
Abstract
Background
Hypoxia and inflammation tumor microenvironment (TME) play a crucial role in tumor development and progression. Although increased understanding of TME contributed to gastric cancer (GC) progression and prognosis, the direct interaction between macrophage and GC cells was not fully understood.
Methods
Hypoxia and normoxia macrophage microarrays of GEO database was analyzed. The peripheral blood mononuclear cell acquired from the healthy volunteers. The expression of C-X-C Motif Chemokine Ligand 8 (CXCL8) in GC tissues and cell lines was detected by quantitative reverse transcription PCR (qRT-PCR), western-blot, Elisa and immunofluorescence. Cell proliferation, migration, and invasion were evaluated by cell counting kit 8 (CCK8), colony formation, real-time imaging of cell migration and transwell. Flow Cytometers was applied to identify the source of cytokines. Luciferase reporter assays and chromatin immunoprecipitation were used to identify the interaction between transcription factor and target gene. Especially, a series of truncated and mutation reporter genes were applied to identify precise binding sites. The corresponding functions were verified in the complementation test and in vivo animal experiment.
Results
Our results revealed that hypoxia triggered macrophage secreted CXCL8, which induced GC invasion and proliferation. This macrophage-induced GC progression was CXCL8 activated C-X-C Motif Chemokine Receptor 1/2 (CXCR1/2) on the GC cell membrane subsequently hyperactivated Janus kinase 1/ Signal transducer and activator of transcription 1 (JAK/STAT1) signaling pathway. Then, the transcription factor STAT1 directly led to the overexpression and secretion of Interleukin 10 (IL-10). Correspondingly, IL-10 induced the M2-type polarization of macrophages and continued to increase the expression and secretion of CXCL8. It suggested a positive feedback loop between macrophage and GC. In clinical GC samples, increased CXCL8 predicted a patient’s pessimistic outcome.
Conclusion
Our work identified a positive feedback loop governing cancer cells and macrophage in GC that contributed to tumor progression and patient outcome.
Similar content being viewed by others


Background
Gastric cancer (GC) is a prevalent yet incurable malignancy of the digestive system. It is the 3rd leading cause of cancer-related mortalities across the globe [1]. China has a high GC incidence and a heavy disease burden, with an estimated 320,000 annual deaths, accounting for 45% of all GC deaths globally [2]. Despite the constant iterations of comprehensive therapy based on surgical resection, GC prognosis remains poor, with a 5-year survival rate of not more than 30% [3]. On one hand, this is attributed to the malignant phenotype of the tumor; while on the other hand, the tumor microenvironment plays an essential role as an “accomplice” [4]. Hypoxia and inflammation are essential components of the tumor microenvironment (TME) [5, 6]. Our previous studies confirmed that hypoxia promotes GC progression [7]. Moreover, the hypoxia-related core element, hypoxia-inducible factor-1α (HIF-1α) were confirmed as transcription factors that could broadly regulate the transcription of downstream genes, promoting GC proliferation [8] and invasion [9, 10]. Therefore, it is essential to further explore the crosstalk between hypoxia and inflammation in the GC-TME.
Macrophages are the most abundant inflammatory cells in TME. They exhibit significant plasticity and can freely switch from one phenotype to another; however, this depends on the signals received from their surrounding microenvironment [11, 12]. The process is called macrophage polarization. Based on the activation state, macrophages can be classified into classically activated macrophages (M1 macrophages) and alternatively activated macrophages (M2 macrophages) [13]. Although this dichotomy is somewhat arbitrary, it remains the most popular macrophage definition [14, 15]. It is generally believed that the M1 type has an anti-tumor property, whereas the M2 type is a pro-cancer factor. Also, the cancer cell is an “educator” that converts M1 to M2 [16]. M2 macrophages promote tumor metastasis and angiogenesis by secreting various cytokines and exerting immunosuppressive effects [17]. Macrophages recruited into TME by cytokines are referred to as tumor-associated macrophages (TAMs) [18]. TAMs predominantly exhibit an M2 phenotype, indicating a poor prognosis of solid tumors in TME [19, 20]. In particular, M2 macrophages could promote GC peritoneal metastasis [21]; extracellular vesicles secreted by M2 macrophages promoted GC progression [22]. Correspondingly, hypoxia GC cells induced the M2-type polarization in the TME [23]. The complex interaction mechanism of the two cells in the TME remains to be explored. Since hypoxia could promote immune evasion, angiogenesis, proliferation, and metastasis, research suggests that it can as well facilitate TAMs in tumor development. Additionally, the TAMs in tumor hypoxic regions mediate treatment resistance and promote cancer recurrence [24].
This paper focuses on the effect of hypoxia on macrophages then analyzes the role of hypoxic microenvironment and inflammatory microenvironment in GC development. CXCL8 (C-X-C Motif Chemokine Ligand 8, also called IL-8) was overexpressed in hypoxic macrophages. Moreover, we discovered that the macrophage-derived CXCL8 could activate the JAK/STAT1 signaling pathway and promote GC invasion as well as proliferation. Besides, as a transcription factor, STAT1 could upregulate IL-10 (Interleukin 10) expression in GC; the latter could naturally trigger macrophage M2-type polarization. The M2 macrophages increased the release of CXCL8. The hypoxia-activated positive feedback loop CXCL8/CXCR1/2 (C-X-C Motif Chemokine Receptor 1/2)/STAT1/IL-10/NFKB1/CXCL8 could cascade and amplify the interaction between GC and macrophages leading to uncontrolled progressive signaling in GC.
Materials and methods
Bioinformatics
Hypoxic and normoxic cultured human monocyte-derived macrophage dataset GSE4630 was downloaded from the GEO (Gene Expression Omnibus) database (https://www.ncbi.nlm.nih.gov/geo/). They included two hypoxic-cultured samples and two normoxic-cultured samples. The GC RNA sequencing data were downloaded from the TCGA (The Cancer Genome Atlas, TCGA-STAD, 375 tumor samples vs. 31 normal samples, https://portal.gdc.cancer.gov/). CIBERSORT was used to determine the relative frequencies of immune cells in each sample. The GC and normal sample microarray data were also obtained from the GEO (GSE54129, 111 tumor samples vs. 21 normal samples). The differentially expressed genes were identified by the “limma” package (P < 0.05, and |FoldChange| ≥ 2). JASPAR (http://jaspar.genereg.net/) and UCSC (http://genome.ucsc.edu/) were performed to predict the transcription factor.
Cell culture and human PBMCs isolation
The GC (HGC-27, NUGC3) cells, human normal gastric epithelial cell line (GES-1) were acquired from China Medical University (Shenyang, China). Mouse GC cancer cell (GAN-KP cells) was acquired from International Research Center for Medical Sciences, Kumamoto University [25]. Peripheral blood mononuclear cells (PBMCs) were obtained from healthy donors in Liaoning Cancer Hospital & Institute. CD14+ cells were enriched by depleting CD8+, CD19+, CD56+ and CD14+ cells by MACS (magnetic-activated cell sorting, EasySep, STEMCELL Technologies, 19,059) columns with negative selection. PBMCs were differentiated into immature macrophages (M0 macrophages) using 50 ng/mL human macrophage colony-stimulating factor (hM-CSF, Sigma, 81,627–83-0) for 7 days (Fig. 1C). All cells were cultured in RPMI-1640 (FUJIFILM Wako Pure Chemical Corporation, 189–02145) supplemented with 10% fetal bovine serum (FBS, Gibco 26,140–079) under 1% O2 or 20% O2 conditions as described previously [8, 10, 26]. In the co-culture model, macrophages and GC cells were added in a 1:1 ratio into a 10 cm dish (1 × 106 cells for each) and analyzed after 7 days of co-culture.

