy for benefit.
Methods
This phase II, single-center, double-blinded, pilot randomized placebo-controlled trial included patients aged 16–64 years with severe TBI (intracranial hemorrhage and Glasgow Coma Scale score ≤ 8) within 24 h of ICU admission. Patients received propranolol and clonidine or double placebo for 7 days. The primary outcome was ventilator-free days (VFDs) at 28 days. Secondary outcomes included catecholamine levels, hospital length of stay, mortality, and long-term functional status. A planned futility assessment was performed mid-study.
Results
Dose compliance was 99%, blinding was intact, and no open-label agents were used. No treatment patient experienced dysrhythmia, myocardial infarction, or cardiac arrest. The study was stopped for futility after enrolling 47 patients (26 placebo, 21 treatment), per a priori stopping rules. There was no significant difference in VFDs between treatment and control groups [0.3 days, 95% CI (− 5.4, 5.8), p = 1.0]. Other than improvement of features related to sympathetic hyperactivity (mean difference in Clinical Features Scale (CFS) 1.7 points, CI (0.4, 2.9), p = 0.012), there were no between-group differences in the secondary outcomes.
Conclusion
Despite the safety and feasibility of adrenergic blockade with propranolol and clonidine after severe TBI, the intervention did not alter the VFD outcome. Given the widespread use of these agents in TBI care, a multi-center investigation is warranted to determine whether adrenergic blockade is of therapeutic benefit in patients with severe TBI.
Trial Registration Number NCT01322048.
Similar content being viewed by others

Background
Severe traumatic brain injury (TBI) is associated with increased intracranial pressure, activation of the sympathetic nervous system and catecholamine response, and major morbidity and mortality [1,2,3,4,5,6]. This increased catecholamine response is predictive of length of stay, mechanical ventilation, neurologic outcome, and mortality [7]. Prior retrospective studies [8,9,10,11,12], including two from our group [13, 14], link adrenergic blockade to survival after severe TBI, possibly mediated by dampened sympathetic hyperactivity.
These findings have resulted in an increase in β-blocker use in our institution to greater than 40% in young, severe TBI patients [14]. β-blockade is just one pharmacologic strategy to reduce sympathetic hyperactivity; centrally acting α2-agonists also serve as sympatholytic agents [15,16,17]. The prototypical centrally acting α2-agonist, clonidine, decreases plasma catecholamines and improves outcomes in a rat model of incomplete cerebral ischemia [18]. Clonidine decreases plasma catecholamines and cerebral vasoconstriction without altering cerebral blood flow in patients with severe TBI [19, 20].
Despite available cohort and open-label data, rigorous blinded prospective trial evidence remains lacking on the feasibility, safety, and efficacy of β-blockers and α2-agonists after severe TBI [21,22,23,24]. A 2017 meta-analysis specifically highlighted the poor quality of the literature on this question and the need for high-quality prospective trials investigating beta blockade in TBI patients [20]. We conducted a single-center, double-blind, pilot randomized placebo-controlled trial to test the hypothesis that sympathetic blockade with propranolol and clonidine improves clinical outcomes in severe TBI patients by increasing ventilator-free days, defined as days alive and not on the ventilator, in addition to demonstrating the safety and feasibility of such a trial.
Methods
Trial design
We conducted a phase II, single-center, randomized, double-blinded, placebo-controlled pilot trial to test whether sympathetic blockade with propranolol and clonidine within 48 h of severe TBI improves clinical outcomes. Our protocol was approved by our Institutional Review Board and registered (NCT01322048). Enrollment took place between August 2011 through January 2015. One group received centrally acting sympatholytic drugs, propranolol and clonidine, and the other group received double placebo with identical routes of administration. To maintain group balance for this small trial, patients were assigned to treatment groups using a Bayesian weighted 1:1 adaptive co-variate randomization scheme with a random element based on neuroradiologist-rated computed tomography (CT) Marshall Class and age (double-weighted). At 50% accrual, two-stage stopping rules were applied on efficacy with Type I error of 3% and futility with conditional power < 50%.
