
Abstract
Infectious diseases may affect brain function and cause encephalopathy even when the pathogen does not directly infect the central nervous system, known as infectious disease-associated encephalopathy. The systemic inflammatory process may result in neuroinflammation, with glial cell activation and increased levels of cytokines, reduced neurotrophic factors, blood–brain barrier dysfunction, neurotransmitter metabolism imbalances, and neurotoxicity, and behavioral and cognitive impairments often occur in the late course. Even though infectious disease-associated encephalopathies may cause devastating neurologic and cognitive deficits, the concept of infectious disease-associated encephalopathies is still under-investigated; knowledge of the underlying mechanisms, which may be distinct from those of encephalopathies of non-infectious cause, is still limited. In this review, we focus on the pathophysiology of encephalopathies associated with peripheral (sepsis, malaria, influenza, and COVID-19), emerging therapeutic strategies, and the role of neuroinflammation.
Graphic abstract

Similar content being viewed by others

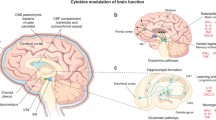

Background
Encephalopathy is an umbrella term which refers to brain dysfunction, regardless of etiology and pathophysiology. A broad range of diseases are capable of causing encephalopathy, including infections (whether or not the underlying pathogen is able to invade the central nervous system, CNS) (Table 1). Encephalopathies are characterized as temporary or permanent disturbances of brain functions, and the clinical picture is widely variable depending on the etiology [1].
Peripheral infections caused by viruses, bacteria, or parasites may lead to a secondary inflammatory response in the brain, commonly known as neuroinflammation [27], through the action of inflammatory mediators which affect the brain endothelium and parenchyma, and a response of brain cells to these mediators [28]. Therefore, this type of encephalopathy is not considered to be due to direct neurotropism, i.e., invasion of the CNS by the infectious agent. Numerous variables, such as intensity, duration, and immunological imprinting [29], play relevant roles in defining each patient’s outcome; neuroinflammation has been causally linked to long-term neurological damage and to a range of cognitive and behavioral symptoms, including memory loss, cognitive impairment, anxiety, and depression. Indeed, neurological consequences associated with infectious diseases may even influence the future incidence and prognosis of neurodegenerative disorders [30], thus making their proper management a meaningful way of reducing the burden on public health systems. To date, however, there is no established treatment or prevention strategy for the neurological damage associated with peripheral inflammation.
Peripheral immune responses can crosstalk with the brain through several pathways. Afferent nerves, including the vagal nerves and trigeminal nerves, respond to circulating interleukin (IL)-1β [31,32,33]. In addition, vagotomized animals do not exhibit sickness behavior after lipopolysaccharide (LPS) or IL-1β injection, despite increased peripheral cytokines levels [34, 35]. The humoral pathway involves macrophage-like cells present in the circumventricular organs and the choroid plexus, which express innate immune receptors that recognize pathogen-associated molecular patterns (PAMP), damage-associated molecular patterns (DAMP), and cytokines. The circumventricular organs do not appear to have an intact blood–brain barrier (BBB); therefore, inflammatory mediators are able to access the brain by volume diffusion, and the cytokine-saturable transporters in the BBB allow the overflowing cytokines present in the peripheral circulation to enter the cerebral parenchyma [31, 36]. The last pathway involves the activation of IL-1 receptors expressed in perivascular macrophages and endothelial cells located in brain microvasculature that initiate a local immune response with local synthesis of prostaglandin E2 [37]. Furthermore, systemic inflammation often leads to an increase in BBB permeability, and, in some cases, frank disruption. The loss of BBB integrity allows cytokines and immune cells to invade the brain parenchyma and directly affect neurons and glial cells [38] (Fig. 1). Glial activation is associated with cognition, memory, and mood disorders, and is a hallmark of neuroinflammation [39, 40].

Inflammatory signaling pathways to the brain. Systemic inflammation caused by pathogens, including viruses, bacteria, and parasites, leads to neuroinflammation with consequent cognitive and behavior impairments. The central nervous system is able to recognize systemic inflammation through (1) BBB dysfunction, with activation and apoptosis of endothelial cells, allowing cytokines and immune cells to invade the brain parenchyma; (2) the humoral pathway and saturable transport system in the blood–brain barrier (BBB), which involves the circumventricular organs (CVOs) and the choroid plexus, as local macrophage-like cells express innate immune receptors that recognize pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs), and cytokines, allowing inflammatory mediators to access the brain by volume diffusion and through cytokine-saturable transporters, since the CVOs do not have an intact BBB; (3) through activation of the afferent nerves (including the vagal nerves in abdominal/visceral infections and the trigeminal nerve in oro-lingual infections) by cytokines; and (4) IL-1β pathway signaling, through activation of IL-1 receptors expressed in perivascular macrophages and endothelial cells located in the brain microvasculature, initiating a local immune response
Neuroinflammation
Neuroinflammation, an inflammatory condition in the CNS, is a common feature of infectious disease-associated encephalopathies, which is mediated by cytokines, chemokines, reactive oxygen species, among others. These mediators are mainly produced by microglia and astrocytes, endothelial cells, and peripherally derived immune cells. Within the brain, cytokines are able to activate glial cells, modulate neurotransmitter metabolism, and lead to neurotoxic mechanisms [27, 39, 41]. After exposure to pro-inflammatory stimuli, microglia undergo morphological and functional changes, and orchestrate an immune response in the CNS. A pro-inflammatory milieu also leads to several pathological alterations in astroglia. This reactive astrogliosis is characterized by hypertrophy, a modified secretome, and increased expression of intermediate-filament proteins, especially glial fibrillary acidic protein (GFAP) and vimentin [42].
Cytokines exert deleterious effects on the brain, especially the hippocampus. IL-1β inhibits synaptic strength and long-term potentiation in the rodent hippocampus, impacting neuronal morphology, synaptic plasticity [43, 44], and memory and learning processes [45, 46]. Cytokines also affect brain function by modulating neurotrophins. Brain-derived neurotrophic factor (BDNF) signaling is impaired by cytokines, particularly IL-1β [47]. Moreover, systemic injection of LPS has been shown to reduce BDNF, nerve growth factor (NGF), and neurotrophin-3 levels [48], and changes in levels of neurotrophins are known to impact synaptic plasticity, memory, and neuronal survival.
Neuronal cells are also affected by glial reactivity and the subsequent loss of the supportive function of glial cells. Astrocytes regulate the concentration of neurotransmitters, such as gamma-aminobutyric acid (GABA), glutamate, and glycine at the synaptic cleft [49]. One of the major consequences of astrogliosis is loss of this function, resulting in glutamate toxicity [39]. Toxicity by glutamatergic activation are also mediated by indoleamine-2,3 dioxygenase (IDO), an enzyme expressed by microglial cells [50]; in the presence of inflammatory mediators, including interferon (IFN)-γ and tumor necrosis factor (TNF)-α, IDO activity is modulated. Moreover, IDO is also involved in tryptophan-serotonin availability suggesting that pro-inflammatory cytokines causes neurotransmitter disbalance [50, 51] (Fig. 2).

Molecular and cellular mechanisms of neuroinflammation. Blood–brain barrier (BBB) dysfunction contributes to the process of neuroinflammation. After losing its integrity, the BBB allows circulating leukocytes (e.g., monocytes and neutrophils) and proinflammatory mediators, such as cytokines, to enter the brain parenchyma. Microglia and astrocytes proliferate, become reactive, and undergo functional and morphological changes. Microglial cells increase the release of reactive oxygen species, cytokines, chemokines, and indoleamine 2,3-dioxygenase (IDO) expression/activity, as well as decrease brain-derived neurotrophic factor (BDNF) expression. Astrocytes increase the expression of glial fibrillary acidic protein (GFAP) and vimentin, which cause morphological changes, losing their function as supportive glial cells and developing impairment of neurotransmitter recycling. Neuroinflammation also impacts neurons and synaptic transmission, leading to impairments in long-term potentiation (LTP) and neurotransmitter system dysfunctions
Sepsis-associated encephalopathy
Definition and diagnosis
The brain is among the multiple organs affected by sepsis [52, 53]. Neurological complications associated with sepsis in the absence of CNS infection fall under the umbrella term sepsis-associated encephalopathy (SAE), which affects 70% of septic patients. It represents a risk factor for mortality, and survivors often face long-term disabilities [54, 55]. In its acute stage, SAE involves sickness behavior, lethargy, delirium, memory impairment, mood disorders, and, in the most severe cases, coma. The diagnosis includes several clinical features such as disturbances in sleep–wake cycles, level of consciousness in disagreement with the dose of sedative received, hallucinations, agitation, and other symptoms of delirium. Moreover, SAE may also lead to paratonic rigidity, and, in 70% of advanced cases, neuromyopathy. Despite these features, SAE is basically a diagnosis of exclusion, with no specific clinical manifestations; it can be inferred and should be suspected after meningitis, encephalitis, and septic emboli from endocarditis have been ruled out. Thus, the final diagnosis relies on the clinical context and evidence of infection in some part of the body [53].
