
Abstract
Background
Moebius syndrome (MBS) is rare disease characterized by nonprogressive congenital uni- or bi-lateral facial (i. e. VII cranial nerve) and abducens (i. e. VI cranial nerve) palsy. Although the neurological and ophthalmological findings are quite well-known, data concerning the attendant functional difficulties and their changes over time are seldom addressed.
In this study we attempt to estimate the prevalence of clinical and functional data in an Italian cohort affected by MBS.
Methods
The study included 50 children, 21 males and 29 females, aged 1 month to 14 years. The patients entered into a multidisciplinary diagnostic and follow-up protocol that had the specific purpose of detecting clinical and developmental deficits related to MBS.
Results
Involvement of the VII cranial nerve (total/partial, bilateral or unilateral) was present in 96 % of patients, and of the VI nerve in 85 %. Two patients were without impairment of the VII nerve and seven patients had no involvement of the VI nerve and were thus classified as Moebius-like because of the involvement of other CNs. Additional affected CNs were numbers III-IV in 16 %, V in 11 %, VIII and X each in 8 %, the XI in 6 %, the IX, most often partially, in 22 %, and the XII in 48 % of cases. Their development was characterized by global delay at one year of age, motor, emotional and speech difficulties at two years of age, a trend toward normalization at three years of age but with weakness in hand-eye coordination, and achieving average results at five years of age. Overall 90 % of children had a normal developmental quotient whereas only 10 % manifested cognitive deficits.
Conclusion
Early rehabilitation may enhance the recovery of normal function, particularly in vulnerable areas of development. It is possible that early intervention that integrates sensory and visual information with emotional difficulties can improve the prognosis of the child with MBS.
Similar content being viewed by others

Background
Moebius syndrome (MBS) is a rare disease characterized by unilateral or bilateral nonprogressive congenital facial palsy (VII cranial nerve) with impairments of ocular abduction (VI cranial nerve); it can also be associated with other cranial nerve (CN) palsies, orofacial anomalies and limb defects [1]. This condition was originally described by Von Graefe in 1880 and by Moebius in 1888. Since then, more than 300 cases have been described and reported in the literature by a number of authors [2–8].
The prevalence of MBS is estimated to be 1/250.000 live births with equal incidence in both sexes. Most cases are sporadic, but familial cases, representing about 2 % of all affected individuals, have been documented.
The diagnosis of MBS is based exclusively on clinical criteria, although recent studies are beginning to document causative genetic patterns [9]. The lack of diagnostic criteria complicates the clinical assessment, definition of prognosis, and genetic analysis of patients with MBS. To address this concern, a group of clinicians and researchers met in 2007 at the biannual research meeting of Moebius Syndrome Foundation in Bethesda (MD USA) and defined the MBS as “congenital, uni- or bilateral, nonprogressive facial weakness and limited abduction of the eye(s)” [3].
The cause and pathogenesis of MBS remain unclear and controversial since the initial descriptions by von Graefe and Moebius, and there has been a decade long debate whether MBS has a genetic aetiology or not. Fetal toxic exposure, genetically determined vascular rhombencephalic disturbances in development, or an acquired ischemic event occurring after the fifth week of pregnancy have been proposed as determinants.
It has been supposed that a primary insult leads to a sequence of events involving one or more focal areas of damage perhaps in the brainstem where the neurons of the facial, abducens, and lacrimal (salivary) nuclei, are anatomically coincident during this phase of the embryogenesis.
On the other hand, de novo PLXND1 and REV3L mutations have recently been identified in a a number of of MBS patients. However, PLXND1 and REV3L represent totally unrelated pathways involved, respectively, neural migration during hindbrain development, and DNA translesion synthesis, essential for the replication of endogenously damaged DNA [9].
MBS can be recognized and diagnosed early during the neonatal period. Poor or absent sucking due to incomplete closure of the lips, lack of facial mimicking (especially while crying), fixed gaze, incomplete eyelid closure during sleep and ptosis have all been observed. Hypotonia and developmental delay can also be present.