Hypoxia promoted macrophage-derived CXCL8 secretion. A, B Differential genetic analysis of the transcription data of macrophages cultured under hypoxia and normoxia showed that hypoxia could promote CXCL8 expression. (A. volcano; B. heatmap). C MACS obtained human CD14+ monocytes and induced them into macrophages through M-CSF. D, E Hypoxia promoted human-derived macrophage CXCL8 expression at both transcription and protein levels. F. Hypoxia promoted the secretion of macrophage-derived cytokine CXCL8 secretion. G An evident co-localization of macrophages and CXCL8 in human GC tissue specimens. H Schematic of gating strategy of flow cytometry analysis. GC tissue were dissociated to obtain a single-cell suspension and stained with antibodies. Cells were first gated to exclude debris and dead cells (Sup Fig. 1D), then GC cells and macrophages were selected. Cells were further gated by cluster of CXCL8 expression. I GC cells (green) and macrophages (red) were co-cultured under hypoxia to mimics the microenvironment. J CXCL8 was mainly derived from macrophages and IL-10 was expressed by GC cells in the co-culture system
GC tissues and ethical approval
Between January 2009 and December 2012, gastric cancer and adjacent non-cancerous tissue samples were obtained from 526 patients subjected to adequate complete surgical resection (R0) of locally advanced GC surgical resection in Liaoning Cancer Hospital & Institute. All recruited patients had not received preoperative chemotherapy or radiotherapy. They signed a written informed consent before surgery. The follow-up of patients was closed on December 31, 2020. The study was approved by the Ethics Committee of Liaoning Cancer Hospital and Research Institute (20181226).
Immunohistochemistry
The immunohistochemistry (IHC) was performed based on previously published protocols [27]. Immunohistochemistry was scored based on the intensity of staining and the proportion of positive cells. The blinded review was performed by two pathologists. Ki67 (1:1000, Abcam, ab15580), Caspase-3 (1:1000, Abcam, ab184787) CXCL8 (5 μg/mL, R&D Systems, AF-208-NA).
Immunofluorescence
The slices were overnight incubated with primary antibody at 4 °C. Then, they were incubated with species-appropriate rabbit/mouse secondary antibodies coupled with AlexaFluor dyes (488, 594, 1:200, Invitrogen, A32814, A32723, A32740), DAPI (1:1000, Dojindo, KT013) at room temperature for 1 h. CXCL8 (5 μg/mL, R&D Systems, AF-208-NA), CD68, CD163, CD86 (1:200, Abcam, ab213363, ab182422, ab270719). Olympus Fluoview FL1200 confocal microscope was used to capture 20x and 60x images.
ELISA test
ELISA assay was used to measure the CXCL8 in the supernatant of macrophages cultured in hypoxic or normoxic conditions and IL-10 in the medium of GC cells. The procedure followed the instructions of the Human CXCL8 kit (R&D Systems, D8000C) and Human IL-10 ELISA Kit (Abcam, ab185986).
qRT-PCR
The quantitative real-time polymerase chain reaction (qRT-PCR) was performed as previously documented [8]. Primer sequences were designed by Sangon (China, Supplementary Table 1).
Western blot analysis
The RIPA lysis buffer was used to extract total proteins from cells and tissues (Beyotime, Shanghai, China). Then, 10% SDS-PAGE gel was used to separate the proteins and transferred using the PVDF membrane. The membranes were overnight incubated with the primary antibodies (Supplementary Table 2) at 4 °C. Subsequently, they were co-cultured with the secondary antibody for 1 h. The ECL system (Amersham Imager 600) and ImageJ software were applied to observe and calculate grayscale values [8].
Chromatin immunoprecipitation (ChIP)
ChIP enzymatic chromatin IP kit protocol (Cell Signaling Technology, China) was used to perform the ChIP assay, where 1 × 107 logarithmic phase cells were subjected to ChIP. The chromatin was immunoprecipitated with an anti-STAT1 (Santa Cruz Biotechnology, USA, 1:50) and mouse IgG (Santa Cruz Biotechnology, USA, 1:100) on rotators at 4 °C for 16 h.
Promoter-luciferase reporter assay
Promoter-Luciferase reporter assay confirmed the combination of STAT1 and IL-10 promoter sequence, NFKB1, and CXCL8 promoter sequence, respectively. The recombinant pGL-3 basic-plasmid contained truncated human IL-10 (or CXCL8) promoter (wild type, wt, − 2000 ~ + 99) or mutant IL-10 (or CXCL8) promoter (mutant type, mut). The plasmids were transfected with the POLO3000 transfection reagent (Research and science, China). After 48 h, the luciferase activity of each group was evaluated following the manufacturer’s instructions.
Colony formation assay
A total of 1 × 105 GC cells/each group were seeded into 6-well plates for 2 weeks. The colonies were washed three times using PBS, fixed with 4% paraformaldehyde, and stained with Diff-Quik III Kit (Muto Chemical, ZS0003). The stained colonies were observed under a light microscope (IX81 Olympus).
Cell counting assay
Cell proliferation was evaluated using the Cell counting kit (Cell counting kit 8, CCK-8). The cells were incubated at 37 °C for 24, 48, 72, and 96 h. Subsequently, the medium was discarded then the chromogenic solution at 10:1 was prepared. For incubation, a color-developing solution (10 μl) was added to the 96 well plates at 37 °C for 2 h. The optical density (OD) was detected using the UV spectrophotometer (BioTek Synergy H1) at 450 nm.
Transwell
Gastric cancer cells were seeded in Transwell upper chambers coated with gelatin. The lower chambers with 600 μL RPMI-1640 with 20% FBS. After 24 h, the cells were fixed and stained using methanol, hematoxylin, and eosin (Sigma-Aldrich, St. Louis, MO, USA). The upper chamber was removed and the lower layer migrating cells counted under the microscope (IX81 Olympus).
Real-time imaging of cell migration
A total of 200 μl Matrigel (BD Biosciences, 356,235) pre-coated the bottom of the six-well plates, the cells were then inoculated for 24 h. Thereafter, a 6-well plate was cultured and imaged using KEYENCE BZ-X700 (KEYENCE, Japan), equipped with CO2 and temperature control chamber as well as a time-lapse tracking system. BZ-X Viewer software (KEYENCE) was used to capture phase-contrast images at intervals of every 10 min for 48 h, while the BZ-X Analyzer software (KEYENCE) was used to convert the continuous images into movie files. Furthermore, KEYENCE video editing and analysis software was used to analyze cell migration in the movies. Microsoft Excel 2010 was used to process the trace data to create XY coordinate graphs and distance measurements.
Flow cytometers
The cell concentration was adjusted to 1 × 106 cells/ml in PBS containing 2% FBS. The cell suspensions were incubated with antibodies (BioLegend, Cat# 137005, Cat# 333805, Cat# 506804, Cat# 505007; abcam, ab289967, ab52460) for 30 min on ice, washed with PBS containing 2% FBS, centrifuged twice, and suspended in PBS. Flow cytometry was performed with a FACSVerse instrument (BD Biosciences). The flow cytometry data were analyzed using FlowJo 3.3 software (Tree Star).
Xenograft mouse model
For subcutaneous tumor xenografts, an estimated 1 × 106 mouse GC cells/0.2 ml PBS (with or without CXCL8 which injected every 3 days) were subcutaneously injected into the right axillary region of a 5-week-old female BALB/ C mouse. After 28 days of injection, tumors were collected, measured volumes weighed, and photographed. The volume was calculated using the following formula (tumor volume = L*W*W/2).
