
Abstract
Background
Contiguous gene deletion syndrome at Xp22.3 resulting in nullisomy in males or Turner syndrome patients typically encompasses the steroid sulfatase gene (STS) and contiguously located other genes expanding the phenotype. In large deletions, that encompass also the Kallmann syndrome 1 gene (KAL1), occasionally infantile hypertrophic pyloric stenosis (IHPS) and congenital anomalies of the kidney and urinary tract (CAKUT) have been reported.
Patient presentation
We report on a male newborn with family history in maternal uncle of renal abnormalities and short stature still without ichthyosiform dermatosis. The baby presented CAKUT with kidney failure and progressive vomiting. Renal bicarbonate loss masked hypochloremic and hypokalemic metabolic alkalosis classically present in IHPS and delayed its diagnosis. Antropyloric ultrasound examination and cystourethrography were diagnostic. After Fredet-Ramstedt extramucosal pyloromyotomy feeding and growing was regular and he was discharged home. Comparative whole-genome hybridization detected a maternal inherited interstitial deletion of 1.56 Mb on Xp22.31(6,552,712_8,115,153) × 0 involving the STS gene, but not the KAL1 gene.
Conclusions
Aberrant cholesterol sulfate storage due to STS deletion as the underlying pathomechanism is not limited to oculocutaneous phenotypes but could also lead to co-occurrence of both IHPS and kidney abnormalities, as we report. Thus, although these two latter pathologies have a high incidence in the neonatal age, their simultaneous association in our patient is resembling not a chance but a real correlation expanding the clinical spectrum associated with Xp22.31 deletions.
Similar content being viewed by others


Background
Contiguous gene deletion syndrome at Xp22.3 resulting in nullisomy in males or Turner syndrome patients is characterized by the combination of one or more monogenic disorders and clinical findings as short stature (short stature homeobox gene, SHOX), chondrodysplasia punctata (arylsulfatase genes - ARSD, ARSE, ARSF), X-linked ichthyosis (arylsulfatase C or steroid sulfatase gene, STS), ocular albinism type I (OA1) and elements of X-linked neurodevelopmental disorders and Kallmann syndrome (KAL1; reduced hypothalamic and pituitary function with resulting hypogonadotropic hypogonadism and hypoplasia of the olfactory bulb) [1], whereas the term Rud’s syndrome should no longer be used [2]. FG syndrome 3 is also mapped to this region [3]. In large deletions, occasionally cardiac arrhythmia [4], periventricular nodular heterotopia [5], acute lymphoblastic leukemia [6], end-stage renal failure [7] and infantile hypertrophic pyloric stenosis (IHPS) [3, 4, 8,9,10,11,12] were also reported. The most critical region of deletion breakpoints, characterized by a low frequency of interspersed repeats and a low GC content [13], encompasses the STS gene (MIM*300747) resulting in microsomal enzyme deficiency with an incidence about 1:1500 in males [14]. This membrane-bound enzyme is ubiquitously expressed and hydrolyzes several 3-beta-hydroxysteroid sulfates, which serve as metabolic precursors for estrogens, androgens, and cholesterol [15]. Despite the widespread enzyme deficiency, patients apparently have abnormalities only of the stratum corneum where increased cholesterol sulphate concentrations are causing abnormal desquamation, decreased corneodesmosomal degradation and retention hyperkeratosis of the skin mostly a few weeks after birth, but conatal collodion is also reported [12]. There might be associated cardiac arrhythmia and benign Pre-Descemet corneal dystrophy characterized by cholesterol sulfate accumulation and punctiform opacities without vision impairment on the one side, as well as cryptorchidism and neurobehavioral disorders due to deficient (neuro-) steroids on the other side [4, 16]. Despite the escape of lyonization, some female deletion carriers also have corneal opacities and can present parturition disturbances and cervical dystocia due to lacking placental production of estriol [4, 14]. Congenital anomalies of the kidney and urinary tract (CAKUT) have been reported rarer in STS limited microdeletions or point mutations than in larger deletions of Xp22.3 that encompass also the KAL1 gene, a neighboring gene important for urogenital development [7, 12, 13, 17, 18].
We report on a male newborn with family history in maternal uncle of renal abnormalities and short stature still without ichthyosiform dermatosis. The baby presented CAKUT with kidney failure and progressive vomiting. Renal bicarbonate loss masked hypochloremic and hypokalemic metabolic alkalosis classically present in IHPS and delayed its diagnosis. This report of associated STS deletion and IHPS further define and expand the clinical spectrum associated with CNV in this region and provide support for the role of modifiers contributing to phenotypic variability.
