
Abstract
Background
In this study, we used targeted next-generation sequencing (NGS) to investigate the genetic basis of congenital hypothyroidism (CH) in a 19-year-old Tunisian man who presented with severe hypothyroidism and goiter.
Case presentation
The propositus reported the appearance of goiter when he was 18. Importantly, he did not show signs of mental retardation, and his growth was proportionate. A partial organification defect was detected through the perchlorate-induced iodide discharge test. NGS identified a novel homozygous mutation in exon 18 of the SLC26A7 gene (P628Qfs*11), which encodes for a new iodide transporter. This variant is predicted to result in a truncated protein. Notably, the patient's euthyroid brother was heterozygous for the same mutation. No renal acid–base abnormalities were found and the administration of 1 mg of iodine failed to correct hypothyroidism.
Conclusions
We described the first case of goitrous CH due to a homozygous mutation of the SLC26A7 gene diagnosed during late adolescence.
Similar content being viewed by others


Background
Congenital hypothyroidism (CH) may result from thyroid dysgenesis (80%) or dyshormonogenesis (20%), the latter due to mutations in genes involved in thyroid hormone synthesis, namely NIS, DUOX2, DUOXA2, TG, TPO, SLC26A4, and DEHAL1 [1,2,3]. SLC26A7, also known as a Cl-/HCO3- exchanger in the kidney [4,5,6,7], is part of the SLC26 transporter family, which includes multiple anion exchangers such as pendrin, with an affinity for iodide and chloride. This molecule has been identified as a new iodide transporter expressed at the apical membrane of thyroid follicular cells, playing a significant role in thyroid hormone biosynthesis. Recent exome sequencing studies have implicated SLC26A7 gene mutations as a new cause of CH [8,9,10]. In the study, we used targeted Next Generation Sequencing (NGS) to investigate a young man with goiter and hypothyroidism. This led to the discovery of a novel homozygous mutation in exon 18 of the SLC26A7 gene (P628Qfs*11), predicted to translate into a truncated protein.
Case presentation
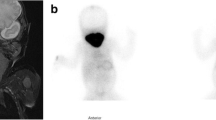
The patient was a 19-year-old Tunisian man who moved to Italy in 2021. Two months after his arrival, he experienced symptomatic neck enlargement, which was confirmed as a diffuse thyroid goiter by ultrasound (US). Initial blood tests revealed severe primary hypothyroidism (FT4 0.06 ng/dL n.v. 0.90–1.70, FT3 0.50 pg/mL n.v 2.10–4.30, TSH 396.80 µIU/mL n.v. 0.27–4.20) and the patient was promptly referred to our Center for further investigations. Notably, he exhibited no signs of mental retardation or explicit hypothyroidism symptoms. He reported no personal or familial history of thyroid disease or medication use. At the time of admission, his height was 1,76 m, his weight 62,5 kg, and his body mass index 20,2 kg/m2, indicative of normal harmonious growth. Physical examination showed a myxedematous face, with periorbital and labial edema, and neck examination identified a large diffuse goiter. No other signs of hypothyroidism, such as macroglossia, umbilical hernia or cutaneous annexes abnormalities were detected. The patient presented with severe primary hypothyroidism, as indicated by thyroid function tests (Table 1), and high levels of transaminases and lactate dehydrogenase, compatible with the state of hypothyroidism (data not shown). Ultrasound confirmed a diffuse goiter (thyroid volume 94 ml). A tru-cut biopsy showed a micro-macrofollicular thyroid with hyperplastic features. Autoimmune and infiltrative thyroid diseases were excluded, and no iatrogenic/toxic implication was detected. Thyroid scintigraphy with 131I showed increased uptake (81.6% and 98.3% after 3 and 24 h, respectively) (Fig. 1a) and the perchlorate-induced iodide discharge test indicated a 31.8% discharge rate (Fig. 1b), suggesting a partial organification defect. L-thyroxine (LT4) replacement therapy was initiated and titrated, resulting in euthyroidism and goiter reduction (thyroid volume 41 ml). Urinary parameters were also evaluated to assess potential kidney-related fluids and electrolytes alterations, but no significant abnormalities were observed (Table 2). The patient’s brother, the only relative available for examination, was clinically and biochemically euthyroid (FT4 1.06 ng/dL; FT3 5.16 pg/mL; TSH 1.1 mIU/mL; AbTPO and AbTg undetectable).