For the intraperitoneally model, approximately 5 × 106 GC cells/0.1 ml PBS (with or without CXCL8) were injected into the mouse as mentioned above. Similarly, the mice were euthanized 28 days after injection, then the volume of ascites and the number of visible (> 0.1 cm) metastatic nodules in the peritoneal cavity were measured.
Other reagent or resource
Recombinant Human IL-8 (CXCL8, PeproTech, 200–08), Recombinant Human IL-10 (PeproTech, 200–10), Recombinant Human M-CSF (PeproTech, 300–25), Repertaxin (Sigma-Aldrich, 266,359), Fludarabine (Tocris, 3495/10), Sarsasapogenin (Selleck, S3607), Bay11–7082 (Selleck, S2913), Anemoside B4 (Selleck, S9081).
Statistical analysis
GraphPad Prism 9.0 (GraphPad Software Inc) and SPSS 24.0 statistical software (IBM) were used for statistical analyses of data. The student’s t-test was utilized to perform statistical analysis. P < 0.05 was considered statistically significant.
Results
Hypoxia promotes macrophage-derived CXCL8 secretion
Relevant expression profile data were downloaded from the GEO database (GSE4630) to identify the reflection of macrophages under hypoxic conditions. The dataset included four PBMCs-derived macrophages samples. Two of them were cultured in 0% O2, while the other two were cultured in 20% O2. The “limma” package was used to identify the differentially expressed genes (DEGs, Supplementary Table 3). As a type of immune cell, macrophages could disrupt the microenvironment through cytokines. Thus, this work assessed the effect of hypoxia on cytokine-related gene expression. Among them, CXCL8 was significantly upregulated in the hypoxic-cultured macrophage (Fig. 1A, B). This implied that hypoxic TME might stimulate CXCL8 secretion by macrophages; nevertheless, this warrants further validation. CD14+ positive monocytes by MACS (magnetic-activated cell sorting) negative selection were obtained from healthy volunteers to accurately reflect the in vivo features of macrophages. The PBMCs were induced into immature macrophages (M0 macrophages) for 7 days by M-CSF (Fig. 1C). Subsequently, incubation of M0 for 72 h was continued under hypoxic (1% O2) and normoxic (20% O2) conditions, respectively. Both qRT-PCR and western-blot showed that CXCL8 was overexpressed in hypoxic-cultured M0 (Fig. 1D, E). Similarly, the upregulated CXCL8 in the supernatant of hypoxic-culture M0 was confirmed through the ELISA test (Fig. 1F).
On the other hand, we also examined the effect of hypoxia on GC-derived CXCL8 expression. We found that hypoxia slightly promoted the expression and release of CXCL8 in GCs, but not as dramatically as in macrophages (Sup Fig. 1A, B). At the same time, we reviewed the microarray data obtained in previous studies [8]. The data indicated that the knockdown of HIF-1α had no effect on the expression of CXCL8 in GC cells (Sup Fig. 1C). Immunofluorescence analysis in human GC tissues showed extensive co-localization of CD68 and CXCL8, suggesting that macrophages could secrete CXCL8 (Fig. 1G). To determine the cells responsible for CXCL8 and IL-10 production in GC tissue, we examined CXCL8-expressing cells by flow cytometry (Sup Fig. 1D, Fig. 1H). We observed that CXCL8-expressing cells were expressed of predominantly macrophage (CD68+, Fig. 1H, middle, down), but not GC cells (Ctyokeratin+, Fig. 1H, middle, up). Subsequently, we co-cultured GC cells and macrophages under hypoxic conditions (Fig. 1I). CXCL8 was mainly derived from macrophages in vitro (Fig. 1J, up; Red: macrophage; Green: GC cells).
CXCL8 correlates with poor prognosis and tumor progression of GC
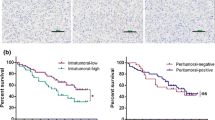
CXCL8 expression in TCGA and GEO (GSE54129) was assessed to further elucidate its role in GC. CXCL8 expression was significantly upregulated in GC tissues compared to that in normal tissues (Fig. 2A, B). The CIBERSORT analysis showed that the expression of CXCL8 was positively correlated with macrophage infiltration in GC-TME (Sup Fige. 1E). Further, the samples were divided into CXCL8-high group and CXCL8-weak group according to the median of CXCL8 expression. The infiltration ratio of macrophages in the TME in the CXCL8-high group was significantly higher than that in the CXCL8-weak group (Sup Fig. 1F). Subsequently, we assessed the clinical significance of CXCL8 based on the Liaoning cancer hospital cohort. CXCL8 in cancerous tissues was remarkably higher than that in paired paracancerous tissues (Fig. 2C). Figure 2D reveals the representative images of high expression and low expression CXCL8 in GC tissue. Moreover, high CXCL8 expression promoted poor DFS (disease-free survival, Fig. 2E) and OS (overall survival, Fig. 2F). The cohort information is listed in Supplementary Table 4.

CXCL8 overexpression in GC promotes poor survival. A, B CXCL8 was up-regulated in GC tissue both in TCGA-GC and GEO-GC cohort. C In matched GC and adjacent tissues, CXCL8 was highly expressed in cancer tissues. D Representative image of CXCL8 expression in GC tissue. (Left: high-expression; Right: weak-expression). E, F CXCL8 promoted poor DFS and OS in the GC cohort. (E. DFS; F. OS)
CXCL8 activates the CXCL8-CXCR1/2 axis of GC and exacerbates the malignant phenotype
In vitro and in vivo assays were performed to assess the function of CXCL8 on GC cells. The GC cells were seeded on the six-well plates coated with Matrigel. The real-time imaging recorded the motion state of the cells. Migration of GC cells was significantly improved when co-culture with recombinant human CXCL8. CXCL8 accelerated cells migration (50 ng/ml, Fig. 3A-C, Sup movie 1, 2). Nonetheless, the enhanced motility was inhibited by adding CXCL8-CXCR1/2 inhibitors (Repertaxin, 50 μg/ml, Fig. 3A-C, Sup movie 1, 2). The promoting impact of CXCL8 on GC invasion ability was achieved by the CXCL8-CXCR1/2 axis. In keeping with this result, the Transwell assay also exhibiting CXCL8 caused an increase in the number of cells penetrating the membrane, but was blocked by repertaxin (Fig. 3D). On cell proliferation, the CCK-8 and colony formation assay demonstrated the promotion of cell proliferation by CXCL8 (Fig. 3E, F); this was also achieved through the CXCL8-CXCR1/2 axis (Fig. 3E, F). Likewise, we knockdown the expression of CXCR1/2 in GC cells (Sup Fig. 2A, B). After inhibited the expression of CXCR1/2, the promoting effect of CXCL8 on cell motility was significantly attenuated (Sup Fig. 2C, D, Sup movie 3, 4). The number of invasive cells in the Transwell assay also decreased accordingly (Sup Fig. 2E). Colony formation and CCK-8 assays also confirmed that with the deletion of CXCR1/2, CXCL8 had almost no effect on cell proliferation (Sup Fig. 2F-H).