Paralysis of the VII CN, is responsible for the absence of mimicry, the lack of smile and the suction deficit. Three specific patterns of ocular motility alterations have been identified [10]: pattern A consists of orthotropia in the primary position with a complete defect in both abduction and adduction ocular movements, found in 41 % of cases; pattern B, with large-angle esotropia (convergent strabismus), crossed fixation, documented in 50 % of cases; and pattern C, characterized by a large-angle exotropia (divergent strabismus) with torticollis, absence of convergence, and vertical eye misalignment with involvement of the III and IV CNs, seen in a minority of patients (9 %).
In a few patients other CNs can be compromised including paralysis of the XII CN giving rise to lingual palsy and hypoplasia; damage of the V CN affecting endo- and perioral sensitivity while damage to the IX CN impairs palatine and pharyngeal motility.
Impairment of the cochlear branch of the VIII CN can contribute to language delay [10–13].
Among MBS’s patients club foot, hand anomalies (syndactyly, brachydactyly, ectrodactyly) and agenesis of the pectoral muscle and dysmorphisms are occasionally observed. Association with other syndromes like Poland syndrome, Pierre Robin sequence, Carey-Fineman-Ziter, Klippel-Feil anomaly has also been reported [4].
In 1998 Abramson first proposed the acronym CLUFT: C, cranial nerve, L, lower limb; U, upper limb, F, face, T, thorax to define grading of disease, to describe the heterogeneity of the clinical features and establish a level of impairment (Table 1) [4].
Methods
Setting
The Centre Moebius (CMM) in Fondazione IRCCS Ca’ Granda, Milan (The Center for the diagnosis and treatment of Moebius syndrome) was established in June 2003 as one of the programs of the Italian Moebius Syndrome Association Onlus (AISMo Onlus), aimed at developing referral centers for early diagnosis. The goals of the Center are: early identification of developmental abnormalities in newborns and infants with suspected MBS; supporting the parents with diagnostic, educational and rehabilitative interventions; creating a network with specialized services and personalized rehabilitation; disseminating knowledge about the neurobehavioral characteristics of the MBS child, for pediatricians and rehabilitation professionals; investigating the psychomotor and functional pathways of MBS subjects.
Study Population
We report the clinical and functional analysis of a group of 50 Italian children (mean age 38 months, range 1 month to 14 years) with a suspected diagnosis of MBS, recruited from the AISMo Onlus, from the MBS Referral Center of the Hospital of Parma and from individual pediatricians [14–16]. To diagnose MBS, the major criteria developed by the the First Scientific Conference of Moebius Syndrome in 2007 were used, i.e., the presence of a congenital unilateral or bilateral nonprogressive paresis of the sixth and/or the seventh CNs. Children with additional involvement of other CNs and/or, motor, musculoskeletal and neurodevelopmental disorders were also included [10].
Patients who did not meet both major criteria were classified as having a Moebius-like syndrome and were considered separately.
Statement regarding ethics committee approval
The study focuses on the observational description of patients followed in the period between 2003 and 2015. Patients underwent a protocol of assessments, which were normally administered to all children referred to the outpatients department. Data were obtained from clinical files and were retrospectively reported. Ethics committee approval is not required in Italy for this type of reports.
Clinical evaluation and intervention
All MBS children were followed by a series of clinical and functional evaluations, as listed in Table 2. The multidisciplinary diagnostic and follow-up protocol included interviews with parents and caregivers (medical and genetic history), and physiatrist assessment to define the skills and functional development of the child for prognostic purposes and intervention. Patient care includes not only the child but takes into account the expectations and strengths of the family. Outpatient service provides physical rehabilitation for the movement disorders; feeding and speech therapy for the oral-motor functions deficits; psychomotor intervention because of communication difficulties and visuomotor coordination.
Physical therapy, speech therapy, psychomotor treatments sessions are held from one to three times a week, depending on whether functions are merely delayed or can be improved by treatment. Psychological assistance was also offered to parents if needed.