Thyroid scintigraphy with 131I (a) and perchlorate discharge test (b)
Thyroid morphology and function were evaluated using thyroid ultrasound, perchlorate discharge test as well as measurements of serum FT4, FT3, TSH, anti-TG and anti-TPO antibodies, as described [11]. A twenty-four-hour urine collection was performed to determine urine ionic composition via potentiometric system. Written informed consent for genetic analyses and for the scientific use of data were obtained from the proband and his brother in accordance with the Declaration of Helsinki and subsequent amendments (Good Clinical Practice guidelines).
Targeted NGS was conducted to investigate the cause of the goitrous hypothyroidism in our patient. We designed a custom panel targeting 34 genes involved in primary CH pathogenesis [10, 12,13,14,15,16] (Table 3). Genomic DNA was isolated from peripheral blood cells. For library preparation, we used the SureSelect QXT Reagent Kit (Agilent Technologies Inc., Santa Clara, CA, USA). Custom capture probes were then hybridized to the target sequences of the library for sequence enrichment. The enriched library was amplified using dual indexing primers. Equimolar amounts of multiple libraries were pooled into a single sample and sequenced on an Illumina MiSeq Dx System platform (Illumina Inc, San Diego, CA, USA) using the MiSeq Reagent Nano Kit V2 300 Cycles (2 × 151 bp paired-end run). The MiSeq platform generated a pair of FastQ files per sample suitable for secondary analysis with SureCall NGS software version 4.2.1 (Agilent Technologies Inc., Santa Clara, CA, USA). We performed in silico analysis of clinically relevant variants using the free web-based softwares Mutation Taster, PROVEAN and MutPred to estimate the variant's impact on the gene product. The selected variants were validated by Sanger sequencing [17]. Segregation analysis was subsequently performed.
SureCall software identified 6 pathogenic gene variants, five of which were UTR or non-coding transcript variants for the IYD and HOXB3 genes. The sixth variant, identified as chr8:92406214 AC > A, was located in the coding region of the SLC26A7 gene. This variant, registered as dbSNP ID rs768718640, affected transcripts NM_001282356, NM_001282357, NM_052832, and NM_134266, leading to a frameshift mutation with a high impact. Specifically, the homozygous deletion of the C at position 1883 of the exon 18 of the SLC26A7 gene caused a proline to glutamine substitution at position 628 of the protein, a frameshift, and a formation of a stop codon (TAA) after 11 amino acids. GnomAD Genome indicated a minor allele frequency of 0.00002833 suggesting that the variant is exceptionally rare in the general population. The new variant called c.1883delC or p.P628Qfs*11 was not present in ClinVar and was predicted to be pathogenic by MutationTaster, PROVEAN and MutPred. The Exome Aggregation Consortium accessed in January 2023 did not report homozygous cases (ExAC, https://gnomad.broadinstitute.org/) for this truncating variant. Sanger sequencing confirmed the presence of the homozygous and heterozygous P628Qfs*11 mutation in the proband and the brother, respectively. The proband family tree and sequence electropherograms were illustrated in Fig. 2.

Sanger sequencing and segregation analysis of the pathogenic variant of the SLC26A7 gene. Patient’s sequence electropherogram showing the homozygous deletion of the C (a), brother’s sequence electropherogram showing the heterozygous deletion of the C (b) and family pedigree (c). Empty symbols with a “?” inside represent family members not investigated
Given that the 30 μg iodide/day supplementation partially reversed hypothyroidism in Slc26a7-null male mice [8], we suggested to the patient a treatment trial with 1 mg iodide/day after LT4 suspension (TSH 5,72 mIU/L; FT3 5,25 ng/L; FT4 1,02 ng/dL). However, after 13 days, we ceased this treatment and reintroduced LT4 due to the decline of thyroid hormonal profile and the onset of an acute myopericarditis (TSH 17,4 mIU/L; FT4 0,65 ng/dL).