CXCL8-CXCR1/2 deteriorates the malignant phenotype of GC. A Migration patterns of GC cells on Matrigel compared to cells cultured with CXCL8 and/or CXCL8-CXCR1/2 inhibitor repertaxin. B Migration trail of GC cells cultured with CXCL8 or CXCL8 and CXCL8-CXCR1/2 inhibitor. C Migration average distances of GC cells in each group. D Transwell exhibited the differences in the number of permeable cells in each group. E, F CCK-8, and colony formation assay confirmed CXCL8 could promote GC proliferation which could be inhibited by CXCL8-CXCR1/2 inhibitor. G CXCL8 increased the subcutaneous tumor size and weight. H IHC representative image of subcutaneous tumors showing that CXCL8 could promote Ki67 and inhibit the expression of Capcase3; the inhibitors could counteract this effect. I CXCL8 induced ascites formation and intra- metastasis of GC
In the in vivo assays, we confirmed that CXCL8 promoted the formation of the subcutaneous tumor. After injection of mouse-GC cells stimulated by CXCL8, the subcutaneous tumors had a larger size and heavier weight than the control. In contrast, no significant difference was noted in tumor size between the control and the inhibitor-added groups (Fig. 3G). At the same time, the expression of Ki67 increased and the expression of Caspase3 decreased in the CXCL8 group; after adding the inhibitor, Ki67 decreased and Caspase3 increased (Fig. 3H). This implied that CXCL8 could promote GC malignant phenotype; this was achieved through the CXCL8-CXCR1/2 axis. Furthermore, unlike other digestive system cancers metastasizing primarily through the vasculature, GC cells had a greater tendency to develop peritoneal metastases. Therefore, we evaluated the effect of CXCL8 on these types of metastases. After intraperitoneal GC cells co-culturing with CXCL8, the mice evolved peritoneal metastases (Fig. 3I).
JAK/STAT1 pathway is downstream of CXCL8
After clarifying the oncogenic effect of CXCL8 on GC, it is essential to explore the possible mechanisms involved. Since JAK/STAT1 pathway is a canonical downstream of the chemokine signaling pathway [28, 29], the effect of the CXCL8-CXCR1/2 axis on the JAK/STAT1 pathway activation was assessed. The protein expression of p-JAK, p-STAT1 was significantly upregulated after stimulation of GC with CXCL8, while the expression of JAK, STAT1 did not significantly change (Fig. 4A). Similarly, the CXCL8-CXCR1/2 inhibitors, repertaxin, inhibited the activation of the JAK/STAT1 pathway by CXCL8 (Fig. 4A). JAK/STAT1 pathway might be the downstream effector of the CXCL8-CXCR1/2 axis. To validate the result, CXCL8-induced GC cells were co-cultured with STAT1 specific inhibitor (Fludarabine). As expected, the activation effect of CXCL8 on JAK/STAT1 disappeared when cells were treated with Fludarabine (50 μM, Fig. 4A). This confirmed that the JAK/STAT1 is the downstream effector of CXCL8. Unsurprisingly, the JAK/STAT1 pathway inhibitors also limited the effects of CXCL8 on invasion (Fig. 4B-E, Sup movie 5, 6) and proliferation (Fig. 4F, G). Thus, a preliminary conclusion would be exogenous CXCL8 continuity activated the CXCL8/CXCR1/2-JAK/STAT1 pathway and triggered invasion and proliferation of GC.

CXCL8-CXCR1/2 activated JAK/STAT1 signal pathway and promoted GC progression. A CXCL8 activated the JAK/STAT1 signal pathway. The STAT1 specific inhibitor (Fludarabine) could antagonize the effects of CXCL8. B-D Migration patterns, migration trail, and average traveled distances of GC cells treated with CXCL8 or CXCL8 and Fludarabine together. E Fludarabine limited the GC invasion. F, G CCK-8, and colony formation assay revealed that the Fludarabine could resist CXCL8’s promotion of GC proliferation
STAT1 regulates the expression of IL-10 directly but not HIF-1α
STAT1 belongs to the STAT protein family that forms homo- or hetero-dimers and translocates to the nucleus to exert regulatory effects as a transcriptional activator. Naturally, we sought to identify the downstream regulatory molecules of STAT1. Through the JAPAR database, the target genes of STAT1 were predicted. Among them, HIF-1α and IL-10 attracted our attention. The former was the hypoxic associated core effector molecule, while the latter was closely related to the polarization of macrophages. It should assess the influence of STAT1 on HIF-1α and IL-10. The shRNA and OE-vector of STAT1 could up-or down-regulated STAT1 expression (Fig. 5A). STAT1 inhibition was followed by a downregulation in HIF-1α and IL-10 expressions both at the transcriptional and protein levels (Fig. 5B-E). In contrast, both were upregulated with STAT1 overexpression (Fig. 5B-E). With the promising results, luciferase reporter assay and ChIP were performed to assess the interaction of STAT1 with HIF-1α and IL-10 promoters. Therefore, full-length HIF-1α and IL-10 promoters were cloned into luciferase reporter plasmids, respectively. The luciferase reporter assay revealed that the up-regulated STAT1 could enhance the luciferase activity of IL-10-wt, but not IL-10-mut (Fig. 5F). Unfortunately, STAT1 did not affect the fluorescence intensity of HIF-1α, whether HIF-1α-wt or HIF-1α-mut (Fig. 5G). This suggestd that as a transcription factor, STAT1 could directly activate the transcription of IL-10, however, HIF-1α promotion might rely on other indirect pathways. Subsequently, the ChIP assay confirmed that the STAT1 antibody could dismantle the IL-10 sequence but not the control IgG (Fig. 5H). A series of reporter genes comprising truncated IL-10 promoter sequences were constructed to further refine the target sequence of the promoter. The luciferase reporter assay showed that deleting the region between − 1811 and − 1821 bases of IL-10 promoter severely abolished IL-10 by STAT1 activation (Fig. 5I).

Transcription factor STAT1 could directly upregulate IL-10 expression. A The shRNA and plasmic regulated STAT1 expression. B, C STAT1 regulated HIF-1α expression at transcription and protein levels. D, E STAT1 could also influence IL-10 expression at transcription and protein levels. F, G The promoter-luciferase reporter assay, showing that STAT1 directly promoted IL-10 expression but not HIF-1α. H The interaction of STAT1 with IL-10 shown using ChIP assays with control (IgG) or anti-STAT1 antibody. I Deletion and selective mutation analysis identified STAT1-responsive regions in the IL-10 promoter. Luciferase reporter plasmids containing serially truncated or mutated IL-10 promoter constructs were co-transfected with OE-STAT1 into NUGC3 cells, and relative luciferase activities were detected. The − 1811 ~ − 1821 sequence was the binding site
Unsurprisingly, CXCL8 could significantly promote the expression of IL-10 in GCs, but it did not alter HIF-1α expression significantly (Sup Fig. 3A, B). Following the increase of IL-10 in the TME, we further assessed the effect of IL-10 and hypoxia on the JAK/STAT1 pathway. Consistent with previous studies [30], both IL-10 and hypoxia were also activators of the JAK/STAT1 pathway (Sup Fig. 3C, D). That was to say, CXCL8 overexpressed due to hypoxia could promote the expression of IL-10 together with the hypoxic microenvironment, and the IL-10 could amplify this effect through positive feedback (Fig. 7). Further, we also compared the differences in the effects of hypoxic microenvironment and CXCL8 on IL-10 expression. The results showed that both tumor hypoxia and CXCL8 could increase the expression of IL-10 in GC, but the gain effect of CXCL8 was more significant (Sup Fig. 3E).CXCL8 remained the dominant factor in inducing IL-10 expression. In addition, both of in vitro and in vivo analysis confirmed that IL-10 was mainly expressed in GC cells (Fig. 1H, right, up; Fig. 1J, down). In subcutaneous tumors, the tumor size (Sup Fig. 3F), the expression of IL-10 and CXCL8 also decreased with the addition of Repertaxin (Sup Fig. 3G, H). The CXCL8/CXCR1/2-JAK/STAT1/IL-10 axis was an indispensable oncogenic factor in the TME.
