
Abstract
Cancer cells can evade immune responses by exploiting inhibitory immune checkpoints. Immune checkpoint inhibitor (ICI) therapies based on anti-CTLA-4 and anti-PD-1/PD-L1 antibodies have been extensively explored over the recent years to unleash otherwise compromised anti-cancer immune responses. However, it is also well established that immune suppression is a multifactorial process involving an intricate crosstalk between cancer cells and the immune systems. The cancer glycome is emerging as a relevant source of immune checkpoints governing immunosuppressive behaviour in immune cells, paving an avenue for novel immunotherapeutic options. This review addresses the current state-of-the-art concerning the role played by glycans controlling innate and adaptive immune responses, while shedding light on available experimental models for glycoimmunology. We also emphasize the tremendous progress observed in the development of humanized models for immunology, the paramount contribution of advances in high-throughput single-cell analysis in this context, and the importance of including predictive machine learning algorithms in translational research. This may constitute an important roadmap for glycoimmunology, supporting careful adoption of models foreseeing clinical translation of fundamental glycobiology knowledge towards next generation immunotherapies.
Similar content being viewed by others

Introduction
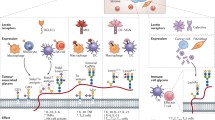
Over the last decade, the introduction of T cell targeted immunomodulators blocking immune checkpoints CTLA-4 and PD1 or PDL1 has been unprecedented, with immune checkpoint inhibitors (ICI) being used as single agents or in combination with chemotherapies in about 50 cancer types [1]. Despite its efficiency in high mutational load tumours, such as melanoma and lung cancer [2, 3], ICI does not meet its promise for most cancer patients. The overall response rate to ICI is approximately 12% in low mutational burden tumours, which increases the demand for biomarkers of response and alternative therapeutic strategies to overcome therapy resistance [4]. Furthermore, patients non-responding to immune checkpoint inhibitors experience a vast panoply of immune-mediated iatrogenic effects [5], leading to treatment discontinuation, or become refractory [6]. Initial resistance to ICI has been associated with the lack of T cell infiltration (cold tumour phenotype) (Fig. 1). Recently, the main mechanisms governing poor T cell infiltration in cold tumours were described, including lack of tumour antigens, defects in antigen presentation, absence of T cell activation and deficit of immune homing into the tumour bed [7], all of these factors being highly dependent on the tumour microenvironment (TME) [8] and likely enhanced by aberrant tumour glycosylation [9] (Fig. 1).

Currently accepted immune checkpoint inhibitors resistance mechanisms. Resistance to currently used immune checkpoint inhibitors, namely anti-PD1, anti-PD-L1 and anti-CTLA-4 therapeutic antibodies, has been associated with the lack of effective tumour T cell infiltration. This could be due to lack of tumour antigens, defects in antigen presentation, and absence of T cell activation. Of note, cancer-associated glycosignatures, (e.g. Tn, STn, lewis antigens, among others) that are distinct from those found on healthy cells, can interact with glycan-binding receptors (GBR) in immune cells, driving immunosuppression. Glycan-mediated immunosuppression is promoted by non-classical pathways, including altered antigen uptake, processing, and presentation by antigen presenting cells, ultimately conditioning T-cell priming. Moreover, GBRs engagement alters immune cell signalling, differentiation, and cytokine responses toward anti-inflammatory or immunosuppressive phenotypes. Furthermore, the abnormal glycosylation of tumour-associated glycoproteins generates neoantigens that can serve as targets for tumour-specific T cells
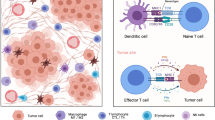
Over 40 years of cancer glycobiology studies support the notion that the glycome at the cell-surface of cancer cells, extracellular vesicles and secreted/released molecules is fundamentally altered, generating cancer-associated glycosignatures that are distinct from those of healthy cells [10,11,12,13,14]. However, while their contribution to cancer aggressiveness has been subject of much interest, the interplay with the immune system has remained mostly overlooked. Nevertheless, it is rather consensual that alterations in glycosylation reshape tumour recognition by the immune system and induce immunosuppressive signalling through glycan-binding receptors (GBR) [15] (Fig. 1). Namely, altered glycosylation directly impacts on antigen presentation by modulating antigen presenting cells (APC) uptake of abnormally glycosylated proteins, its proteolytic processing and presentation by Major Histocompatibility Complexes (MHC), ultimately governing subsequent T-cell priming [16]. Moreover, glycan interactions with GBRs, namely galectins, C-type lectins, and siglecs on APCs is known to alter immune cell signalling, differentiation, and cytokine responses toward anti-inflammatory or immunosuppressive phenotypes [9, 15, 17] (Fig. 2). Furthermore, the abnormal glycosylation of tumour-associated glycoproteins generates neoantigens that can serve as targets for tumour-specific T cells [15]. Collectively, these findings support the relevance of engaging in a thorough understanding of the glycan-immune system interplay towards next generation immunotherapies. The election of appropriate models is critical to fully disclose this intricate crosstalk as well as support therapeutic design and pre-clinical trials. As such, the present review addresses recent studies on this field, emphasizing advantages and disadvantages of available and emerging pre-clinical models in the road towards translational glycoimmunology.