Pathophysiology and biological alterations
The pathophysiology of SAE is complex and involves several mechanisms, including neuroinflammation, ischemic processes, neurotransmitter imbalances, and mitochondrial dysfunction [56, 57]. The challenge in defining SAE pathophysiology is the involvement of nonspecific mechanisms and the lack of specific biomarkers. The systemic cytokine storm of sepsis increases BBB permeability and leads to dysfunctions in microcirculation due to exacerbated endothelial-cell activation, resulting in microvascular tone impairment, coagulation activation, and ischemic lesions [58] (Fig. 3). In addition, SAE leads to increased expression and activity of endothelial nitric oxide synthase in neurons and glial cells [59, 60], resulting in augmented NO levels and, consequently, tissue edema and NO-mediated cell death [61, 62].

Mechanisms implicated in neurological complications after infection. In COVID-19, SARS-CoV-2 can access the brain by a trans-synaptic route and also through endothelial and lymphocyte invasion, resulting in neuroinflammation. Lower thrombin, higher D-dimer, fibrin/fibrinogen degradation products, and fibrinogen levels are frequent in COVID-19, and activation of the coagulation cascade may contribute to the development of stroke and cerebrovascular accidents. Brain-lung crosstalk is an axis involved in brain hypoxia due to systemic oxygenation reduction and, subsequently, secondary brain oxygenation damage. In sepsis-associated encephalopathy, the cytokine storm leads to endothelial activation and increased eNOS activity, which results in nitric oxide (NO) production, leading to hypotension and ischemic lesions. Cytokines trigger glial reactivity, reactive oxygen species (ROS) production, mitochondrial dysfunction, and neurotransmitter imbalances, with consequent glutamate excitotoxicity. In malaria infection, there is an exacerbated inflammatory response to the parasite and activation of multiple cell death pathways leading to microcirculatory damage. Endothelial dysfunction, platelet activation, cytoadherence, and a downregulation of normal endogenous anticoagulant pathways are hallmarks. Dysregulation of the coagulation pathway leads to microvascular lesions; thrombin may be implicated. In the process of hemoglobin digestion, the malaria parasite releases heme and aggregates it into hemozoin, a highly toxic and proinflammatory signaling molecule. Hemozoin and free heme released into the bloodstream lead to exacerbated inflammation, tissue damage, apoptosis of microvascular brain endothelial cells through activation of STAT3, and loss of BBB integrity through binding to the metalloproteinase MMP3. The proinflammatory milieu leads to microglial M1 phenotype activation, release of proinflammatory cytokines, astrogliosis, axonal injury, and increase in synapsin I. In influenza infection, there is a peripheral inflammatory response and release of several proinflammatory mediators, including interferons (IFs), interleukins (ILs), tumor necrosis factor (TNF), and chemokines. Both neurotropic and non-neurotropic strains of influenza are able to induce neuroinflammation, with microglial activation, decrease in neurotrophin levels, and increase in IFN-α and other proinflammatory cytokines
A decrease in brain volume, especially in the cortex and hippocampus, has been observed in clinical and experimental models of sepsis [63,64,65]. Damage to these brain areas are associated with impairments in long-term potentiation, affecting learning and memory in models of SAE [66]. Imaging changes can occur in the cortex, subcortical regions, and white matter. Magnetic resonance imaging (MRI) changes are, in the most consistently reported cases, due to cytotoxic edema (caused by hypoxia/ischemia) and vasogenic edema (due to BBB disruption) [67]. In patients diagnosed with some degree of SAE, mortality was directly related to the electroencephalogram (EEG) severity [68].
Changes in neurotransmitter pathways (acetylcholine, GABA, dopamine, norepinephrine, serotonin and glutamate) are considered a hallmark of SAE and are closely related to delirium [69,70,71,72,73,74,75]. These changes are in part induced by increased IDO activity [70], but also due to increased plasma levels of the aromatic amino-acid precursors of neurotransmitters, such as tyrosine, tryptophan, and phenylalanine, due to muscular proteolysis and liver failure [71]. These alterations enhance CNS amino acid uptake, which directly impacts neurotransmitter synthesis, leading to abnormalities in neurotransmission [71,72,73].
During neuroinflammation, several changes occur in cellular metabolism, resulting in mitochondrial dysfunction [76,77,78,79], which involve ROS production, increased superoxide dismutase activity [80], energy deficit due to a decrease in adenosine triphosphate (ATP) generation, and cellular apoptosis triggered by the release of cytochrome c [76].
Therapeutic tools
Sepsis survivors are a complex and heterogeneous group, making it difficult to find a specific therapeutic target. To date, there are still no approaches to prevent or treat the neurological consequences of SAE or the subsequent cognitive decline. In clinical SAE, treatment is primarily symptomatic, despite the fact that these neurological deficits may persist for many years after hospital discharge [77]. Investigation of SAE therapies is a necessary and promising field.
Treatment of delirium requires identification and cessation of any medication with anticholinergic, histaminergic, and other psychotropic properties [78]. Sedatives and neuroleptics must be used with caution, and potent benzodiazepines such as lorazepam must be avoided. In some cases, low doses of neuroleptics may be administered to improve sleep cycles. Dexmedetomidine was associated with shorter duration of clinical encephalopathy, shorter ventilator time, and lower rates of mortality when compared to lorazepam [79]. Considering that the incidence of seizures in SAE is relatively low (10%), antiepileptic drugs should be avoided and only used when justified [81] (Table 2). Immunotherapy with an anti-TNF-α monoclonal antibody reduces mortality in patients with septic shock or high levels of circulating cytokines [82]. Despite this promising finding, there is no evidence that anti-TNF therapy can lead to clinical improvement of SAE.
In experimental sepsis, mesenchymal stromal cells (MSC) mitigate BBB dysfunction and neuroinflammation, reduce astrogliosis, and lead to long-term improvements in cognition and anxiety-like behavior [83], as well as resulting in better memory retrieval and decreased sepsis scores at acute time points [84]. Treatment with statins [85], antidepressants [86], and resveratrol [87] reduces microglial activation and prevents long-term cognitive dysfunction [85] and attenuates cognitive and behavioral impairments [86, 87]. Mitochondria is a possible therapeutic target [88] and the use of the mitochondrial division inhibitor Mdivi-1 attenuates oxidative stress and reduces cell death in the hippocampus [89].
Malaria
Definition and diagnosis
Malaria is caused by parasites of the gender Plasmodium [90]. In 2019, there were an estimated 229 million malaria cases in the world and 409,000 deaths [90]. The proper diagnosis of malaria is essential because identification of the causative Plasmodium species is decisive for disease prognosis and choice of therapy. Diagnosis is simple and involves microscopic visualization of parasites in a blood sample or rapid diagnostic tests that detect enzymes or antigens from Plasmodium. In countries that have a high prevalence of P. falciparum, the causative agent of cerebral malaria (CM), the rapid test for Plasmodium falciparum histidine-rich protein 2 (PfHRP2) is commonly used. Severe malaria carries high mortality rates [91, 92] due to complications as metabolic disorders, kidney failure, liver and lung disorders, anemia, and CM [93,94,95,96]. Cerebral malaria may lead to neurological complications (seizures, delirium, and coma) as well as cognitive deficits in survivors [97] and is the leading cause of non-traumatic encephalopathy in endemic regions. Non-cerebral malaria may also impact the brain, leading to cognitive and behavioral deficits [17, 98,99,100,101].
Pathophysiology and biological alterations
The pathophysiology of CM involves apoptosis of endothelial cells, BBB rupture, and subsequent neuroinflammation [97], related to an exacerbated systemic inflammation associated with parasite presence and release of toxic molecules, such as heme and hemozoin [101,102,103,104,105]. Additionally, the neurological complications of CM suggest abnormalities in neurotransmitter release. Axonal injury has been observed, thus interrupting neural integrity, distribution of neurosecretory granules, and the transport of enzymes and chemicals involved in the formation of neurotransmitters [103]. Pre-synaptic excitation and activation of synapsin I, a neuronal phosphoprotein that regulates exocytosis of synaptic vesicles and the release of neurotransmitters, have also been reported [104] (Fig. 3).
