
Abstract
Background
Duplications of the long arm of chromosome 3 are rare, and associated to a well-defined contiguous gene syndrome known as partial trisomy 3q syndrome. It has been first described in 1966 by Falek et al., and since then around 100 patients have been reported. Clinical manifestations include characteristic facial dysmorphic features, microcephaly, hirsutism, congenital heart disease, genitourinary anomalies, hand and feet abnormalities, growth disturbances and intellectual disability. Most of cases are due to unbalanced translocations, inherited from a parent carrying a balanced aberration (reciprocal translocation or inversion), and rarely the genomic anomaly arises de novo. Very few studies report on the prenatal identification of such rearrangements.
Case presentation
Hereby, we report on a newborn with a rare pure duplication of the long arm of chromosome 3. Noninvasive prenatal test (cell free fetal DNA analysis on maternal blood), performed for advanced parental age and use of assisted reproductive technique, evidenced a partial 3q trisomy. Then, invasive cytogenetic (standard and molecular) investigations, carried out through amniocentesis, confirmed and defined a 3q27.1-q29 duplication spanning 10.9 Mb, and including about 80 genes. Our patient showed clinical findings (typical facial dysmorphic features, esotropia, short neck, atrial septal defect, hepatomegaly, mild motor delay) compatible with partial trisomy 3q syndrome diagnosis, in addition to pre- and postnatal overgrowth.
Conclusions
Advanced parental age increases the probability of chromosomal and/or genomic anomalies, while ART that of epigenomic defects. Both conditions, thus, deserve more careful prenatal monitoring and screening/diagnostic investigations. Among the latter, cell free fetal DNA testing can detect large segmental aneuploidies, along with chromosomal abnormalities. It identified in our patient a wide 3q rearrangement, then confirmed and defined through invasive molecular cytogenetic analysis. Neonatologists and pediatricians must be aware of the potential risks associated to duplication syndromes. Therefore, they should offer to affected subjects an adequate management and early and careful follow-up. These may be able to guarantee to patients satisfactory growth and development profiles, prevent and/or limit neurodevelopmental disorders, and timely recognition of complications.
Similar content being viewed by others


Background
Duplications of the long arm of chromosome 3 are rare, and usually diagnosed after birth [1]. They are associated to well-defined conditions known as partial trisomy 3q syndrome [2]. Its clinical manifestations include typical dysmorphic facial features (synophrys, broad nasal bridge, anteverted nares, micrognathia, low-set dysplastic ears), microcephaly, hirsutism, congenital heart disease, genitourinary disorders, growth disturbances and cognitive deficits [2]. In around 70% of cases, the genomic anomaly is the result of a balanced translocation of a parent, who is carrier of a concomitant deletion of another chromosome, while in the others it arises de novo [3]. In 1966, Falek et al. [4] described the first subject affected with partial duplication 3q, and since then around 100 patients have been reported [5]. The critical region associated with the syndrome phenotype was later mapped to 3q26.3q27.7 [6, 7]. Cases with pure and more distal duplications have been rarely reported, and even more uncommon are those identified through prenatal diagnostic tests [8]. Hereby, we report on a newborn with prenatal finding of a de novo 3q27.1q29 duplication of 10.9 Mb, identified and defined with array comparative gemomic hybridization (a-CGH) analysis, performed through amniocentesis due to advanced parental age and use of assisted reproductive technique (ART, namely egg donation and intracytoplasmatic sperm injection, ICSI), and clinical manifestations compatible with partial trisomy 3q syndrome diagnosis, in addition to pre- and postnatal overgrowth.