Participants
Inclusion criteria were severe TBI defined as Glasgow Coma Score (GCS) less than 8 with hemorrhage on head CT in patients between the ages of 16 and 64. Participants may or may not have had associated extracranial injuries. By definition, patients with TBI without hemorrhage on CT (i.e., diffuse axonal injury visible on magnetic resonance imaging [MRI] only) were not included. Exclusion criteria included pre-existing heart disease, study drug contraindication, penetrating TBI, pre-injury brain dysfunction, spinal cord injury, current β-blocker or α2-agonist use, and impending herniation, craniotomy, or death. Prisoners, pregnant women, and those unable to complete all follow-up visits were also excluded.
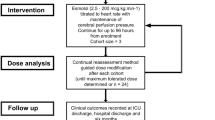
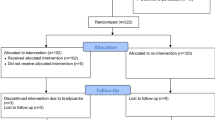
Surrogates of patients who met inclusion criteria and no exclusion criteria were approached for enrollment, and informed consent was obtained from the designated surrogate before any study procedures were performed. Participants were re-consented if they regained capacity. Patients without surrogates were not enrolled. Potential participants had to be screened within 24 h of injury, with study drug initiation within 48 h of injury. Figure 1 shows the study CONSORT diagram [25].

Screening, enrollment, and randomization for propranolol and clonidine versus double placebo after severe traumatic brain injury
Intervention
Patients were administered either both propranolol and clonidine (treatment arm), or placebo. All patients and study personnel, except for the pharmacist and biostatistician, were blinded to treatment assignment. Treatment (or placebo) delivery started within 48 h of injury but after plasma and 24-h urine catecholamine measurements were obtained. Both drugs (or double placebos) were administered at staggered times for 7 days with drug holds for hemodynamic parameters [26]. Propranolol was administered intravenously at a dose of 1 mg every 6 h, and doses were held for heart rate less than 60 bpm, mean arterial pressure less than 60 mmHg, and/or cerebral perfusion pressure less than 60 mmHg. Placebo was intravenous administration of sterile 0.9% normal saline. Clonidine was administered at a dose of 0.1 mg per tube every 12 h and was held for mean arterial pressure < 60 mmHg and/or cerebral perfusion pressure < 60 mmHg. Placebo was per tube administration of microcrystalline cellulose compounded by the investigational drug service. If hemodynamic parameters were met while a patient was on vasopressors, drug delivery still occurred. We documented compliance with treatment delivery and any reasons for missed administrations.
Clinicians were allowed to use β-blockers at any point if there was a myocardial infarction or need for heart rate control refractory to calcium channel blockers and anti-arrhythmic medications. Dexmedetomidine, a prototypical α2-agonist, was allowed if standard sedation regimens, such as propofol, lorazepam, and midazolam, failed to achieve adequate sedation. In the case of vasopressors, phenylephrine and vasopressin were preferred agents, with additional support from inotropes dobutamine and milrinone. Norepinephrine, epinephrine, and dopamine agents were avoided unless increased cardiac output was needed or further systemic vascular resistance was required beyond that produced by preferred vasopressors. We tracked the use of non-study β-blockers and α2-agonists. Ventilators were weaned utilizing clinical judgment by an interdisciplinary team of respiratory therapists, critical care fellows, and attending physicians, who conducted twice-daily simultaneous spontaneous awakening and breathing trials, a practice developed at our institution [27].
Safety monitoring
This phase II study focused on safety and efficacy. Safety parameters collected throughout the study included cardiac complications such as dysrhythmia (e.g., symptomatic bradycardia), myocardial infarction, cardiac arrest, and neuro-worsening. All serious, unexpected, and study-related adverse events were reported in a blinded fashion to the Data Safety Monitoring Board within 24 h.