In patients with CM, brain autopsy shows: (1) cerebral edema, with blood vessels blocked by red blood cells and leukocytes, (2) malarial pigment hemozoin within the vessels, (3) petechial hemorrhages in the white matter, and (4) an abrupt transition from white to gray matter [105]. MRI in CM has revealed: (1) lesions mainly in the frontoparietal lobe, corpus callosum, and internal capsule [106], (2) vasogenic and cytotoxic edema mainly in posterior areas of the brain [107], and (3) focal or diffuse lesions in the centrum semiovale, corpus callosum, thalamus, and cortex [106, 107]. Notably, cases of non-cerebral malaria also show brain changes on MRI [108].
Therapeutic tools
Although its global impact remains high, malaria is a treatable disease. The main objective of current treatment is to ensure elimination of the parasite. The development of drug-resistant Plasmodium strains is a major obstacle to malaria control [109]. Chemoprophylaxis and chemotherapy are currently the only alternatives capable of controlling malaria. Rapid treatment prevents transmission and the progression to severe forms of the disease, including death. The choice of antimalarial drug regimen is largely dependent on the causative species of Plasmodium, the severity of the disease, and whether the patient is part of a high-risk group (children, pregnant women, and immunosuppressed individuals) (Table 2). The current first-line treatment for cases of complicated malaria is combination therapy based on intravenous artesunate, artemisinin and its derivatives. Adjuvant therapies such as administration of antipyretics, anticonvulsants, anti-inflammatories, vasodilators, glucose infusion, and blood transfusion are also used in complicated malaria [110]. Routine seizure prophylaxis and induced coma are not recommended in patients with CM. Likewise, the empirical administration of mannitol to reduce intracranial pressure [111] or phenobarbital or fosphenytoin [112] is not recommended. Dexamethasone and other corticosteroids have been shown not to improve vasogenic edema, coma, or recovery, and are therefore not recommended [113, 114].
There are still no therapies to treat the neurological sequelae of CM. Several adjuvant therapies for severe malaria have been tested, such as: rosiglitazone [115,116,117,118], statins [119], fasudil, and curcumin [120, 121]. In experimental cerebral malaria (ECM), several therapies have been studied with controversial results [122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139].
Influenza
Definition and diagnosis
Influenza is an extremely contagious disease caused by a single-stranded RNA virus and a leading cause of illness and death worldwide, with an estimated of 1 billion cases, and 290,000–650,000 influenza-related respiratory deaths occurring every year [140]. Influenza A and B viruses lead to an acute respiratory infection with fever, cough, chills, myalgia, and headache [141]. Although most patients recover completely from influenza infection, there are short- and long-term consequences in the CNS. The most common extra-respiratory complications are encephalopathies, presenting as delirium, myelopathy, seizures, and ataxia, among other manifestations which usually occur one week after the first symptoms of influenza [4]. Since 1918, various neurological and cognitive effects have been associated with influenza infection. During the 1918 pandemic, several cases of post-influenza psychosis were reported in Europe and the U.S. [122], followed by a nearly decade-long global epidemic of encephalitis lethargica, a complex condition which involves Parkinsonism, lethargy, and sleep disorders [4]. In addition, several cases of other CNS disorders were reported in flu patients, suggesting that influenza may affect the brain and lead to long-term consequences [123]. Influenza-associated encephalopathies and other neurological complications were described in Japan and in several countries following the 2009 pandemic [142]. Fifty percent of patients infected with H1N1 presented neurological symptoms, such as headache, and 9% presented several neurological complications [143]. Moreover, recent outbreaks of seasonal flu have confirmed that neurological complications may arise as a consequence of influenza infections [124]. Nevertheless, the causal link between encephalitis lethargica and influenza remains controversial [125].
The diagnosis of influenza-associated encephalopathy is challenging due to a lack of specific criteria. Detection of influenza RNA in the cerebrospinal fluid, blood samples, and nasopharynx can confirm infection. EEG, brain computed tomography (CT) scan and/or MRI findings, this may suffice to confirm influenza encephalopathy [126, 127]. The major symptoms are headache, numbness, drowsiness, seizures, and, in some cases, coma. Other symptoms such as focal or generalized weakness, vertigo, ataxia, dystonia, and speech disorders have been reported [128, 143].
Pathophysiology and biological alterations
Some influenza virus strains are considered neurotropic/neurovirulent because they are able to enter the CNS through infection of microvascular endothelial cells or through the olfactory, vagus, trigeminal, and sympathetic nerves. Nevertheless, neurological complications have been reported after infection with neurotropic [129] and non-neurotropic [130, 131] virus strains alike. As most influenza virus strains are considered non-neurotropic, the neurological complications associated with influenza infection likely occur as a consequence of systemic inflammation rather than direct viral invasion [123, 131]. High levels of pro-inflammatory cytokines and chemokines are released into the circulation [144, 145] (Fig. 3). All viral infections, including influenza, elicit a type-I interferon response in the host, which is essential to control the infection [132, 133]. However, increased levels of IFN-α in the brain may contribute to cerebral damage, resulting in memory impairment and depression in humans [134]. In rodents, increased expression of IFN-α leads to neurodegeneration, neuroinflammation, and changes in cognitive function [135]. The non-neurotropic H1N1 influenza strain has been associated with an increase in the hippocampus cytokine levels after infection [130], and spatial memory deficits associated with changes in hippocampal neuron morphology, increased microglial reactivity, and a decrease in neurotrophin expression levels have been reported [131].
In up to 50–55% of individuals with influenza-associated encephalopathy, brain CT scans are normal. MRI may show lesions in the corpus callosum, cerebellum, brain stem, and thalamus bilaterally. Changes in white matter, deep grey matter, and cortical areas may also be seen [127, 136,137,138,139].
Therapeutic tools
There are few studies about therapeutic approaches to treat the neurological complications associated with influenza; in clinical practice, treatment is essentially symptomatic. The main recommendation is to use antiviral treatment as soon as possible to prevent the development of neurological damage [127] (Table 2). The specific mechanism behind this effect remains unclear, but it is presumed that antiviral drugs inhibit viral expression and replication, which results in a diminished inflammatory response [146,147,148].
There are few reports of a combination of high-dose oseltamivir with glucocorticoids, such as methylprednisolone [149], and dexamethasone [150] with promising results. However, whether oseltamivir reaches sufficient concentrations to inhibit viral replication in the cerebrospinal fluid is unknown [151].
SARS-CoV-2 infection
Definition and diagnosis
Severe acute respiratory disease coronavirus 2 (SARS-CoV-2) is a novel coronavirus that has rapidly disseminated worldwide, causing the coronavirus disease 2019 (COVID-19) pandemic [152]. COVID-19 presents a very heterogeneous clinical spectrum from no symptoms to multiple organ dysfunction syndrome (MODS) [12, 153] Neurological symptoms can be present early in the course of the disease [154]; thus, the use of blood biomarkers for diagnosis, such as proteins that have been described to be predictive of brain injury (e.g., S100B), could be helpful [154].
Pathophysiology and biological alterations
SARS-CoV-2 infects host cells by using its structural proteins—spike (particularly S1), envelope, matrix, and nucleocapsid—to bind angiotensin-converting enzyme-2 (ACE2), a transmembrane protein widely disseminated in the respiratory tract, heart, lung, vessels, kidney, gut, and nervous system. Once bound to ACE2, SARS-CoV-2 is primed by the transmembrane serine protease-2 (TMPRSS2) in two subunits (S1 and S2). The resulting SARS-CoV-2/S1/ACE2 complex is translocated into the target cell, the S2 domain is cleaved, and the genome is released into the cytoplasm. Viral RNA is newly synthetized and replicated, and new viral particles are then assembled and released to infect other cells [155]. Although SARS-CoV-2 enters host cells by endocytosis, three key hypotheses have been proposed for cerebral involvement: (1) direct viral neurotropism; (2) hyperinflammation and hypercoagulation [156]; and (3) brain-lung cross-talk [157, 158] (Fig. 3).
Viral neurotropism may involve binding of SARS-CoV-2 to ACE2 at peripheral nerve terminals, followed by retrograde trans-synaptic passage into the CNS [159]. Other mechanisms include leukocyte migration across the BBB or binding to endothelial cells, allowing the virus to cross the BBB via the microcirculation [157]. Clinically, neurological manifestations of neuro-invasion include smell and taste disorders, which occur in 39.2% of infected individuals [160], as confirmed by MRI findings of cortical hyperintensity in the olfactory bulb and right gyrus rectus [144, 145]. SARS-CoV-2 has also been found in the brain parenchyma at autopsy, as has evidence of a lymphocytic panencephalitis and meningitis [161]. Moreover, infection of the CNS by coronaviruses may be associated with demyelinating, multiple sclerosis-like lesions [162]. However, the majority of cerebrospinal fluid samples are negative for SARS-CoV-2, limiting this hypothesis to few cases of COVID-19-related cerebral involvement [157, 163].