Case presentation
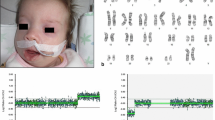
A female newborn was the first child of healthy and nonconsanguineous parents (46 year-old mother and 42 year-old father). She was delivered preterm at 35 + 2 weeks of gestation (WG) from the third pregnancy, by cesarean section due to metrorrhagia complicating placenta previa. Family history was negative for genetic diseases and/or malformation syndromes. Two previous pregnancies ended with miscarriages occurred during the first trimester. Current gestation was obtained by medically ART (in-vitro fertilization treatment by egg donation and intracytoplasmatic sperm injection, ICSI). Obstetric history revealed increased nuchal translucency (4.7 mm) on the first trimester screening examination. Soon after, non-invasive prenatal testing (NIPT), using cell free fetal DNA analysis in maternal blood, evidenced a duplication of the long arm of chromosome 3. Therefore, invasive diagnostic investigations through amniocentesis, including standard and molecular cytogenetic examinations, were suggested by obstetricians [9] and performed by the mother, along with subsequent genetic counselling. Karyotype test identified a normal female set, and a partial duplication involving the long arm of chromosome 3 (46,XX,dup(3) (q27.1q29), which was then confirmed by a-CGH analysis. The latter further defined the anomaly, spanning 10.9 Mb from position 182.989.731 to 193.854.071 (Oligo array platform 8 × 60 K, mean resolution 200–250 kb, genome assembly GRCh37.p13). The genomic abnormality comprises about 80 genes, some of which are disease causing and included in the minimal critical region responsible for 3q duplication syndrome (i.e. DVL3, EPHB3) (Fig. 1). a-CGH and fluorescent in situ hybridization (FISH) were, then, also performed in the father (the normal standard and molecular cytogenetic profiles of the biological mother were ascertained before egg donation) and evidenced no segmental aneuploidies and/or structural chromosomal rearrangements at the 3q27.1q29 region. Genetic consultation, then, confirmed that the observed genetic profile was compatible with the indication (NIPT detection of chromosome 3 abnormality) for which the fetal diagnostic test had been requested. Pregnancy follow-up through second level prenatal ultrasound (US) investigations was also suggested. Finally, the genetic tests performed in the father and the mother and resulted normal, allowed to provide a reproductive counselling to the family, and to establish the recurrence risk as low. Subsequent US investigations performed during the second trimester (21 WG), revealed increased fetal growth (main anthropometric measures around the 90th centile), in addition to lumbosacral lordosis and bilateral first degree hydronephrosis. Apgar scores were 7 and 7, at 1 and 5 min. Postnatally, our patient needed primary resuscitation, which was briefly conducted through non-invasive positive pressure ventilation. Due to dysmorphic features, along with prenatal findings of genetic and renal abnormalities, she was transferred on the first day of life to our Neonatal Intensive Care Unit. At admission, anthropometric measures were as follows: weight 2900 g (92nd centile, + 1.43 standard deviations, SD), length 47 cm (74th centile, + 0.64 SD), occipitofrontal circumference (OFC) 33.5 cm (89th centile, + 1.23 SD). Physical examination disclosed high frontal hairline, bushy eyebrows, long eyelashes, down slanting palpebral fissures, telecanthus, epicanthus, broad nasal bridge, bulbous tip, anteverted nares, long philtrum, large maxilla, carp shaped mouth with thin lips, downturned corners and tendency to keep it open (Fig. 2a). Short and wide neck, low-set, dysplastic and anteriorly rotated ears with prominent helix, and microretrognathia completed the craniofacial profile (Fig. 2c). Abdominal evaluation disclosed palpable liver, 3 cm under the costal arch. Bilateral brachydactyly of fingers, with proximally placed thumbs and clinodactyly of the fifth ones, as well as feet brachydactyly with bilateral hallux varus and crowded toes (overlapping of the second and fourth toes on the third and fifth ones, respectively) were also observed (Fig. 3a/b). Neurological findings were a mild axial central type hypotonia, as well as decreased osteotendinous and archaic reflexes. Due to the mild perinatal hypoxic injury and the following respiratory distress, she performed non-invasive ventilatory support during the first 72 h of life, and then oxygen supplementation for further two days. Owing to lack