
Abstract
Background
15q26 deletion is a relatively rare chromosomal disorder, and it is described only in few cases. Patients with this aberration show many signs and symptoms, particularly pre- and postnatal growth restriction, developmental delay, microcephaly, intellectual disability and various congenital malformations.
Case presentation
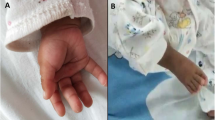
We report on a girl, 4 years old, of consanguineous parents, with a 15q26 deletion. Clinical manifestations included failure to thrive, developmental delay, microcephaly, dysmorphic facies with broad forehead, hypertelorism, narrowed eyelid slits and protruding columella. The patient also showed skeletal abnormalities, especially clinodactyly of the 5th finger, varus equine right foot and left club foot. Additionally, she had teething delay and divergent strabismus. Heart ultrasound displayed two atrial septal defects with left-to-right shunt, enlarging the right cavities. Routine cytogenetic analysis revealed a shortened 15q chromosome. Subsequent array analysis disclosed a terminal 9.15 Mb deletion at subband 15q26.1-q26.3. Four candidate genes associated with 15q26 deletion phenotype were within the deleted region, i.e. IGF1R, NR2F2, CHD2 and MEF2A.
Conclusion
We report on an additional case of 15q26 monosomy, characterized by array-CGH. Molecular cytogenetic analysis allowed us to identify the exact size of the deletion, and four candidate genes for genotype-phenotype correlation. 15q26 monosomy should be considered when growth retardation is associated with hearing anomalies and congenital heart defect, especially atrioventricular septal defects (AVSDs) and/or aortic arch anomaly (AAA).
Similar content being viewed by others


Introduction
15q26 monosomy can occur either as a de novo event leading to a pure deletion or as a consequence of ring chromosome 15 formation and unbalanced translocation. Up to now, 58 cases of pure deletion have been documented in the literature [1, 2]. The sub-bands involved in this rearrangement include many candidate genes responsible for common symptoms, especially pre and postnatal growth retardation [3], developmental delay, microcephaly. Other genes were involved in more particular features like congenital heart disease (CHD) [4], skeletal anomalies [5], diaphragmatic hernia [6], kidney anomalies [1] and seizures [7]. This variability could be assigned to the difference in breakpoint location and the size of the deleted fragment. Recently, several authors focused on some particular regions especially the report of Klasseens et al., which restricted the critical region for congenital diaphragmatic hernia (CDH) to 4 Mb at 15q26.1-q26.3 band. Thus, two annotated genes, namely NR2F2 (MIM 107773) and CDH2 (MIM 602119), were considered relevant for CDH [8]. Other genes have been reported as playing crucial role in pathogenesis of 15q26 deletions, particularly IGF1R, CHD2, NR2F2, involved respectively in growth restriction [3], neurodevelopmental disorders [9], and CHD [4].
Herein, we report a further patient with CHD, intellectual disability and failure to thrive. Array-CGH displayed a terminal 9.15 Mb deletion spanning 15q26.1-q26.3, four relevant disease genes, i.e. IGF1R, NR2F2, MEF2A, and CHD2 were involved and are directly related to the clinical presentation of our case.
Until now, no patient had a deletion of this exact size, without similar works in the literature that already focused on the types of CHD assigned to the 15q26 deletion, or on other possible genotype-phenotype correlations.
Case report
The proband, a four-year-old girl, came to our attention because of dysmorphic face and heart malformation. She was the only child of healthy, consanguineous parents. There were no health problems in the family or a history of miscarriages. She was born at term by cesarean section because of intrauterine growth restriction (IUGR) associated with oligoamnios. IUGR was noted since the fourth month of pregnancy without that maternal or placental causes have been identified. Her birth weight was 950 g (< 3rd centile). Clinical history was suggestive of and congenital hypotonia. Upon clinical examination, her weight was 8 kg (< 3rd centile), height 81 cm (< 3rd centile) and head circumference 43 cm (< 3rd centile). She had dysmorphic features including broad forehead, hypertelorism, narrowed eyelid slits, low set ears, protruding columella, and short neck. She also presented with skeletal abnormalities, especially clinodactyly of the 5th finger, right foot varus equine, left club foot, biphalangeal fifth finger, and widely-spaced toes. Additionally, she had teething delay and divergent strabismus. Chest X-ray showed dorsal scoliosis and enlarged cardiac silhouette with a cardiothoracic ratio of 70%. An echocardiogram displayed a left-to-right shunt with significant flow, presence of two atrial septal defects (ASD) of 10 mm and 7 mm in width, dilating the right heart cavities and the pulmonary artery trunk with normal right pressures no aortic arch anomalies were evidenced. Her bone age was 2 years at a chronological age of 4 years. Hematologic investigations at the age of 4 and half years old showed normal serum concentrations of calcium, T4, TSH, GH and IGF1. Ocular assessment and brainstem acoustic potential evaluation revealed divergent strabismus and sensorineural hearing loss, respectively. Further investigations including brain MRI, computed tomography of the brain and abdominal ultrasound were normal.