Other instrumental and specific evaluations as reported in Table 3 were carried out by other Departments of the same institution. Psychomotor evaluation was formally assessed at 1, 2, 3 and 5 years of age according to the Griffiths Mental Development Scale Revised (GMDS-R) [17].
Finally, we attempted to further classify the syndrome according to the involvement of other CNs by identifying the incidence of additional neuro-specific symptoms.
Results
Clinical findings
Fifty children were evaluated in our Centre. Among these, 34 satisfied the above mentioned major criteria for MBS diagnosis while 9 patients missing one of the two major criteria were classified as having a Moebius-like syndrome. Five children exhited complex conditions aggravating their clinical expression with Carey-Fineman-Ziter syndrome diagnosed in two, severe myopathy in one, one child had neonatal asphyxia and severe cerebral palsy, and one had psychiatric symptoms. Two patients with a doubtful diagnosis were excluded.
Thirty one percent of the children (15/48) were assessed between 1 to 6 months of age, 15 % between 6 and 12 months of age (7/48), and 21 % between 12 and 36 months of age (10/48). The remaining 16 children (33 %) were older than 3 years. The mean age at the time of the first examination was 38 months.
All cases were sporadic with negative family histories for genetic disorders. Most of these MBS patients were born at term after an uneventful pregnancy. In four cases the pregnancy was complicated by ultrasound evidence of clubfoot and in one case was associated with ventriculomegaly and intrauterine growth retardation. Cerebral magnetic resonance imaging (MRI) was carried out in 16 patients 11 of which were found to be normal. In the remaining five cases, cerebral anomalies such as sixth and seventh CN hypoplasia were evident. Brainstem hypoplasia and bilateral ventricle enlargement was detected in one child and hypophysis hypoplasia was seen in another.
In all cases karyotyping was performed and was normal. Array-CGH was carried out only in cases affected by Carey-Fineman-Ziter syndrome and severe myopathy and the results were normal.
The VII CN (total/partial, bilateral or unilateral) was involved in 96 % of cases and the VI CN in 85 %. Two patients with no impairment of the VII CN and seven patients without involvement of the VI nerve, but with deficiencies of other CNs were defined as being Moebius-like. Other affected CNs were the III-IV in 8 subjects (16 %), the V in 5 (11 %), the VIII and X in 4 (8 %), the XI in 3 (6 %), the IX but usually partially in 10 (22 %) of cases, and the XII in 18/48 (37 %). One case hospitalized in our Neonatal Intensive Care Unit, with involvement of the X CN and calcifications pontine at MRI, died at one month of age due to the absence of respiratory drive. In 19 % of patients Pierre Robin syndrome was evident; 9 % of children showed Poland syndrome; clubfeet were found in 24 %; 11 % showed skeletal anomalies, especially of the hands, feet, or spine (scoliosis); hearing loss or deafness were found in 8 % of children; 8 % were being fed by nasogastric tube until 1 year of age; 5 % demonstrated myopathic features; and 13 % were intellectually disabled.
Functional findings
Due to the multifactoriality of the disorders, we separated the clinical features according to their functional involvement into the following three major domains as determined by their CN involvement: feeding, language and hearing and visual functions. In the feeding and oral domain (involvement of V, VII, IX, XI, XII CNs) we found poor neonatal sucking and swallowing in 37,8 % of our patients, need for nasogastric tubes and gastrostomy in 5,5 %, nutritional problems in 16 %, dental problems in 17 % and palatal problems or micrognathia in 7,4 %. Problems in language and hearing (involvement of VII, VIII, IX, X, XI, XII CNs) caused hearing loss in 6,8 % of patients, language delay in 31 %, speech deficit in 42 %.
Disturbance of the visual domain (involvement of III, IV, VI, VII CNs) included deficits of both ocular motility and neurovisual function, was evident in 89,8 % and in 19,3 % of children, respectively. Photophobia was also observed in 15 % (Table 4) [18–25].