Glycan binding receptors (GBR) interactions with tumour cell aberrant glycosylation as a novel class of immune checkpoints. Cancer cell associated glycans modulate immune cell responses by interacting with various classes of GBR, as galectins (A), most C-type lectins (B) and siglecs (C). Galectins (Gal) are secreted lectins that bind terminal β-galactoside-containing carbohydrates, including the T antigen. Tumour secreted galectins can induce apoptosis of activated human T cells, while antagonizing Type 1 T helper (Th1) and Type 17 T helper (Th17) cells survival. Furthermore, exhausted T cells frequently express galectin ligands, as the T cell immunoglobulin and mucin domain 3 (TIM3), regulating T cell exhaustion while modulating the inhibitory action of CD4 + CD25 + regulatory T cells (Tregs). Furthermore, galectins endow dendritic cells (DCs) with a tolerogenic phenotype capable of promoting interleukin 10 (IL-10)-mediated T cell tolerance. In turn, C-type lectin-like domain superfamily members as DC-SIGN, that avidly recognizes fucosylated lewis antigens on O- and N-glycans, and MGL, with affinity for terminal GalNAc (Tn antigen), are widely expressed by DCs and macrophages. Particularly, DC-SIGN engagement in the tumour microenvironment enhances interleukin -10 and -27, as well as Th2-attracting chemokine expression, shifting Thelper polarization from Th1 to Th2. MGL interaction with Tn antigens, and possibly STn, is also known to partially abrogate Th1 cell responses by promoting IL-17 and IL-10 expression, reducing effector T cell proliferation, and inducing T cell apoptosis. Siglecs are sialic acid-binding immunoglobulin-type lectins expressed by immune cells which selectively recognize tumour cell sialic acids, including STn and ST antigens. By interacting with tumour associated sialoglycans, siglecs modulate tolerogenic functions in DCs, preventing expansion of effector CD4 + and CD8 + T cells and increasing Treg cell numbers. In particular, STn and ST antigens seem to be potent agonists of inhibitory siglecs, as macrophage Siglec-15 and -9, respectively, inducing tumour-associated macrophage (TAM)-like phenotypes and permitting immune scape
Cancer associated glycosylation
Aberrant glycosylation is a prominent feature of advanced stage solid tumours [14, 18,19,20]. These changes are mainly driven by altered expression/activity of glycosyltransferases and glycosidases [21], mislocalization of glycosyltransferases throughout the protein secretory pathways [22], epigenetic silencing of molecular chaperones, as COSMC [23], and variations in the bioavailability of sugar donors [21]. Several tumour microenvironmental factors have been suggested to be upstream of glycan-associated phenotypic changes, including variations in oxygen [11], nutrients [24], and inflammatory cytokines levels [25]. The underlying structural alterations contribute to all accepted cancer hallmarks [9], highlighting the critical functional implications of glycans in cancer cells behaviour and fate [11, 26, 27], with considerable negative impact in clinical outcomes [10, 28, 29]. The most well-known glycome alterations result from premature stop in protein O-GalNAc glycosylation (occurring in Ser/Thr residues of proteins), yielding immature glycans, such as the Tn, sialyl-Tn (STn) and T antigens, rather than more extended and complex glycosidic chains. The functional and clinical implication of such glycoepitopes has been extensively revised by us and other authors [9, 14, 29, 30]. In brief, the simplest O-GalNAc glycan, the Tn antigen, is expressed by 10–90% of human epithelial tumours, including bladder, ovary, lung, breast, cervix, colon, stomach, and prostate cancer, while being mostly absent form healthy tissues [31,32,33]. In cancer, Tn antigen expression has been correlated to poor prognosis and metastasis [34]; however the molecular mechanisms through which it modulates tumour progression remains poorly understood and appears to be dependent on the microenvironment, requiring comprehensive systems biology approaches for elucidation. Notwithstanding, Tn antigen expression seems to significantly impact the tumour-associated immune cell repertoire, which translates in reduced levels of cytotoxic CD8 + T cells and enhanced accumulation of myeloid-derived suppressor cells [35, 36]. Furthermore, Tn antigen is involved in the adhesion of tumour cells to the endothelium via a mechanism recruiting Galectin-3 and MUC-1, which is one of the first steps of metastasis formation [34]. This processes ultimately accelerate tumour growth in vivo, providing pathways for therapeutical intervention. Based on these observations, several therapeutic approaches against Tn antigen are in pre-clinical development or in clinical trials, including glycovaccines [37,38,39], cellular immunotherapies, as chimeric antigen receptor T cells (CAR-T) [40, 41], and monoclonal antibodies [38, 42]. The sialylated Tn antigen, or sialyl-Tn (STn), is also frequently expressed in tumour tissues, including breast, gastrointestinal, lung, prostate, oesophagus, and bladder cancers [43]. It modulates key aspects of cancer progression, being an independent predictor of poor prognosis [14, 29, 43] frequently found in glycoproteins intimately linked to cancer aggressiveness [10, 27]. Numerous reports describe STn as a driver of decreased cell adhesion [11, 44], increased migratory and invasive [11, 45] capacity of tumour cells, and decreased chemotherapy-induced apoptosis [46]. Its presence in highly undifferentiated circulating tumour cells and metastases also suggests a potential role in metastasis [28]. Finally, STn is a potent inducer of cancer tolerogenecity in immune cells, impairing both dendritic cell maturation and anti-tumour T cell responses [47], while inducing multipotent growth factor production by tumour associated macrophages [48]. Given such tremendous body of evidence supporting STn as a key modulator of tumour growth and progression, several STn targeted therapeutics are in pre-clinical stage of development, including humanized antibodies [49,50,51,52], glycovaccines [53, 54] and glycomimetics for immunization [55, 56]. In turn, the core 1 O-glycan T antigen is also expressed by 90% of human carcinomas, including precancerous lesions [57] and disseminated tumour cells [58]. In cancer, T antigen serves as a specific ligand of galectin-3, providing a pivotal interaction for metastatic cell adhesion to endothelial cells in several cancer models [59,60,61,62]. Moreover, T antigen overexpression was associated with increased invasive capacity and stem-like properties of cancer cells [63], as well as with cancer cells metastatic potential irrespectively of galectin-3 assessment [64]. In line with this, targeted pre-clinical therapeutics against T antigen include clinical immunolocalization antibodies [65], humanized antibodies for immunotherapy [66], and cancer vaccines [67, 68]. Sialylated T antigens (ST) are also a common post-translational modification of membrane glycoproteins. Despite its non-cancer specific nature, it has been found overexpressed in numerous cancers in comparison to healthy tissues [69]. Namely, overexpression of ST3Gal-I, responsible for ST biosynthesis, is a biomarker of metastatic potential [70], while ST antigen was demonstrated to impact on tumour cell proliferation, migration, and apoptosis. Of note, ST antigen has also been identified in several key cancer glycoproteins, some of which displaying prognostic potential [27, 69].
Polylactosamine structures present on tri and tetra-antennary N-glycans are also commonly found in cancer, especially as result of β1–6 Mannose branching, whose synthesis is under control of GlcNAcT-V [71]. This originates functionally diverse N-glycosidic chains in relevant cancer-associated proteins that are profoundly involved in cancer growth, invasion, metastasis, and immune recognition [68, 71,72,73], while being correlated with poor prognosis [74]. The negative functional impact of this class of N-glycans has been thereby targeted in pre-clinical studies exploiting glycosyltransferases silencing towards potentiated immune recognition [73] and cell based immunotherapies [75]. Polylactosamines are further modified by the addition of different carbohydrate antigens such as Lewis antigens and their sialylated counterparts. Accordingly, another typical glycome-related alteration in cancer cells is the overexpression of sialyl lewis antigens, namely sialyl lewis A (SLeA) and sialyl lewis X (SLeX). These are terminal epitopes of extended N- and O-glycosidic chains of glycoproteins and glycolipids. Due to their affinity for selectins expressed on endothelial cells (E-selectin), platelets (P-selectin) and leukocytes (L-selectin), these glycans are key mediators of cancer cells recruitment to activated endothelial cells in primary tumour sites, intravasation into the blood stream and homing to distant locations. Moreover, their circulating levels are frequently used in the clinics as disease monitoring tools [14, 76, 77]. Besides adhesion, SLe antigens also influence angiogenesis [78,79,80] and immune recognition of cancer cells [81, 82]. For instance, SLe-P-selectin interactions contribute to protect circulating tumour cells against shear forces and immune recognition by forming shielding platelet cloaks [83]. This interaction also contributes to tumour cell adhesion to metastasis sites, as recently reviewed [83]. Given this rational, several therapeutic tools have been developed against such structures, including therapeutic antibodies [84, 85], gene therapy [86], and CAR T-cell immunotherapy [87].
These findings reinforce the remarkable contribution of the glycome to disease progression and dissemination in the context of well-known cancer hallmarks [9]. Furthermore, several recent studies have put glycans in the spotlight as novel checkpoints in compromised immune responses that should be accounted for in terms of patient stratification and therapeutic development. This rationale has also served as basis for glycoproteomics studies, supported by dedicated and progressively more standardized mass spectrometry workflows [88], which have now started to unveil the nature of the proteins carrying many of the described post-translational modifications. Blueprints regarding the existence of cancer unique and context-dependent glycoproteomes have already been provided [13, 27, 89], paving the way towards precise cancer targeting and personalization. These advances will be of major importance for decoding the crosstalk between cancer cells and the immune system towards clinical intervention. However, it becomes pressing that this effort is followed by the election and refinement of cellular and animal models for translational clinical research, crucial for both functional studies and development of glycan-based immunotherapies. Therefore, subsequent sections will explore and summarize glycan mediated immune modulation of innate and adaptative responses known to date, while shedding light on the potentialities and hurdles associated to the most used disease models.