Methods and results
Cytogenetics
Chromosome slides were prepared from cultured peripheral blood lymphocytes of the proband and her parents after obtaining informed consent. RHG-banding and high resolution R-banded chromosome analysis was performed on the three samples according to standard procedures. Both parents displayed normal karyotypes. Cytogenetic studies of the child showed an abnormal female karyotype with an apparently terminal deletion of the long arm of one chromosome 15 (Fig. 1). The patient’s karyotype was designated as 46,XX,?del (15q)dn.

High-resolution R-banded karyogram (a,c), partial RHG (b) karyogram (c) showing deletion 15q chromosome with an abnormally short q-arm. (Red arrow)
Array analysis
After disclosing the chromosomal aberration in the child, a genome wide array analysis was performed using the CytoScan HD SNP-based array platform (Affymetrix, Inc., Santa Clara, CA, USA) with an average resolution of approximately 20 kb following the manufacturer’s protocols. Inherent to the structure of the human genome, this resolution is not achieved for all regions such as the centromeric regions and heterochromatic parts.
Genome wide array analysis confirmed the cytogenetic results and mapped the terminal deletion to a 9.15 Mb region encompassing 36 annotated genes with the proximal breakpoint at 93,275,228 Mb in band q26.1. (Fig. 2).

A. CGH-array data showing a terminal loss of 9.15 Mb in 15q26.1-q26.3 involving 93,275,228-102,429,113 breakpoints. B. UCSC genome browser assembly (GRCh37/hg19) highlighting genes involved in the loss segment
Based on these results, the final karyotype was designated as 46,XX,del [10](q26.1) dn.arr [GRCh37] 15q26.1q26.3(93,275,228-102,429,113) × 1.
In addition to this terminal loss of chromosome 15, several homozygous regions (ROH) were detected (164 Mb of the autosomal genome (~ 5.6%)), which is in agreement with the indicated consanguinity of the parents. The analysis of ROH using Genomic Oligoarray and SNP array evaluation tool (http://firefly.ccs.miami.edu/cgi-bin/ROH/ROH_analysis_tool_for_trial.cgi) did not reveal any candidate gene with recessive inheritance pattern.
Discussion and conclusion
Here we report a further case of “pure” terminal deletion 15q26 associated with complex CHD. Fifteen other cases with such aberration and CHD were previously described. Table 1 summarizes clinical and cytogenetic data in these patients and ours.
Our proband shares many relevant signs and symptoms with other patients, especially pre- and postnatal growth retardation, developmental delay, skeletal anomalies, microcephaly, and hearing defects. In the other patients eye anomalies were observed less frequently. Less common features were found in some cases including kidney anomaly, CDH and lung hypoplasia; however, these are lacking in our patient. Through this table, we also note that the CHD was most often described as complex, with several concomitant abnormalities like in our patient. Among the major cardiac defect there are ASD/VSD and aortic arch anomaly. Valvulopathy, patent ductus arteriosus, cardiac shunt and hypoplastic heart were rarely described. Our patient shared some of these anomalies, namely ASD and cardiac shunt. However, she lacked VSD, AAA and valvular defect. Cardiomegaly was an unusual feature reported exclusively in our patient. Indeed, the atrial septal defect resulted in the formation of significant shunts, which led to volume overload of the right atrium and ventricle and consequently our patient developed cardiomegaly.