of the sucking reflex, nasogastric tube feeding along with parenteral nutrition were initially required and continued until the third day of life. Then, gradual improvement of enteral nutrition allowed adequate and complete bottle feeding, with standard infant formula [10], from day 14 of life. Newborn metabolic screening along with blood and urine biochemical tests showed normal results. Head US detected asymmetric ventricular system due to mild enlargement of the left lateral ventricle. Abdomen US revealed a bilateral II degree hydronephrosis (according with radiology grading system), with renal pelvis dilation, in the anteroposterior diameter, of 20 and 18 mm of the left and right kidney respectively, as well as hepatomegaly with homogenous echo structure. Echocardiogram documented an ostium secundum type atrial septal defect (ASD), in addition to patent foramen ovale. Ophthalmological evaluation documented no abnormalities. The following clinical course was regular, and only marked by global overgrowth. She was then discharged, and included in a multidisciplinary (auxological, neurodevelopmental, cardiological, ophthalmological, audiological, urological/surgical) follow-up. Serial auditory brainstem response (ABR) evaluations, at 2 and 4 months of age, ruled out hypoacusis. In the following months, heart US evaluations excluded the persistence of foramen ovale, while renal US documented a progressive decrease of hydronephrosis, lasting only in the left kidney (anteroposterior diameter of the renal pelvis 15 mm). Head US no longer showed abnormalities, while ophthalmological evaluation conversely disclosed bilateral esotropia. No renal and/or urinary tract infections have been registered to date. The patient currently is 8 months and 6 days old (7 months and 3 days of corrected age), and shows, according to World Health Organization growth standards for neonatal and infant close monitoring [11], global overgrowth: weight 10.1 kg (99th centile, + 2.2 SD), length 73 cm (99th centile, + 2.3 SD), and OFC 46.2 cm (99th centile, + 2.5 SD) (Fig. 2b/d). Neurodevelopmental assessment is at present normal in relational, communication, fine and gross motor areas with normal righting reactions, apart a mild delay involving upper limb lifting in prone position. She presently shows no further abnormalities.

Overview of chromosome 3, showing present patient’s q27.1-q29 duplication and involved genes, spanning 10.9 Mb of genomic DNA from position 182,989,731 to 193,854,071, according with DECIPHER Genome Browser (GRCh37/hg19 assembly) [12]

a/b/c/d a and b Patient’s front view at birth and age 8 months: high frontal hairline, bushy eyebrows, down slanting palpebral fissures, broad nasal bridge, bulbous tip, anteverted nares, long philtrum, large maxilla, carp shaped mouth with thin lips and downturned corners. c and d Lateral view at birth and age 8 months: short and wide neck, low-set, dysplastic and anteriorly rotated ears with prominent helix, and microretrognathia

a/b a Brachydactyly of fingers, with proximally placed thumb and clinodactyly of the fifth one. b Feet brachydactyly with bilateral hallux varus and crowded toes (overlapping of the second and fourth toes on the third and fifth ones)
Discussion and conclusions
Partial duplication 3q syndrome is a rare but well-defined clinical entity [1, 2]. Its phenotype includes abnormalities of the central nervous system, typical facial dysmorphic features, congenital heart disease, urogenital tract, hands and feet abnormalities, growth disturbances and intellectual disability [1]. It must be distinguished from other genetic diseases, mostly from Cornelia de Lange syndrome [5]. It is usually diagnosed after birth [1]. The present case is among the very few reported ones identified in utero [1, 8]. The critical genomic region responsible for dup(3q) syndrome phenotype has been narrowed down to 3q26.3-q27.7 regions [1, 3, 5, 13]. However, ours is among the rarely described cases with more distal duplication including chromosomal bands telomeric than q27 [2]. Indeed, the bands from 3q26.3 to 3q27 are spared in our patient, who however shows most of the clinical manifestations of the syndrome, suggesting that a role for the related critical genes might be narrowed down between 3q27.1 and 3q27.3. Actually, the comparison of our patient with those, among the few reported in literature, with even rarer “pure” 3q duplications and available precise genomic and clinical characterization reveals typical common findings, which are summarized in Table 1.