Three additional important functional areas, neurological, musculoskeletal and cognitive-emotional are also summarized in Table 5. It is noteworthy that neurological findings included hypotonia in 26 % of patients and balance impairment in 12 %. Musculoskeletal involvement included thoracic anomalies observed in 5 %, absence or hypoplasia of pectoralis major muscle in 3 %, chest, arms and hand deformities in 14 %, clubfeet in 32 %, and scoliosis in 6 %. In the cognitive emotional area, we found attention deficit in 17 % of children, cognitive impairment and developmental delay in 15 %, sleep and regulation disturbances in 28 % and stereotypies in 4,2 % (Table 5).
Neuropsychological findings
Excepting the Moebius-like, the complex cases and the unclassified ones, GMDS-R was carried out in 34 children.
Data showed a delay in the General Quotient (GQ) at 1 year of age (i.e., GQ 82) with a homogeneous profile in all the subscales. At 2 years we noted a modest improvement of the GQ to 89, with delay most evident in motor, behavioral and language subscales. At 3 years of age the mean GQ was 90 with deficiencies in specific cognitive subscales, particularly in hand-eye coordination while at 5 years, the average GQ was 103, with lowest results in the motor subscale due to clumsiness (Table 6).
Discussion
MBS consists of complete or partial facial diplegia, often accompanied by other CN deficiencies.
Most of the previous reviews of MBS tend to focus on the neuroradiological and systemic features of the syndrome, but comprehensive developmental evaluations were not specifically investigated [1–13].
We interpreted the neurofunctional data of the psychometric assessments, taking into account the children’s dysfunctional nutrition (arising from difficulties in feeding), communication and visual-motor function.
The suffering of the children caused by difficulties in management of feeding tubes and the consequent frustration of many of the mothers because they were unable to directly feed their children reduced the pleasure of the food interaction, which plays a large role in building a secure attachment and sense of competency of the mother-child dyad. The lack of facial communicative expressions and the difficulties in speech sounds that negatively affect the reward of the parent-child feedback interaction also can lead to failure of the normal mother-child attachment.
Furthermore, oculomotor dysfunction has serious negative consequences in motor, perceptual and cognitive development. The visual system, provides the function of the "what", the recognition of objects and shapes, but the ocular movements provide the functions of the "where", that is, the location of objects, providing data on spatial orientation, the perception of movement and the third dimension (across saccades, optokinetic nystagmus, smooth pursuit, scanning and visual exploration).
Our experience has suggested that MBS children exhibit not only the above main morpho-functional deficits involving movement, food, vision and language, but also secondary developmental problems.
Primary motor problems consisted of hypotonia, musculo-skeletal dysmorphism, delay in righting, locomotion and clumsiness. Feeding disorders consisted of sucking-swallowing difficulty, breathing problems, lack of the sensitivity of the orofacial area, dysphagia with the need, in severe cases, for a gastrostomy. Language difficulties such as dyslalia, mechanical disorders of phonological coding and speech structure and writing were common. Visual impairment consisted of scanning, exploration and visual-perceptual deficits, and, in some cases, corneal ulcers.
Secondary problems included visual exploration deficits, oral-motor difficulties as well as lack of the categorization of facial expressions affecting cognitive strategies in early development. Pain and other difficulties during eating, maternal concerns, and lack of recognition of emotions, can form the basis of significant emotional distress. From the perspective of social maturity, self-image and communication with peers can be affected adversely both in childhood and in adolescence.
Equally important is the peculiar functional profile of MBS children. In general, they usually present with delay at 1 year of age, with motor emotional and language difficulties persisting until 2 years of age and reduced hand-eye coordination at 3 years of age. Children generally achieve average levels of GQ at five years of age although with retained aspects of clumsiness. Overall 90 % of children reached a normal GQ with only 10 % maintaining cognitive deficits.
The gradual recovery in fragile areas and the progressive harmonization of functions might be explained by early rehabilitation and by support by and of parents. Improving the methods of early intervention, enhancing motor ability and sensory information, and the timely support of parents having emotional difficulties, can improve the prognosis of the child with MBS.