IL-10 induces M2 polarization through the NF-κB pathway and promotes CXCL8 expression
As evidenced above, hypoxic-macrophage-derived CXCL8 could promote IL-10 expression by activating JAK/STTA1 in GC. Besides, studies have shown that cytokine IL-10 can induce M2 polarization [31]. Therefore, the effect of IL-10 on macrophages and the potential underlying mechanisms should be confirmed. Unsurprisingly, immunofluorescence confirmed that IL-10 significantly promoted CD163 expression in PBMCs induced macrophage, and M2 polarization (Fig. 6A). Abnormally activated NF-κB pathway exists in TAMs [32]. Thus, it is necessary to evaluate the effect of IL-10 on the NF-κB pathway in macrophages. The addition of IL-10 upregulated p50 and p65 expression in the nucleus and promoted p65 phosphorylation (Fig. 6B). Subsequently, IL-10 and BAY 11–7082 (10 μM) or Sarsasapogenin (50 μM) were used to inhibit the NF-κB pathway and co-cultured macrophages. Sarsasapogenin, a steroidal sapogenin, is purified from the Chinese Materia Medica Anemarrhena asphodeloides Bunge (rhizome) [33]. It can provoke the generation of reactive oxygen species and activate unfolded protein response (UPR) signaling pathways. It specifically targeting IκB, potently inhibits NF-κB activation [34]. Both sarsasapogenin and Bay11–7082 attenuated CD163 expression (Fig. 6C, Sup Fig. 4A). It inhibited the M2 polarization. This demonstrates that the effect of IL-10 on macrophage M2 polarization was partly achieved through the NF-κB pathway. This implied that the IL-10-NF-κB axis was implicated in CXCL8 regulation at the transcriptional level. Besides, we analyzed the effects of IL-10 and NF-κB pathways on CXCL8 expression. IL-10 upregulated CXCL8 expression at the transcriptional and protein levels (Fig. 6D, E) and intensified the secretion of the latter (Fig. 6F). Correspondingly, the NF-κB pathway could antagonize this effect (Fig. 6D-F, Sup Fig. 4B-D). Subsequently, we examined the effects of CXCL8 and hypoxia on macrophage polarization, IL-4 was set as a positive control. CXCL8 had no significant effect on the M2 polarization of macrophages, and hypoxia could induce the M2 polarization, but the polarized level was not as intensity as that of IL-4 (Sup Fig. 4E). In the macrophages and GC cells co-culture system, the expression of CXCL8 decreased with IL-10 inhibitor added (Sup Fig. 4F). In vivo assay, after inhibited the expression of IL-10, the tumor contracted (Sup Fig. 4G) and the expression of CXCL8 in the tumor tissue also decreased (Sup Fig. 4H). With the inhibition of IL-10 expression, the expression of macrophage-derived CXCL8 in the tissue was also suppressed (Sup Fig. 4I).

Cytokines IL-10 positive feedback induces macrophage M2 polarization and upregulates macrophage-derived CXCL8 expression. A IL-10 promoted macrophages CD163 expression and induced M2 polarization. B IL-10 activated NF-κB signaling pathway. C The NF-κB pathway inhibitor (Sarsasapogenin) could limit IL-10 induced M2 polarization. D E. IL-10 upregulated CXCL8 expression, and it could be prevented by NF-κB pathway inhibitor. F. IL-10 could promote CXCL8 secretion, and Sarsasapogenin could antagonize the effect of IL-10. G Deletion and selective mutation analysis recognized the NFKB1-responsive regions in the CXCL8 promoter. Luciferase reporter plasmids containing serially truncated or mutated CXCL8 promoter constructs were co-transfected with OE-NFKB1 into macrophage cells, and relative luciferase activities were detected. The − 1829 ~ − 1819 and − 91 ~ − 81 sequences were all the binding sites
The subsequent transcription factor luciferase reporter assay and ChIP confirmed our suspicions. The gene (p50) was bound to the promoter of CXCL8 (Fig. 6G, H). A series of truncated CXCL8 promoter sequences were constructed, which included at least one potential binding site (in red). Nevertheless, every sequence could promote CXCL8 expression. Mutated the mut1 (in blue, − 1829 ~ − 1819) and mut2 (in green, − 91 ~ − 81) sequences simultaneously, the fluorescence intensity significantly decreased. This confirmed the presence of two NFKB1 binding sites in the promoter sequence and up-regulated CXCL8 expression. In this way, a positive feedback loop was formed between macrophage and GC cells that exacerbated tumor deterioration under the catalysis of hypoxic TME.
Discussion
TME of most solid tumors is characterized by hypoxia and inflammation [35]. The crosstalk between TME and cancer cells promotes tumor growth and progression [36]. Cytokines are the critical bridge that maintains this intercellular communication; besides, they can favorably shape the TME for tumor cells [37]. Gastric cancer cells rely on this complex environment to achieve sustainable development, invasion, and metastasis. In our previous study, we continuously focused on the effect of hypoxia on the progression of GC cells [8]. Nevertheless, this effect on TME stromal cells and inflammatory cells has been overlooked.
Of note, tumor-infiltrating macrophages release the strongest signal in inflammatory TME [38]. Thus, we concentrated on the effect of hypoxia on macrophage cytokines i.e., the communication tools. Among them, CXCL8 exhibited the most potent response to the hypoxic signal. CXCL8 is one of the earliest and most comprehensively studied chemokines. It was discovered, purified, and sequenced in monocytes in the 1980s [39]. CXCL8 is secreted by different cells, including monocytes, alveolar macrophages, fibroblasts, endothelial cells, and epithelial cells [40, 41]. Additionally, cytokines (interleukin-1, interleukin-6, CXCL12, TNFα), hypoxia, reactive oxygen species (ROS), bacterial particles could stimulate CXCL8 expression. The transcription factor NF-κB and activator protein-1 (AP-1) were also implicated in CXCL8 activation [42, 43]. Notably, CXCL8 was nearly undetectable in unstimulated cells. In contrast, the above stimulation would upregulate CXCL8 expression by 10 to 100 times [44]. Besides, this type of explosive growth provides a basis for its role in cancers. CXCL8 binds G protein-coupled receptors or CXCR1/2 to transmit its signal. CXCL8-CXCR1/2 axis stimulates endothelial cells and promotes tumor angiogenesis [45]. Blocked CXCL8-CXCR2 signal transmission between TAMs and cancer cells could improve the effect of anti-PD1 treatment [46, 47]. This study found that the CXCL/8-CXCR1/2 axis activated the JAK/STAT1 pathway in GC, thereby promoting the malignant phenotype. Further, STAT1 acted as a transcription factor directly regulating IL-10 expression, this was closely associated with macrophages. In addition, it could not ignore that STAT1 also affected the HIF-1α expression, which was the most crucial element for the organism responding to hypoxia. The indirect regulatory pattern of hypoxia/CXCL8/CXCR1/2/JAK/STAT1/HIF-1α might be a non-classical adaptation pattern of GC cells to the hypoxic microenvironment.