In the attempt to establish if a genotype–phenotype correlation exists, comparing our patient with the previously reported ones carrying more proximal duplications, brain abnormalities leading to cognitive disability and epilepsy have been more commonly observed in these subjects [1, 3]. The duplicated chromosomal fragment 3q26.3-q27.7 contains different genes (e.g., NLGN1, BCHE, TNIK, SOX2 and Map6D1 ) highly expressed during the embryonic development of the brain [1, 5], many of which are spared in the rearrangement of our proband. This may explain the more expressed neurological alterations of these patients [1, 3], compared with the minimal neurodevelopmental involvement of the proposita, mildly affecting only the psychomotor domain. Moreover, some patients with dup(3q) and caudal defects (one patient affected by Currarino syndrome) have been described [5], suggesting that these anomalies may be included in the phenotype of the syndrome. However, no caudal abnormalities have been observed in the present patient. Also, prenatal growth retardation is more frequently present in cases with more proximal breakpoints in 3q [2]. Conversely, normal postnatal growth, or even increased as in our case and in those reported by Grossmann et al. [2] and Lisi et al. [17], seems to be associated with a more distal breakpoint. Therefore, a chromosomal region associated to growth delay is likely located proximal to band 3q27.1, while genes responsible for growth excess, macrocephaly and obesity may harbor within the rearranged chromosomal region of our patient, according to literature data [2, 17]. Finally, the copy number variation (CNV) here reported involves the 3q29 region associated with moderate cognitive deficits, and other abnormalities (defining the 3q29 microduplication syndrome, MIM #611,936) [5, 16, 17, 20], which could also contribute to the phenotype of our proband.
An overall careful comparison of the clinical features described in other 3q duplications can help clarify the influence of specific genomic regions on the phenotype. Then, the phenotypic variability between the patients reported to date can be explained by variations of the number (and type) of active genes present in chromosomal fragments of different sizes, according to contiguous gene syndromes [3, 21,22,23,24,25,26,27]. Indeed, the genes included in the present duplicated chromosome are around 80, and some of them are associated with well-known phenotypes of the 3q duplication syndrome [2]. Among them, there are some disease causing ones, including DVL3 (MIM #601,368), EPHB3 (MIM #601,839), IGF2BP2 (MIM #608,289), AHSG (MIM #138,680), ADIPOQ (MIM #605,441), LPP (MIM #600,700), TP63 (MIM #603,273), P3H2 (MIM #610,341), FGF12 (MIM #601,513), and OPA1 (MIM #605,290) (Fig. 1) [28]. Two (DVL3 and EPHB3) are those comprised within the partial trisomy 3q syndrome critical region [5], while the others are involved in many processes, including cellular growth, brain, heart, skeletal, skin tissues and urorectal development [5], and their mutations are mainly associated with neurological, cardiac, and ocular diseases, as well as to some types of cancer [29].
Advanced parental age increases the probability of chromosomal and/or genomic anomalies, while ART that of epigenomic defects [30, 31]. Both conditions, thus, deserve more careful prenatal monitoring and screening/diagnostic investigations. Among the latter, cell free fetal DNA analysis may have a key role. Actually, it allowed in our patient the identification before birth of a large rearrangement of chromosome 3q, then confirmed and defined (precise determination of size and gene content) through molecular karyotyping technique. The present case shows how minor phenotypic effects may be due to large chromosomal rearrangements, especially if these are duplications compared to deletions. The genetic characterization of such conditions, as occurs in other chromosomal and/or genomic diseases, may address clinicians towards more individualized follow-up [32,33,34,35,36,37,38,39,40,41], focusing on definite issues (e.g., auxological and nutritional aspects, ophthalmological assessment, oncologic surveillance), in relation to the involvement of genes potentially linked to the development of specific diseases (i.e., obesity, strabismus, cancers) [5, 14, 42, 43].