Immune modulation promoted by cancer-associated glycans
The influence of cell surface glycosylation on innate and adaptive immune responses has been consistently explored in the context of inflammation, autoimmunity and cancer, opening an avenue for the new field of glycoimmunology [15, 90]. More recently, several tumour associated glycans have been suggested as novel immune checkpoint intermediates [15], as their negative impact on immune cell differentiation and antigen-presenting cell functions [91] starts to be unveiled. Briefly, cancer cell associated glycans modulate immune cell responses by interacting with various classes of secreted or membrane-bound glycan-binding receptors, as galectins, most C-type lectins and siglecs [92,93,94] (Fig. 2). The carbohydrate specificity of these receptors and their immune cell subpopulation biodistribution is well documented and summarized elsewhere [15, 90]. Herein, we will focus on the glycan binding receptor-glycan interactions in the control of innate and adaptive immune responses. Briefly, galectins (Gal) are soluble lectins that are secreted via non-classical pathways to interact with cell surface glycoproteins and extracellular matrix (ECM) ligands [95]. Galectins typically bind terminal β-galactoside-containing carbohydrates with high specificity [96], including the short core 1 O-glycan T antigen and polylactosamine structures present on tri and tetra-antennary N-glycans (Fig. 2). Notwithstanding, the glycan-binding specificity of each galectin is governed by sulfation, sialylation, fucosylation, repeating N-acetyllactosamine units and β1,6 GlcNAc branching of the glycan moiety, differing between individual family members [97]. Found in a wide variety of immune cells, galectins as Gal-1 can induce apoptosis of activated human T cells [98], while antagonizing Type 1 T helper (Th1) and Type 17 T helper (Th17) cells survival [99]. Promotion of T-cell receptor (TCR)-induced Type 2 T helper (Th2) cytokine production [100] towards immunosuppressive microenvironments has also been described. Galectin ligands, as the T cell immunoglobulin and mucin domain 3 (TIM3), can be found in exhausted T cells concomitantly expressing PD-1, negatively regulating T cell exhaustion and immunotherapy efficacy in a glycan dependent manner [101, 102] (Fig. 2). On the same note, the TIM3-Gal-9 pathway has consistently been implicated in the inhibitory action of CD4 + CD25 + regulatory T cells (Tregs), reinforcing its immunosuppressive role [103]. Furthermore, galectins can endow dendritic cells (DCs) with a tolerogenic phenotype capable of promoting interleukin 10 (IL-10)-mediated T cell tolerance [104]. On the same note, galectins expressed by innate immune cells, such as polymorphonuclear neutrophils [105], macrophages [106], NK cells [107, 108], and B cells [109, 110] can also interact with glycoconjugates at the surface of tumour cells and ultimately generate a local microenvironment that is permissive to tumour growth.
In turn, the C-type lectin-like domain superfamily is a calcium depend receptor class that often comprises carbohydrate recognition domains [111]. Depending on glycan recognition and calcium coordination [112], C-type lectin receptors (CLRs) are classified as group I mannose-specific (with affinity to terminal mannose and fucose) or group II galactose-specific (with affinity to terminal galactose and N-acetylgalactosamine) [94]. In particular, group II receptors are important pattern recognition receptors expressed by APCs, such as DCs and macrophages [94]. These include, the myeloid DC receptor DC-SIGN (CD209) that avidly recognizes fucosylated Lewis antigen structures, and MGL (CLEC10A, CD301), with affinity for terminal GalNAc (Tn antigen), among others [94] (Fig. 2). As previously mentioned, malignant transformation is often accompanied by overexpression of these receptors’ ligands, tailoring suppressive immune responses on tumour sites. Particularly, DC-SIGN engagement in the tumour microenvironment by fucose-based pathogen-associated molecular patterns (PAMPs) enhances interleukin -10 and -27 (IL-10, IL-27), as well as Th2-attracting chemokine expression, shifting Thelper polarization from Th1 to Th2 [113,114,115]. Reduction of T cell proliferation [116] has also been described upon DC-SIGN engagement. In turn, MGL interaction with shortened glycan moieties, as Tn antigens (GalNAc), strongly affects B and T cell immunogenicity, partially abrogating Th1 cell responses, promoting IL-17 and IL-10 expression [117, 118], reducing effector T cell proliferation and inducing T cell apoptosis [119] (Fig. 2). Interestingly, MGL has been demonstrated to bind STn antigen with a similar dead adhesion force than Tn [120, 121]. However, the impact of such interaction in immune responses is still poorly explored. Notwithstanding, it may, in part, contribute to STn associated immune tolerance [47, 122].
Siglecs are sialic acid-binding immunoglobulin-type lectins widely expressed in immune cells. These receptors selectively recognize α[2, 3], α[2,3,4,5,6] or α[2,3,4,5,6,7,8] sialic acids on the cell surface of cancer cells [123], including the broadly studied STn, ST antigens and sialylated N-glycans (Fig. 2). These Ig-type lectins are subdivided into CD33-related siglecs (-3, 5, 6, 7, 8, 9, 10, 11 and 14), showing high degree of sequence homology, and Siglec-1 (Sialoadhesin, CD169), Siglec-2 (CD22), Siglec-4 (MAG) and Siglec-15, which show low sequence similarity [123]. Siglec-2 and most CD33-related siglecs (except Siglec-14 and Siglec-15) have one or more cytosolic immunoreceptor tyrosine-based inhibitory motifs (ITIMs), actively supressing signals coming from receptors associated with immunoreceptor tyrosine-based activation motifs (ITAMs) [124]. In this context, siglecs are able to modulate tolerogenic functions in DCs, preventing expansion of effector CD4( +) and CD8( +) T cells and increasing Treg-cell numbers [125]. In particular, STn and ST antigens seem to be potent agonists of inhibitory siglecs, as macrophage Siglec-15 [48] and -9 [126], respectively, inducing tumour-associated macrophage (TAM)-like phenotypes and permitting immune scape (Fig. 2). Altogether, these glycan binding receptors interactions with tumour cell aberrant glycosylation constitute a novel class of immune checkpoints, offering potential for clinical intervention, including APC targeted glycovaccines envisaging T cell anti-tumour responses [127, 128].
Interestingly, immune cells also change their glycophenotype upon maturation and activation [90, 129]. For instance, mature DCs upregulate glycosyltransferases involved in the expression of LacNAc, core 1 and sialylated structures, while downregulating genes involved in the synthesis of core 2 O-glycans [130]. On the same note, murine activated CD4 and CD8 T cells experience a significant reduction in sialylated biantennary N-glycans with terminal NeuGcα[2,3,4,5,6]Gal, while overexpressing Galα[1,2,3]Gal terminal sequences in response to glycosyltraferase ST6Gal I downregulation and α1-3GalT overexpression [131]. Moreover, TCR expression of β1-6GlcNAc N-glycans promotes Th2 cells polarization over Th1 responses [132]. Since tumour cells also express GBRs with immunomodulatory potential that can bind these glycoepitopes, these findings are expected to also have implications in tumour cell recognition and tumour clearance responses [133]. In a very clear example, the glycophenotypes of T helper cells can directly modulate their susceptibility to Gal-1, which is frequently secreted by tumour cells [133, 134]. Namely, while Th1- and Th17-differentiated cells display a repertoire of cell surface glycans critical for Gal-1-induced cell death, Th2 cells present a different set of cell surface sialoglycoproteins that protects them from Gal-1 binding, thereby preventing inflammatory responses [135] (Fig. 2). In line with these observations, blockage of tumour Gal-1 interactions with activated T cells has been demonstrated to potentiate effective immune responses against tumour cells [134].
