
Abstract
Background
Tigecycline is a vital antibiotic treatment option for infections caused by multiresistant bacteria in the intensive care unit (ICU). Acute kidney injury (AKI) is a common complication in the ICU requiring continuous renal replacement therapy (CRRT), but pharmacokinetic data for tigecycline in patients receiving CRRT are lacking.
Methods
Eleven patients mainly with intra-abdominal infections receiving either continuous veno-venous hemodialysis (CVVHD, n = 8) or hemodiafiltration (CVVHDF, n = 3) were enrolled, and plasma as well as effluent samples were collected according to a rich sampling schedule. Total and free tigecycline was determined by ultrafiltration and high-performance liquid chromatography (HPLC)-UV. Population pharmacokinetic modeling using NONMEM® 7.4 was used to determine the pharmacokinetic parameters as well as the clearance of CVVHD and CVVHDF. Pharmacokinetic/pharmacodynamic target attainment analyses were performed to explore the potential need for dose adjustments of tigecycline in CRRT.
Results
A two-compartment population pharmacokinetic (PK) model was suitable to simultaneously describe the plasma PK and effluent measurements of tigecycline. Tigecycline dialysability was high, as indicated by the high mean saturation coefficients of 0.79 and 0.90 for CVVHD and CVVHDF, respectively, and in range of the concentration-dependent unbound fraction of tigecycline (45–94%). However, the contribution of CRRT to tigecycline clearance (CL) was only moderate (CLCVVHD: 1.69 L/h, CLCVVHDF: 2.71 L/h) in comparison with CLbody (physiological part of the total clearance) of 18.3 L/h. Bilirubin was identified as a covariate on CLbody in our collective, reducing the observed interindividual variability on CLbody from 58.6% to 43.6%. The probability of target attainment under CRRT for abdominal infections was ≥ 0.88 for minimal inhibitory concentration (MIC) values ≤ 0.5 mg/L and similar to patients without AKI.
Conclusions
Despite high dialysability, dialysis clearance displayed only a minor contribution to tigecycline elimination, being in the range of renal elimination in patients without AKI. No dose adjustment of tigecycline seems necessary in CRRT.
Trial registration
EudraCT, 2012–005617-39. Registered on 7 August 2013.
Similar content being viewed by others


Background
Tigecycline is the first example of a glycylcycline, a new derivative of tetracyclines, and an important option for the treatment of infections caused by multiresistant bacteria in the intensive care unit (ICU) [1, 2]. Acute kidney injury (AKI) is a frequent complication in ICU patients and may require renal replacement therapy (RRT). Continuous veno-venous hemodialysis (CVVHD) and continuous veno-venous hemodiafiltration (CVVHDF) are well established and very effective continuous RRT (CRRT) methods that can cause enhanced elimination of drugs, including antibiotics. Accordingly, higher doses of antibiotics may be necessary in patients with AKI during CRRT [3, 4]. No dosage adjustment of tigecycline is considered necessary in patients with renal impairment or in patients undergoing hemodialysis, but data on the pharmacokinetics (PK) during CRRT are lacking [5]. The aim of this study was to provide pharmacokinetic data in ICU patients undergoing CVVHD or CVVHDF, and to explore the potential need for dose adjustments by a probability of target attainment analysis.
Methods
Setting and study population
The study was performed in an anesthesiological ICU (40 beds) of a tertiary care hospital. Patients were included when they required RRT for AKI and were treated with tigecycline (loading dose of 100 mg followed by 50 mg twice daily). Major exclusion criteria were age > 85 or < 18 years, severe liver insufficiency (Child-Pugh C), acute pancreatitis, concomitant anti-coagulation therapy, or patients with a history of allergy to tigecycline. Written informed consent was obtained from either the patient or the legal representative.
Continuous renal replacement therapy
All the equipment and solutions for CRRT were obtained from Fresenius Medical Care, Bad Homburg, Germany, except for calcium solutions which were provided by Serag-Wiessner, Naila, Germany. Patients were treated with CVVHD or CVVHDF using the MultiFiltrate system equipped with an Ultraflux AV 1000 S polysulfone membrane. For CVVHD, Ci-Ca® Dialysate K2, sodium citrate 4% and 0.5 M CaCl2 solution were used. Blood flow and dialysate flow were adjusted to body weight (< 90 kg/> 90 kg; 100/120 mL/min and 2000/2500 mL/h, respectively) [6]. The pre-filter dose of sodium citrate was adjusted to obtain a target concentration of ionized calcium post-filter of 0.25–0.35 mmol/L with a median flow rate of 176 mL/h (< 3% of blood flow) [7]. For CVVHDF, the multiBic® fluid was used for both dialysis and post-filter fluid replacement (post-dilution). Ultrafiltration rate (QFil) was 1 L/h. Anticoagulation was achieved with unfractionated heparin, targeting a 1.5-times greater activated clotting time compared with baseline.