The present study may be helpful to a better characterization of both clinical and genomic features of 3q duplication syndrome. It underlines how neonatologists and pediatricians must be aware of the potential risks associated to such conditions. Therefore, they should offer to affected subjects an adequate management and early and careful follow-up. These may be able to guarantee to patients satisfactory growth and development profiles, prevent and/or limit neurodevelopmental disorders, and timely recognition of complications [44,45,46,47,48,49,50].
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- a-CGH:
-
Array-Comparative Genomic Hybridization
- ART:
-
Assisted Reproductive Techniques
- ASD:
-
Atrial Septal Defect
- CNV:
-
Copy Number Variation
- FISH:
-
Fluorescent In Situ Hybridization
- ICSI:
-
Intracytoplasmic Sperm Injection
- NIPT:
-
Non-Invasive Prenatal Testing
- OFC:
-
Occipitofrontal Circumference
- TEOAEs:
-
Transient-Evoked Otoacoustic Emissions
- US:
-
Ultrasound
- WG:
-
Weeks of Gestation
References
Pasińska M, Adamczak R, Repczyńska A, Łazarczyk E, Iskra B, Runge AK, Haus O. Prenatal identification of partial 3q duplication syndrome. BMC Med Genomics. 2019;12(1):85.
Grossmann V, Müller D, Müller W, Fresser F, Erdel M, Janecke AR, Zschocke J, Utermann G, Kotzot D. “Essentially” pure trisomy 3q27 –> qter: further delineation of the partial trisomy 3q phenotype. Am J Med Genet A. 2009;149A(11):2522–6.
Abreu-González M, García-Delgado C, Cervantes A, Aparicio-Onofre A, Guevara-Yáñez R, Sánchez-Urbina R, et al. Clinical, cytogenetic, and biochemical analyses of a family with a t(3;13)(q26.2;p11.2): further delineation of 3q duplication syndrome. Case Rep Genet. 2013;2013:895259.
Falek A, Schmidt R, Jervis GA. Familial de Lange syndrome with chromosome abnormalities. Pediatrics. 1966;37(1):92–101.
Dworschak GC, Crétolle C, Hilger A, Engels H, Korsch E, Reutter H, Ludwig M. Comprehensive review of the duplication 3q syndrome and report of a patient with Currarino syndrome and de novo duplication 3q26.32-q27.2. Clin Genet. 2017;91(5):661–71.
Aqua MS, Rizzu P, Lindsay EA, Shaffer LG, Zackai EH, Overhauser J, Baldini A. Duplication 3q syndrome: molecular delineation of the critical region. Am J Med Genet. 1995;55(1):33–7.
Imataka G, Watabe Y, Kajitani S, Watanabe S, Ichikawa J, Drago F, Suzumura H, Yoshihara S. Rare de novo inversion-duplication case with pure 3qter duplication syndrome including an overlap of the dup(3q) critical region: a case report. Rare Exp Ther Med. 2017;13(6):3494–6.
Arıkan DC, Coşkun A, Arıkan I, Kıran G, Ceylaner G. Prenatally diagnosed partial trisomy 3q case with an omphalocele and less severe phenotype. J Turk Ger Gynecol Assoc. 2010;11(4):228–32.
Petersen OB, Smith E, Van Opstal D, Polak M, Knapen MFCM, Diderich KEM, Bilardo CM, Arends LR, Vogel I, Srebniak MI. Nuchal translucency of 3.0-3.4 mm an indication for NIPT or microarray? Cohort analysis and literature review. Acta Obstet Gynecol Scand. 2020;99(6):765–74.
Dipasquale V, Serra G, Corsello G, Romano C. Standard and specialized infant formulas in Europe: making, marketing, and health outcomes. Nutr Clin Pract. 2020;35(2):273–81.
World Health Organization. Child growth standards. 2021. https://www.who.int/tools/child-growth-standards/standards.
DECIPHER (DatabasE of genomiC varIation and Phenotype in Humans using Ensembl Resources). https://www.deciphergenomics.org/browser#q/MCF2L2/location/3:182870771-193854071.