Efficacy outcomes
The primary outcome was ventilator-free days (VFD), defined as days alive and not requiring mechanical ventilation during a 28-day period, chosen to avoid the competing risk of mortality [28, 29]. Secondary outcomes included change in plasma and urine catecholamine levels following treatment (measured at enrollment/pre-randomization and study day 8), coma-free days, ICU length of stay (LOS), hospital LOS, in-hospital agitation, in-hospital mortality, and functional status at discharge. Arousal and agitation were measured twice daily with both the Richmond Agitation-Sedation Score (RASS) [30], Glasgow Coma Scale (GCS), and the Agitation Behavior Scale (ABS) for TBI until hospital discharge. Paroxysmal sympathetic hyperactivity (PSH) was measured using the validated Clinical Features Scale (CFS) [31]. At the time the study protocol was developed in 2011, the Paroxysmal Sympathetic Hyperactivity-Assessment Measure (PSH-AM) [31], which contains the CFS and the Diagnostic Likelihood Tool (DLT), had not yet been developed. However, at the time, the components of the CFS were well known as important diagnostic criteria for PSH [32] and thus, were prospectively collected. As the PSH-AM, including the CFS, was published during the conduct of the trial, these components of the CFS, identified a priori were used to calculate the CFS.
Additional secondary outcomes included neuropsychological and quality of life metrics obtained at in-person long-term follow-up. Study personnel obtained a baseline neuropsychological evaluation at hospital discharge using the Rancho Los Amigos level of cognitive functioning scale [33]. Follow-up visits at 3 and 12 months included neuropsychological testing for global executive function and processing speed using the Trail Making Test Part B [34]. Quality of life and functional status were assessed with the Quality of Life after Brain Injury (QOLIBRI) [35] and Extended Glasgow Outcome Scale (GOSE) [36]. The Social Security Death Index was monitored monthly to assess out-of-hospital mortality.
Sample size calculation
Our sample size was calculated based on our primary outcome of ventilator-free days (VFD) after adrenergic blockade in severe TBI patients. As this was a pilot study that is the first of its kind, we utilized a clinically relevant difference between the experimental and control mean VFD of 2 days, with a standard deviation of 3 days; we calculated that 48 experimental and 48 control subjects were required to achieve 90% power, plus accounting for 4 patient withdrawals. A planned interim analysis (described in detail in our “Interim Analysis Plan”) accounts for the uncertainty in the true difference in VFD [37]. The Type I error rate was set at 5%. With independent biostatistical input, this calculation was performed using PS, Power and Sample Size Calculations program [38]. The primary outcome was VFD with a planned enrollment of 100 subjects. At 50% accrual, the two-stage stopping rules were based on efficacy with Type I error of 3% and futility with conditional power < 50%, utilizing an alpha spending approach to account for multiple tests [37]. This a priori planned futility assessment was performed mid-way through participant accrual, while maintaining the blind, to determine either overwhelming evidence of efficacy or futility.
Randomization
In order to maintain group balance, patients were assigned to treatment groups using a Bayesian weighted adaptive co-variate randomization scheme with a random element based on CT Marshall Class and age, with age weighted twice the CT Marshall Class [39, 40]. This allocation scheme allowed a dominant element of randomness while achieving balance between the two groups. Randomization was performed by investigational pharmacists using a password-protected computer program.
Blinding
Apart from investigational pharmacy and study biostatisticians, all clinical and research personnel were blinded to each patient’s treatment group. Placebo, both injections and tablets, were indistinguishable from active treatments when sent from the investigational pharmacy.
Statistical methods
Data were analyzed with an intention-to-treat approach. Continuous data were summarized using means and standard deviations, while categorical data were summarized using frequencies and proportions. Differences between the treatment groups on continuous variables were assessed using Wilcoxon rank-sum tests, and for categorical variables, difference in proportions was estimated with the Pearson method. In-hospital mortality was compared using Fisher’s exact test. Survival proportions were estimated with Kaplan–Meier method, and log-rank test was used to assess the difference in survival function. A significance level of 0.05 was used to indicate statistical significance. Secondary neurocognitive outcomes were assessed in survivors who successfully completed long-term follow-up. All analyses were completed using R statistical software version 3.4.