Discussion and conclusions
We report the case of a 19-year-old man with late-onset dyshormonogenic goiter and hypothyroidism. Thyroid ultrasound revealed diffuse goiter, tracer thyroid uptake was elevated, and the perchlorate-induced iodide discharge test showed a partial organification defect. NGS analysis detected a novel homozygous mutation (P628Qfs*11) in exon 18 of the SLC26A7 gene, resulting in a truncated protein. In particular, the P628Qfs*11 mutation determines a stop codon at the same position of F631Lfs*8 mutation described by Cangul et al. [8] that, by homology modeling, was predicted to destabilize the carboxyterminal sulfate transporter and anti-sigma factor antagonists (STAS) domain, which is required for membrane localization and exchanger function. The P628Qfs*11 mutant is predicted to remain trapped inside the cells as the F631Lfs*8 mutant, being not able to migrate on the cell membrane.
The first SLC26A7 gene mutations were described in 2018, identified through NGS in Arabian patients affected by dyshormonogenic CH [10]. A study by Cangul et al. examined 6 families [8], in which at least one case of CH due to SLC26A7 homozygous mutations was present. All 13 homozygous carriers of SLC26A7 mutations had CH, with goiter in 8 cases. Heterozygous carriers were euthyroid. Three of these families harbor the F631Lfs*8 mutation and CH was recognized by neonatal screening (NS) in homozygous subjects for the variant. In our case NS data were not available, but the absence of intellectual retardation and the normal growth suggest late onset hypothyroidism. Nevertheless, we can not exclude an early initiation of LT4 replacement therapy for CH, which was then interrupted and denied by the patient.
Similarly, two Japanese siblings with goitrous CH were reported to have a homozygous nonsense mutation in the SLC26A7 gene (p.Gln500Ter) [9]. In the 15-day-old male neonate CH was diagnosed by NS, while his younger sister was diagnosed with hypothyroidism and goiter at 5 years old. These cases, along with ours, suggest that the same genotype may be associated with phenotypic heterogeneity.
SLCA26A7 null mice exhibit goitrous hypothyroidism but differ from humans in radioiodine studies, showing reduced tracer uptake but normal discharge after perchlorate administration [8]. Cangul et al. [8] demonstrated that 30 µg iodide/day supplementation partially reversed hypothyroidism in Slc26a7-null mice. However, the effect of iodine supplementation in patients with SLC26A7 homozygous mutations remains unexplored. Our trial of 1 mg/die of iodine supplementation for 2 weeks after LT4 suspension did not correct hypothyroidism, but it was interrupted due to concomitant myopericarditis.
Given that Slc26a7-null mice also exhibit distal renal tubular acidosis [8], we investigated acid–base status and urinary electrolytes in our patient, finding normal renal function.
In conclusion, while from literature all CH patients with homozygous mutations of SLC26A7 gene were detected by neonatal screening or within the first years of life, we described the first case of CH due to a homozygous mutation of SLC26A7 diagnosed during late adolescence. Although the NS test was unavailable, the absence of intellectual retardation and the normal growth suggested a late onset of hypothyroidism. We suppose that other environmental factors and genetic polymorphisms of other genes involved in thyroid hormone synthesis may influence iodine transport into the lumen of the thyroid follicles and have a role in the timing and severity of hypothyroidism. No overt renal acid–base abnormalities in the healthy state were observed, but a role of the mutation when homeostasis is perturbed is possible.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- CH:
-
Congenital hypothyroidism
- NGS:
-
Next generation sequencing
- FT4:
-
Free thyroxine
- FT3:
-
Free triiodothyronine
- rT3:
-
Reverse free triiodothyronine
- TSH:
-
Thyroid stimulating hormone
- Tg:
-
Thyroglobulin
- anti-Tg antibodies:
-
Anti-thyroglobulin antibodies
- anti-TPO antibodies:
-
Anti-thyroid peroxidase antibodies
- n.v.:
-
normal values
- LT4:
-
L-thyroxine
- STAS:
-
Anti-sigma factor antagonists
- NS:
-
Neonatal screening
References
Grasberger H, Refetoff S. Genetic causes of congenital hypothyroidism due to dyshormonogenesis. Curr Opin Pediatr. 2011;23(4):421–8. https://doi.org/10.1097/MOP.0b013e32834726a4.