Faas BHW, De Vries BBA, Van Es-Van GJ, Merkx G, Draaisma JMT, Smeets DFCM. A new case of dup(3q) syndrome due to a pure duplication of 3qter. Clin Genet. 2002;62(4):315–20.
Meins M, Hagh JK, Gerresheim F, Einhoff E, Olschewski H, Strehl H, Epplen JT. Novel case of dup(3q) syndrome due to a de novo interstitial duplication 3q24-q26.31 with minimal overlap to the dup(3q) critical region. Am J Med Genet A. 2005;132A(1):84–9.
Wilson GN, Hieber VC, Schmickel RD. The association of chromosome 3 duplication and the Cornelia de Lange syndrome. J Pediatr. 1978;93(5):783–8.
Ballif BC, Theisen A, Coppinger J, Gowans GC, Hersh JH, Madan-Khetarpal S, Schmidt KR, Tervo R, Escobar LF, Friedrich CA, McDonald M, Campbell L, Ming JE, Zackai EH, Bejjani BA, Shaffer LG. Expanding the clinical phenotype of the 3q29 microdeletion syndrome and characterization of the reciprocal microduplication. Mol Cytogenet. 2008;1:8.
Lisi EC, Hamosh A, Doheny KF, Squibb E, Jackson B, Galczynski R, Thomas GH, Batista DA. 3q29 interstitial microduplication: a new syndrome in a three-generation family. Am J Med Genet A. 2008;146A(5):601–9.
Azar GM, Conte RA, Kleyman SM, Logush AZ, Verma RS. Probing the human genome in search for a new 3q syndrome. Ann Genet. 1999;42(2):95–100.
European cytogeneticists association register of unbalanced chromosome aberrations (ECARUCA): http://agserver01.azn.nl:8080/ecaruca/ecaruca.jsp.
Willatt L, Cox J, Barber J, Cabanas ED, Collins A, Donnai D, FitzPatrick DR, Maher E, Martin H, Parnau J, Pindar L, Ramsay J, Shaw-Smith C, Sistermans EA, Tettenborn M, Trump D, de Vries BB, Walker K, Raymond FL. 3q29 microdeletion syndrome: clinical and molecular characterization of a new syndrome. Am J Hum Genet. 2005;77(1):154–60.
Rosenfeld W, Verma RS, Jhaveri RC, Estrada R, Evans H, Dosik H. Duplication 3q: severe manifestations in an infant with duplication of a short segment of 3q. Am J Med Genet. 1981;10(2):187–92.
Serra G, Antona V, Giuffrè M, Piro E, Salerno S, Schierz IAM, Corsello G. Interstitial deletions of chromosome 1p: novel 1p31.3p22.2 microdeletion in a newborn with craniosynostosis, coloboma and cleft palate, and review of the genomic and phenotypic profiles. Ital J Pediatr. 2022;48(1):38.
Piro E, Serra G, Giuffrè M, Schierz IAM, Corsello G. 2q13 microdeletion syndrome: report on a newborn with additional features expanding the phenotype. Clin Case Rep. 2021;9:e04289.
Serra G, Memo L, Antona V, Corsello G, Favero V, Lago P, Giuffrè M. Jacobsen syndrome and neonatal bleeding: report on two unrelated patients. Ital J Pediatr. 2021;47:147.
Schierz IAM, Giuffrè M, Cimador M, D’Alessandro MM, Serra G, Favata F, Antona V, Piro E, Corsello G. Hypertrophic pyloric stenosis masked by kidney failure in a male infant with a contiguous gene deletion syndrome at Xp22.31 involving the steroid sulfatase gene: case report. Ital J Pediatr. 2022;48:19.
Serra G, Felice S, Antona V, Di Pace MR, Giuffrè M, Piro E, Corsello G. Cardio-facio-cutaneous syndrome and gastrointestinal defects: report on a newborn with 19p13.3 deletion including the MAP2K2 gene. Ital J Pediatr. 2022;48:65.