Results
Over a four-year period, we screened 302 consecutive patients with severe TBI admitted to our center. Enrollment was stopped at 50% accrual at 48 patients as the futility stopping rule determined that, even at its accrual goal of 96, the trial would not achieve conditional power to detect a significant difference in the primary outcome. A single patient was withdrawn from the study by their surrogate prior to randomization, leaving 47 patients randomized to placebo or treatment. Figure 1 shows our screening, enrollment, randomization, and follow-up. A total of 26 patients were allocated to the placebo group and 21 to the treatment group. A total of 11 patients died in-hospital, 6 in the placebo group and 5 in the treatment group, leaving a total of 20 and 16 in the placebo and treatment groups, respectively, for follow-up. Two patients in the placebo group died before their 3-month follow-up. Two patients in the treatment group completed the 3-month assessment but were lost to follow-up at the 12-month assessment. All other patients completed the study and were observed for their entire hospitalization or until study day 28. Among 47 randomized, the age, sex, race, injury and imaging severity, and organ failure score are listed in Table 1. Total hospital doses of fentanyl and propofol were comparable between groups. In terms of sedatives other than fentanyl or propofol, two patients received phenobarbital (both < 60 mg per day), and one patient received midazolam (< 40 mg per day).
Protocol adherence
Compliance and protocol adherence were high (99%, 1854 of 1872 doses possible), and blinding was intact. Reasons for non-administration of the 18 doses were as follows: hemodynamic parameters not met (11 cases), nursing error (4), pharmacy error (1), and problems with enteral access for clonidine (1). Study drug was discontinued in one patient due to rapid neurologic decline with plans for no further escalation of care. Study drug was never held due to physician preference, physician error, lack of IV access, or the patient not being present in the ICU (e.g., receiving MRI). Patients received non-study β-blockers or α2-agonists during the study period in two instances outside of the ICU, where β-blockers were administered intraoperatively by anesthesia personnel.
Safety evaluation
Neither cardiac complications nor other serious adverse events occurred. A single patient in the treatment arm had a temporary asymptomatic junctional bradyarrhythmia that required no intervention, and the study drug was continued. This event was noted, and the patient remained in the study. No patients were removed from the study due to safety concerns. Study drug was never held due to concerns over clinical care. Non-study propranolol or clonidine were never required by clinical personnel in the ICU during the treatment period.
Primary and secondary outcomes
The mean ventilator-free days (VFD) did not differ between treatment and placebo groups (Treatment: 13.4 ± 9.2 vs. Placebo: 13.6 ± 9.9; mean difference 0.3 days, 95% CI − 5.4–5.8, p = 1.0). Similarly, we did not detect a significant difference in the secondary outcomes of agitation, LOS, in-hospital mortality (OR 1.4, 95% CI 0.3–6.7, p = 0.7), or improvements in long-term neuropsychological status despite > 90% follow-up (Table 1). Survival probability is shown in Fig. 2, which was similar between the treatment groups (p = 0.6). Sympathetic hyperactivity, as measured by the Clinical Features Scale (CFS), was significantly more severe in the placebo group (mean of 9.3 vs. 7.6, mean difference 1.7 points, 95% CI 0.4–2.9, p = 0.012). However, there was no significant difference in the secondary outcomes of plasma or urine catecholamine levels between pre-enrollment and study day 8 (Table 2).

Kaplan–Meier survival curves of propranolol and clonidine (treatment) versus double placebo after severe traumatic brain injury
Discussion
This study is the only prospective double-blinded, placebo-controlled randomized trial of sympathetic blockade using propranolol and clonidine in patients with severe TBI. We found that it is safe and feasible to administer combined adrenergic blockade in these critically ill patients. However, we identified no significant differences in our primary outcome of ventilator-free days. The mean ventilator-free days were 13.6 in the placebo group and 13.4 in the intervention, whereas study drug administration only ran from up to 7 to 9 days (starting between 0 and 48 h after injury). This discrepancy may have resulted in a rebound effect of sympathetic hyperactivity that may have resulted in an inability to detect a difference. Although we did not identify significant differences in our primary clinical endpoint, we did find that despite this small sample, the treatment group receiving adrenergic blockade had improved scores on the Clinical Features Scale (CFS), a scale designed to assess symptoms of paroxysmal sympathetic hyperactivity (PSH), also known as “neurostorming”, “sympathetic storming”, and by other synonyms [31, 41,42,43,44,45]. Although findings regarding secondary outcomes such as this may have been due to failure to correct for multiplicity in our analysis, it is a signal to be further explored in future work, and also aligns with the treatment mentality of clinicians who use these pharmacologic interventions to decrease agitation after TBI.