Patient presentation
This male term newborn is the second son of healthy non consanguineous Caucasian parents. His maternal uncle suffering from nephropathy had undergone a kidney transplant. Fetal sonographic assessment revealed hydronephrosis bilaterally, and oligohydramnios inducted to Caesarean section. At birth baby’s weight was 2710 g (− 1.07 SDS/10th centile), length 46 cm (− 1.82 SDS/3rd centile), and head circumference 33 cm (− 1.09 SDS/14th centile). During the first week of life, he developed severe acidosis and was referred to our department. Physical examination was unremarkable except for pale skin and hyporeactive aspect; male genitals were normal. There were no edemas. Diuresis, and blood pressure were normal. Laboratory investigations diagnosed renal insufficiency by low bicarbonates 15 mmol/l, augmented creatininemia 3.12 mg/dl, urea 89 mg/dl, chlor 120 mEq/l, moderate proteinuria 327 mg/l, glucosuria 500 mg/l and microhematuria, while anion gap, albuminemia, proteinemia and uric acid were preserved. Abdominal ultrasound and subsequent voiding cystourethrography showed renal hypoplasia on the left and renal dysplasia on the right as well as moderate hydronephrosis due to grade IV vesicoureteral reflux. X-ray, cranial and cardiac ultrasounds and electrocardiogram were normal. He started intravenous rehydration and bicarbonate supplementation. Refeeding by breast milk and a special powdered feed with low levels of potassium for renal impairment (Kindergen® 1 g in 5 ml water) was initiated after 12 h. He tended to have regurgitations attributed to a urinary infection and treated on the fifth day of the hospital stay with oral amoxicillin switched to oral cefixime on day 14 (sensitive to Escherichia coli) until negative urinary cultures were reported. Persistent regurgitation did not ameliorate by trials of smaller, more frequent feeds, thickened formula, and anti-Trendelenburg positional management. At 1 month of age, intermittent nonbilious vomiting increased markedly, he weighed 3110 g (< 0.4 centile), creatininemia and urea were halved, bicarbonates kalium and chlor were normal.
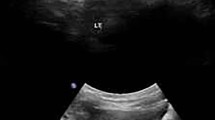
Antropyloric ultrasound examination revealed hypertrophied muscular layer of 4.6 mm and elongation of the pyloric canal of 19 mm (diameter 14 mm). In retrospect, some frame of the cystourethrogram had already shown an air-filled stomach with undulating contours known as “Caterpillar sign” (Fig. 1). Fredet-Ramstedt extramucosal longitudinal pyloromyotomy was performed. Afterwards, feeding and growing was regular and he was discharged home 45 days old. All treatment options have been discussed with both parents. Erythropoietin treatment and clinical multidisciplinary follow-up are ongoing. At 6 month of age, large polygonal, brownish scales appeared particularly on the anterior aspect of the lower extremities.

At 24 days of life, voiding cystourethrogram is showing absence of bladder (B) and urethral abnormalities, but right-sided vesicoureteral reflux with moderate dilatation of the ureter (U), renal pelvis (R) and calyces and blunting of fornices as well as accidental finding of persistent distended stomach (S) with undulating contours known as Caterpillar sign, virtually pathognomonic for hypertrophic pyloric stenosis
Comparative whole-genome hybridization was performed during hospitalization using the Agilent® 8x60K microarray and detected a maternal inherited interstitial deletion of 1.56 Mb on arr [hg19] Xp22.31(6,552,712_8,115,153) × 0 involving the genes STS, variable charge X-linked (VCX; *300229), pseudouridine 5′-phospatase (PUDP; *306480), Patatin like phospholipase domain containing-4 (PNPLA4; *300102) and microRNA MIR4767 and MIR651.
Discussion and conclusion
We report on a male newborn with nonaccidental association of IHPS, kidney failure and maternal Xp22.3 deletion involving the STS gene.