In summary, the crosstalk between GBRs and glycans on tumour and immune cells frequently leads to poor tumour specific responses. This is mainly driven by immune cell tolerogenecity, increased immune cell death and arrested proliferation, as well as promotion of non-effector T cell phenotypes as Tregs and Thelper cells. These findings highlight mechanistic interactions that are suitable for therapeutic intervention, which could be of major importance for immune checkpoint inhibitors non-responders that are currently faced with very limited therapeutic options.
Models for cancer glycobiology and glycoimmunology
Models for glycobiology research
Cancer glycobiology and glycoimmunology have extensively explored cancer cell models has primary research tools, building on its easy manipulation under co-culture with immune cells in controlled in vitro settings. Notably, most relevant cancer-associated glycome alterations, e.g. the overexpression of immature O-GalNAc glycans, are the result of poorly understood and, therefore, difficult to reproduce microenvironment cues. Advances in gene editing technologies have enabled the precise and stable modulation of glycan biosynthesis pathways in mammalian cells, inducing homogeneous cancer glycophenotypes for exploring glycosylation biological functions [136, 137]. Currently, mammalian cells glycoengineering relies mostly on nuclease-based gene-editing methods, including CRISPR-Cas9 technology [136, 138, 139]. The generalization of these approaches has been greatly facilitated by the pioneer work at Wandall’s lab [136, 138, 140, 141], which delivered a validated gRNA library for CRISPR/Cas9 targeting of the human glycosyltransferase genome [142]. This technology has been used to generate a wide number of different cancer cells expressing immature O-GalNAc glycosylation [143,144,145] which, to great extent, support our current knowledge on their role in heath and disease [146]. However, cellular glycoengineering for modulation of protein N- and O-glycosylation is being explored beyond its functional dimension, constituting an important tool for producing therapeutic glycoproteins [147, 148] (Fig. 3). Cell factory technologies have been used for glycovaccines production [149], surpassing low yields, scalability challenges and costs of glycopeptides synthesis using chemical approaches [128, 150] (Fig. 3). Currently, several pre-clinical glycovaccines for cancer and infectious diseases rely on these models as primary sources of antigens [149, 151, 152].

Current research tools and models for glycobiology studies. Cancer cell models have been extensively explored as primary research tools. Advances in gene editing technologies have enabled the precise and stable modulation of glycan biosynthesis pathways in mammalian cells, allowing the interrogation of glycosylation functional impact and the production of therapeutic glycoproteins. Cell factory technologies have been used for glycovaccines production, surpassing glycoproteins scalability challenges. The focus is now on progressing beyond 2D genetic glycoengineering towards 3D organotypic models, including high-throughput 3D spheroid cultures employing cell lines without or without genetic manipulation. Notwithstanding, the costly and time-consuming nature of these models remain major challenges. The introduction of computationally supported methods for glycoengineering constitutes the next logical cornerstone to address these limitations. Several transgenic mouse models reflecting the systemic impact of glycan deficiencies have also been developed and extensively employed as pre-clinical tools. Humanized models as patient-derived cancer xenografts (PDX) may add additional translational value. However, the loss of the immune system component poses as a major limitation. Alternatively, the adoption of syngeneic animal models developed from allografted glycoengineered cell lines, which can retain intact immune systems and provide the necessary means to address impact of glycosylation, could pose a valuable tool
The focus is now on progressing beyond 2D genetic glycoengineering towards 3D organotypic models [153] (Fig. 3). As a result, the goal of establishing tissue libraries with altered glycosylation for therapy testing as well as cell differentiation, morphogenesis, cell–cell interactions, and cell–matrix interactions studies is currently at close range. However, the time consuming and costly nature of such models associated with the need for cell immortalization for long term studies, poses as a major operational limitation. Interestingly, high-throughput 3D spheroid cultures employing cell lines without genetic manipulation have been found to better mimic in vivo tumour differentiation and glycosylation features when compared to their 2D counterparts [154] (Fig. 3). However, a comprehensive characterization of the glycome, ideally supported by single-cell resolution, should be undertaken to fully disclose the potential of these approaches. Nevertheless, the new generation of 3D cellular models has already demonstrated potential for glycosylation studies [155] and may be of great interest for glycan-based therapy development. Notwithstanding, the costly and time-consuming nature of these models associated with challenges reproducing the complex and heterogeneous nature of the human glycome remain major challenges. The introduction of computationally supported methods for glycoengineering may constitute the next logical cornerstone to address these limitations. Genetic algorithms, as the Markov model, may be used to identify cell lines and clones requiring minimal intervention to achieve the desired glycophenotypes (Fig. 3). These strategies may also help defining operational parameters (e.g. amount of starting material; microenvironmental cues) towards the best possible predictable outcome [156, 157]. Ultimately, these approaches also offer a flexible and user-friendly platform to optimize mammalian cell factories production, optimizing biopharmaceutical production efforts.
Less explored, but far more informative approaches imply the use of cancer animal models derived from different approaches, e.g. glycogene glycoengineering, grafting of cell models and tumours, or induction of lesions by either chemical of physical methods. Several transgenic mouse models reflecting the systemic impact of glycan deficiencies have also been developed and extensively employed as pre-clinical tools. The most frequently used mouse models display induced deficiency of core 1-derived O-glycans [158, 159], branched N-glycans [160], O-GlcNAcylation [161], and multiple enzymes determining congenital disorders of glycosylation (CDGs) [162]. These models have been used for investigating CDGs progression and associated therapeutic options [163], susceptibility to intestinal inflammation [160], gut microbial ecology [164], host physiology [165], T cell development [166, 167] and cancer progression [168] (Fig. 3). However, the net abrogation of a glycophenotype in an animal may not fully reflect the context-dependent nature exhibited by subpopulations of cells in tumours. As such, humanized models may add additional translational value to current models. For instance, patient-derived cancer xenografts (PDX) were demonstrated to preserve many of the original tumour molecular features, including glycosylation signatures, providing a suitable model for therapeutics testing [169], while being a relevant tool for soluble glycobiomarker discovery [170] (Fig. 3). However, the loss of the immune system component poses as a major limitation. Alternatively, the adoption of syngeneic animal models developed from allografted glycoengineered cell lines, which retain intact immune systems and provide the necessary means to address impact of glycosylation [171, 172], could pose a valuable tool. The induction of tumours in animal models also poses as a valuable alternative for glycobiology, as we have previously demonstrated building on chemically induced bladder tumours [173]. However, these solutions are yet to be generalized and require careful consideration of intrinsic molecular and immune system differences between humans and animal models in experimental design.