In contrast to the lack of clinical benefit demonstrated in this study, several translational animal-model, retrospective, prospective and non-placebo-controlled trials support a link between adrenergic blockade with survival and even long-term cognitive benefit in severe TBI [21, 24, 46, 47]. The potential reasons for the conclusions of these non-randomized, non-blinded, and non-placebo-controlled trials are several. No studies to date have been able to effectively address potential unmeasured confounding of unknown or pre-existing cardiovascular disease in a diverse patient population, and thus, survival benefit of adrenergic blockade seen in these larger retrospective and prospective studies may be due to these unmeasured factors. In addition to excluding patients with known cardiovascular disease, we intentionally excluded patients > 65 years old to avoid confounding by undiagnosed cardiac risk factors that are prevalent in older patients with TBI, which may be the confounding mechanism by which other studies have found a survival benefit of adrenergic blockade.
Limitations of our study include small sample size and single-center design. Despite being the largest double-blinded placebo-controlled randomized trial to date of sympathetic blockade in patients with severe TBI, the power of the study was limited due to it being a small single-center pilot. Demographic and injury diversity was limited, with most patients being very young (< 30 years of age), male, white, and with enrollment criteria of severe TBI defined as GCS < 8. Marshall CT Class does not account for diffuse axonal injury (DAI), a characteristic seen on advanced neuroimaging, however, not usually typical in the acute phase of injury, and with an unclear relationship to the intervention delivered [48]. We used a critical care outcome, ventilator-free days (VFD), which may not best reflect the benefits of adrenergic blockade in these patients. However, we did measure secondary outcomes such as agitation and features of sympathetic hyperactivity using a validated clinical scale, which are common clinically cited reasons for these drugs. Mortality has been the primary outcome reported in the existing retrospective or observational studies; however, given the nature of this study as a pilot single-center double-blinded randomized placebo-controlled trial, it was not feasible to achieve the participant number needed to identify a statistically significant difference in mortality. Candidates for outcomes in future studies include mortality, severity of sympathetic hyperactivity, and ventilator-free days. Future trials may also utilize dexmedetomidine, with its more favorable pharmacokinetic profile and more widespread contemporary use in the ICU, in lieu of clonidine as an α2-agonist.
Conclusions
It is feasible to conduct a rigorous double-blinded placebo-controlled trial of adrenergic blockade in young severe TBI patients without major adverse cardiovascular effects despite a complex ICU environment and drug administration schedule, while achieving long-term functional and neurocognitive follow-up. The practical execution of this single-center trial should support multi-center investigation powered for mortality and balanced for unmeasured confounders to determine whether the widespread practice of adrenergic blockade has any benefit after severe TBI.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
(CDC) CfDCaP: CDC grand rounds: reducing severe traumatic brain injury in the United States. MMWR Morb Mortal Wkly Rep. 2013:62(27):549–552.
Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21(5):375–8.
National Academies of Sciences E, and Medicine 2022: Traumatic Brain Injury: A Roadmap for Accelerating Progress. In. Washington, DC; 2022.
Traumatic Brain Injury Related Emergency Department Visits, Hospitalizations and Deaths [https://www.cdc.gov/traumaticbraininjury/data/tbi-edhd.html].
Maas AI, Marmarou A, Murray GD, Teasdale SG, Steyerberg EW. Prognosis and clinical trial design in traumatic brain injury: the IMPACT study. J Neurotrauma. 2007;24(2):232–8.
Menon DK, Zahed C. Prediction of outcome in severe traumatic brain injury. Curr Opin Crit Care. 2009;15(5):437–41.
Woolf PD, Hamill RW, Lee LA, Cox C, McDonald JV. The predictive value of catecholamines in assessing outcome in traumatic brain injury. J Neurosurg. 1987;66(6):875–82.
Arbabi S, Campion EM, Hemmila MR, Barker M, Dimo M, Ahrns KS, Niederbichler AD, Ipaktchi K, Wahl WL. Beta-blocker use is associated with improved outcomes in adult trauma patients. J Trauma. 2007;62(1):56–61 (discussion 61–52).