IHPS is the most common form of gastrointestinal obstruction in infancy (1:700), five times more frequent in males than in females and hereditably is high as 87% [19, 20]. Isolated and syndromic IHPS are described [21]. The exact etiology of isolated IHPS is unknown, although neuronal nitric oxide synthase (NOS) upregulation and an extracellular matrix abnormality have been reported in subsets [21]. Various potential genetic loci have been investigated, as well as various environmental factors (maternal smoking or young age, firstborn, feeding practice, post-natal erythromycin use) without producing conclusive data. Interestingly, reducing erythromycin indications and increasing dietary intake of omega-3 fatty acids in Western countries during the last decade probably decreased the incidence of isolated IHPS [20, 22]. By studying syndromic IHPS (Table 1) it was evidenced that the lipid metabolism plays a fundamental role in etiopathogenesis [23]. The risk of IHPS is inversely and significantly associated with total cholesterol level with an Odds ratio of 0.77 (95% CI, 0.64–0.92; p = 0.005) per 10 mg/dL [24]. Indeed, there are higher incidence of IHPS in syndromes affecting the lipid metabolism. A classic example is the Smith-Lemli-Opitz syndrome, an autosomal recessive congenital disorder caused by mutations in the 7-dehydrocholesterol reductase (DHCR7) gene at 11q13. Affected individuals are unable to complete the final step in cholesterol biosynthesis with accumulation of aberrant 7-dehydrocholesterol in developing tissues causing a wide range of metabolic and developmental abnormalities, including IHPS in 10–15% of cases [21]. In congenital generalized lipodystrophy type IV (CAVIN1 gene at 17q21) diffuse skeletal and smooth muscle hypertrophy are leading to cardiac arrythmia and IHPS [21, 25]. In syndromes associated with hypotonia, as in FG syndrome 3 (Xp22.3) or Down syndrome (critical region 21q22.3), the IHPS incidence is about 7% [3, 21]. Other syndromes frequently associated with IHPS are connective tissue disorders in which abnormal or excess of connective tissue in the pylorus gradually develop mechanic obstruction [21]. Furthermore, biopsies have shown not only muscle layer hypertrophy but also accumulation of extracellular matrix molecules (chondroitin-sulfate proteoglycan and fibronectin) [26]. This is also the underlying cause for unsuccessful non-surgical conservative treatment with oral or intravenous administration of atropine, leaving the surgical extramucosal pyloromyotomy as the gold standard [27].
STS alterations as in our case report, can lead to disturbed intracellular metabolism of cholesterol and to storage phenomenon of cholesterol sulphate. It was evidenced that age of onset of ichthyosis or absent/mild forms of XLI, frequently found in Southern European countries, are not related to width of Xp22.3 deletion [12, 18]. The late-onset of cutaneous presentation in our newborn is possible and clinical follow up have to direct dermatological, nephrological, endocrinological and neurobehavioral care as well as infection surveillance. VCX, PUDP and mitochondria-related PNPLA4 have been implicated in neurocognitive development, although the functional significance of these genes remains under debate [11, 13]. KAL1 gene, implicated in urogenital development, is not deleted in our case.
Vomiting and growth failure present a clinical challenge in neonatal age. Major causes are severe gastroesophageal reflux, neonatal sepsis, anatomical and functional gastrointestinal obstructions including IHPS and pylorospasm; less frequent are food allergy, inborn errors of metabolism, congenital adrenal hyperplasia, intracerebral abnormalities such as subdural hemorrhage or hydrocephalus, drugs or toxic agents and/or renal tubular acidosis. This spectrum widens in case of CAKUT, as in our patient, including renal impairment, risk of urosepsis and renal adapted diet. A concomitant edema could involve also the antropyloric region and cases of IHPS have been described [28]. Interestingly, a frequent recurrence linked polycystic kidney disease (PKD) and IHPS to NOS deficiency [29, 30]. Renal neuronal NOS and inducible NOS in cystic epithelium are suppressed or lost in PKD rats [31]. Thus, NOS deficiency leads to lack of locally available nitric oxide which may cause pyloric stenosis as a result of failure of smooth muscle relaxation. Downregulation of nitric oxide production may also be involved in the pathogenesis of pyloric stenosis in this subset. On the other hand, it was shown that deficiency of STS in kidneys results in increased cholesterol sulfate accumulation which interferes with normal functioning of transglutaminase 1, responsible for maintaining the integrity of cadherin-based adherens junctions between epithelial cells. The slit diaphragm of glomerular visceral epithelial cells is a modified adherens junction and, therefore, disruption of its structure by the above mechanism can result in proteinuria [7].
In conclusion, aberrant cholesterol sulfate storage due to STS deletion as the underlying pathomechanism is not limited to oculocutaneous phenotypes but could also lead to co-occurrence of both IHPS and kidney abnormalities, as we report. Thus, although these two latter pathologies have a high incidence in the neonatal age, their simultaneous association in our patient is resembling not a chance but a real correlation expanding the clinical spectrum associated with Xp22.31 deletions.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ARSD :
-
Arylsulfatase gene D
- CAKUT:
-
Congenital anomalies of the kidney and urinary tract
- CAVIN1 :
-
Caveolae associated protein 1
- DHCR7 :
-
7-dehydrocholesterol reductase
- IHPS:
-
Infantile hypertrophic pyloric stenosis
- KAL1 :
-
Kallmann syndrome 1 gene
- NOS:
-
Nitric oxide synthase
- OA1 :
-
Ocular albinism type I
- PKD:
-
Polycystic kidney disease
- PNPLA4 :
-
Patatin like phospholipase domain containing-4
- PUDP :
-
Pseudouridine 5′-phospatase
- SHOX :
-
Short stature homeobox gene
- STS :
-
Steroid sulfatase gene
- VCX :
-
Variable charge X-linked
References
Davies W. The contribution of Xp22.31 gene dosage to turner and Klinefelter syndromes and sex-biased phenotypes. Eur J Med Genet. 2021;64(4):104169.