SARS-CoV-2 may pass to the systemic circulation, enhancing the local inflammatory response. Inflammation is a main activator of the coagulation cascade, promoting hypercoagulability, vascular dysfunction, immunothrombosis, and diffuse endotheliitis [157]. The activation of hypercoagulability and pro-inflammation may induce an immune-mediated neuropathology with spontaneous or post-traumatic hemorrhages due to consumption coagulopathy, which can be enhanced by disseminated intravascular coagulation [157]. This hyperinflammatory state can lead to a cytokine storm extending to the nervous system, with possible acute necrotizing encephalitis (ANE) [164]. Stroke can also occur secondary to altered coagulative status in COVID-19 [165,166,167,168].
Finally, the brain-lung crosstalk axis is an underestimated mechanism that suggests implications for ventilatory management in the pathogenesis of COVID-19 brain involvement. Reduced systemic oxygenation may affect brain tissue oxygenation, followed by secondary brain damage. Lung derangement may alter the fine balance between oxygen and carbon dioxide [169], an important determinant of cerebral homeostasis because of the changes in cerebral blood flow with consequent brain ischemia or hyperemia [157], eventually causing cerebral edema and loss of cerebral autoregulation [157]. Brain autopsies reported that acute brain hypoxic damage to the cerebrum and cerebellum was present in 100% of COVID-19 deaths, without evidence of encephalitis or brain invasion [170].
Therapeutic tools
Emerging therapies for COVID-19 include antivirals, immunomodulators, and other agents. No specific therapies have been identified for SARS-CoV-2 brain involvement [12], although general principles regarding neuro-ICU management (such as maintenance of appropriate mean arterial pressure and oxygenation) are warranted. Most of the drugs used against SARS-CoV-2 are currently in clinical trials, and definitive evidence is urgently needed. Direct antiviral activity remains elusive. However, all these drugs do not have a specific effect on the CNS. Dexamethasone has been shown to decrease mortality in patients requiring ventilatory support [171]. The efficacy of corticosteroids in neurological disorders depends on the pathophysiology of the underlying condition. In case of encephalitis or demyelinating lesions, corticosteroids may improve the clinical response, while no recommendation can be made in neurological disorder associated with COVID-19.
The hypercoagulative state characteristic of COVID-19 can be modulated with anticoagulants (e.g., heparin) which have been associated with better prognosis in those with markedly elevated D-dimer [172,173,174,175]. In non-ventilated COVID-19 patients, tocilizumab has been included among the medications able to reduce the likelihood of progression to mechanical ventilation or death, but it does not improve survival [176].
Finally, since COVID-19 is characterized by hypoxia, maintaining optimal oxygen delivery by modulating the hemoglobin concentration, the cardiac output, and optimizing ventilator strategies in patients requiring mechanical ventilation may be essential to preventing hypoxic and ischemic brain damage [157, 177] (Table 2).
Conclusions
To date, no literature review has focused on infectious disease-associated encephalopathies. In this paper, we focused on four infectious diseases known to cause encephalopathy: sepsis, malaria, influenza, and, COVID-19. Observing these infectious diseases caused by different pathogens (bacteria, viruses, and parasites), which present different diagnostic challenges, distinct pathophysiology and different therapeutic approaches allows us to compare the different processes (e.g., cytokine storm, ischemia, alterations in amino acid metabolism) involved in the development of an encephalopathy. Importantly, observing common points shared by these different diseases may help develop new or emerging therapies. Further studies focusing on the treatment of encephalopathies are urgently needed, as therapy remains largely supportive and most experimental studies have yet to reach clinical trials. Lastly, neuroinflammation is a key and common factor between several CNS disorders, including infectious diseases from different etiologies. Thus, the search for therapeutic approaches to address infectious disease-associated encephalopathies must be prioritized to prevent and mitigate additional strain on already overburdened health systems.
Availability of data and materials
Not applicable.
Abbreviations
- ATP:
-
Adenosine triphosphate
- ACE2:
-
Angiotensin-converting enzyme-2
- BBB:
-
Blood–brain barrier
- BDNF:
-
Brain-derived neurotrophic factor
- CLP:
-
Cecal ligation and puncture
- CNS:
-
Central nervous system
- CM:
-
Cerebral malaria
- CT:
-
Computed tomography
- DAMPs:
-
Damage-associated molecular patterns
- eNOS:
-
Endothelial nitric oxide synthase
- ECM:
-
Experimental CM
- GABA:
-
Gamma-aminobutyric acid
- GFAP:
-
Glial fibrillary acidic protein
- HHV:
-
Human herpesviruses
- IDO:
-
Indoleamine-2,3 dioxygenase
- IFN:
-
Interferon
- IL:
-
Interleukin
- LPS:
-
Lipopolysaccharide
- MRI:
-
Magnetic resonance imaging
- MSC:
-
Mesenchymal stromal cells
- MPTP:
-
Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NGF:
-
Nerve growth factor
- NO:
-
Nitric oxide
- PAMPs:
-
Pathogen-associated molecular patterns
- PfHRP-2:
-
Plasmodium falciparum Histidine-rich protein 2
- pLDH:
-
Plasmodium Lactate dehydrogenase
- ROS:
-
Reactive oxygen species
- SAE:
-
Sepsis-associated encephalopathy
- SARS-CoV-2:
-
Severe acute respiratory syndrome-coronavirus-2
- STAT:
-
Signal transducers and activators of transcription
- TMPRSS2:
-
Transmembrane serine protease-2
- TNF:
-
Tumor necrosis factor
References
Erkkinen MG, Berkowitz AL. A clinical approach to diagnosing encephalopathy. Am J Med. 2019;132:1142–7.
Turner L, Lavstsen T, Berger SS, Wang CW, Petersen JEV, Avril M, et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. 2013;498(7455):502–5.
Gilden D, Nagel MA. Neurological complications of human herpesvirus infections. Int Neurol. 2016. https://doi.org/10.1002/9781118777329.ch81.
Ludlow M, Kortekaas J, Herden C, Hoffmann B, Tappe D, Trebst C, et al. Neurotropic virus infections as the cause of immediate and delayed neuropathology. Acta Neuropathol. 2016;131:159–84.
Koyuncu OO, Hogue IB, Enquist LW. Virus infections in the nervous system. Cell Host Microbe. 2013;13:379–93.
McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurol. 2005;4(9):543–55.
Tanajura D, Castro N, Oliveira P, Neto A, Muniz A, Carvalho NB, et al. Neurological manifestations in human T-cell lymphotropic virus type 1 (HTLV-1)-infected individuals without HTLV-1-associated myelopathy/tropical spastic paraparesis: a longitudinal cohort study. Clin Infect Dis. 2015;61(1):49–56.
Mehta R, Gerardin P, de Brito CAA, Soares CN, Ferreira MLB, Solomon T. The neurological complications of chikungunya virus: a systematic review. Rev Med Virol. 2018;28(3):e1978.
Maschke M, Kastrup O, Diener HC. CNS manifestations of cytomegalovirus infections: diagnosis and treatment. CNS Drugs. 2002;16(5):303–15.
Carod-Artal FJ, Wichmann O, Farrar J, Gascón J. Neurological complications of dengue virus infection. Lancet Neurol. 2013;12(9):906–19.
Albe JR, Boyles DA, Walters AW, Kujawa MR, McMillen CM, Reed DS, et al. Neutrophil and macrophage influx into the central nervous system are inflammatory components of lethal rift valley fever encephalitis in rats. PLoS Pathog. 2019;15(6):e1007833.
Robba C, Battaglini D, Pelosi P, Rocco PRM, Robba C, Battaglini D, et al. Multiple organ dysfunction in SARS-CoV-2: MODS. Expert Rev Respir Med. 2020. https://doi.org/10.1080/17476348.2020.1778470.
Alvarado-Esquivel C, Rico-Almochantaf YR, Hernández-Tinoco J, Quiñones-Canales G, Sánchez-Anguiano LF, Torres-González J, et al. Toxoplasma gondii exposure and neurological disorders: an age- and gender-matched case-control pilot study. Eur J Microbiol Immunol. 2017;7(4):303–9.
Py MO. Neurologic manifestations of chagas disease. Curr Neurol Neurosci Rep. 2011;11(6):536–42.
Sorrell TC, Chen SC-A, Phillips P, Marr KA. Clinical perspectives on Cryptococcus neoformans and Cryptococcus gattii: implications for diagnosis and management. Cryptococcus. 2014. https://doi.org/10.1128/9781555816858.ch44.
Kihara M, Carter JA, Newton CRJC. The effect of Plasmodium falciparum on cognition: a systematic review. Trop Med Int Heal. 2006;11(4):386–97.
Brasil LMBF, Vieira JLF, Araújo EC, Piani PPF, Dias RM, Ventura AMRS, et al. Cognitive performance of children living in endemic areas for Plasmodium vivax. Malar J. 2017;16:370.