Inaba K, Teixeira PG, David JS, Chan LS, Salim A, Brown C, Browder T, Beale E, Rhee P, Demetriades D. Beta-blockers in isolated blunt head injury. J Am Coll Surg. 2008;206(3):432–8.
Chen Z, Tang L, Xu X, Wei X, Wen L, Xie Q. Therapeutic effect of beta-blocker in patients with traumatic brain injury: a systematic review and meta-analysis. J Crit Care. 2017;41:240–6.
Ahl R, Thelin EP, Sjolin G, Bellander BM, Riddez L, Talving P, Mohseni S. beta-Blocker after severe traumatic brain injury is associated with better long-term functional outcome: a matched case control study. Eur J Trauma Emerg Surg. 2017;43(6):783–9.
Tran TY, Dunne IE, German JW. Beta blockers exposure and traumatic brain injury: a literature review. Neurosurg Focus. 2008;25(4):E8.
Cotton BA, Snodgrass KB, Fleming SB, Carpenter RO, Kemp CD, Arbogast PG, Morris JA Jr. Beta-blocker exposure is associated with improved survival after severe traumatic brain injury. J Trauma. 2007;62(1):26–33 (discussion 33–25).
Riordan WP Jr, Cotton BA, Norris PR, Waitman LR, Jenkins JM, Morris JA Jr. Beta-blocker exposure in patients with severe traumatic brain injury (TBI) and cardiac uncoupling. J Trauma. 2007;63(3):503–10 (discussion 510–501).
Chan AK, Cheung CW, Chong YK. Alpha-2 agonists in acute pain management. Expert Opin Pharmacother. 2010;11(17):2849–68.
Pandharipande PP, Pun BT, Herr DL, Maze M, Girard TD, Miller RR, Shintani AK, Thompson JL, Jackson JC, Deppen SA, et al. Effect of sedation with dexmedetomidine vs lorazepam on acute brain dysfunction in mechanically ventilated patients: the MENDS randomized controlled trial. JAMA. 2007;298(22):2644–53.
Riker RR, Shehabi Y, Bokesch PM, Ceraso D, Wisemandle W, Koura F, Whitten P, Margolis BD, Byrne DW, Ely EW, et al. Dexmedetomidine vs midazolam for sedation of critically ill patients: a randomized trial. JAMA. 2009;301(5):489–99.
Hoffman WE, Cheng MA, Thomas C, Baughman VL, Albrecht RF. Clonidine decreases plasma catecholamines and improves outcome from incomplete ischemia in the rat. Anesth Analg. 1991;73(4):460–4.
Asgeirsson B, Grande PO, Nordstrom CH, Berntman L, Messeter K, Ryding E. Effects of hypotensive treatment with alpha 2-agonist and beta 1-antagonist on cerebral haemodynamics in severely head injured patients. Acta Anaesthesiol Scand. 1995;39(3):347–51.
Payen D, Quintin L, Plaisance P, Chiron B, Lhoste F. Head injury: clonidine decreases plasma catecholamines. Crit Care Med. 1990;18(4):392–5.
Alali AS, Mukherjee K, McCredie VA, Golan E, Shah PS, Bardes JM, Hamblin SE, Haut ER, Jackson JC, Khwaja K, et al. Beta-blockers and traumatic brain injury: a systematic review, meta-analysis, and eastern association for the surgery of trauma guideline. Ann Surg. 2017;266(6):952–61.
Ahl R, Thelin EP, Sjölin G, Bellander BM, Riddez L, Talving P, Mohseni S. β-Blocker after severe traumatic brain injury is associated with better long-term functional outcome: a matched case control study. Eur J Trauma Emerg Surg. 2017;43(6):783–9.
Ley EJ, Leonard SD, Barmparas G, Dhillon NK, Inaba K, Salim A, O’Bosky KR, Tatum D, Azmi H, Ball CG, et al. Beta blockers in critically ill patients with traumatic brain injury: Results from a multicenter, prospective, observational American Association for the Surgery of Trauma study. J Trauma Acute Care Surg. 2018;84(2):234–44.