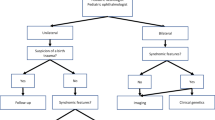
For all of these reasons, the International Classification of functioning Children and Youth (ICF-CY), published by the WHO can be considered the gold standard for describing functioning, disability and health. It appears to be a suitable framework for interpreting functions (vs impairment), activities (vs disability) and participation (vs handicap), in children with MBS and for evaluation and intervention of the familial and social environment [26]. In particular, the ICF-CY framework allows the health care practitioners to investigate and integrate the complexity of signs and functions in children with MBS such as motor skills (musculoskeletal features, posture, posture changes, gait and coordination), feeding (sucking patterns, quality of food, eating habits, sucking-swallowing coordination), communication and language (phonological, articulatory, oral motor skills and functional aspects), and cognitive features. The frequency and uniqueness of medical problems found in patients with MBS suggest the need for such a protocol to carefully define guidelines for the current and continuing care of these patients (Fig. 1).

MBS’ Interpretation ICF
Conclusion
The main finding of this study is the observation that the patients with MBS exhibit development characterized by global delay at 1 year of age, motor, emotional and speech difficulties at 2 years of age, a trend toward normalization at 3 years of age but with weakness in hand-eye coordination, and finally achieving average results at 5 years of age.
It is important that diagnosis and rehabilitation start simultaneously and that the rehabilitative intervention is updated over time in response to functional assessments. It is also essential that any intervention address both the child and the mother-child relationship. The approach must also be multidisciplinary, involving professionals with different skills and with specific training in cooperative therapies.
Lastly, the analyzed population requires larger numbers and needs a more prolonged follow-up to assess the developmental outcomes and the complexity of these children.
References
Bienvenu Broussard A, Borazjani JB. The faces of moebius syndrome: recognition and anticipatory guidance. Am J Matern Child Nurs. 2008;33(5):272–8.
Kumar D. Moebius syndrome. J Med Genet. 1990;27:122–6.
Miller G. The mystery of the missing smile. Science. 2007;316:826–7.
Abramson DL, Cohen Jr MM, Mulliken JB. Moebius syndrome: classification and grading system. Plast Reconstr Surg. 1998;102:961–7.
Pedraza S, Gamez J, Rovira A, et al. MRI findings in Möbius syndrome: correlation with clinical features. Neurology. 2000;55:1058–60.
Verzijl HTFM, van der Zwaag B, Lammens M, et al. The neuropathology of hereditary congenital facial palsy vs Möbius syndrome. Neurology. 2005;64:649–53.
Verzijl HTFM, van der Zwaag B, Cruysberg JRM, Padberg GW. Möbius syndrome redefined: a syndrome of rhombencephalic maldevelopment. Neurology. 2003;61:327–33.
Ventura BV, Tiller Miller M, Danda D, Carta A, Teixeira Brandt C, Oliveira Ventura L. Profile of ocular and systemic characteristics in Möbius sequence patients from Brazil and Italy. Arq Bras Oftalmol. 2012;75(3):202–6.
Tomas-Roca L, Tsaalbi-Shtylik A, Jansen JG, Singh MK, Epstein JA, Altunoglu U, Verzijl H, Soria L, van Beusekom E, Roscioli T, Iqbal Z, Gilissen C, Hoischen A, de Brouwer AP, Erasmus C, Schubert D, Brunner H, Pérez Aytés A, Marin F, Aroca P, Kayserili H, Carta A, de Wind N, Padberg GW, van Bokhoven H. De novo mutations in PLXND1 and REV3L cause Möbius syndrome. Nat Commun. 2015;6:7199.
Carta A, Mora P, Neri A, Favilla S, Sadun AA. Ophthalmologic and Systemic Features in Möbius Syndrome An Italian Case Series. Ophthalmology. 2011;118:1518–23.