Tapajós R, Castro D, Melo G, Balogun S, James M, Pessoa R, et al. Malaria impact on cognitive function of children in a peri-urban community in the Brazilian Amazon. Malar J. 2019;18(1):173.
Barichello T, Simões LR, Valvassori SS, Generoso JS, Aveline PEDV, Dominguini D, et al. Klebsiella pneumoniae meningitis induces memory impairment and increases pro-inflammatory host response in the central nervous system of Wistar rats. J Med Microbiol. 2013;63:111–7.
Korman TM, Turnidge JD, Lindsay Grayson M. Neurological complications of Chlamydial infections: case report and review. Clin Infect Dis. 1997;25(4):847–51.
Wang N, Han YH, Sung JY, Sen LW, Ou TY. Atypical leptospirosis: an overlooked cause of aseptic meningitis. BMC Res Notes. 2016;9:154.
Disson O, Lecuit M. Targeting of the central nervous system by Listeria monocytogenes. Virulence. 2012;3(2):213–21.
Rock RB, Olin M, Baker CA, Molitor TW, Peterson PK. Central nervous system tuberculosis: pathogenesis and clinical aspects. Clin Microbiol Rev. 2008;21(2):243–61.
Decaux G, Szyper M, Ectors M, Cornil A, Franken L. Central nervous system complications of Mycoplasma pneumoniae. J Neurol Neurosurg Psychiatry. 1980;43(10):883–7.
Walker MJ, Barnett TC, McArthur JD, Cole JN, Gillen CM, Henningham A, et al. Disease manifestations and pathogenic mechanisms of group A Streptococcus. Clin Microbiol Rev. 2014;27(2):264–301.
Oyanguren B, Esteban L, Guillán M, de Felipe A, Alonso Cánovas A, Navas E, et al. Central nervous system involvement in adult patients with invasive infection caused by Streptococcus agalactiae. Neurologia. 2015;30:158–62.
Dantzer R, Connor JCO, Freund GG, Johnson RW, Kelley KW. From inflamation to sickness. Nat Rev Neurosci. 2008;9:46–56.
Papadopoulos MC, Davies DC, Moss RF, Tighe D, Bennett ED. Pathophysiology of septic encephalopathy: a review. Crit Care Med. 2000;28(8):3019–24.
Wendeln A, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. 2018;556(7701):332–8.
Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14(7):463–77.
Vitkovic L, Konsman JP, Bockaert J, Dantzer R, Homburger V, Jacque C. Cytokine signals propagate through the brain. Mol Psychiatry. 2000;5(6):604–15.
Romeo HE, Tio DL, Rahman SU, Chiappelli F, Taylor AN. The glossopharyngeal nerve as a novel pathway in immune-to-brain communication: relevance to neuroimmune surveillance of the oral cavity. J Neuroimmunol. 2001;115(1–2):91–100.
Ek M, Kurosawa M, Lundeberg T, Ericsson A. Activation of vagal afferents after intravenous injection of interleukin-1β: role of endogenous prostaglandins. J Neurosci. 1998;18(22):9471–9.
Bluthé RM, Michaud B, Kelley KW, Dantzer R. Vagotomy attenuates behavioural effects of interleukin-1 injected peripherally but not centrally. NeuroReport. 1996;7(9):1485–8.
Bluthé RM, Michaud B, Kelley KW, Dantzer R. Vagotomy blocks behavioural effects of interleukin-1 injected via the intraperitoneal route but not via other systemic routes. NeuroReport. 1996;7(15–17):2823–7.
Quan N, Whiteside M, Herkenham M. Time course and localization patterns of interleukin-1β messenger RNA expression in brain and pituitary after peripheral administration of lipopolysaccharide. Neuroscience. 1998;83(1):281–93.
Konsman JP, Vigues S, Mackerlova L, Bristow A, Blomqvist A. Rat brain vascular distribution of interleukin-1 type-1 receptor immunoreactivity: relationship to patterns of inducible cyclooxygenase expression by peripheral inflammatory stimuli. J Comp Neurol. 2004;472(1):113–29.
Banks WA, Erickson MA. The blood-brain barrier and immune function and dysfunction. Neurobiol Dis. 2010;37:26–32.
Heneka MT, Carson MJ, El Khoury J, Gary E, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405.
Santos LE, Beckman D, Ferreira ST. Microglial dysfunction connects depression and Alzheimer’s disease. Brain Behav Immun. 2016;55:151–65.
DiSabato D, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139:136–53.
Shulyatnikova T, Verkhratsky A. Astroglia in sepsis associated encephalopathy. Neurochem Res. 2020;45:83–99. https://doi.org/10.1007/s11064-019-02743-2.
Milatovic D, Zaja-Milatovic S, Montine KS, Horner PJ, Montine TJ. Pharmacologic suppression of neuronal oxidative damage and dendritic degeneration following direct activation of glial innate immunity in mouse cerebrum. J Neurochem. 2003;87(6):1518–26.
Richwine AF, Parkin AO, Buchanan JB, Chen J, Markham JA, Juraska JM, et al. Architectural changes to CA1 pyramidal neurons in adult and aged mice after peripheral immune stimulation. Psychoneuroendocrinology. 2008;33(10):1369–77.
Lynch MA. Interleukin-1β exerts a myriad of effects in the brain and in particular in the hippocampus: analysis of some of these actions. Vitam Horm. 2002;64:185–219.
Pickering M, O’Connor JJ. Pro-inflammatory cytokines and their effects in the dentate gyrus. Prog Brain Res. 2007;163:339–54.
Tong L, Balazs R, Soiampornkul R, Thangnipon W, Cotman CW. Interleukin-1β impairs brain derived neurotrophic factor-induced signal transduction. Neurobiol Aging. 2008;29(9):1380–93.
Guan Z, Fang J. Peripheral immune activation by lipopolysaccharide decreases neurotrophins in the cortex and hippocampus in rats. Brain Behav Immun. 2006;20(1):64–71.
Seifert G, Schilling K, Steinhäuser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7(3):194–206.
Myint AM, Kim YK. Network beyond IDO in psychiatric disorders: revisiting neurodegeneration hypothesis. Prog Neuro-Psychopharmacol Biol Psychiatry. 2014;48:304–13.
Schmidt SK, Ebel S, Keil E, Woite C, Ernst JF, Benzin AE, et al. Regulation of IDO activity by oxygen supply: inhibitory effects on antimicrobial and immunoregulatory functions. PLoS ONE. 2013;8:e63301.
Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the global burden of disease study. Lancet. 2020;395:200–11. https://doi.org/10.1016/S0140-6736(19)32989-7.
Gofton TE, Bryan YG. Sepsis-associated encephalopathy. Nat Rev Neurol. 2012;8:557–66.
Needham DM, Davidson J, Cohen H, Hopkins RO, Weinert C, Wunsch H, et al. Improving long-term outcomes after discharge from intensive care unit: report from a stakeholders’ conference. Crit Care Med. 2012;40(2):502–9.
Robba C, Crippa IA, Taccone FS. Septic encephalopathy. Curr Neurol Neurosci Rep. 2018;18:82.
Heming N, Mazeraud A, Verdonk F, Bozza FA, Chrétien F, Sharshar T. Neuroanatomy of sepsis-associated encephalopathy. Crit Care. 2017;21:65.
Singer M, Deutschman CS, Seymour C, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA J Am Med Assoc. 2016;315:801–10.
Kuperberg SJ, Wadgaonkar R. Sepsis-associated encephalopathy: the blood–brain barrier and the sphingolipid rheostat. Front Immunol. 2017;8:1–10.
Satta MA, Jacobs RA, Kaltsas GA, Grossman AB. Endotoxin induces interleukin-1β and nitric oxide synthase mRNA in rat hypothalamus and pituitary. Neuroendocrinology. 1998;67(2):109–16.
Korcok J, Wu F, Tyml K, Hammond RR, Wilson JX. Sepsis inhibits reduction of dehydroascorbic acid and accumulation of ascorbate in astroglial cultures: intracellular ascorbate depletion increases nitric oxide synthase induction and glutamate uptake inhibition. J Neurochem. 2002;81:185–93.
Kim PKM, Zamora R, Petrosko P, Billiar TR. The regulatory role of nitric oxide in apoptosis. Int Immunopharmacol. 2001;1(8):1421–41.
Heneka MT, Löschmann P-A, Gleichmann M, Weller M, Schulz JB, Wüllner U, et al. Induction of nitric oxide synthase and nitric oxide-mediated apoptosis in neuronal PC12 cells after stimulation with tumor necrosis FActor-α/lipopolysaccharide. J Neurochem. 2002;71(1):88–94.
Semmler A, Hermann S, Mormann F, Weberpals M, Paxian SA, Okulla T, et al. Sepsis causes neuroinflammation and concomitant decrease of cerebral metabolism. J Neuroinflamm. 2008;5:1–10.