Targovnik HM, Citterio CE, Rivolta CM. Iodide handling disorders (NIS, TPO, TG, IYD). Best Pract Res Clin Endocrinol Metab. 2017;31(2):195–212. https://doi.org/10.1016/j.beem.2017.03.006.
De Marco G, Agretti P, Montanelli L, Di Cosmo C, Bagattini B, De Servi M, Ferrarini E, Dimida A, Freitas Ferreira AC, Molinaro A, Ceccarelli C, Brozzi F, Pinchera A, Vitti P, Tonacchera M. Identification and functional analysis of novel dual oxidase 2 (DUOX2) mutations in children with congenital or subclinical hypothyroidism. J Clin Endocrinol Metab. 2011;96(8):E1335–9. https://doi.org/10.1210/jc.2010-2467.
Petrovic S, Barone S, Xu J, Conforti L, Ma L, Kujala M, Kere J, Soleimani M. SLC26A7: a basolateral Cl-/HCO3- exchanger specific to intercalated cells of the outer medullary collecting duct. Am J Physiol Renal Physiol. 2004;286(1):F161–9. https://doi.org/10.1152/ajprenal.00219.2003.
Petrovic S, Ju X, Barone S, Seidler U, Alper SL, Lohi H, Kere J, Soleimani M. Identification of a basolateral Cl-/HCO3- exchanger specific to gastric parietal cells. Am J Physiol Gastrointest Liver Physiol. 2003;284(6):G1093–103. https://doi.org/10.1152/ajpgi.00454.2002.
Kosiek O, Busque SM, Föller M, Shcheynikov N, Kirchhoff P, Bleich M, Muallem S, Geibel JP. SLC26A7 can function as a chloride-loading mechanism in parietal cells. Pflugers Arch. 2007;454(6):989–98. https://doi.org/10.1007/s00424-007-0254-y.
Xu J, Song P, Nakamura S, Miller M, Barone S, Alper SL, Riederer B, Bonhagen J, Arend LJ, Amlal H, Seidler U, Soleimani M. Deletion of the chloride transporter slc26a7 causes distal renal tubular acidosis and impairs gastric acid secretion. J Biol Chem. 2009;284(43):29470–9. https://doi.org/10.1074/jbc.M109.044396.
Cangul H, Liao XH, Schoenmakers E, Kero J, Barone S, Srichomkwun P, Iwayama H, Serra EG, Saglam H, Eren E, Tarim O, Nicholas AK, Zvetkova I, Anderson CA, Frankl FEK, Boelaert K, Ojaniemi M, Jääskeläinen J, Patyra K, Löf C, Williams ED, UK10K Consortium, Soleimani M, Barrett T, Maher ER, Chatterjee VK, Refetoff S, Schoenmakers N. Homozygous loss-of-function mutations in SLC26A7 cause goitrous congenital hypothyroidism. JCI Insight. 2018;3(20):e99631. https://doi.org/10.1172/jci.insight.99631.
Ishii J, Suzuki A, Kimura T, Tateyama M, Tanaka T, Yazawa T, Arimasu Y, Chen IS, Aoyama K, Kubo Y, Saitoh S, Mizuno H, Kamma H. Congenital goitrous hypothyroidism is caused by dysfunction of the iodide transporter SLC26A7. Commun Biol. 2019;2:270. https://doi.org/10.1038/s42003-019-0503-6.
Zou M, Alzahrani AS, Al-Odaib A, Alqahtani M, Babiker O, Al-Rijjal RA, BinEssa HA, Kattan WE, Al-Enezi AF, Al Qarni A, Al-Faham MSA, Baitei EY, Alsagheir A, Meyer BF, Shi Y. Molecular analysis of congenital hypothyroidism in Saudi Arabia: SLC26A7 mutation is a novel defect in thyroid dyshormonogenesis. J Clin Endocrinol Metab. 2018;103(5):1889–98. https://doi.org/10.1210/jc.2017-02202.