Array analysis allowed us to characterize a 9.15 Mb deletion within the 15q26.1-q26.3 region. Comparable aberrations are often reported as de novo. Most often, terminal 15q deletions are found in combination with a terminal duplication of another chromosome due to an unbalanced translocation. To the best of our knowledge and according to the DECIPHER database, a deletion of this specific size has not been reported previously. Based on the Genome Data viewer (https://www.ncbi.nlm.nih.gov/genome/gdv/), the deleted segment encompasses 36 HGNC genes, 19 of them are referenced in the OMIM database, among which only IGF1R, NR2F2, CHD2 and MEF2A are consistent with the phenotype described in our proband: (Fig. 3).

15q26.1-q26.3 deletions displaying the 36 missing genes together with the four genes involved in the Phenotype in our proband
IGF1R (insulin like growth factor 1 receptor) (MIM 147370) lies on the 15q26.3 locus. It is bound to the growth factor ligands IGF1 and IGF2 to play a key role in pre- and post-natal development [20, 21]. The crucial impact of IGF1R on growth processes was underlined by the growth restriction found in individuals with pathogenic variants in the IGF1R gene [3, 22], in addition to patients with a 15q26 deletion leading to haploinsufficiency [23]. To the best of our knowledge, no heart anomalies have ever seen in patients carrying IGF1 or IGF1R mutations nor in knockout mice lacking these genes. Therefore, it is unlikely that the onset of CHD is only caused by haploinsufficiency of the IGF1R gene [4].
NR2F2 (Nuclear NR2F2 (Nuclear Receptor Subfamily 2, Group F, Member 2) (MIM 107773), located at 15q26.2 locus, is involved in angiogenesis and heart development [24], Indeed, NR2F2 haploinsufficiency in patients with a 15q26 deletion appears to be associated with heart malformations [25]. In addition, variants within the NR2F2 gene were found to cause non-syndromic atrioventricular septal defects (AVSDs) and other heart defects as well [26] . Moreover, this gene has been implicated to be involved in some patients with diaphragmatic hernia [6, 27], but this was not reported in others [28, 29] nor present in our patient.
CHD2 (Chromodaine helicase DNA-binding protein) belongs to a family of ATP-dependant chromatin remodeling proteins involved in chromatin regulation [12]. Mutations in this gene were associated with severe non-syndromic intellectual disability [13], as well as epileptic encephalopathy [14]. Additionally, disruption of CHD2 was associated with scoliosis in murine models [15]. Interestingly, this anomaly was observed in our patient as well as a few in the literature [9, 16, 28]. These findings together highlight the involvement of CHD2 dysfunction in neurodevelopmental disorders and scoliosis. This gene has previously been proposed as a candidate gene for the CDH [8], but this was not evident in our case or elsewhere [7, 9]. MEF2A (Mads Box Transcription Enhancer Factor 2, Polypeptide A) (MIM 600660), mapped to the human chromosome 15q26.3 region, is member of the myocyte enhancer family of transcription factors (MEF2) [17]. The subunit MEF2A is expressed in endothelial and smooth muscle cells of coronary arteries. Subsequently, MEF2A mutations can disturb the growth or differentiation of these cells, increasing the risk of developing coronary artery disease (CAD)/ myocardial infarction (MI) [18, 19]. CAD/MI was not evident in patients with 15q26 deletion involving MEF2A. This could be explained by the relatively young age of these patients compared to others described by Wang and Bhagavatula whose age of diagnosis was between 36 and 80 years [18, 19]. Therefore, regular checking up would be useful from the third decade onwards in these patients.
To sum up, this work focused on the main genes whose haploinsufficiency could explain heart disease in patients with 15q26 monosomy, i.e. the NR2F2 gene involved respectively in AVSDs and AAA/hypoplastic left heart. Scoliosis and psychomotor delay in our patient would be explained by the CHD2 gene disruption. The phenotype in our patient could also be ascribed to the high rate of homozygous regions outlined by the CGH array, without excluding the possible contribution of epigenetic and environmental factors as well. 15q26 monosomy should be considered when growth retardation is associated with congenital heart defect (mainly AVSDs and/or AAA).
Patients with 15q26 deletion need a multidisciplinary management, which includes endocrinological assessment evaluating also possible GH therapy [30], as well as cardiologic, orthopedic and psychomotor follow-up. The genetic counseling in our family was delicate since the parents refused to undergo the array CGH analysis that is important for a complete familiar counseling.