Bozza FA, Garteiser P, Oliveira MF, Doblas S, Cranford R, Saunders D, et al. Sepsis-associated encephalopathy: a magnetic resonance imaging and spectroscopy study. J Cereb Blood Flow Metab. 2010;30:440–8.
Orhun G, Tüzün E, Bilgiç B, Ergin Özcan P, Sencer S, Barburoğlu M, et al. Brain volume changes in patients with acute brain dysfunction due to sepsis. Neurocrit Care. 2020;32:459–68.
Imamura Y, Wang H, Matsumoto N, Muroya T, Shimazaki J, Ogura H, et al. Interleukin-1β causes long-term potentiation deficiency in a mouse model of septic encephalopathy. Neuroscience. 2011;187:63–9.
Stubbs DJ, Yamamoto AK, Menon DK. Imaging in sepsis-associated encephalopathy—insights and opportunities. Nat Rev Neurol. 2013;9:551–61.
Bryan Young G, Bolton CF, Archibald YM, Austin TW, Wells GA. The electroencephalogram in sepsis-associated encephalopathy. J Clin Neurophysiol. 1992;9(1):145–52.
van Gool WA, van de Beek D, Eikelenboom P. Systemic infection and delirium: when cytokines and acetylcholine collide. Lancet. 2010;375:773–5. https://doi.org/10.1016/S0140-6736(09)61158-2.
Dantzer R, Immunology I, Segalen IV, Ii B. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. NIH Public Access. 2009;14:511–22.
Pandharipande P, Morandi A, Adams JR, Girard TD, Thompson JL, Shintani A, et al. Plasma tryptophan and tyrosine levels are independent risk factors for transitioning to delirium in critically ill mechanically ventilated patients: a pilot investigation. Intensive Care Med. 2009;35:1886–92.
Basler T, Meier-Hellmann A, Bredle D, Reinhart K. Amino acid imbalance early in septic encephalopathy. Intensive Care Med. 2002;28:293–8.
Gao R, Kan M, Wang S, Yang R, Zhang S. Disrupted tryptophan metabolism induced cognitive impairment in a mouse model of sepsis-associated encephalopathy. Inflammation. 2016;39:550–60.
Gao J, Wang L, Liu J, Xie F, Su B, Wang X. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants. 2017;6:25.
Gonzalez AS, Elguero ME, Finocchietto P, Holod S, Romorini L, Miriuka SG, et al. Abnormal mitochondrial fusion–fission balance contributes to the progression of experimental sepsis. Free Radic Res. 2014;48:769–83.
Hubbard WJ, Bland KI, Chaudry IH. The role of the mitochondrion in trauma and shock. Shock. 2004;22(5):395–402.
Semmler A, Widmann CN, Okulla T, Urbach H, Kaiser M, Widman G, et al. Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J Neurol Neurosurg Psychiatry. 2013;84(1):62–9.
Chaudhry N, Duggal AK. Sepsis associated encephalopathy. Adv Med. 2014;2014:1–16.
Pandharipande PP, Sanders RD, Girard TD, McGrane S, Thompson JL, Shintani AK, et al. Effect of dexmedetomidine versus lorazepam on outcome in patients with sepsis: an a priori-designed analysis of the MENDS randomized controlled trial. Crit Care. 2010;14(2):R38.
Cepinskas G, Wilson JX. Inflammatory response in microvascular endothelium in sepsis: role of oxidants. J Clin Biochem Nutr. 2008;42:175–84.
Young GB, Doig GS. Continuous EEG monitoring in comatose intensive care patients: epileptiform activity in etiologically distinct groups. Neurocrit Care. 2005;2(1):5–10.
Lv S, Han M, Yi R, Kwon S, Dai C, Wang R. Anti-TNF-α therapy for patients with sepsis: A systematic meta-analysis. Int J Clin Pract. 2014;68:520–8.
Silva AYO, Amorim ÉA, Barbosa-Silva MC, Lima MN, Oliveira HA, Granja MG, et al. Mesenchymal stromal cells protect the blood-brain barrier, reduce astrogliosis, and prevent cognitive and behavioral alterations in surviving septic mice. Crit Care Med. 2020;48:e290–8.
Akhondzadeh F, Kadkhodaee M, Seifi B, Ashabi G, Kianian F. Adipose-derived mesenchymal stem cells and conditioned medium attenuate the memory retrieval impairment during sepsis in rats. Mol Neurobiol. 2020;57(9):3633–45.
Reis PA, Alexandre PCB, D’Avila JC, Siqueira LD, Antunes B, Estato V, et al. Statins prevent cognitive impairment after sepsis by reverting neuroinflammation, and microcirculatory/endothelial dysfunction. Brain Behav Immun. 2017;60:293–303. https://doi.org/10.1016/j.bbi.2016.11.006.
Anderson ST, Commins S, Moynagh P, Coogan AN. Chronic fluoxetine treatment attenuates post-septic affective changes in the mouse. Behav Brain Res. 2016;297:112–5.
Sui DM, Xie Q, Yi WJ, Gupta S, Yu XY, Li JB, et al. Resveratrol protects against sepsis-associated encephalopathy and inhibits the NLRP3/IL-1 β axis in microglia. Mediat Inflamm. 2016;2016:1045657.
de Carvalho LRP, Abreu SC, de Castro LL, Andrade da Silva LH, Silva PM, Vieira JB, et al. Mitochondria-rich fraction isolated from mesenchymal stromal cells reduces lung and distal organ injury in experimental sepsis. Crit Care Med. 2021. https://doi.org/10.1097/CCM.0000000000005056.
Deng S, Ai Y, Gong H, Feng Q, Li X, Chen C, et al. Mitochondrial dynamics and protective effects of a mitochondrial division inhibitor, Mdivi-1, in lipopolysaccharide-induced brain damage. Biochem Biophys Res Commun. 2018;496:865–71.
WHO. World malaria report 2020. Geneva: WHO; 2020.
WHO. World malaria report 2019. Geneva: WHO; 2019.
Idro R, Marsh K, John CC, Newton CRJ. Cerebral malaria: mechanisms of brain injury and strategies for improved neurocognitive outcome. Pediatr Res. 2010;68(4):267–74.
Taylor WRJ, Hanson J, Turner GDH, White NJ, Dondorp AM. Respiratory manifestations of malaria. Chest. 2012;142(2):492–505.
Anstey NM, Handojo T, Pain MCF, Kenangalem E, Tjitra E, Price RN, et al. Lung injury in vivax malaria: pathophysiological evidence for pulmonary vascular sequestration and posttreatment alveolar-capillary inflammation. J Infect Dis. 2007;195(4):589–96.
Akinyemi RO, Owolabi MO, Ihara M, Damasceno A, Ogunniyi A, Dotchin C, et al. Stroke, cerebrovascular diseases and vascular cognitive impairment in Africa. Brain Res Bull. 2019;145:97–108.
Conroy AL, Opoka RO, Bangirana P, Idro R, Ssenkusu JM, Datta D, et al. Acute kidney injury is associated with impaired cognition and chronic kidney disease in a prospective cohort of children with severe malaria. BMC Med. 2019;17(1):98.
Schiess N, Villabona-Rueda A, Cottier KE, Huether K, Chipeta J, Stins MF. Pathophysiology and neurologic sequelae of cerebral malaria. Malar J. 2020;19:266.
Guha SK, Tillu R, Sood A, Patgaonkar M, Nanavaty IN, Sengupta A, et al. Single episode of mild murine malaria induces neuroinflammation, alters microglial profile, impairs adult neurogenesis, and causes deficits in social and anxiety-like behavior. Brain Behav Immun. 2014;42:123–37.
Nankabirwa J, Wandera B, Kiwanuka N, Staedke SG, Kamya MR, Brooker SJ. Asymptomatic Plasmodium infection and cognition among primary schoolchildren in a high malaria transmission setting in Uganda. Am J Trop Med Hyg. 2013;88:1102–8.
John CC, Bangirana P, Byarugaba J, Opoka RO, Idro R, Jurek AM, et al. Cerebral malaria in children is associated with long-term cognitive impairment. Pediatrics. 2008;122:e92–9.
Fernando SD, Rodrigo C, Rajapakse S. The, “hidden” burden of malaria: cognitive impairment following infection. Malar J. 2010;9:366.
Starkl Renar K, Iskra J, Križaj I. Understanding malarial toxins. Toxicon. 2016;119:319–29.
Medana IM, Day NP, Hien TT, Mai NTH, Bethell D, Phu NH, et al. Axonal injury in cerebral malaria. Am J Pathol. 2002;160(2):655–66.