Sampling and drug analysis
Arterial blood samples were collected using Lithium Heparin Monovettes (Sarstedt, Nümbrecht, Germany) on day 4 of treatment with tigecycline after at least 24 h on CRRT. Samples were collected immediately before the start of infusion (time 0), after 1 h (i.e., the end of infusion), and then at 1.25, 1.5, 1.75, 2, 4, 6, 8, and 12 h. At the same time points, effluent was collected into polypropylene tubes from the effluent port of the CRRT circuit.
The blood was centrifuged (10 min, 3800 g), and the plasma as well as the effluent were stored at −70 °C until analysis. Tigecycline was determined by a validated high-performance liquid chromatography (HPLC)-UV method [8]. The free concentrations of tigecycline were measured in plasma after 1, 2, and 12 h. The limit of quantification in plasma was 0.05 mg/L tigecycline, and the intra- and interassay imprecision was < 6%. The respective values in effluent were 0.025 mg/L and < 9%, respectively.
Pharmacometric analysis
For population PK modeling, NONMEM™ 7.4 (ICON Development Solutions, Hanover, MD, USA, FOCEI method) was utilized and executed via PsN (V 4.5.16) [9]. Interindividual variability was implemented on the structural PK parameters as follows:
where Pk,i represents the estimated kth PK parameter for the ith individual calculated from the population PK parameter θk of the typical patient whilst ηk, i represents the deviation from the typical PK parameter assuming log-normal distribution.
The residual variability in an individual patient at each time point (i.e., the difference between individual model predicted (YPRED, i, j) and the observed tigecycline concentration (YOBS, i, j) for the ith subject at the jth time point) was estimated by a combined proportional (εp, i, j) and/or additive (εa, i, j) residual variability model:
One- and two-compartment PK models with first-order disposition and elimination processes were fitted to the plasma data to determine the compartmental structure and the interindividual error model structure using ADVAN1 and ADVAN3 routines of NONMEM™. Allometric scaling models using total body weight with fixed [10] and freely estimated scaling parameters were evaluated.
The plasma PK model was extended to estimate the dialysis clearance. First principle dialysis modeling [11, 12] was used for this purpose as cumulated effluent concentration and volume measurements were not available.
Dialysis clearance (CLDial) of CVVHD was calculated as follows using the effluent concentrations:
where QDial represents the dialysate flow rate, Ceff represents the concentration of tigecycline in the effluent, and CPla represents the pre-filter plasma concentration.
For the CVVHDF method, the ultrafiltrate flow rate (QFil) needed to be considered in addition to QDial:
Mean saturation coefficients (Ceff/Cpla) for CVVHD indicating diffusion and for CVVHDF indicating convection, respectively, were calculated for each patient in the population PK model. Due to the very low citrate flow in relation to blood flow, no predilution correction of the CRRT clearance [13] was performed.
Age, sex, serum creatinine, creatinine clearance (Cockcroft-Gault), and bilirubin were tested as covariates on the body clearance (i.e., remaining total clearance separated from the dialysis clearance). All population PK parameters were modeled simultaneously.
Model selection was based on the likelihood ratio test (alpha = 0.05, change in degree of freedom = 1, i.e., drop in objective function value (dOFV) > 3.84) for nested models, Akaike information criterion (AIC) for non-nested models, and graphical criteria (goodness of fit plots (GOF) of population and individual prediction vs. observed concentrations, residual analyses, visual predictive checks (VPCs), n = 1000).
A mass balance analysis over 24 h under steady state was performed with the final model to investigate the impact of CVVHD; the low number of CVVHDF patients did not allow further investigation.
Probability of target attainment analysis
The final pharmacometric model was used for clinical trial simulations (n = 500 simulations from the original study design) to investigate the impact of CRRT in the present population and to compare the present patient population to patients without renal failure and to healthy volunteers. Therefore, simulations from published models for patients with complicated skin and skin structure infections (cSSSI)/intra-abdominal infections (cIAI) [14] and healthy volunteers, respectively [15], were performed. Steady-state 24-h area under the concentration-time curve (AUC24h) and probability of target attainment (PTA) were calculated for the AUC24h/minimal inhibitory concentration (MIC) target for intra-abdominal infections of 6.96 [16].