Happle R. Rud syndrome does not exist. Eur J Dermatol. 2012;22(1):7.
Dessay S, Moizard MP, Gilardi JL, Opitz JM, Middleton-Price H, Pembrey M, et al. FG syndrome: linkage analysis in two families supporting a new gene localization at Xp22.3 [FGS3]. Am J Med Genet. 2002;112(1):6–11.
Brcic L, Underwood JF, Kendall KM, Caseras X, Kirov G, Davies W. Medical and neurobehavioural phenotypes in carriers of X-linked ichthyosis-associated genetic deletions in the UK biobank. J Med Genet. 2020;57(10):692–8.
Ozawa H, Osawa M, Nagai T, Sakura N. Steroid sulfatase deficiency with bilateral periventricular nodular heterotopia. Pediatr Neurol. 2006;34(3):239–41.
Mallory SB, Kletzel M, Turley CP. X-linked ichthyosis with acute lymphoblastic leukemia. Arch Dermatol. 1988;124(1):22–4.
Matsukura H, Fuchizawa T, Ohtsuki A, Higashiyama H, Higuchi O, Higuchi A, et al. End stage renal failure in a child with X- linked ichthyosis. Pediatr Nephrol. 2003;18:297–300.
Garcia Perez A, Crespo M. X-linked ichthyosis associated with hypertrophic pyloric stenosis in three brothers. Clin Exp Dermatol. 1981;6(2):159–61.
Weissörtel R, Strom TM, Dörr HG, Rauch A, Meitinger T. Analysis of an interstitial deletion in a patient with Kallmann syndrome, X-linked ichthyosis and mental retardation. Clin Genet. 1998;54(1):45–51.
Bruno L, Bocanegra O, Magnelli N. Recessive X-linked ichthyosis associated with hypertrophic pyloric stenosis: a chance occurrence? Clin Exp Dermatol. 2003;28(1):74–6.
Puri PK, Reddi DM, Spencer-Manzon M, Deak K, Steele SU, Mikati MA. Banding pattern on polarized hair microscopic examination and unilateral polymicrogyria in a patient with steroid sulfatase deficiency. Arch Dermatol. 2012;148(1):73–8.
Diociaiuti A, Angioni A, Pisaneschi E, Alesi V, Zambruno G, Novelli A, et al. X-linked ichthyosis: clinical and molecular findings in 35 Italian patients. Exp Dermatol. 2019;28(10):1156–63.
Nagai K, Shima H, Kamimura M, Kanno J, Suzuki E, Ishiguro A, et al. Xp22.31 microdeletion due to microhomology-mediated break-induced replication in a boy with contiguous gene deletion syndrome. Cytogenet Genome Res. 2017;151(1):1–4.
Craig WY, Roberson M, Palomaki GE, Shackleton CH, Marcos J, Haddow JE. Prevalence of steroid sulfatase deficiency in California according to race and ethnicity. Prenat Diagn. 2010;30(9):893–8.
Mueller JW, Gilligan LC, Idkowiak J, Arlt W, Foster PA. The regulation of steroid action by Sulfation and Desulfation. Endocr Rev. 2015;36(5):526–63.
Rudolf M, Grösch S, Geerling G. Rezidivierende bilaterale Erosio corneae und Trübungen im Hornhautstroma. Predescemet-Dystrophie bei X-chromosomaler rezessiver ichthyosis [recurrent bilateral corneal erosions and opacities in corneal stroma. Pre-Descemet dystrophy in X chromosome recessive ichthyosis]. Ophthalmologe. 2002;99(12):962–3.
Martul P, Pineda J, Levilliers J, Vazquez JA, Rodriguez-Soriano J, Loridan L, et al. Hypogonadotrophic hypogonadism with hyposmia, X-linked ichthyosis, and renal malformation syndrome. Clin Endocrinol. 1995;42(2):121–8.