Agretti P, De Marco G, Di Cosmo C, Ferrarini E, Montanelli L, Bagattini B, Vitti P, Tonacchera M. Congenital hypothyroidism caused by a novel homozygous mutation in the thyroglobulin gene. Eur J Pediatr. 2013;172(7):959–64. https://doi.org/10.1007/s00431-013-1976-9.
Stoupa A, Kariyawasam D, Polak M, Carré A. Genetics of congenital hypothyroidism: Modern concepts. Pediatr Investig. 2022;6(2):123–34. https://doi.org/10.1002/ped4.12324.
Yamaguchi T, Nakamura A, Nakayama K, Hishimura N, Morikawa S, Ishizu K, Tajima T. Targeted Next-generation sequencing for congenital hypothyroidism with positive neonatal TSH screening. J Clin Endocrinol Metab. 2020;105(8):dgaa308. https://doi.org/10.1210/clinem/dgaa308.
Bernal Barquero CE, Geysels RC, Jacques V, Carro GH, Martín M, Peyret V, Abregú MC, Papendieck P, Masini-Repiso AM, Savagner F, Chiesa AE, Citterio CE, Nicola JP. Targeted next-generation sequencing of congenital hypothyroidism-causative genes reveals unexpected thyroglobulin gene variants in patients with iodide transport defect. Int J Mol Sci. 2022;23(16):9251. https://doi.org/10.3390/ijms23169251.
Fu C, Luo S, Zhang Y, Fan X, D’Gama AM, Zhang X, Zheng H, Su J, Li C, Luo J, Agrawal PB, Li Q, Chen S. Chromosomal microarray and whole exome sequencing identify genetic causes of congenital hypothyroidism with extra-thyroidal congenital malformations. Clin Chim Acta. 2019;489:103–8. https://doi.org/10.1016/j.cca.2018.11.035.
Nettore IC, Desiderio S, De Nisco E, Cacace V, Albano L, Improda N, Ungaro P, Salerno M, Colao A, Macchia PE. High-resolution melting analysis (HRM) for mutational screening of Dnajc17 gene in patients affected by thyroid dysgenesis. J Endocrinol Invest. 2018;41(6):711–7. https://doi.org/10.1007/s40618-017-0795-7.
Tonacchera M, De Marco G, Agretti P, Montanelli L, Di Cosmo C, Freitas Ferreira AC, Dimida A, Ferrarini E, Ramos HE, Ceccarelli C, Brozzi F, Pinchera A, Vitti P. Identification and functional studies of two new dual-oxidase 2 (DUOX2) mutations in a child with congenital hypothyroidism and a eutopic normal-size thyroid gland. J Clin Endocrinol Metab. 2009;94(11):4309–14. https://doi.org/10.1210/jc.2009-0426.
Acknowledgements
We would like to thank all the clinical staff and the laboratory members of the Endocrine Unit of Pisa for technical support. We would also like to thank Dr. Claudio Urbani for his contribution.
Funding
No funding was perceived in relation with this case report.
Author information
Authors and Affiliations
Contributions
Each person listed as authors participated in the Work in a substantive manner. LM performed clinical evaluation and ultrasound and collected data. AB and BB contributed to clinical evaluation, SC and LP contributed to data collection and writing the manuscript. GDM, EF, CN and MRS provided technical support in laboratory. ES collected data, reviewed literature, and wrote the manuscript. PA supervised laboratory experimentations and genetic analysis and revised the manuscript. MT, CDC, FS and FL critically revised the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable (case report—see consent for publication).
Consent for publication
Consent for publication has been obtained from the patient and his brother.
Competing interests
The authors declare that they have no competing interests in connection with this case report.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sciarroni, E., Montanelli, L., Di Cosmo, C. et al. Late-onset dyshormonogenic goitrous hypothyroidism due to a homozygous mutation of the SLC26A7 gene: a case report. Ital J Pediatr 50, 106 (2024). https://doi.org/10.1186/s13052-024-01672-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13052-024-01672-3