Models for cancer glycoimmunology
The current cancer glycoimmunology state-of-the-art has mainly focused on the immune modulation promoted by cancer-associated glycans, emphasizing the study of glycans as novel immune checkpoints. This quest has been mostly backed by glycobiology models, including molecular simulation methods of docking and protein interactions [96], where glycan and glycan binding receptor interactions probabilities are modulated. Furthermore, glycoengineered cell lines co-culture with immune cells, immunohistochemistry validations in tumour samples, and mouse models have also aided the discovery of immunosuppressive interactions [174]. Notwithstanding, in vitro methods frequently lack the necessary tumour microenvironment context, and mouse models have been filling this gap. However, fundamental physiological differences between mouse and human immune receptors and glycosyltransferases expression could be hampering clinical translation and ICI-based immunotherapy development. For instance, mice only have 9 functional homologues of the 15 human siglecs, with mouse siglec 3 also lacking the ITIM motif found in humans [175]. Furthermore, most research on DC-SIGN has relied on in vitro studies, since there are eight genetic homologs of human DC-SIGN in mice with no clear DC-SIGN ortholog [176]. Accordingly, the physiological role of this receptor in vivo has been hard to address. Also, the phylogeny of glycosyltransferase genes influences the endogenous expression of several ligands of these receptors in mice, with some enzymes being absent from the mouse genome as FUT3, FUT5 and FUT6 that support the expression of lewis antigens in humans [177]. Bioimaging, enzymatic synthesis of relevant glycopeptides [117] and glycoengineered cell lines protein production have been supporting studies of glycopeptide antigenicity and analysis of antibody responses in immunocompetent mice, as well as the induction of specific T cell hybridomas to investigate APC functions [16].
Currently, the field is progressing to organotypic and humanized scale. An elegant example employs patient derived cancer organoids (PDOs) expressing cancer specific Carcino-Embryonic Antigen (CEA) glycoconjugates, which allowed the refinement of immunotherapies based on bispecific antibodies targeting CEA on cancer cells and CD3 on T cells [178]. Glycoengineered transgenic mice have also been extensively used in this context, despite posing limitations regarding incapacity to mimic the context dependent nature of glycosylation. For instance, a human ex vivo pre-clinical study using mucosal T lymphocytes from patients with ulcerative colitis (UC) has highlighted a possible targeted-specific immunomodulatory role for N-acetylglucosamine (GlcNAc) metabolic supplementation [179]. Using transgenic mouse models missing branched N-glycosylation potential (Mgat5 − / −), Dias et al. has demonstrated that GlcNAc supplementation has the potential to enhance T cell receptor branched N-glycosylation, thereby controlling T cell-mediated immune responses at the intestinal mucosa and reducing UC severity and progression [179]. Ultimately, this study proposes a simple rescue therapy approach for patients with UC, with potential to avoid unnecessary toxic effects of mainstay treatments. Using human cancer cell lines and the above mentioned (Mgat5 − / −) mouse model, the immunoediting capacity of complex branched N-glycans and the impact of the removal of such epitopes in cancer cell immunogenicity was also investigated [73]. In brief, the removal of branched N-glycans exposed immunogenic mannose antennas that potentiated immune recognition by DC-SIGN-expressing immune cells, resulting in an effective antitumour immune response [73]. These findings highlight the therapeutic efficacy of glycosylation modulation as a strategy to potentiate immune recognition. On the same note, the (Mgat5 − / −) mouse model was also explored in the context of autoimmunity and the negative impact of branched N-glycans in T-cell activation was reenforced [180]. However, a second mechanism of immunoediting was suggested. Namely, deficiency in Mgat5 was determined to lower T-cell activation thresholds by directly enhancing TCR clustering and potentiating T cell activation [180].
Overall, glycoimmunology is still at its infancy in terms of exploring the full potential of humanized models of the immune system, relying heavily on 2D in vitro co-culture of immune and cancer cells and glycoengineered mice. Of note, transgenic mice presenting glycosylation deficiencies display persistent comorbidities. For instance, Mgat5-deficient mice consistently show kidney autoimmune disease, enhanced delayed-type hypersensitivity, increased susceptibility to experimental autoimmune encephalomyelitis, and reduced depression‐like phenotype [180, 181], suggesting not only autoimmune disease predisposition but also expected behavioural changes. This constitutes a major limitation that should be accounted for facing clinical translation. Given these insights, progress from co-culture systems towards 3D humanized and organotypic settings is expected to pose as a mainstay approach in the future. However, the field could also extensively benefit from the outstanding advances in immune system models and machine learning algorithms already explored by immunologists.
Emerging models for translational immunology: opportunities for glycoimmunology
Murine models of the human immune system
There is growing awareness regarding the need to progress towards human-based systems able to support the translation of findings from fundamental immunology studies to the clinics. We will first focus on novel murine models of the human immune system, given their preferred use in onco-immunology (Fig. 4). In fact, most of our basic understanding on the subject has been clarified by these models, given their manageability and readily available reagents and tools. However, the incapacity of mice to mirror human immune responses poses as a great limitation for clinical translational studies [182,183,184,185,186,187]. This is mainly due to interspecies differences between cytokines and cytokine receptors, incapacity to reflect immune senescence, lack of genetic heterogeneity, inability of many common pathogens to develop in mice, among others [188,189,190,191,192]. Notwithstanding, humanized murine models have been a step forward in mimicking human immune traits. Some of the more broadly used humanized models include the Hu-PBL (peripheral blood leukocyte)-SCID model, SRC (SCID repopulating cell)-Hu model, and the Thy/HSC model, each having its own advantages and disadvantages reviewed elsewhere [193] (Fig. 4). In these examples, immunodeficient mice can be engrafted with functional human cells and tissues, including orthotopic human tumours and human hematopoietic stem cells (HSC) that develop into functional human immune systems [194,195,196]. These models rely on tissue engineering approaches to create a humanized microenvironment, as opposed to simply engrafting cells [197]. Nevertheless, there are substantial costs underlying the development of humanized models, a restrictive supply of animals, and ethical concerns related with animal model research. Moreover, several organs are unable to interact with the grafted human leukocytes due to species specificity, potentiating conflicting results. Furthermore, transplantation is performed using identical batches of stem cells in highly inbred animals, which fails to reflect the necessary human heterogeneity [198]. To address these hurdles, co-engraftment of several human tissues with human hematopoietic stem cells has been proposed to improve on humanization of immune responses [199]. However, these tools will never be fully humanized, urging the constant development and refining of new models.