Results
Patients, infections and pathogens
A total of 11 patients were included in the study (Table 1). The patients were treated with tigecycline due to cIAI (n = 10) or infection caused by Acinetobacter baumanii (n = 1). Relevant co-conditions were: liver failure or cirrhosis (four patients), liver transplantation (two patients), extracorporeal membrane oxygenation (ECMO; one patient). Two patients died during the follow-up period of the study. A total of 109 blood samples and 108 effluent samples were collected and used for the pharmacometric analysis, excluding two 12-h blood samples with very high tigecycline concentrations indicating that blood was taken after the start of the following infusion.
Unbound fraction of tigecycline in plasma
The free plasma concentrations of tigecycline were determined after 1, 2, and 12 h (high, medium, low concentration), as tigecycline is reported to exhibit an inverse concentration-dependent plasma protein binding [17]. The decrease of the unbound fraction with increasing concentration was moderate (mean intraindividual coefficient of variation 4.9%, available as Additional file 1: Figure S1). The median unbound fraction of tigecycline in the patients was 61% (range 45–94%).
Pharmacometric analysis
A two-compartment model with first-order disposition processes described the plasma concentration-time profiles adequately and was superior to a one-compartment model (dOFV of −113.77). Various interindividual variability (IIV) models were assessed. The best model included IIV on clearance, central volume of distribution and intercompartmental clearance, guided by lowest objective function value as well as graphical improvement. Shrinkage of the individual parameters towards the population mean was moderate (≤ 26%). A combined residual variability model (proportional and additive) was not supported (additive error tended to zero), so a proportional residual variability model was chosen.
Bilirubin (normalized by the population median of bilirubin, 2.3 mg/dL) as a covariate on clearance significantly improved the fit (dOFV = -5.71, p = 0.017, interindividual variability on clearance reduced from 58.6% to 43.6%) and was included in the final model (lower bilirubin concentrations corresponded to higher clearances). Allometric scaling with a fixed exponent did not improve the model significantly and was not included. The typical body clearance was 18.3 L/h. Individual clearance values varied from 9.3 L/h (10th percentile) to 19.1 L/h (90th percentile) depending on the bilirubin concentration (24 mg/dL to 1.8 mg/dL). One patient receiving ECMO was investigated by a case deletion study that showed no significant influence of this patient on the model parameters.
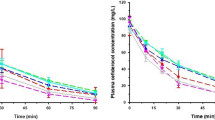
The effluent measurements were added to the model and resulted in an estimated dialysis clearance of 1.69 L/h for CVVHD and 2.71 L/h for the CVVHDF method. The mean (±SD, interindividual variability) saturation coefficient was 0.79 ± 0.36 for CVVHD and 0.90 for CVVHDF (an IIV for this method was not supported by the data (IIV tended to zero during estimation)). The population pharmacokinetic parameters are presented in Table 2. The visual predictive check indicated high agreement between the observed and model-predicted tigecycline concentration-time profiles in plasma and effluent (Fig. 1).

Visual predictive checks for the developed population PK model. Plasma concentrations (left) and effluent concentrations (right). Observed (solid lines) and predicted median (dashed-dotted lines) with 10th to 90th observed (dashed) and predicted (dotted) percentiles. Shaded areas: 95% confidence interval
The mass balance analysis revealed a median proportion of 11.2% (3.8% to 18.3%, 10th and 90th percentile) of tigecycline eliminated by hemodialysis within a dosing interval under steady state whereas the main proportion (88.8%) was eliminated by the body.
Probability of target attainment
The simulated AUC24h under steady state was (median, 10th to 90th percentile) 6.15 mg·h/L (3.39 mg·h/L, 11.13 mg·h/L) in the CVVHD patients in the present study. The simulated AUC24h in cSSSI/cIAI patients was 5.65 mg·h/L (3.46 mg·h/L, 9.79 mg·h/L) and hence similar to the AUC24h determined in our patient collective, but more variable than the AUC24h of 6.24 mg·h/L (5.26 mg·h/L, 8.33 mg·h/L) in healthy volunteers.