Thonsranoi K, Glaharn S, Punsawad C, Chaisri U, Krudsood S, Viriyavejakul P. Increased synapsin I expression in cerebral malaria. Int J Clin Exp Pathol. 2015;8:13996–4004.
Milner DA, Whitten RO, Kamiza S, Carr R, Liomba G, Dzamalala C, et al. The systemic pathology of cerebral malaria in African children. Front Cell Infect Microbiol. 2014;4:1–13.
Rasalkar DD, Paunipagar BK, Sanghvi D, Sonawane BD, Loniker P. Magnetic resonance imaging in cerebral malaria: a report of four cases. Br J Radiol. 2011;84:380–5.
Mohanty S, Benjamin LA, Majhi M, Panda P, Kampondeni S, Sahu PK, et al. Magnetic resonance imaging of cerebral malaria patients reveals distinct pathogenetic processes in different parts of the brain. MSphere. 2017;2:e00193-e217.
Frölich AM, Tober-Lau P, Schönfeld M, Brehm TT, Kurth F, Vinnemeier CD, et al. Brain magnetic resonance imaging in imported malaria. Malar J. 2019;18:74.
Ross LS, Fidock DA. Elucidating mechanisms of drug-resistant Plasmodium falciparum. Cell Host Microbe. 2019;26:35–47.
Newton CRJC, Krishna S. Severe falciparum malaria in children: current understanding of pathophysiology and supportive treatment. Pharmacol Ther. 1998;79(1):1–53.
Namutangula B, Ndeezi G, Byarugaba JS, Tumwine JK. Mannitol as adjunct therapy for childhood cerebral malaria in Uganda: a randomized clinical trial. Malar J. 2007;6:4–9.
Gwer SA, Idro RI, Fegan G, Chengo EM, Mpoya A, Kivaya E, et al. Fosphenytoin for seizure prevention in childhood coma in Africa: a randomized clinical trial. J Crit Care. 2013;28:1086–92.
Warrell DA, Looareesuwan S, Warrell MJ, Kasemsarn P, Intaraprasert R, Bunnag D, et al. Dexamethasone proves deleterious in cerebral malaria. A double-blind trial in 100 comatose patients. N Engl J Med. 1982;306:313–9.
Prasad K, Garner P. Steroids for treating cerebral malaria. Cochrane Database Syst Rev. 1999. https://doi.org/10.1002/14651858.CD000972.
Serghides L, Patel SN, Ayi K, Lu Z, Gowda DC, Liles WC, et al. Rosiglitazone modulates the innate immune response to Plasmodium falciparum infection and improves outcome in experimental cerebral malaria. J Infect Dis. 2009;199(10):1536–45.
Serghides L, McDonald CR, Lu Z, Friedel M, Cui C, Ho KT, et al. PPARγ agonists improve survival and neurocognitive outcomes in experimental cerebral malaria and induce neuroprotective pathways in human malaria. PLoS Pathog. 2014;10:e1003980.
Varo R, Crowley VM, Sitoe A, Madrid L, Serghides L, Bila R, et al. Safety and tolerability of adjunctive rosiglitazone treatment for children with uncomplicated malaria. Malar J. 2017;16(1):215.
Boggild AK, Krudsood S, Patel SN, Serghides L, Taagpukdee N, Katz K, et al. Use of peroxisome proliferator-activated receptor γ agonists as adjunctive treatment for Plasmodium falciparum malaria: a randomized, double-blind, placebo-controlled trial. Clin Infect Dis. 2009;49(6):841–9.
Reis PA, Estato V, da Silva TI, D’Avila JC, Siqueira LD, Assis EF, et al. Statins decrease neuroinflammation and prevent cognitive impairment after cerebral malaria. PLoS Pathog. 2012;8:e1003099.
Waknine-Grinberg JH, McQuillan JA, Hunt N, Ginsburg H, Golenser J. Modulation of cerebral malaria by fasudil and other immune-modifying compounds. Exp Parasitol. 2010;125:141–6. https://doi.org/10.1016/j.exppara.2010.01.005.
Dende C, Meena J, Nagarajan P, Nagaraj VA, Panda AK, Padmanaban G. Nanocurcumin is superior to native curcumin in preventing degenerative changes in experimental cerebral malaria. Sci Rep. 2017;7(1):10062.
Kristensson K. Avian influenza and the brain—comments on the occasion of resurrection of the Spanish flu virus. Brain Res Bull. 2006;68:406–13.
Henry J, Smeyne RJ, Jang H, Miller B, Okun MS. Parkinsonism and neurological manifestations of influenza throughout the 20th and 21st centuries. Park Relat Disord. 2010;16(9):566–71.
Britton PN, Blyth CC, Macartney K, Dale RC, Li-Kim-Moy J, Khandaker G, et al. The spectrum and burden of influenza-associated neurological disease in children: combined encephalitis and influenza sentinel site surveillance from Australia, 2013–2015. Clin Infect Dis. 2017;65(4):653–60.
Dale RC, Church AJ, Surtees RAH, Lees AJ, Adcock JE, Harding B, et al. Encephalitis lethargica syndrome: 20 new cases and evidence of basal ganglia autoimmunity. Brain. 2004;127(Pt 1):21–33.
Ambrozaitis A, Radzišauskiene D, Žagminas K, Kuprevičiene N, Gravenstein S, Jančoriene L. Influenza A(H1N1)pdm09 and postpandemic influenza in Lithuania. Open Med. 2016;11(1):341–53.
Meijer WJ, Linn FHH, Wensing AMJ, Leavis HL, van Riel D, GeurtsvanKessel CH, et al. Acute influenza virus-associated encephalitis and encephalopathy in adults: a challenging diagnosis. JMM Case Rep. 2016;3(6):e005076.
Radzišauskienė D, Vitkauskaitė M, Žvinytė K, Mameniškienė R. Neurological complications of pandemic A(H1N1)2009pdm, postpandemic A(H1N1)v, and seasonal influenza A. Brain Behav. 2021;11:1–11.
Jang H, Boltz D, McClaren J, Pani AK, Smeyne M, Korff A, et al. Inflammatory effects of highly pathogenic H5N1 influenza virus infection in the CNS of mice. J Neurosci. 2012;32:1545–59.
Hosseini S, Wilk E, Michaelsen-Preusse K, Gerhauser I, Baumgärtner W, Geffers R, et al. Long-term neuroinflammation induced by influenza a virus infection and the impact on hippocampal neuron morphology and function. J Neurosci. 2018;38:3060–80.
Jurgens HA, Amancherla K, Johnson RW. Influenza infection induces neuroinflammation, alters hippocampal neuron morphology, and impairs cognition in adult mice. J Neurosci. 2012;32(12):3958–68.
Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. 2012;76:16–32.
Liu Q, Zhou YH, Yang ZQ. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol Immunol. 2016;13:3–10.
Capuron L, Miller AH. Cytokines and psychopathology: lessons from interferon-α. Biol Psychiatry. 2004;56:819–24.
Sas AR, Bimonte-Nelson H, Smothers CT, Woodward J, Tyor WR. Interferon-α causes neuronal dysfunction in encephalitis. J Neurosci. 2009;29:3948–55.
Akins PT, Belko J, Uyeki TM, Axelrod Y, Lee KK, Silverthorn J. H1N1 encephalitis with malignant edema and review of neurologic complications from influenza. Neurocrit Care. 2010;13:396–406.
Goenka A, Michael BD, Ledger E, Hart IJ, Absoud M, Chow G, et al. Neurological manifestations of influenza infection in children and adults: results of a national British surveillance study. Clin Infect Dis. 2014;58(6):775–84.
Algahtani H, Shirah B. Neurological complications of novel influenza A (H1N1) pdm09 Infection: report of two cases and a systematic review of the literature. J Neuroinfectious Dis. 2016. https://doi.org/10.4172/2314-7326.1000201.
Ferrari S, Toniolo A, Monaco S, Luciani F, Cainelli F, Baj A, et al. Viral encephalitis: Etiology, clinical features, diagnosis and management. Open Infect Dis J. 2009;3:1–12.
WHO. Global influenza strategy 2019–2030. Wkly Epidemiol Monit. 2019.
Popescu CP, Florescu SA, Lupulescu E, Zaharia M, Tardei G, Lazar M, et al. Neurologic complications of influenza B virus infection in adults. Romania Emerg Infect Dis. 2017;23:574–81.
Mizuguchi M. Influenza encephalopathy and related neuropsychiatric syndromes. Influenza Other Respi Viruses. 2013;7:67–71.
Asadi-Pooya AA, Yaghoubi E, Nikseresht A, Moghadami M, Honarvar B. The neurological manifestations of H1N1 influenza infection; diagnostic challenges and recommendations. Iran J Med Sci. 2011;36(1):36–9.