Emerging models for translational immunology. Humanized murine models have been a step forward in mimicking human immune traits. Some of the more broadly used humanized models include the Hu-PBL (peripheral blood leukocyte)-SCID model, SRC (SCID repopulating cell)-Hu model, and the Thy/HSC model. In these examples, immunodeficient mice can be engrafted with functional human cells and tissues, including orthotopic human tumours and human hematopoietic stem cells (HSC) that develop into functional human immune systems. Mass and fluorescence cytometry of peripheral blood samples have been key technologies explored for multiparametric human immunophenotyping in oncology settings, revealing interindividual variations and tissue specialization of immune subsets. In parallel, high-throughput sequencing has enabled the unlocking of the extraordinary heterogeneity of the immune repertoire from a single sample of blood or tissues. In this context, sophisticated machine learning algorithms are being used to integrate large sequencing datasets. Regarding in vitro models of the human immune system, some challenging approaches have been proposed, including human modular immune in vitro constructs (MIMIC™) and organoid-like cultures. The MIMIC system technology is a 3D structure composed by a peripheral tissue equivalent (PTE) and a lymphoid tissue equivalent (LTE), allowing multivariate studies. Immuno-engineered organoids have also been used as an alternative to overcome the limitations associated with animal models, 2D systems, and previously existing 3D models
Biopsy-based models
Addressing these difficulties, several studies sought to investigate immune cells from peripheral blood and tissue biopsies of tonsils and lymph nodes (LN) of human patients. Mass and fluorescence cytometry of peripheral blood samples have been key technologies explored for multiparametric human immunophenotyping in oncology settings, revealing interindividual variations and tissue specialization of immune subsets [200,201,202,203] (Fig. 4). In parallel, high-throughput sequencing has enabled the unlocking of the extraordinary heterogeneity of the immune repertoire from a single sample of blood or tissue [204]. In this context, sophisticated machine learning algorithms are being developed to integrate large sequencing datasets, infer on T-cell specificity and B cell receptors heterogeneity as well as vaccine-specific antibody production, which could provide key knowledge regarding adaptive immune responses [204,205,206,207,208]. On the same note, repertoire analysis of antigen specific TCRs has been suggested as an important readout to assess vaccination’s ability to generate memory cells and the necessary clonal expansion for immune protection, reinforcing the immediate clinical translation of peripheral blood analysis [209]. Furthermore, RNA-seq, ChIP-seq, and ATAC-seq have been providing key information on immune genes regulome, offering insights on epigenomic state and functional aspects of human immune cell types [210,211,212,213,214]. Also, single-cell omics approaches, including highly multiplexed simultaneous detection of RNAs, proteins, miRNAs, and gene-specific mRNA, has been elucidating the identity and functional state of critical immune subsets, while opening new avenues for the characterization of cellular metabolism [215,216,217,218]. More importantly, the comprehensive integration of multi omics information has brought us one step closer to identifying global immune signatures associated with clinical outcome, even when patients’ cohorts are small and heterogeneous [219] (Fig. 4). Ultimately, this high-resolution systems-level immune monitoring offers a roadmap for the development and evaluation of immunotherapies.
In turn, LN biopsies have also been employed in human immune repertoire studies as well as vaccine and immune checkpoint inhibitors (ICI) response research. Biopsies of vaccine-draining lymph nodes have allowed the in vitro generation of the same antibodies observed in the blood of vaccinated patients [220], which has been facilitating investigation on human antibody activity and therapeutic antibodies development. Furthermore, high-dimensional mass cytometry of sentinel LNs biopsy samples combined with T cell receptor repertoire sequencing has allowed the interrogation of T follicular helper (Tfh) cells in primary human LNs [221], which is expected to aid in the therapeutic manipulation of cellular functional capacity to improve antibody responses to vaccination [222]. Most of these cellular processes are practically indetectable in peripheral blood, as such, gathering information from fine needle aspirates of human LN, including tumour draining LN, has emerged as a valuable tool for human immunization studies [220, 223,224,225]. In parallel, regional LN immune profiles have been suggested as predictive biomarkers for immune checkpoint inhibitor response, challenging the view that ICI activity occurs primarily at the tumour site [226] and focusing attention on tumour draining lymph nodes [227].
In vitro and lab-on-a-chip models for validation studies
Even though blood and liquid biopsy-based assays have been extensively informative, validation studies have been mainly undertaken building on studies in vitro. Co-cultures between cancer cells with immune cells isolated from blood or immune tissue fractions (human tonsils and lymph nodes) sorted cells are by far the preferred strategies [228,229,230] (Fig. 4). In this context, antigen presenting cells, as DCs and macrophages, interactions with tumour cells and downstream immune effectors, as T cells, can be closely monitored. By exploring lymphocyte–APC contacts, many signalling molecules, including receptors, enzymes, adaptors, and secondary messengers, have been identified and support the current state of the art [231,232,233,234]. Furthermore, imaging of cell–cell contacts has led to an appreciation of the remarkable supramolecular changes, covering molecular structure, conformational changes, mobility of bound species, intracellular activity of proteins, intracellular localization, aggregation state of receptors, mobility at the plasma membrane, and cell morphology [235]. Even though intravital imaging of murine models has been extensively used towards this end, the physiological context may have to be compromised to obtain higher resolution. Given these obstacles, a combination of low-resolution in vivo imaging and high-resolution in vitro experiments could allow advances in the study of APCs engagement, lymphocyte activation and tumour cell elimination or evasion processes.
Regarding in vitro models of the human immune system, some challenging approaches have been proposed, including human modular immune in vitro constructs (MIMIC™) and organoid-like cultures. The MIMIC system technology is a 3D structure composed by a peripheral tissue equivalent (PTE) and a lymphoid tissue equivalent (LTE) [236] (Fig. 4). The PTE is comprised by a monolayer of human umbilical vein endothelial cells (HUVEC) cultured above a 3D extra-cellular collagen matrix, upon which patient peripheral blood mononuclear cells (PBMCs) are seeded. This module can simulate innate immune responses and gives rise to four main mononuclear populations. Namely, immature CD14 + DCs precursors, immature CD14– DCs, and mature DCs. A fourth population is more macrophage-like and is retained in the matrix. The LTE is essentially an artificial lymph node, simulating the adaptive immune response. Dendritic cells, follicular dendritic cells, T- and B-cells are applied in sequential order to mimic the immune response expected in vivo. As such, DC–T-cell interactions, antigen–B-cell interactions, T-cell and B-cell interactions, Th1 or Th2 polarisation bias, antigen-specific antibody production, and cytotoxic T-cells activity, can all be assessed from this in vitro module [226, 237]. In turn, immuno-engineered organoids have been used as an alternative to overcome the limitations associated with animal models, 2D systems, and previously existing 3D models (Fig. 4). Namely, organoids simulate the physiological organ structure, can include important stromal components, can be specifically genetically engineered, do not encompass ethical conflicts, and allow different degrees of malignancy to be cultured. Moreover, patient derived organoids allow tumour and immune cells to be cocultured, which is critical for immuno-oncology studies and immunotherapeutic screening [238,239,240]. For instance, immuno-engineered organoids have been used to accelerate the induction of a germinal centre (GC)-like phenotype in B cells to support a controllable immunoglobulin class-switching reaction [241], surpassing all previously described 2D ex vivo systems for GC-like phenotypes. Furthermore, murine- and patient-derived organotypic tumour spheroids retaining autologous lymphoid and myeloid cell populations have been used to anticipate response to cancer immunotherapy, including antibody-based immunotherapy and ICI, oncolytic virus therapy, and adoptive cell transfer therapy [242, 243]. Accordingly, complex immune–organoid cultures have been providing real time tools for pre-clinical testing of therapeutic combinations and facilitating precision immuno-oncology efforts [244, 245]. Interestingly, immuno-engineered organoids research has been hand-to-hand with dynamic microfluidics and organ-on-a-chip solutions for oncoimmunology [246, 247]. Accordingly, these microfluidics multicellular systems have been employed in the study of soluble immune checkpoint inhibitors [248, 249], sequencing of antibodies secreted during innate immune responses [250], the crosstalk between cancer and immune cells [251], immune cell migration [252, 253], the cytotoxic activity of TCR-engineered T cells [247], and the impact of anti-tumour chemotherapy/ radiation and combination therapies on tumour and immune cells [254,255,256]. Altogether, an encouraging body of literature has demonstrated that OncoImmuno multicellular chips provide a flexible and valuable alternative to animal models, mostly due to their affordability and accuracy in drug testing screenings while recapitulating the relationships between the immune system and cancerous tissues [246].