Availability of data and materials
All data is contained in the manuscript.
Abbreviations
- CHD:
-
Congenital Heart disease
- CNV:
-
Copy number variations
- NR2F2 :
-
Nuclear Receptor Subfamily 2, Group F, Member 2
- AVSDs:
-
Atrioventricular septal defects
- MEF2A :
-
Mads Box Transcription Enhancer Factor 2, Polypeptide A
- CAD/ MI:
-
Coronary artery disease/ myocardial infarction
- CoA:
-
Coarctation of the aorta
- IGF1R :
-
Insulin like growth factor 1 receptor
- ASD:
-
Atrial Septal Defects
- VSD:
-
Atrioventricular Septal Defects
- AAA:
-
Aortic Arch Anomaly
- IUGR:
-
intrauterine growth restriction
- CDH:
-
congenital diaphragmatic hernia
References
Lurie IW. Kidney abnormalities in persons with monosomy 15q26. Am J Med Genet A. 2008;146A(13):1761–4. https://doi.org/10.1002/ajmg.a.32333.
Poot M, Verrijn Stuart AA, van Daalen E, van Iperen A, van Binsbergen E, Hochstenbach R. Variable behavioural phenotypes of patients with monosomies of 15q26 and a review of 16 cases. Eur J Med Genet. 2013;56(7):346–50. https://doi.org/10.1016/j.ejmg.2013.04.001.
Abuzzahab MJ, Schneider A, Goddard A, Grigorescu F, Lautier C, Keller E, et al. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N Engl J Med. 2003;349(23):2211–22. https://doi.org/10.1056/NEJMoa010107.
Nakamura E, Makita Y, Okamoto T, Nagaya K, Hayashi T, Sugimoto M, et al. 5.78 Mb terminal deletion of chromosome 15q in a girl, evaluation of NR2F2 as candidate gene for congenital heart defects. Eur J Med Genet. 2011;54(3):354–6. https://doi.org/10.1016/j.ejmg.2010.12.004.
Veenma DC, Eussen HJ, Govaerts LC, de Kort SW, Odink RJ, Wouters CH, et al. Phenotype-genotype correlation in a familial IGF1R microdeletion case. J Med Genet. 2010;47(7):492–8. https://doi.org/10.1136/jmg.2009.070730.
Slavotinek AM, Moshrefi A, Davis R, Leeth E, Schaeffer GB, Burchard GE, et al. Array comparative genomic hybridization in patients with congenital diaphragmatic hernia: mapping of four CDH-critical regions and sequencing of candidate genes at 15q26.1-15q26.2. Eur J Hum Genet. 2006;14(9):999–1008. https://doi.org/10.1038/sj.ejhg.5201652.
Capelli LP, Krepischi AC, Gurgel-Giannetti J, Mendes MF, Rodrigues T, Varela MC, et al. Deletion of the RMGA and CHD2 genes in a child with epilepsy and mental deficiency. Eur J Med Genet. 2012;55(2):132–4. https://doi.org/10.1016/j.ejmg.2011.10.004.
Klaassens M, van Dooren M, Eussen HJ, Douben H, den Dekker AT, Lee C, et al. Congenital diaphragmatic hernia and chromosome 15q26: determination of a candidate region by use of fluorescent in situ hybridization and array-based comparative genomic hybridization. Am J Hum Genet. 2005;76(5):877–82. https://doi.org/10.1086/429842.
Chenier S, Yoon G, Argiropoulos B, Lauzon J, Laframboise R, Ahn JW, et al. CHD2 haploinsufficiency is associated with developmental delay, intellectual disability, epilepsy and neurobehavioural problems. J Neurodev Disord. 2014;6(1):9. https://doi.org/10.1186/1866-1955-6-9.
Rump P, Dijkhuizen T, Sikkema-Raddatz B, Lemmink HH, Vos YJ, Verheij JB, et al. Drayer's syndrome of mental retardation, microcephaly, short stature and absent phalanges is caused by a recurrent deletion of chromosome 15(q26.2-->qter). Clin Genet. 2008;74(5):455–62. https://doi.org/10.1111/j.1399-0004.2008.01064.x.