Politi LS, Salsano E, Grimaldi M. Magnetic resonance imaging alteration of the brain in a patient with coronavirus disease 2019 (COVID-19) and anosmia. JAMA Neurol. 2020;77(8):1028–9.
Coolen T, Lolli V, Sadeghi N, Rovaï A, Trotta N, Taccone FS, et al. Early postmortem brain MRI findings in COVID-19 non-survivors. Neurology. 2020. https://doi.org/10.1212/WNL.0000000000010116.
Klein EY, Monteforte B, Gupta A, Jiang W, May L, Hsieh YH, et al. The frequency of influenza and bacterial coinfection: a systematic review and meta-analysis. Influenza Other Respi Viruses. 2016;10(5):394–403.
Kwong JC, Vasa PP, Campitelli MA, Hawken S, Wilson K, Rosella LC, et al. Risk of Guillain-Barré syndrome after seasonal influenza vaccination and influenza health-care encounters: a self-controlled study. Lancet Infect Dis. 2013;13:769–76.
Paksu MS, Aslan K, Kendirli T, Akyildiz BN, Yener N, Yildizdas RD, et al. Neuroinfluenza: evaluation of seasonal influenza associated severe neurological complications in children (a multicenter study). Child’s Nerv Syst. 2018;34(2):335–47.
Alsolami A, Shiley K. Successful treatment of influenza-associated acute necrotizing encephalitis in an adult using high-dose oseltamivir and methylprednisolone: case report and literature review. Open Forum Infect Dis. 2017;4:1–5.
Okumura A, Mizuguchi M, Kidokoro H, Tanaka M, Abe S, Hosoya M, et al. Outcome of acute necrotizing encephalopathy in relation to treatment with corticosteroids and gammaglobulin. Brain Dev. 2009;31:221–7.
Jhee SS, Yen M, Ereshefsky L, Leibowitz M, Schulte M, Kaeser B, et al. Low penetration of oseltamivir and its carboxylate into cerebrospinal fluid in healthy Japanese and Caucasian volunteers. Antimicrob Agents Chemother. 2008;52(10):3687–93.
World Health Organization. The coronavirus disease 2019 (COVID-19) Situation Reports. 2020.
François B, Laterre PF, Luyt CE, Chastre J. The challenge of ventilator-associated pneumonia diagnosis in COVID-19 patients. Crit Care. 2020. https://doi.org/10.1186/s13054-020-03013-2.
Dekosky ST, Kochanek PM, Valadka AB, Clark RSB, Chou SHY, Au AK, et al. Blood biomarkers for detection of brain injury in COVID-19 patients. J Neurotrauma. 2021;38(1):1–43.
Braga CL, Silva-Aguiar RP, Battaglini D, Peruchetti DB, Robba C, Pelosi P, et al. The renin-angiotensin-aldosterone system: role in pathogenesis and potential therapeutic target in COVID-19. Pharmacol Res Perspect. 2020. https://doi.org/10.1002/prp2.623.
Robba C, Battaglini D, Ball L, Valbusa A, Porto I, Della Bona R, et al. Coagulative disorders in critically ill COVID-19 patients with acute distress respiratory syndrome: a critical review. J Clin Med. 2021;10:140.
Battaglini D, Brunetti I, Anania P, Fiaschi P, Zona G, Ball L, et al. Neurological manifestations of severe SARS-CoV-2 infection: potential mechanisms and implications of individualized mechanical ventilation settings. Front Neurol Front. 2020;11:845.
Paterson RW, Brown RL, Benjamin L, Nortley R, Wiethoff S, Bharucha T, et al. The emerging spectrum of COVID-19 neurology: clinical, radiological and laboratory findings. Brain. 2020;143:2–37.
Li YC, Zhang Y, Tan BH. What can cerebrospinal fluid testing and brain autopsies tell us about viral neuroinvasion of SARS-CoV-2. J Med Virol. 2021. https://doi.org/10.1002/jmv.26943.
Beltrán-Corbellini Á, Chico-García JL, Martínez-Poles J, Rodríguez-Jorge F, Natera-Villalba E, Gómez-Corral J, et al. Acute-onset smell and taste disorders in the context of COVID-19: a pilot multicentre polymerase chain reaction based case–control study. Eur J Neurol. 2020;27(9):1738–41.
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417–8.
Rodriguez M, Soler Y, Perry M, Reynolds JL, El-Hage N. Impact of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in the nervous system: implications of COVID-19 in neurodegeneration. Front Neurol. 2020;11:583459.
Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, et al. Neurologic features in severe SARS-CoV-2 infection. N Engl J Med. 2020;382:2268–70.
Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S, Griffith B. COVID-19-associated acute hemorrhagic necrotizing encephalopathy: imaging features. Radiology. 2020;296:E119–20. https://doi.org/10.1148/radiol.2020201187.
Han H, Yang L, Liu R, Liu F, Liu F, Wu KL, et al. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin Chem Lab Med. 2020;58:1116–20. https://doi.org/10.1515/cclm-2020-0188.
Aghayari Sheikh Neshin S, Shahjouei S, Koza E, Friedenberg I, Khodadadi F, Sabra M, et al. Stroke in SARS-CoV-2 infection: a pictorial overview of the pathoetiology. Front Cardiovasc Med. 2021;8:193. https://doi.org/10.3389/fcvm.2021.649922
Nannoni S, de Groot R, Bell S, Markus HS. Stroke in COVID-19: a systematic review and meta-analysis. Int J Stroke. 2021. https://doi.org/10.1177/1747493020972922.
Qureshi AI, Baskett WI, Huang W, Shyu D, Myers D, Raju M, et al. Acute ischemic stroke and COVID-19: an analysis of 27 676 patients. Stroke. 2021;52:905–12.
Robba C, Ball L, Battaglini D, Cardim D, Moncalvo E, Brunetti I, et al. Early effects of ventilatory rescue therapies on systemic and cerebral oxygenation in mechanically ventilated COVID-19 patients with acute respiratory distress syndrome: a prospective observational study. Crit Care. 2021;25(1):111.
Solomon IH, Normandin E, Bhattacharyya S, Mukerji SS, Keller K, Ali AS, et al. Neuropathological features of Covid-19. N Engl J Med. 2020. https://doi.org/10.1056/NEJMc2019373.
RECOVERY Collaborative Group, Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, et al. Dexamethasone in hospitalized patients with Covid-19—preliminary report. N Engl J Med. 2020;384(8):1–11.
Horie S, McNicholas B, Rezoagli E, Pham T, Curley G, McAuley D, et al. Emerging pharmacological therapies for ARDS: COVID-19 and beyond. Intensive Care Med. 2020. https://doi.org/10.1007/s00134-020-06141-z.
Pugin D, Vargas MI, Thieffry C, Schibler M, Grosgurin O, Pugin J, et al. COVID-19-related encephalopathy responsive to high-dose glucocorticoids. Neurology. 2020;95:543–6.
Powers WJ, Rabinstein AA, Ackerson T, Adeoye OM, Bambakidis NC, Becker K, et al. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of acute ischemic stroke a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2019. https://doi.org/10.1161/STR.0000000000000211.
Lancaster E. The diagnosis and treatment of autoimmune encephalitis. J Clin Neurol. 2016. https://doi.org/10.3988/jcn.2016.12.1.1.
Salama C, Han J, Yau L, Reiss WG, Kramer B, Neidhart JD, et al. Tocilizumab in patients hospitalized with covid-19 pneumonia. N Engl J Med. 2021;384:20–30. https://doi.org/10.1056/NEJMoa2030340.
Robba C, Battaglini D, Ball L, Patroniti N, Loconte M, Brunetti I, et al. Distinct phenotypes require distinct respiratory management strategies in severe COVID-19. Respir Physiol Neurobiol. 2020;279:103455.
Acknowledgements
Figures created with BioRender.com and MindTheGraph. The authors thank Filippe Vasconcellos (São Paulo, Brazil) for editing assistance.
Funding
Dr. Maron-Gutierrez was supported by the Brazilian Council for Scientific and Technological Development [Grant Number 406110/2016-6] and Inova Fiocruz/Oswaldo Cruz Foundation [Grant Number VPPCB-008-FIO-18-2-56-30]. Dr. Rocco was supported by the Brazilian Council for Scientific and Technological Development (CNPq) [Grant Numbers 403485/2020-7, 401700/2020-8].
Author information
Authors and Affiliations
Contributions
MBS, MNL, TM-G, PP and PRMR made substantial contributions to the conception and design of the work and revised it critically. MBS, MNL, DB and CR wrote the manuscript and revised it critically. All authors gave final approval of the version to be published.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Barbosa-Silva, M.C., Lima, M.N., Battaglini, D. et al. Infectious disease-associated encephalopathies. Crit Care 25, 236 (2021). https://doi.org/10.1186/s13054-021-03659-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13054-021-03659-6