Schroeppel TJ, Sharpe JP, Shahan CP, Clement LP, Magnotti LJ, Lee M, Muhlbauer M, Weinberg JA, Tolley EA, Croce MA, et al. Beta-adrenergic blockade for attenuation of catecholamine surge after traumatic brain injury: a randomized pilot trial. Trauma Surg Acute Care Open. 2019;4(1): e000307.
Schulz KF, Altman DG, Moher D, Group C. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials. BMC Med. 2010;8:18.
Patel MB, McKenna JW, Alvarez JM, Sugiura A, Jenkins JM, Guillamondegui OD, Pandharipande PP. Decreasing adrenergic or sympathetic hyperactivity after severe traumatic brain injury using propranolol and clonidine (DASH After TBI Study): study protocol for a randomized controlled trial. Trials. 2012;13:177.
Girard TD, Kress JP, Fuchs BD, Thomason JW, Schweickert WD, Pun BT, Taichman DB, Dunn JG, Pohlman AS, Kinniry PA, et al. Efficacy and safety of a paired sedation and ventilator weaning protocol for mechanically ventilated patients in intensive care (Awakening and Breathing Controlled trial): a randomised controlled trial. Lancet. 2008;371(9607):126–34.
Schoenfeld DA, Bernard GR, Network A. Statistical evaluation of ventilator-free days as an efficacy measure in clinical trials of treatments for acute respiratory distress syndrome. Crit Care Med. 2002;30(8):1772–7.
Toft P, Olsen HT, Jorgensen HK, Strom T, Nibro HL, Oxlund J, Wian KA, Ytrebo LM, Kroken BA, Chew M. Non-sedation versus sedation with a daily wake-up trial in critically ill patients receiving mechanical ventilation (NONSEDA Trial): study protocol for a randomised controlled trial. Trials. 2014;15:499.
Sessler CN, Gosnell MS, Grap MJ, Brophy GM, O’Neal PV, Keane KA, Tesoro EP, Elswick RK. The Richmond Agitation-Sedation Scale: validity and reliability in adult intensive care unit patients. Am J Respir Crit Care Med. 2002;166(10):1338–44.
Baguley IJ, Perkes IE, Fernandez-Ortega JF, Rabinstein AA, Dolce G, Hendricks HT, Consensus Working G. Paroxysmal sympathetic hyperactivity after acquired brain injury: consensus on conceptual definition, nomenclature, and diagnostic criteria. J Neurotrauma. 2014;31(17):1515–20.
Perkes IE, Menon DK, Nott MT, Baguley IJ. Paroxysmal sympathetic hyperactivity after acquired brain injury: a review of diagnostic criteria. Brain Inj. 2011;25(10):925–32.
Gouvier WD, Blanton PD, LaPorte KK, Nepomuceno C. Reliability and validity of the Disability Rating Scale and the Levels of Cognitive Functioning Scale in monitoring recovery from severe head injury. Arch Phys Med Rehabil. 1987;68(2):94–7.
Reitan RM. Validity of the trail making test as an indicator of organic brain damage. Percept Mot Skills. 1958;8:271–6.
von Steinbuchel N, Wilson L, Gibbons H, Hawthorne G, Hofer S, Schmidt S, Bullinger M, Maas A, Neugebauer E, Powell J, et al. Quality of Life after Brain Injury (QOLIBRI): scale development and metric properties. J Neurotrauma. 2010;27(7):1167–85.
Wilson JT, Pettigrew LE, Teasdale GM. Structured interviews for the Glasgow Outcome Scale and the extended Glasgow Outcome Scale: guidelines for their use. J Neurotrauma. 1998;15(8):573–85.
Meurer WJ, Tolles J. Interim analyses during group sequential clinical trials. JAMA. 2021;326(15):1524–5.
Dupont WD, Plummer WD Jr. Power and sample size calculations. A review and computer program. Control Clin Trials. 1990;11(2):116–28.
Pocock SJ, Simon R. Sequential treatment assignment with balancing for prognostic factors in the controlled clinical trial. Biometrics. 1975;31(1):103–15.
Marshall LF, Marshall SB, Klauber MR, Van Berkum CM, Eisenberg H, Jane JA, Luerssen TG, Marmarou A, Foulkes MA. The diagnosis of head injury requires a classification based on computed axial tomography. J Neurotrauma. 1992;9(Suppl 1):S287-292.