High-throughput single cell technologies for translational immunology: opportunities for glycoimmunology
There has been an increasing awareness that solid tumours are not discrete bodies but an interconnected network of cell populations, which can drive tumour progression and therapy resistance in a clonal dependent manner [257, 258]. However, traditional molecular profiling techniques have focused on the tumour bulk disregarding discrete contributions from relevant subpopulations. In fact, cancer cells in a tumour display distinct cellular morphologies, gene expression patterns, proliferation rates, metastatic potential and, sensitivity to treatment, which poses a major obstacle to understanding and treating cancer [257, 258]. Accordingly, the pursue for single-cell resolution tools has opened new avenues for single-cell characterization, including genomics [258], transcriptomics [259], epigenomics [260], proteomics [261] and metabolomics [262] sequencing. This has been mostly aiding therapy resistant clones’ phenotyping and immunotyping of immune cells.
Currently, several high-throughput and high resolution techniques are becoming standard practice in deciphering tumour heterogeneity in a multiplexed manner. For instance, cytometry by time of flight (CyTOF) allows non-targeted and comprehensive cellular characterization, offering a broader vision on cell signalling pathways at various cell stages [263]. Cell staining in CyTOF protocols are fairly similar to standard flow cytometry procedures, taking advantage of antibodies labelled with stable metal isotopes and culminating in charged plasma vaporization of cells prior to TOF unit analysis [264]. CyTOF offers several advantages to traditional flow cytometry, including highly multiplexed target detection, allowing the scale up to more than 40 intracellular and extracellular targets in a single run [265], while surpassing fluorescence compensation issues. However, mass compensation and oxidative processes need to be considered, similarly to other widespread mass spectrometry techniques [266]. On the same note, full spectrum flow cytometry (SFC) allows the same multiplexed analysis at a single cell level than CyTOF, with similar data readouts [267]. However, SFC offers the possibility of using all conventional flow cytometry reagents, from FACS buffers to fluorophore-tagged antibodies, further driving the adoption of SFC as a mainstay of single-cell phenotyping. Briefly, full spectrum flow cytometers detect the entire spectral signature of a fluorophore through various tandem detectors, allowing the multiplexed acquisition of more than 30 cellular targets at a time, even when using highly overlapping fluorophores [268]. Moreover, full spectrum flow cytometers are able to use autofluorescence values to improve data quality, surpassing conventional flow cytometers both in resolution as well as in compensation issues [269]. Notwithstanding, the search for automated and extremely multiplexed approaches has led to the development of cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq), which brings together surface protein phenotyping and single-cell RNA sequencing (scRNA-seq) [270]. This technique takes advantage of antibodies conjugated to oligonucleotides, namely a unique antibody oligonucleotide barcode that identifies the antibody/marker and a terminal poly(A) sequence. When cells stained by these antibodies are lysed, the marker poly(A) tail is captured by hybridizing with beads covered in poly(dT) oligonucleotides, following scRNA-seq [271]. Since CITE-seq relies on oligonucleotides instead of fluorophores and has sequencing as a readout, there is no known limit for the number of simultaneous markers that can be analysed on a single run, allowing a scale up to more than 200 multiplexed indicators. Accordingly, CITE-seq allows measurement of a potentially unlimited number of protein markers in parallel to transcriptomes, enables sample multiplexing, robust multiplet detection and super-loading of scRNA-seq platforms. Furthermore, some high-throughput technologies have been developed to address specific questions. For instance, the need to pair B cell receptor sequence to antigen specificity at a single-cell level is currently addressed through LIBRA-seq [272]. LIBRA-seq (linking B cell receptor to antigen specificity through sequencing) enables high-throughput mapping of paired heavy- and light-chain BCR sequences to their cognate antigen specificities. B cells are mixed with a panel of oligonucleotides conjugated to recombinant antigens so that both the antigen and BCR sequence are recovered during paired-chain BCR sequencing experiments and bioinformatically mapped to single cells [272]. Ultimately, LIBRA-seq enables mapping of monoclonal antibody sequences, theoretically unlimited in number, and facilitates rapid identification of cross-reactive antibodies, being an integral tool for antibody discovery as well as vaccine and immunotherapy development.
In addition to the above-mentioned technologies, advances in multilayer microscopy have been aiding the accurate assessment of tissue architectures and microenvironments, while allowing single cell resolution. In this context, co-detection by indexing (CODEX) multiplexed immunofluorescence was created to address the need for multi-marker spatial analysis. This need derives from the notion that not only the composition of tissues but also the spatial proximity of cell subtypes affects biological outcomes. CODEX can be applied to human and mouse fresh frozen or FFPE tissues, which are incubated with a cocktail of oligonucleotide tagged primary antibodies without fluorophores. During the automated processing of tissues in the CODEX platform, dye-labelled oligonucleotides (CODEX reporters) bind to their complementary antibodies and give out fluorescence signals, following the successive removal and addition of new reporters for up to 16 cycles. This can generate spatial information for up to 46 protein biomarkers [273]. As such, CODEX technology automates whole tissue imaging, allowing complex single cell phenotyping and discovery of novel phenotypes, while disclosing spatial interactions within tissues [274]. In a similar way, spatial transcriptomics has also been extensively pursued through NanoString technology, enabling researchers to locate transcripts down to the subcellular level and providing an unbiased map of RNA targets throughout tissue sections. Herein, target RNA is directly tagged with specific capture and reporter probes, creating unique target-probe complexes for each target. These complexes are automatically immobilized in an imaging surface and the sample is scanned by an automated fluorescence microscope, enabling digital quantitation of hundreds of unique targets in a single reaction.
Adding to the advances brought by CyTOF, mass spectrometry (MS) has reached the technological readiness to enable more detailed unbiased proteome characterization close to single cell resolution [275,276,277,278,279]. Opportunities, limitations, and key enabling milestones towards clinical translation regarding this technology have been recently revised [280,281,282,283] and therefore, will not be discussed in detail in this review. However, improvements in sample processing, separation and MS instrumentation have made possible to quantify over 1000 proteins from individual mammalian cells, which until very recently could only be achieved with an input of thousands of cells. The challenge is now set on expanding the dynamic range of identified proteins, also enabling the identification of low abundance proteins and post-translational modifications, including glycosylation. In parallel, there have been significant advances in terms of spatially resolved proteomics at single cell resolution [284,285,286,287]. Nevertheless, single cell glycomics and glycoproteomics remains a challenging enterprise that has just began to be tackled. Namely, imaging mass spectrometry is currently being employed for glycomic characterization of tumour sections, allowing identification of glycome signatures according to tissue distribution and underlying associations with relevant histopathological features [288,289,290]. Furthermore, innovative workflows such as the SUrface-protein Glycan and RNA-seq (SUGAR-seq) allowed the combo analysis of glycans, extracellular epitopes, and the transcriptome at the single-cell level, progressing over existing technologies in terms of mapping cellular transcriptional and phenotypic features [291]. Integrated SUGAR-seq and glycoproteome analysis has led to the characterization of tumour-infiltrating T cells glycophenotypes, mirroring their epigenetic and functional state and holding potential for advances in cancer glycoimmunology. Very recently, we have also materialized the concept of glycoproteogenomics, which builds on comprehensive integration of RNAseq-data to customized databases used in protein annotation [89, 292]. This has allowed deeper access to the glycoproteome, now requiring translation to single cell analysis. Collectively, the combination of genomics, transcriptomics, and proteomics in the context of single cell analysis will provide tremendous advances in terms of understanding how gene products interact to produce a cellular phenotype. The integration of glycomics and glycoproteomics will also be key to identify protein functional targets. The exploitation of these concepts and technologies in glycoimmunology will allow to gain knowledge on the role played by glycans in the crosstalk between cancer cells and the immune system and the relevance of the immune cells glycome in health and disease.