Tigecycline standard dosing provided a high PTA of ≥ 0.9 for pathogens with MIC ≤ 0.5 mg/L to attain the PK/pharmacodynamic (PD) target for cIAI (AUC24h/MIC of 6.96) in our patient collective undergoing CRRT being comparable to cSSSI/cIAI patients (Fig. 2).

Probability of PK/PD target attainment analysis. Complicated intra-abdominal infections (AUC24h/MIC of 6.96) for dialysis patients of the present study compared with patients with cSSSI/cIAI (simulated from [15]) and healthy volunteers (simulated from [14]). Horizontal dashed line indicates PTA ≥ 0.9 considered as reliable target attainment. cSSSI complicated skin and skin structure infections, MIC minimal inhibitory concentration
Discussion
The present study investigated the steady-state pharmacokinetics of tigecycline 50 mg twice daily in ICU patients with AKI during CVVHD or CVVHDF. The pharmacometric analysis was performed using a population pharmacokinetic model employing effluent measurements for estimation of CRRT clearance. The clearance of the CVVHDF method was estimated to be more efficient (2.71 L/h) than the CVVHD method (1.69 L/h).
The typical PK parameters in our collective of CRRT patients were in close approximation to those determined in healthy volunteers [15] and cSSSI/cIAI patients [14]. This was also reflected in the clinical trial simulations, where the obtained mean AUC24h under a steady state was similar between our CRRT patients, cSSSI/cIAI patients [14], and healthy volunteers [15]. Notably, the variability in AUC24h (10th to 90th percentile) in healthy volunteers was markedly lower (5.26 mg·h/L, 8.33 mg·h/L) than in our patients (3.39 mg·h/L, 11.13 mg·h/L).
The covariate inclusion of bilirubin as a marker for hepatic function seemed reasonable and a previous pharmacokinetic analysis also identified bilirubin as a significant covariate, even though it was not included in their final model [14]. The use of ECMO in one patient had no significant influence on the model, which is in agreement with the case report of Veinstein et al. [18]. Additionally, the inclusion of the effluent into the pharmacometric model allowed us to distinguish between body (hepatic) clearance and dialysis clearance. The mass balance analysis using the pharmacometric model clarified that CRRT clearance is a minor factor for drug elimination (11.2%) and on a comparable level to renal elimination (13% according to the summary of product characteristics (SPC)).
According to the manufacturer’s SPC (SPC Tygacil 50 mg powder for solution for infusion, Pfizer Limited, Sandwich, UK), the in-vitro plasma protein binding of tigecycline ranges from 71% to 89% at concentrations observed in clinical studies (0.1 to 1.0 mg/L), corresponding to an unbound fraction (fu) of only 11–29%. These protein binding data have been suggested as an explanation for the poor elimination of tigecycline by intermittent hemodialysis [19]. However, the mean saturation coefficient of 0.79 for CVVHD or 0.90 for CVVHDF as found in the present study indicates good transfer of tigecycline through the dialysis membrane, and is a strong argument against a high plasma protein binding of tigecycline. Indeed, recent in-vitro investigations revealed that the protein binding of tigecycline in human plasma is rather moderate with a fu of about 50–70% at therapeutic concentrations [8]. Using this ultrafiltration method, which (in contrast to other methods) mimicked physiological conditions during ultrafiltration, the mean unbound fraction of tigecycline in the plasma of the study patients was determined to be 45–94%, in line with the saturation coefficient for CVVHD. In conclusion, the small extracorporeal clearance of tigecycline of only 11.2% of the administered dose is due to the high volume of distribution of tigecycline and not due to high plasma protein binding.
The PK/PD target attainment in our patients was similar to that in cSSSI/cIAI patients using the published model by Van Wart et al. [14]. Considering the reported PK/PD target for AUC24h/MIC of 6.96 (cIAI) [16], the standard dose of tigecycline (100 mg followed by 50 mg b.i.d.) would be considered appropriate for pathogens with a MIC value of up to 0.5 mg/L.