Hengstschlager M, Mittermayer C, Repa C, Drahonsky R, Deutinger J, Bernaschek G. Association of deletions of the chromosomal region 15q24-ter and diaphragmatic hernia: a new case and discussion of the literature. Fetal Diagn Ther. 2004;19(6):510–2. https://doi.org/10.1159/000080164.
Lamar KJ, Carvill GL. Chromatin remodeling proteins in epilepsy: lessons from CHD2-associated epilepsy. Front Mol Neurosci. 2018;11:208. https://doi.org/10.3389/fnmol.2018.00208.
Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380(9854):1674–82. https://doi.org/10.1016/S0140-6736(12)61480-9.
Carvill GL, Heavin SB, Yendle SC, McMahon JM, O'Roak BJ, Cook J, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45(7):825–30. https://doi.org/10.1038/ng.2646.
Kulkarni S, Nagarajan P, Wall J, Donovan DJ, Donell RL, Ligon AH, et al. Disruption of chromodomain helicase DNA binding protein 2 (CHD2) causes scoliosis. Am J Med Genet A. 2008;146A(9):1117–27. https://doi.org/10.1002/ajmg.a.32178.
Courage C, Houge G, Gallati S, Schjelderup J, Rieubland C. 15q26.1 microdeletion encompassing only CHD2 and RGMA in two adults with moderate intellectual disability, epilepsy and truncal obesity. Eur J Med Genet. 2014;57(9):520–3. https://doi.org/10.1016/j.ejmg.2014.06.003.
Black BL, Olson EN. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol. 1998;14(1):167–96. https://doi.org/10.1146/annurev.cellbio.14.1.167.
Wang L, Fan C, Topol SE, Topol EJ, Wang Q. Mutation of MEF2A in an inherited disorder with features of coronary artery disease. Science. 2003;302(5650):1578–81. https://doi.org/10.1126/science.1088477.
Bhagavatula MR, Fan C, Shen GQ, Cassano J, Plow EF, Topol EJ, et al. Transcription factor MEF2A mutations in patients with coronary artery disease. Hum Mol Genet. 2004;13(24):3181–8. https://doi.org/10.1093/hmg/ddh329.
Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75(1):73–82. https://doi.org/10.1016/S0092-8674(05)80085-6.
Li J, Choi E, Yu H, Bai XC. Structural basis of the activation of type 1 insulin-like growth factor receptor. Nat Commun. 2019;10(1):4567. https://doi.org/10.1038/s41467-019-12564-0.
Forbes BE, Blyth AJ, Wit JM. Disorders of IGFs and IGF-1R signaling pathways. Mol Cell Endocrinol. 2020;111035.
Roback EW, Barakat AJ, Dev VG, Mbikay M, Chretien M, Butler MG. An infant with deletion of the distal long arm of chromosome 15 (q26.1----qter) and loss of insulin-like growth factor 1 receptor gene. Am J Med Genet. 1991;38(1):74–9. https://doi.org/10.1002/ajmg.1320380117.
Al Turki S, Manickaraj AK, Mercer CL, Gerety SS, Hitz MP, Lindsay S, et al. Rare variants in NR2F2 cause congenital heart defects in humans. Am J Hum Genet. 2014;94(4):574–85. https://doi.org/10.1016/j.ajhg.2014.03.007.
Tumer Z, Harboe TL, Blennow E, Kalscheuer VM, Tommerup N, Brondum-Nielsen K. Molecular cytogenetic characterization of ring chromosome 15 in three unrelated patients. Am J Med Genet A. 2004;130A(4):340–4. https://doi.org/10.1002/ajmg.a.30035.
Upadia J, Philips JB 3rd, Robin NH, Lose EJ, Mikhail FM. A case report of chromosome 17q22-qter trisomy with distinct clinical presentation and review of the literature. Clin Case Rep. 2018;6(4):612–6. https://doi.org/10.1002/ccr3.1298.
Dateki S, Fukami M, Tanaka Y, Sasaki G, Moriuchi H, Ogata T. Identification of chromosome 15q26 terminal deletion with telomere sequences and its bearing on genotype-phenotype analysis. Endocr J. 2011;58(3):155–9. https://doi.org/10.1507/endocrj.K10E-251.