Georgopoulos NA, Koika V, Galli-Tsinopoulou A, Spiliotis BE, Adonakis G, Keramida MK, et al. Renal dysgenesis and KAL1 gene defects in patients with sporadic Kallmann syndrome. Fertil Steril. 2007;88(5):1311–7.
Krogh C, Fischer TK, Skotte L, Biggar RJ, Øyen N, Skytthe A, et al. Familial aggregation and heritability of pyloric stenosis. JAMA. 2010;303(23):2393–9.
Oetzmann von Sochaczewski C, Muensterer OJ. The incidence of infantile hypertrophic pyloric stenosis nearly halved from 2005 to 2017: analysis of German administrative data. Pediatr Surg Int. 2021;37(5):579–85.
Peeters B, Benninga MA, Hennekam RC. Infantile hypertrophic pyloric stenosis--genetics and syndromes. Nat Rev Gastroenterol Hepatol. 2012;9:646–60.
Paran M, Freud E, Samuk I, Steiner Z. Does maternal omega 3 supplementation protect against infantile hypertrophic pyloric stenosis? J Pediatr Gastroenterol Nutr. 2020;70(5):652–6.
Chung E. Infantile hypertrophic pyloric stenosis: genes and environment. Arch Dis Child. 2008;93:1003–4.
Feenstra B, Geller F, Carstensen L, Romitti PA, Körberg IB, Bedell B, et al. Plasma lipids, genetic variants near APOA1, and the risk of infantile hypertrophic pyloric stenosis. JAMA. 2013;310(7):714–21.
Rajab A, Straub V, McCann LJ, Seelow D, Varon R, Barresi R, et al. Fatal cardiac arrhythmia and long-QT syndrome in a new form of congenital generalized lipodystrophy with muscle rippling (CGL4) due to PTRF-CAVIN mutations. Plos Genet. 2010;6(3):e1000874.
Pueyo Gil C, Oshiro K, Elías Pollina J, Esteban Ibarz JA, Puri P. Aumento de la expresión del proteoglicano condroitín-sulfato, fibronectina y fibroblastos en la estenosis hipertrófica de píloro [Increase of the chondroitin-sulfate proteoglycan, fibronectin and fibroblasts in infantile hypertrophic pyloric stenosis]. Cir Pediatr. 2001;14(3):103–7.
Chiarenza SF, Bleve C, Escolino M, Esposito C, Beretta F, Cheli M, Scuderi MG, Di Benedetto V, Casadio G, Marzaro M, Gambino M, Conforti A, Pini Prato A, Molinaro F, Gerocarni Nappo S, Caione P, Mendoza-Sagaon M. Guidelines of the Italian Society of Videosurgery (SIVI) in infancy for the minimally invasive treatment of hypertrophic pyloric stenosis in neonates and infants. Pediatr Med Chir. 2020;42(1):16–24. https://doi.org/10.4081/pmc.2020.243.
Takahashi T, Sato Y, Yamazaki T, Hayashi A, Okamoto T. Vomiting in an infant with congenital nephrotic syndrome: questions. Pediatr Nephrol. 2017;32(9):1519–20.
Tennakoon J, Koh TH, Alcock G. Pyloric stenosis in a newborn baby with polycystic kidneys. J Perinatol. 2007;27(2):125–6.
Mishra K, Batra VV, Basu S, Rath B, Saxena R. Steroid-resistant nephrotic syndrome associated with steroid sulfatase deficiency-x-linked recessive ichthyosis: a case report and review of literature. Eur J Pediatr. 2012;171(5):847–50.
Wang D, Braendstrup OB, Larsen S, Horn T, Strandgaard S. The expression and activity of renal nitric oxide synthase and circulating nitric oxide in polycystic kidney disease rats. APMIS. 2004;112:358–68.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
IAMS contributed in all parts of the study, concepted, and wrote the paper. MG contributed to clinical management and consulting and revised the manuscript. MC performed surgical consulting and instrumental investigation. MMDA performed the nephrological assessment. GS contributed to clinical management and follow-up. FF collected the patient data and revised the literature. VA contributed to the interpretation of genetic data. EP performed data analysis and interpretation, and critically revised the manuscript. GC performed genetical consulting, coordinated and supervised all parts of the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Parent’s informed written consent was provided.
Consent for publication
Not applicable.
Competing interests
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Schierz, I.A.M., Giuffrè, M., Cimador, M. et al. Hypertrophic pyloric stenosis masked by kidney failure in a male infant with a contiguous gene deletion syndrome at Xp22.31 involving the steroid sulfatase gene: case report. Ital J Pediatr 48, 19 (2022). https://doi.org/10.1186/s13052-022-01218-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13052-022-01218-5