Serra G, Giambrone C, Antona V, Cardella F, Carta M, Cimador M, Corsello G, Giuffrè M, Insinga V, Maggio MC, Pensabene M, Schierz IAM, Piro E. Congenital hypopituitarism and multiple midline defects in a newborn with non-familial Cat Eye syndrome. Ital J Pediatr. 2022;48(1):170.
National Center for Biotechnology Information (NCBI)-Genome data viewer: https://www.ncbi.nlm.nih.gov/genome/gdv/browser/genome/?id=GCF_000001405.25.
National Center for Biotechnology Information (NCBI)-Gene: https://www.ncbi.nlm.nih.gov/gene.
du Fossé NA, van der Hoorn MLP, van Lith JMM, le Cessie S, Lashley EELO. Advanced paternal age is associated with an increased risk of spontaneous miscarriage: a systematic review and meta-analysis. Hum Reprod Update. 2020;26(5):650–69.
Chen W, Peng Y, Ma X, Kong S, Tan S, Wei Y, Zhao Y, Zhang W, Wang Y, Yan L, Qiao J. Integrated multi-omics reveal epigenomic disturbance of assisted reproductive technologies in human offspring. EBioMedicine. 2020;61: 103076.
Zhu H, Hu Y, Zhu R, Yang Y, Zhu X, Wang W. A boy with partial trisomy of chromosome 3q24-q28 from paternal balanced insertion and multiple congenital anomalies. Am J Med Genet. 2013;161A:327–30.
Coelho Molck M, Simioni M, Paiva Vieira T, Paoli Monteiro F, Gil-da-Silva-Lopes VL. A Pure 2-Mb 3q26.2 Duplication Proximal to the Critical Region of 3q Duplication Syndrome. Mol Syndromol. 2018;9(4):197–204.
Serra G, Antona V, Schierz M, Vecchio D, Piro E, Corsello G. Esophageal atresia and Beckwith-Wiedemann syndrome in one of the naturally conceived discordant newborn twins: first report. Clin Case Rep. 2018;6(2):399–401.
Piro E, Schierz IAM, Antona V, Pappalardo MP, Giuffrè M, Serra G, Corsello G. Neonatal hyperinsulinemic hypoglycemia: case report of kabuki syndrome due to a novel KMT2D splicing-site mutation. Ital J Pediatr. 2020;46:136.
Serra G, Corsello G, Antona V, D’Alessandro MM, Cassata N, Cimador M, Giuffrè M, Schierz IAM, Piro E. Autosomal recessive polycystic kidney disease: case report of a newborn with rare PKHD1 mutation, rapid renal enlargement and early fatal outcome. Ital J Pediatr. 2020;46:154.
Piro E, Serra G, Antona V, Giuffrè M, Giorgio E, Sirchia F, Schierz IAM, Brusco A, Corsello G. Novel LRPPRC compound heterozygous mutation in a child with early-onset Leigh syndrome French-Canadian type: case report of an Italian patient. Ital J Pediatr. 2020;46(1):140.
Serra G, Antona V, Giuffré M, Li Pomi F, Lo Scalzo L, Piro E, Schierz IAM, Corsello G. Novel missense mutation of the TP63 gene in a newborn with Hay-Wells/Ankyloblepharon-Ectodermal Defects-Cleft Lip/Palate (AEC) syndrome: clinical report and follow-up. Ital J Pediatr. 2021;47:196.
Serra G, Antona V, D’Alessandro MM, Maggio MC, Verde V, Corsello G. Novel SCNN1A gene splicing-site mutation causing autosomal recessive pseudohypoaldosteronism type 1 (PHA1) in two Italian patients belonging to the same small town. Ital J Pediatr. 2021;47:138.
Serra G, Memo L, Cavicchioli P, Cutrone M, Giuffrè M, La Torre ML, Schierz IAM, Corsello G. Novel mutations of the ABCA12, KRT1 and ST14 genes in three unrelated newborns showing congenital ichthyosis. Ital J Pediatr. 2022;48(1):145.