Mirhoseini MF, Hosay MA, McPherson M, Patel MB. Paroxysmal sympathetic hyperactivity: diagnostic criteria, complications, and treatment after traumatic brain injury. Curr Phys Med Rehabil Rep. 2018;6(1):81–8.
Lv LQ, Hou LJ, Yu MK, Qi XQ, Chen HR, Chen JX, Hu GH, Luo C, Lu YC. Prognostic influence and magnetic resonance imaging findings in paroxysmal sympathetic hyperactivity after severe traumatic brain injury. J Neurotrauma. 2010;27(11):1945–50.
Lv LQ, Hou LJ, Yu MK, Qi XQ, Chen HR, Chen JX, Hu GH, Luo C, Lu YC. Risk factors related to dysautonomia after severe traumatic brain injury. J Trauma. 2011;71(3):538–42.
Meyfroidt G, Baguley IJ, Menon DK. Paroxysmal sympathetic hyperactivity: the storm after acute brain injury. Lancet Neurol. 2017;16(9):721–9.
Samuel S, Allison TA, Lee K, Choi HA. Pharmacologic management of paroxysmal sympathetic hyperactivity after brain injury. J Neurosci Nurs J Am Assoc Neurosci Nurses. 2016;48(2):82–9.
Zeeshan M, Hamidi M, O’Keeffe T, Bae EH, Hanna K, Friese RS, Kulvatunyou N, Zakaria ER, Gries L, Tang A, et al. Propranolol attenuates cognitive, learning, and memory deficits in a murine model of traumatic brain injury. J Trauma Acute Care Surg. 2019;87(5):1140–7.
Ley EJ, Leonard SD, Barmparas G, Dhillon NK, Inaba K, Salim A, O’Bosky KR, Tatum D, Azmi H, Ball CG, et al. Beta blockers in critically ill patients with traumatic brain injury: results from a multicenter, prospective, observational American Association for the Surgery of Trauma study. J Trauma Acute Care Surg. 2018;84(2):234–44.
Humble SS, Wilson LD, Wang L, Long DA, Smith MA, Siktberg JC, Mirhoseini MF, Bhatia A, Pruthi S, Day MA, et al. Prognosis of diffuse axonal injury with traumatic brain injury. J Trauma Acute Care Surg. 2018;85(1):155–9.
Acknowledgements
Not applicable.
Funding
This project was supported by the NINDS Clinical Trials Methodology Course (R25 NS088248), the Vanderbilt Faculty Research Scholar Program, and the Eastern Association for the Surgery of Trauma Research Scholarship. We used REDCap, a secure online database, supported in part by the National Institutes of Health (UL1TR000445) and VICTR CTSA support (UL1TR002243). MRC was supported by the Vanderbilt Medical Scholars Program. MFN, MBP, AWM, ELR, SR and PPP have been supported by National Institutes of Health (F32AG062045, R01GM120484, 1K23GM150110, R01HL111111, R01AG058639, I01RX002992, T32GM135094).
Author information
Authors and Affiliations
Contributions
Conception and design, and/or acquisition of data, and/or analysis and interpretation of data were contributed by MFN, AWM, LDW, TK, ELR, SR, MDR, CLM, JCJ, WJM, RJL, PPP, MBP. Drafting article and/or revising critically for important intellectual content were contributed by all authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All methods were approved by the Institutional Review Board (IRB) at Vanderbilt University Medical Center. The research was carried out in accordance with IRB standards. Written informed consent was obtained from eligible patients.
Consent for publication
Not applicable.
Competing interests
Pfizer, Inc. (previously Hospira Inc.) has provided unrelated research support to PPP. Haemonetics, Inc. has provided unrelated research support to MBP. For the remaining authors, no competing interests were declared.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Nordness, M.F., Maiga, A.W., Wilson, L.D. et al. Effect of propranolol and clonidine after severe traumatic brain injury: a pilot randomized clinical trial. Crit Care 27, 228 (2023). https://doi.org/10.1186/s13054-023-04479-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13054-023-04479-6