Poot M, Eleveld MJ, Van 't slot R, van Genderen MM, Verrijn Stuart AA, Hochstenbach R, et al. Proportional growth failure and oculocutaneous albinism in a girl with a 6.87 Mb deletion of region 15q26.2-->qter. Eur J Med Genet. 2007;50(6):432–440, DOI: https://doi.org/10.1016/j.ejmg.2007.08.003.
Davidsson J, Collin A, Bjorkhem G, Soller M. Array based characterization of a terminal deletion involving chromosome subband 15q26.2: an emerging syndrome associated with growth retardation, cardiac defects and developmental delay. BMC Med Genet. 2008;9:2.
Ho SC, Clayton P, Vasudevan P, Greening J, Wardhaugh B, Shaw N, et al. Recombinant human growth hormone therapy in children with chromosome 15q26 deletion. Hormone Res Paediatrics. 2015;83(6):424–30. https://doi.org/10.1159/000380949.
Tonnies H, Schulze I, Hennies H, Neumann LM, Keitzer R, Neitzel H. De novo terminal deletion of chromosome 15q26.1 characterised by comparative genomic hybridisation and FISH with locus specific probes. J Med Genet. 2001;38(9):617–21. https://doi.org/10.1136/jmg.38.9.617.
Bhakta KY, Marlin SJ, Shen JJ, Fernandes CJ. Terminal deletion of chromosome 15q26.1: case report and brief literature review. J Perinatol: official journal of the California Perinatal Association. 2005;25(6):429–32. https://doi.org/10.1038/sj.jp.7211301.
Choi JH, Kang M, Kim GH, Hong M, Jin HY, Lee BH, et al. Clinical and functional characteristics of a novel heterozygous mutation of the IGF1R gene and IGF1R haploinsufficiency due to terminal 15q26.2->qter deletion in patients with intrauterine growth retardation and postnatal catch-up growth failure. J Clin Endocrinol Metab. 2011;96(1):E130–4. https://doi.org/10.1210/jc.2010-1789.
Biggio JR Jr, Descartes MD, Carroll AJ, Holt RL. Congenital diaphragmatic hernia: is 15q26.1-26.2 a candidate locus? Am J Med Genet A. 2004;126A(2):183–5. https://doi.org/10.1002/ajmg.a.20464.
Okubo Y, Siddle K, Firth H, O'Rahilly S, Wilson LC, Willatt L, et al. Cell proliferation activities on skin fibroblasts from a short child with absence of one copy of the type 1 insulin-like growth factor receptor (IGF1R) gene and a tall child with three copies of the IGF1R gene. J Clin Endocrinol Metab. 2003;88(12):5981–8. https://doi.org/10.1210/jc.2002-021080.
O'Riordan AM, McGrath N, Sharif F, Murphy NP, Franklin O, Lynch SA, et al. Expanding the clinical spectrum of chromosome 15q26 terminal deletions associated with IGF-1 resistance. Eur J Pediatr. 2017;176(1):137–42. https://doi.org/10.1007/s00431-016-2802-y.
Lopez I, Bafalliu JA, Bernabe MC, Garcia F, Costa M, Guillen-Navarro E. Prenatal diagnosis of de novo deletions of 8p23.1 or 15q26.1 in two fetuses with diaphragmatic hernia and congenital heart defects. Prenat Diagn. 2006;26(6):577–80. https://doi.org/10.1002/pd.1468.
Acknowledgements
We thank the patients parents for their gracious participation and support.
Funding
Study has no funding source.
Author information
Authors and Affiliations
Contributions
YB and SA carried out the cytogenetic study and drafted the manuscript. NL and DS carried out the molecular cytogenetic study and revising the work critically for important intellectual content. AS participated in the cytogenetic study and revised the manuscript. KS and AS participated in the design of the study and in the draft of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval is considered unnecessary according to national provisions. This report was not presented as a research study as all family members were seen in a medical consultation for diagnostic purposes and they gave their written consent to participate and benefit from this analysis.
Consent for publication
This family gave written consent for clinical data to be published.
Competing interests
The authors declare that there are no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Benbouchta, Y., De Leeuw, N., Amasdl, S. et al. 15q26 deletion in a patient with congenital heart defect, growth restriction and intellectual disability: case report and literature review. Ital J Pediatr 47, 188 (2021). https://doi.org/10.1186/s13052-021-01121-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13052-021-01121-5