Schierz IAM, Serra G, Antona V, Persico I, Corsello G, Piro E. Infant developmental profile of Crisponi syndrome due to compound heterozygosity for CRLF1 deletion. Clin Dysmorphol. 2020;29(3):141–3.
Maier B, Beck JD. Dup 3(q) syndrome and neuroblastoma. Eur J Pediatr. 1992;151(9):715–6.
Dundar M, Uzak A, Erdogan M, Saatci C, Akdeniz S, Luleci G, Keser I, Karauzum S. Partial trisomy 3q in a child with sacrococcygeal teratoma and Cornelia de Lange syndrome phenotype. Genet Couns. 2011;22(2):199–205.
Serra G, Giuffrè M, Piro E, Corsello G. The social role of pediatrics in the past and present times. Ital J Pediatr. 2021;47(1):239.
Piro E, Serra G, Schierz IAM, Giuffrè M, Corsello G. Fetal growth restriction: a growth pattern with fetal, neonatal and long-term consequences. Euromediterranean Biomedical Journal. 2019;14(09):038–44.
Serra G, Memo L, Coscia A, Giuffrè M, Iuculano A, Lanna M, Valentini D, Contardi A, Filippeschi S, Frusca T, Mosca F, Ramenghi LA, Romano C, Scopinaro A, Villani A, Zampino G, Corsello G, on behalf of their respective Scientific Societies and Parents’ Associations. Recommendations for neonatologists and pediatricians working in first level birthing centers on the first communication of genetic disease and malformation syndrome diagnosis: consensus issued by Italian scientific societies and parents’ associations. Ital J Pediatr. 2021;47(1):94.
Piro E, Schierz IAM, Serra G, Puccio G, Giuffrè M, Corsello G. Growth patterns and associated risk factors of congenital malformations in twins. Ital J Pediatr. 2020;46(1):73.
Pensabene M, Di Pace MR, Baldanza F, Grasso F, Patti M, Sergio M, La Placa S, Giuffre’ M, Serra G, Casuccio A, Cimador M. Quality of life improving after propranolol treatment in patients with Infantile Hemangiomas. Ital J Pediatr. 2022;48(1):140.
Piro E, Serra G, Schierz IAM, Antona V, Giuffrè M, Corsello G. Large for gestational age, macrosomia, overgrowth: an update on definitions and determinants. Euromediterr Biomed J. 2020;15(29):116–20.
Piro E, Serra G, Schierz IAM, Giuffrè M, Corsello G. Neonatal ten-year retrospective study on neural tube defects in a second level University Hospital. Ital J Pediatr. 2020;46(1):72.
Acknowledgements
Not applicable.
Funding
No funding was granted for this research.
Author information
Authors and Affiliations
Contributions
GCor conceptualized the report, revised the manuscript, and gave final approval of the version to be submitted. GS took care of the patient and drafted the paper. VA contributed to the acquisition of genetical data. MC performed urological assessment. GCol and MGu reviewed the literature, collected the clinical data, and drafted the first version of the manuscript. EP performed neurological and developmental assessment and follow-up. IAMS took care of the patient, performed cardiological evaluation and contributed to draft the manuscript. VV performed instrumental investigations. MGi revised the paper. All authors approved the final manuscript as submitted.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from both parents at admission of their newborn. The study was approved by the Mother and Child Department of the University of Palermo, ethics committee Palermo 1 (Palermo, Italy). All procedures performed in this report were in accordance with the ethical standards of the institutional and national research committee, and with the 1964 Helsinki declaration and its later amendments, or comparable ethical standards.
Consent for publication
Written informed consent was obtained from patient’s parents for publication of this case report and accompanying images.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Serra, G., Antona, V., Cimador, M. et al. New insights on partial trisomy 3q syndrome: de novo 3q27.1-q29 duplication in a newborn with pre and postnatal overgrowth and assisted reproductive conception. Ital J Pediatr 49, 17 (2023). https://doi.org/10.1186/s13052-023-01421-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13052-023-01421-y