Some limitations of our study have to be acknowledged. The data have a pilot character, as they are based on eight patients receiving CVVHD and three patients receiving CVVHDF. However, it should be acknowledged that the studied collective represents a vulnerable population and pharmacokinetic data are lacking, and the chosen approach of a pharmacometric analysis maximized the information content drawn from the population. Still, it would be desirable to study longer time periods across several dosing occasions in future studies to detect potential time-dependencies in the pharmacokinetics. Protein binding of tigecycline is affected by divalent cations such as calcium [8]; hence, citrate anticoagulation within the extracorporeal circulation might theoretically affect its transfer through the dialysis membrane. Moreover, tigecycline can adsorb to plastic labware [8] and apparently also to dialysis membranes [19]. In one patient, we observed a time delay in the effluent concentrations which may have been caused by adsorption losses, e.g., after changing the filter, and which would have resulted in an underestimated dialysis clearance. Since the delay indicated a reversible or saturable binding, probable adsorption losses did not impact the estimated dialysis clearance significantly, and certainly did not influence systemic drug exposure. However, potential adsorption of tigecycline to other membrane types and tigecycline dialysis clearance in other RRT systems should be investigated in future studies. For the PK/PD target attainment analysis, our analysis focused on total rather than unbound AUC24h due to a lack of reliable clinical breakpoints for fAUC24h/MIC. Future clinical studies considering unbound concentrations for PK/PD target attainment are highly warranted. The use of reliable techniques in these trials will be crucial to ensure that the determined fAUC24h/MIC will be not biased by the methodology utilized to determine the unbound fraction.
Conclusions
The pharmacokinetic parameters of tigecycline are not significantly influenced by CRRT. The probability of target attainment was similar in the present patient collective receiving CRRT compared with patients with no AKI, indicating that no dose adjustment seems necessary in CRRT.
Abbreviations
- AIC:
-
Akaike information criterion
- AKI:
-
Acute kidney injury
- AUC24h :
-
Area under the concentration-time curve in 24 h
- Ceff :
-
Effluent concentration of tigecycline
- cIAI:
-
Complicated intra-abdominal infections
- CL:
-
Clearance
- CLDial :
-
Dialysis clearance
- CPla :
-
Plasma concentration of tigecycline
- CRRT:
-
Continuous renal replacement therapy
- cSSSI:
-
Complicated skin and skin structure infections
- CVVHD:
-
Continuous veno-venous hemodialysis
- CVVHDF:
-
Continuous veno-venous hemodiafiltration
- dOFV:
-
Drop in objective function value
- ECMO:
-
Extracorporeal membrane oxygenation
- fu:
-
Unbound fraction
- HPLC:
-
High performance liquid chromatography
- ICU:
-
Intensive care unit
- IIV:
-
Interindividual variability
- MIC:
-
Minimal inhibitory concentration
- NONMEM:
-
Nonlinear mixed effects modeling
- PK :
-
Pharmacokinetics
- PTA:
-
Probability of target attainment
- QDial :
-
Dialysate flow rate
- QFil :
-
Ultrafiltration flow rate
- RRT:
-
Renal replacement therapy
- SPC:
-
Summary of product characteristics
- VPC:
-
Visual predictive check
References
Giamarellou H, Poulakou G. Pharmacokinetic and pharmacodynamic evaluation of tigecycline. Expert Opin Drug Metab Toxicol. 2011;7:1459–70.
Morfin-Otero R, Dowzicky MJ. Changes in MIC within a global collection of Acinetobacter baumannii collected as part of the Tigecycline Evaluation and Surveillance Trial, 2004 to 2009. Clin Ther. 2012;34:101–12.
Lewis SJ, Mueller BA. Antibiotic dosing in critically ill patients receiving CRRT: underdosing is overprevalent. Semin Dial. 2014;27:441–5.
Eyler RF, Mueller BA. Antibiotic dosing in critically ill patients with acute kidney injury. Nat Rev Nephrol. 2011;7:226–35.
Korth-Bradley JM, Troy SM, Matschke K, Muralidharan G, Fruncillo RJ, Speth JL, et al. Tigecycline pharmacokinetics in subjects with various degrees of renal function. J Clin Pharmacol. 2012;52:1379–87.
Morgera S, Schneider M, Slowinski T, Vargas-Hein O, Zuckermann-Becker H, Peters H, et al. A safe citrate anticoagulation protocol with variable treatment efficacy and excellent control of the acid–base status. Crit Care Med. 2009;37:2018–24.
Brandenburger T, Dimski T, Slowinski T, Kindgen-Milles D. Renal replacement therapy and anticoagulation. Best Pract Res Clin Anaesthesiol. 2017;31:387–401 Available from: https://doi.org/10.1016/j.bpa.2017.08.005.
Dorn C, Kratzer A, Liebchen U, Schleibinger M, Murschhauser A, Schlossmann J, et al. Impact of experimental variables on the protein binding of tigecycline in human plasma as determined by ultrafiltration. J Pharm Sci. 2018;107:739–44. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0022354917306299.