IL-10 is one of the most famous molecules causing macrophage polarization [48]. It triggers M2 polarization by regulating the NF-κB and JAK/STAT pathway [49]. Generally speaking, NF-κB pathway activation is often regarded as a marker of M1 macrophages because of its pro-inflammatory effects. However, various tumor-associated cells appeare to exhibit complex characteristics as the TME forms. Even inflammatory responses are involved in tumor immunity while inducing maturation of tumor-associated fibroblasts [50]. TAMs also deviate from the traditional classification. Although the simple dichotomy of macrophage (M1 and M2) is widely used, researchers continue to express doubts. With the advancement of single-cell technology, further sub-classification of macrophage by markers on the surface of the macrophage membrane deepen the understanding of the TME [51]. Specifically, tissue-resident macrophages and monocyte-derived macrophages have entirely different biological functions, however, the two co-exist in TME [19]. The subclassification theory of M2 macrophages indicates that the M2d subtype is the closest to TAMs, and the activation of NF-κB pathway is also a sign event of M2d activation [52, 53]. This means that the activation of the NF-κB pathway in the complex TME is still a double-edged sword. Then, it was natural for us to further evaluate its effect on macrophage and macrophage-derived CXCL8. Our results demonstrated that IL-10 regulated M2 polarization via the NF-κB pathway and affected CXCL8 expression. Subsequent luciferase reporter assays and ChIP assays confirmed the regulatory role of the transcription factor NFKB1 (p50) on CXCL8. This was also consistent with previous findings [54, 55]. Thus, macrophage-derived CXCL8 could further promote the deterioration of GC and cause IL-10 expression, recurrence of the cycle.
The above results illustrate a novel paradigm of GC-TAMs’ interaction: hypoxia spurs the release of overexpressed CXCL8 by macrophages which then activates the CXCL8/CXCR1/2-JAK/STAT1 pathway and promotes GC progression. Moreover, this pathway directly promotes the expression of GC-derived IL-10, further accelerating the release of macrophage-derived CXCL8 under the IL-10/NF-κB/CXCL8 axis. In this process, the M2-type polarization is simultaneously induced. This causes ordinary macrophages involved in immune surveillance to “betray” into TAMs implicated in immune escape and suppression. Hypoxia/CXCL8/CXCR1/2-JAK/STAT1/IL-10/NF-κB/CXCL8 forms a positive feedback loop between GC and macrophage (or TAMs). As the core molecule of this circuit, CXCL8 is a high-risk factor for GC prognosis. This suggests that the signaling loop is a potential therapeutic target. Also, research on anti-CXCL-8 therapy in solid tumors recruits or collates data (NCT01831310, NCT02536469, NCT04347226), and we have reason to expect these corresponding results.
Conclusion
In conclusion, our work identifies a hypoxic-activated, inflammation-associated molecular network; which involve CXCL8, CXCR1/2, JAK/STAT1, IL-10, and NF-κB signaling pathways that regulate a positive feedback loop between GC cells and TAMs, tumor progression, macrophage polarization, as well as patient outcomes (Fig. 7). These results indicate the potential of this feedback loop as a therapeutic target for GC.

The mechanism graph of the positive feedback loop between GC cells and TAMs. Hypoxia-induced macrophage-derived CXCL8 secretion activated CXCL8- CXCR1/2-JAK/STAT1 signaling pathway and deteriorated the GC malignant phenotype. The transcription factor STAT1 could directly promote IL-10 expression. The latter activated the macrophage IL-10/ NF-κB signaling pathway and induced M2 polarization. The transcription factor NFKB1 directly upregulated CXCL8 expression
Availability of data and materials
The data are available from the sources listed in the manuscript—the TCGA and GEO data portal. The data used to support the findings of this study are available from the corresponding author upon request.
Change history
26 March 2023
Author requested to capture Affiliation 7, his corresponding affiliation, in the first page.
Abbreviations
- GC:
-
Gastric cancer
- TME:
-
Tumor microenvironment
- HIF-1α:
-
Hypoxia-inducible factor-1α
- TAMs:
-
Tumor-associated macrophages
- CXCL8/IL-8:
-
C-X-C Motif Chemokine Ligand 8
- IL-10:
-
Interleukin 10
- CXCR1/2:
-
C-X-C Motif Chemokine Receptor 1/2
- JAK:
-
Janus Kinase 1
- STAT1:
-
Signal Transducer And Activator Of Transcription 1
- GEO:
-
Gene Expression Omnibus
- TCGA:
-
The Cancer Genome Atlas
- PBMC:
-
Peripheral blood mononuclear cell
- FBS:
-
Fetal bovine serum
- IHC:
-
Immunohistochemistry
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- ChIP:
-
Chromatin immunoprecipitation
- CCK8:
-
Cell counting kit 8
- OD:
-
Optical density
- DEGs:
-
Differentially expressed genes
- MACS:
-
Magnetic-activated cell sorting
- DFS:
-
Disease-free survival
- OS:
-
Overall survival
References
Siegel RL, Miller KD, Fuchs HE, et al. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33.
Yang L, Zheng R, Wang N, et al. Incidence and mortality of stomach cancer in China, 2014. Chin J Cancer Res. 2018;30(3):291–8. https://doi.org/10.21147/j.issn.1000-9604.2018.03.01 [published Online First: 2018/07/27].
Galletti G, Zhang C, Gjyrezi A, et al. Microtubule engagement with Taxane is altered in Taxane-resistant gastric Cancer. Clin Cancer Res. 2020;26(14):3771–83. https://doi.org/10.1158/1078-0432.Ccr-19-3018 [published Online First: 2020/04/24].
Zheng ZQ, Chen JT, Zheng MC, et al. Nestin +/CD31 + cells in the hypoxic perivascular niche regulate glioblastoma Chemoresistance by upregulating JAG1 and DLL4. Neuro-oncology. 2020. https://doi.org/10.1093/neuonc/noaa265 [published Online First: 2020/11/30].
Massara M, Bonavita O, Savino B, et al. ACKR2 in hematopoietic precursors as a checkpoint of neutrophil release and anti-metastatic activity. Nat Commun. 2018;9(1):676. https://doi.org/10.1038/s41467-018-03080-8 [published Online First: 2018/02/16].
Lee KE, Spata M, Bayne LJ, et al. Hif1a deletion reveals pro-neoplastic function of B cells in pancreatic neoplasia. Cancer Discov. 2016;6(3):256–69. https://doi.org/10.1158/2159-8290.Cd-15-0822 [published Online First: 2015/12/31].
Zhang J, Wu Y, Lin YH, et al. Prognostic value of hypoxia-inducible factor-1 alpha and prolyl 4-hydroxylase beta polypeptide overexpression in gastric cancer. World J Gastroenterol. 2018;24(22):2381–91. https://doi.org/10.3748/wjg.v24.i22.2381 [published Online First: 2018/06/16].
Piao H-y, Liu Y, Kang Y, et al. Hypoxia associated lncRNA HYPAL promotes proliferation of gastric cancer as ceRNA by sponging miR-431-5p to upregulate CDK14. Gastric Cancer. 2021;25:1–20.
Zhang J, Guo S, Wu Y, et al. P4HB, a novel hypoxia target gene related to gastric Cancer invasion and metastasis. Biomed Res Int. 2019;2019:9749751. https://doi.org/10.1155/2019/9749751 [published Online First: 2019/08/31].
Zhang J, Jin HY, Wu Y, et al. Hypoxia-induced LncRNA PCGEM1 promotes invasion and metastasis of gastric cancer through regulating SNAI1. Clin Transl Oncol. 2019;21(9):1142–51. https://doi.org/10.1007/s12094-019-02035-9 [published Online First: 2019/01/29].
Murray PJ. Macrophage polarization. Annu Rev Physiol. 2017;79:541–66. https://doi.org/10.1146/annurev-physiol-022516-034339 [published Online First: 2016/11/05].
Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell. 2014;157(4):832–44. https://doi.org/10.1016/j.cell.2014.04.016 [published Online First: 2014/05/06].
Hallowell RW, Collins SL, Craig JM, et al. mTORC2 signalling regulates M2 macrophage differentiation in response to helminth infection and adaptive thermogenesis. Nat Commun. 2017;8:14208. https://doi.org/10.1038/ncomms14208 [published Online First: 2017/01/28].
Stein M, Keshav S, Harris N, et al. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176(1):287–92.
Mills CD, Kincaid K, Alt JM, et al. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164(12):6166–73.
Elinav E, Nowarski R, Thaiss CA, et al. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13(11):759–71. https://doi.org/10.1038/nrc3611 [published Online First: 2013/10/25].
Ramesh A, Brouillard A, Kumar S, et al. Dual inhibition of CSF1R and MAPK pathways using supramolecular nanoparticles enhances macrophage immunotherapy. Biomaterials. 2020;227:119559. https://doi.org/10.1016/j.biomaterials.2019.119559 [published Online First: 2019/11/02].
Lu SW, Pan HC, Hsu YH, et al. IL-20 antagonist suppresses PD-L1 expression and prolongs survival in pancreatic cancer models. Nat Commun. 2020;11(1):4611. https://doi.org/10.1038/s41467-020-18244-8 [published Online First: 2020/09/16].
DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19(6):369–82. https://doi.org/10.1038/s41577-019-0127-6 [published Online First: 2019/02/06].
Cao Q, Yan X, Chen K, et al. Macrophages as a potential tumor-microenvironment target for noninvasive imaging of early response to anticancer therapy. Biomaterials. 2018;152:63–76. https://doi.org/10.1016/j.biomaterials.2017.10.036 [published Online First: 2017/11/08].
Yamaguchi T, Fushida S, Yamamoto Y, et al. Tumor-associated macrophages of the M2 phenotype contribute to progression in gastric cancer with peritoneal dissemination. Gastric Cancer. 2016;19(4):1052–65. https://doi.org/10.1007/s10120-015-0579-8 [published Online First: 2015/12/02].
Zhang Y, Meng W, Yue P, et al. M2 macrophage-derived extracellular vesicles promote gastric cancer progression via a microRNA-130b-3p/MLL3/GRHL2 signaling cascade. J Exp Clin Cancer Res. 2020;39(1):134. https://doi.org/10.1186/s13046-020-01626-7 [published Online First: 2020/07/15].
Wang X, Luo G, Zhang K, et al. Hypoxic tumor-derived Exosomal miR-301a mediates M2 macrophage polarization via PTEN/PI3Kγ to promote pancreatic Cancer metastasis. Cancer Res. 2018;78(16):4586–98. https://doi.org/10.1158/0008-5472.Can-17-3841 [published Online First: 2018/06/09].
Henze AT, Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Invest. 2016;126(10):3672–9. https://doi.org/10.1172/jci84427 [published Online First: 2016/08/03].
Okazaki S, Shintani S, Hirata Y, et al. Synthetic lethality of the ALDH3A1 inhibitor dyclonine and xCT inhibitors in glutathione deficiency-resistant cancer cells. Oncotarget. 2018;9(73):33832–43. https://doi.org/10.18632/oncotarget.26112 [published Online First: 2018/10/20].
Piao HY, Guo S, Wang Y, et al. Exosome-transmitted lncRNA PCGEM1 promotes invasive and metastasis in gastric cancer by maintaining the stability of SNAI1. Clin Transl Oncol. 2020. https://doi.org/10.1007/s12094-020-02412-9 [published Online First: 2020/06/11].
Piao H-y, Guo S, Jin H, et al. LINC00184 involved in the regulatory network of ANGPT2 via ceRNA mediated miR-145 inhibition in gastric cancer. J Cancer. 2021;12(8):2336.
Li HS, Watowich SS. Innate immune regulation by STAT-mediated transcriptional mechanisms. Immunol Rev. 2014;261(1):84–101. https://doi.org/10.1111/imr.12198 [published Online First: 2014/08/16].
Mellado M, Rodríguez-Frade JM, Mañes S, et al. Chemokine signaling and functional responses: the role of receptor dimerization and TK pathway activation. Annu Rev Immunol. 2001;19:397–421. https://doi.org/10.1146/annurev.immunol.19.1.397 [published Online First: 2001/03/13].
Hu X, Li J, Fu M, et al. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. 2021;6(1):402. https://doi.org/10.1038/s41392-021-00791-1 [published Online First: 2021/11/27].
Lee JH, Lee GT, Woo SH, et al. BMP-6 in renal cell carcinoma promotes tumor proliferation through IL-10-dependent M2 polarization of tumor-associated macrophages. Cancer Res. 2013;73(12):3604–14. https://doi.org/10.1158/0008-5472.Can-12-4563 [published Online First: 2013/05/02].
Khalife J, Ghose J, Martella M, et al. MiR-16 regulates crosstalk in NF-κB tolerogenic inflammatory signaling between myeloma cells and bone marrow macrophages. JCI Insight. 2019;4(21). https://doi.org/10.1172/jci.insight.129348 [published Online First: 2019/10/09].
Zhang YM, Zheng T, Huang TT, et al. Sarsasapogenin attenuates Alzheimer-like encephalopathy in diabetes. Phytomedicine. 2021;91:153686. https://doi.org/10.1016/j.phymed.2021.153686 [published Online First: 2021/08/02].
Peng J, Zhao K, Zhu J, et al. Sarsasapogenin suppresses RANKL-induced Osteoclastogenesis in vitro and prevents lipopolysaccharide-induced bone loss in vivo. Drug Des Devel Ther. 2020;14:3435–47. https://doi.org/10.2147/dddt.S256867 [published Online First: 2020/09/19].
Riera-Domingo C, Audigé A, Granja S, et al. Immunity, hypoxia, and metabolism-the Ménage à trois of Cancer: implications for immunotherapy. Physiol Rev. 2020;100(1):1–102. https://doi.org/10.1152/physrev.00018.2019 [published Online First: 2019/08/16].
Iriondo O, Liu Y, Lee G, et al. TAK1 mediates microenvironment-triggered autocrine signals and promotes triple-negative breast cancer lung metastasis. Nat Commun. 2018;9(1):1994. https://doi.org/10.1038/s41467-018-04460-w [published Online First: 2018/05/20].
Hamarsheh S, Osswald L, Saller BS, et al. Oncogenic Kras(G12D) causes myeloproliferation via NLRP3 inflammasome activation. Nat Commun. 2020;11(1):1659. https://doi.org/10.1038/s41467-020-15497-1 [published Online First: 2020/04/05].
Hakimi AA, Voss MH, Kuo F, et al. Transcriptomic profiling of the tumor microenvironment reveals distinct subgroups of clear cell renal cell Cancer: data from a randomized phase III trial. Cancer Discov. 2019;9(4):510–25. https://doi.org/10.1158/2159-8290.Cd-18-0957 [published Online First: 2019/01/10].
Peveri P, Walz A, Dewald B, et al. A novel neutrophil-activating factor produced by human mononuclear phagocytes. J Exp Med. 1988;167(5):1547–59. https://doi.org/10.1084/jem.167.5.1547 [published Online First: 1988/05/01].
Strieter RM, Kunkel SL, Showell HJ, et al. Monokine-induced gene expression of a human endothelial cell-derived neutrophil chemotactic factor. Biochem Biophys Res Commun. 1988;156(3):1340–5. https://doi.org/10.1016/s0006-291x(88)80779-4 [published Online First: 1988/11/15].