Verzijl HTFM, Padberg GW, Zwarts MJ. The spectrum of Moebius syndrome: an electrophysiological study. Brain. 2005;128:1728–36.
MacKinnon S, Oystreck DT, Andrews C, Chan WM, Hunter DG, Engle EC. Diagnostic distinctions and genetic analysis of patients diagnosed with moebius syndrome MDC for moebius syndrome. Am Acad Ophthalmol. 2014;121:1461–8. doi:10.1016/j.ophtha.2014.01.006.
Möbius PJ. Ueber angeborene doppelseitige Abducens-Facialis-Lahmung. MMW Munch Med Wochenschr. 1888;35(91–4):108–11 [In German].
Bianchi B, Copelli C, Ferrari S, Ferri A, Sesenna E. Successful salvage surgery after treatment failures with cross graft and free muscle transplant in facial reanimation. J Craniomaxillofac Surg. 2012;40:185–9.
Bianchi B, Copelli C, Ferrari S, Ferri A, Sesenna E. Free flaps: outcomes and complications in head and neck reconstructions. J Craniomaxillofac Surg. 2009;37(8):438–42.
Bianchi B, Copelli C, Ferrari S, Ferri A, Bailleul C, Sesenna E. Free animation with free-muscle transfer innervated by the masseter motor nerve in unilateral facial paralysis. J Oral Maxillofac Surg. 2010;68(7):1524–9.
Griffiths R. The abilities of young children: a comprehensive system of mental measurement for the first 8 years of life, Young and Son. London: Child Development Research Centre London; 1970.
Momtchilova M, Pelosse B, Rocher F, Renault F. Laroche Le Syndrome de Möbius : manifestations ophtalmologiques et cliniques. J Fr Ophtalmol. 2007;30(2):177–82.
Briegel W. Neuropsychiatric findings of Moebius sequence – a review. Clin Genet. 2006;70:91–7.
Briegel W. Psychopathology and personality aspects of adults with Moebius sequence. Clin Genet. 2007;71:376–7.
Cattaneo L, Chierici E, Bianchi B, Sesenna E, Pavesi G. The localization of facial motor impairment in sporadic Moebius syndrome. Neurology. 2006;66:1907–12.
Gondipalli P, Tobias JD. Anesthetic implications of Mo¨bius syndrome. J Clin Anesth. 2006;18:55–9.
Jennings JE, Costigan C, Reardon W. Moebius Sequence and Hypogonadotrophic Hypogonadism. Am J Med Genet. 2003;123A:107–10.
Lopez de Lara D, Cruz-Rojo J, Sánchez del Pozo J, Gallego Gómez ME, Valera GL. Moebius-Poland syndrome and hypogonadotropic hypogonadism. Eur J Pediatr. 2008;167:353–4.
Sensat ML. Moebius syndrome: a dental hygiene case study and review of the literature. Int J Dent Hygiene. 2003;1:62–7.
World Health Organization. The international classification of functioning, disability and health for children and youth: ICF-CY. Geneva: World Health Organization; 2007.
Acknowledgements
The authors are grateful to the patients and their parents for their collaboration. The authors would also like to thank Mrs Pina Gallotto, Mrs Caterina De Grandi e Mr Renzo De Grandi of AISMo for their support to this work. English was revised by Steve Tint, Philadelphia, USA.
Funding
The authors did not receive any specific funding for this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
OP performed the data analysis, designed the study, wrote the main manuscript, approved the final manuscript and submitted it. MP and SC participated in the clinical evaluation of patients. EC contributed to collect data and to carry out the clinical follow up of patients. GM designed the study and approved the final manuscript. FM critically reviewed the manuscript, approved the final version. MFB performed the data analysis, participated in its design and coordination and helped to draft the manuscript, approved and submitted the final manuscript. All authors have read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Picciolini, O., Porro, M., Cattaneo, E. et al. Moebius syndrome: clinical features, diagnosis, management and early intervention. Ital J Pediatr 42, 56 (2016). https://doi.org/10.1186/s13052-016-0256-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13052-016-0256-5