Lindbom L, Pihlgren P, Jonsson EN, Jonsson N. PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79:241–57.
Anderson BJ, Holford NHG. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol. 2008;48:303–32 Available from: http://www.annualreviews.org/doi/10.1146/annurev.pharmtox.48.113006.094708.
Lee CS, Marbury TC, Benet LZ. Clearance calculations in hemodialysis: application to blood, plasma, and dialysate measurements for ethambutol. J Pharmacokinet Biopharm. 1980;8:69–81.
Li AMMY, Gomersall CD, Choi G, Tian Q, Joynt GM, Lipman J. A systematic review of antibiotic dosing regimens for septic patients receiving continuous renal replacement therapy: do current studies supply sufficient data? J Antimicrob Chemother. 2009;64:929–37 Available from: https://academic.oup.com/jac/article-lookup/doi/10.1093/jac/dkp302.
Choi G, Gomersall CD, Tian Q, Joynt GM, Freebairn R, Lipman J. Principles of antibacterial dosing in continuous renal replacement therapy. Crit Care Med. 2009;37:2268–82 Available from: https://insights.ovid.com/crossref?an=00003246-200907000-00021.
Van Wart SA, Owen JS, Ludwig EA, Meagher AK, Korth-Bradley JM, Cirincione BB. Population pharmacokinetics of tigecycline in patients with complicated intra-abdominal or skin and skin structure infections. Antimicrob Agents Chemother. 2006;50:3701–7 Available from: http://aac.asm.org/cgi/doi/10.1128/AAC.01636-05.
Van Wart SA, Cirincione BB, Ludwig EA, Meagher AK, Korth-Bradley JM, Owen JS. Population pharmacokinetics of tigecycline in healthy volunteers. J Clin Pharmacol. 2007;47:727–37 Available from: http://doi.wiley.com/10.1177/0091270007300263.
Passarell JA, Meagher AK, Liolios K, Cirincione BB, Van Wart SA, Babinchak T, et al. Exposure-response analyses of tigecycline efficacy in patients with complicated intra-abdominal infections. Antimicrob Agents Chemother. 2008;52:204–10 Available from: http://aac.asm.org/cgi/doi/10.1128/AAC.00813-07.
Barbour A, Schmidt S, Ma B, Schiefelbein L, Rand KH, Burkhardt O, et al. Clinical pharmacokinetics and pharmacodynamics of tigecycline. Clin Pharmacokinet. 2009;48:575–84.
Veinstein A, Debouverie O, Gregoire N, Goudet V, Adier C, Robert R, et al. Lack of effect of extracorporeal membrane oxygenation on tigecycline pharmacokinetics. J Antimicrob Chemother. 2012;67:1047–8 Available from: https://academic.oup.com/jac/article-lookup/doi/10.1093/jac/dkr550.
Honore PM, Jacobs R, De Waele E, Van Gorp V, Spapen HD. The blind spot in high-dose tigecycline pharmacokinetics in critically ill patients: membrane adsorption during continuous extracorporeal treatment. Crit Care. 2015;19:24 Available from: http://ccforum.com/content/19/1/24.
Acknowledgments
The technical assistance of the study nurses (Friederike Mezger, Anja Neth, and Kathrin Pfister) is gratefully acknowledged. Part of these data were presented at the 25th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), 25–28 April 2015, Copenhagen, Denmark (P1136).
Funding
This study was supported in part by Pfizer Germany, Berlin, Germany.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
AH and HH conceived, designed, and conducted the clinical study. CD, AK, MS, and FK performed the bioanalysis and analyzed the protein binding experiments. MGK interpreted the clinical and analytical data. AB and SGW performed the pharmacometric analysis. All authors drafted the manuscript and approved the final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the ethics committee at the medical faculty of the University of Tübingen (no. 057/2013AMG1) and the German Federal Institute for Drugs and Medical Devices (Eudra-CT number 2012–005617-39).
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. Unbound fraction of tigecycline in the plasma of 11 patients undergoing CRRT. Solid line = CVVHD, dashed line = CVVHDF. (DOCX 59 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Broeker, A., Wicha, S.G., Dorn, C. et al. Tigecycline in critically ill patients on continuous renal replacement therapy: a population pharmacokinetic study. Crit Care 22, 341 (2018). https://doi.org/10.1186/s13054-018-2278-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13054-018-2278-4