Wanninger J, Neumeier M, Weigert J, et al. Adiponectin-stimulated CXCL8 release in primary human hepatocytes is regulated by ERK1/ERK2, p38 MAPK, NF-kappaB, and STAT3 signaling pathways. Am J Physiol Gastrointest Liver Physiol. 2009;297(3):G611–8. https://doi.org/10.1152/ajpgi.90644.2008 [published Online First: 2009/07/18].
Brat DJ, Bellail AC, Van Meir EG. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro-Oncology. 2005;7(2):122–33. https://doi.org/10.1215/s1152851704001061 [published Online First: 2005/04/16].
Wald O, Shapira OM, Izhar U. CXCR4/CXCL12 axis in non small cell lung cancer (NSCLC) pathologic roles and therapeutic potential. Theranostics. 2013;3(1):26–33. https://doi.org/10.7150/thno.4922 [published Online First: 2013/02/06].
Podolin PL, Bolognese BJ, Foley JJ, et al. A potent and selective nonpeptide antagonist of CXCR2 inhibits acute and chronic models of arthritis in the rabbit. J Immunol. 2002;169(11):6435–44. https://doi.org/10.4049/jimmunol.169.11.6435 [published Online First: 2002/11/22].
Keane MP, Belperio JA, Xue YY, et al. Depletion of CXCR2 inhibits tumor growth and angiogenesis in a murine model of lung cancer. J Immunol. 2004;172(5):2853–60. https://doi.org/10.4049/jimmunol.172.5.2853 [published Online First: 2004/02/24].
Zhang M, Huang L, Ding G, et al. Interferon gamma inhibits CXCL8-CXCR2 axis mediated tumor-associated macrophages tumor trafficking and enhances anti-PD1 efficacy in pancreatic cancer. J Immunother Cancer. 2020;8(1). https://doi.org/10.1136/jitc-2019-000308 [published Online First: 2020/02/14].
Lin C, He H, Liu H, et al. Tumour-associated macrophages-derived CXCL8 determines immune evasion through autonomous PD-L1 expression in gastric cancer. Gut. 2019;68(10):1764–73. https://doi.org/10.1136/gutjnl-2018-316324 [published Online First: 2019/01/21].
Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. https://doi.org/10.1016/j.immuni.2014.06.008 [published Online First: 2014/07/19].
Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117(5):1155–66. https://doi.org/10.1172/jci31422 [published Online First: 2007/05/04].
Bu L, Baba H, Yoshida N, et al. Biological heterogeneity and versatility of cancer-associated fibroblasts in the tumor microenvironment. Oncogene. 2019;38(25):4887–901. https://doi.org/10.1038/s41388-019-0765-y [published Online First: 2019/03/01].
Casanova-Acebes M, Dalla E, Leader AM, et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature. 2021;595(7868):578–84. https://doi.org/10.1038/s41586-021-03651-8 [published Online First: 2021/06/18].
Wang Q, Ni H, Lan L, et al. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. 2010;20(6):701–12. https://doi.org/10.1038/cr.2010.52 [published Online First: 2010/04/14].
Xu F, Wei Y, Tang Z, et al. Tumor-associated macrophages in lung cancer: friend or foe? (review). Mol Med Rep. 2020;22(5):4107–15. https://doi.org/10.3892/mmr.2020.11518 [published Online First: 2020/10/02].
Karakurum M, Shreeniwas R, Chen J, et al. Hypoxic induction of interleukin-8 gene expression in human endothelial cells. J Clin Invest. 1994;93(4):1564–70. https://doi.org/10.1172/jci117135 [published Online First: 1994/04/01].
Frey S, Derer A, Messbacher ME, et al. The novel cytokine interleukin-36α is expressed in psoriatic and rheumatoid arthritis synovium. Ann Rheum Dis. 2013;72(9):1569–74. https://doi.org/10.1136/annrheumdis-2012-202264 [published Online First: 2012/12/27].
Acknowledgements
None.
Informed consent
Informed consent was obtained from patients before surgery at the Liaoning Province Cancer Hospital & Institute.
Funding
Liaoning S&T Project (2020-ZLLH-46), CSCO (Y-HR2019–0337, Y-HR2019–0341).
Author information
Authors and Affiliations
Contributions
Piao HY and Fu LF performed the majority of experiments and analyzed the data and drafted the manuscript; Zhang J designed the research; Zhang J and Wang YX performed the revision; Liu Y and Yang D conducted the molecular biology assays and assisted in writing the manuscript; Wang Y and Meng XY collected and analyzed the data. Xiao X contributed to data visualization. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was reviewed and approved by the Faculty of Science Ethics Committee at Liaoning Cancer Hospital & Institute (Cancer Hospital of China Medical University, 20181226).
Consent for publication
All authors agree with the content of the manuscript.
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1
. Primer sequence. Supplementary Table 2. Antibodies information.
Additional file 4: Supplementary Fig. 1.
A. Hypoxia promoted CXCL8 expression in GC cells at mRNA level. B. Hypoxia induced the secretion of GC-derived cytokine CXCL8 secretion. C. The knockdown HIF-1α did not affect the expression of CXCL8. D. Cells were gated to exclude debris and dead cells. E. CXCL8 was positively correlated the proportion of infiltrating macrophages in TME. F. The infiltration ratio of macrophages in CXCL8-high group was significantly higher than that in CXCL8-weak group in TME. Supplementary Fig. 2. A, B. The siRNAs could effectively inhibit the expression of CXCR1 and CXCR2. C. Migration patterns of GC cells on Matrigel compared to cells cultured with CXCL8 alone and CXCL8 + si-CXCR1/2. D. Migration average distances of GC cells in each group. E. Transwell exhibited the differences in the number of permeable cells in each group. F, G, H. Colony formation assay and CCK-8 confirmed CXCL8 could promote GC proliferation which could be inhibited by si-CXCR1/2. Supplementary Fig. 3. A. CXCL8 promoted IL-10 expression in vitro. B. CXCL8 slightly promotes the expression of HIF-1α. C, D. IL-10 and hypoxia activated the JAK/STAT1 signal pathway. The STAT1 specific inhibitor (Fludarabine) could antagonize the effects. E. CXCL8 was more able to promote the expression of GC-derived IL-10 than hypoxia. F. CXCL8-CXCR1/2 inhibitor limited the growth of subcutaneous tumors. G, H. CXCL8-CXCR1/2 inhibitor restricted the expression of CXCL8 and IL-10 in tumor tissues. Supplementary Fig. 4. A. The NF-κB pathway inhibitor (Bay11–7082) could limit IL-10 induced M2 polarization. B, C. IL-10 upregulated CXCL8 expression, and it could be prevented by NF-κB pathway inhibitor (Bay11–7082). D. IL-10 could promote CXCL8 secretion, and Bay11–7082 could antagonize the effect of IL-10. E. CXCL8 and hypoxia could slightly induce macrophage M2 polarization. F. IL-10 inhibitor restricted CXCL8 expression in co-cultured cells (Red: macrophage; Green: GC cells). G. IL-10 inhibitor limited the growth of subcutaneous tumors. H, I. IL-10 inhibitor restricted the expression of CXCL8.
Additional file 5.
Additional file 6.
Additional file 7.
Additional file 8.
Additional file 9.
Additional file 10.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Piao, H., Fu, L., Wang, Y. et al. A positive feedback loop between gastric cancer cells and tumor-associated macrophage induces malignancy progression. J Exp Clin Cancer Res 41, 174 (2022). https://doi.org/10.1186/s13046-022-02366-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-022-02366-6