Machine learning tools for immunological modelling
Finally, all the disruptive technologies mentioned above provide high resolution molecular and cellular insights, which require potent machine learning tools to integrate information and accelerate the translation of these insights into therapies. Accordingly, several user-friendly computational tools that facilitate immunological modelling have been developed, ultimately providing an additional quality-control mechanism that improves the rigor and reproducibility of immune studies [293]. For instance, deep-learning Generative Adversarial Networks (GAN) can predict how an immune cell migratory path will evolve based on a time lapse microscopy video file [294]. In oncoimmunology settings, GAN provides a meaningful estimation of the probability for each immune cell to physically interact with cancer cells in their vicinity. Moreover, this long-term prediction of cell trajectories may reduce the spatial–temporal burden of video sequences storage, solving a significant bottleneck of the experimental pipeline. On the same note, Complex Pathway Simulator (COPASI) [295], ENteric Immunity Simulator (ENISI) MultiScale Modeling (MSM) [296], Simmune [297], Monte Carlo Cell (MCell) [298], RuleBender [299], and Virtual Cell (VCell) [300] constitute several other comprehensive platforms for modelling that are used by immunologists. These tools provide cellular, spatial, and time-dependent simulations of immune processes [293], all of which holding promise for the systematic interrogation of complex pathways. Furthermore, bioinformatics tools can also be used to integrate publicly available curated data, taking advantage of pre-existing information to advance beyond the current state-of-the-art [301,302,303,304,305,306,307] in a way that often improves reproducibility by integrating data from multiple studies [308].
In summary, as we progress toward increasingly sophisticated humanized models of the immune system, several relevant stromal and microenvironmental cues driving malignancy have been identified. The spotlight is currently on the tremendous potential brought by multicomponent 3D models for personalised oncoimmunology. These approaches supported by multi-omics characterization at single cell resolution [309, 310] are positively contributing to better understand the role played by the cancer microenvironment in disease [311, 312]. The exploitation of bioinformatics and computational tools backed by artificial intelligence has been decisive towards this objective, setting a roadmap ready to be translated to cancer glycobiology.
Concluding remarks
Immune checkpoint inhibitors against PD-1/PD-L1, or CTLA-4 have revolutionized the field of immunotherapy. However, immune suppression is supported by multifactorial intricate molecular networks, building on interplays between cancer and different types of immune cells, which we are only now beginning to understand. Furthermore, there has been an increasing awareness that discrete tumour cell populations can drive tumour progression and therapy resistance in a clonal dependent manner. Thereby, focusing on relevant subpopulations at the single cell level constitutes the next cornerstone in understanding and treating cancer. Multi-omics analysis at single cell resolution has greatly contributed to this objective, being decisive to bring the cancer glycome into the spotlight. As a result, there is growing awareness that glycans classically known to mediate cancer invasion and metastasis are also playing significant suppressive roles of both innate and adaptative immunity. As we continue to unveil the cancer glycome, it becomes pressing to invest on exploring the mechanistic aspects of glycan-GBR interactions towards the rational development of novel interventions. Notably, this knowledge has been successfully prototyped into blocking antibodies, glycoengineered vaccines and cellular immunotherapies, as CAR-T cells, with promising results in pre-clinical settings. However, few therapeutics ever get to surpass early clinical trial phases. We believe that the missing link between fundamental glycoimmunology knowledge and translational medicine may lay in the experimental models adopted by most studies. Currently, the field heavily relies on short term in vitro co-cultures, generally involving glycoengineered cell models and immune cells extracted from peripheral blood of healthy donors and cancer patients. To less extent, transgenic mouse models resulting from glycogenes editing have also been explored (Fig. 4). Even though these tools have successfully supported breakthrough advances in the field, they often lack the necessary humanized features and the underlying tumour microenvironment. Accordingly, the generalization of 3D systems, namely organoids, organs-on-a-chip, and humanized animal models, that are currently used in infectious disease, autoimmunity, oncology, and immunotherapy studies, will be of key importance for the glycobiology field. Finally, it is pressing to invest on the comprehensive molecular characterizations of cancer and immune cells by high-throughput genomics, transcriptomics, glycomics and (glyco)proteomics at single cell resolution. These new approaches will be key for dissecting tumour heterogeneity, including the precise characterization of immune cells constituting the microenvironment. This will translate into a deeper understanding about the role played by glycans and glycoconjugates in immune responses, cancer progression and dissemination, facilitating the identification of targetable molecules and the rationale design of novel and more effective therapies. The generalization of artificial intelligence approaches will also be decisive to integrate massive omics data, including glycomics and glycoproteomics, into comprehensive models (Fig. 5). We anticipate that such roadmap could speed the arrival of much needed novel therapeutic options, constituting valuable alternatives to current immunotherapies.

Roadmap towards novel glycan-based immunotherapy. Humanized in vitro and in vivo models are expected to hasten the translation of fundamental glycoimmunology research into glycan-based tools with theragnostic potential. Currently, the field heavily relies on short term in vitro co-cultures involving glycoengineered cell models and blood derived immune cells. Transgenic mouse models resulting from glycogenes editing have also been explored. The generalization of 3D systems, namely organoids, organs-on-a-chip, and humanized animal models will be of key importance to capture tumour heterogeneity and immune system contributions to tumour progression. The generalization of artificial intelligence approaches will also be decisive to integrate massive omics data into comprehensive models. We anticipate that such roadmap could speed the arrival of much needed novel therapeutic options, constituting valuable alternatives to current immunotherapies.
Availability of data and materials
Data sharing is not applicable to this article. All data analysed during the current study is available in the PubMed repository, https://pubmed.ncbi.nlm.nih.gov.
Abbreviations
- APC:
-
Antigen presenting cells
- ATAC-seq:
-
Assay for transposase-accessible chromatin using sequencing
- BCR B:
-
Cell receptor
- CAR-T:
-
Chimeric antigen receptor T cell
- CDGs:
-
Congenital disorders of glycosylation
- ChIP-seq:
-
Chromatin immunoprecipitation sequencing
- CITE-seq:
-
Indexing of transcriptomes and epitopes by sequencing
- CLR:
-
C-type lectin receptor
- CODEX:
-
Co-detection by indexing
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- CTLA-4:
-
Cytotoxic T lymphocyte-associated antigen 4
- CyTOF:
-
Cytometry by time of flight
- ECM:
-
Extracellular matrix
- Gal:
-
Galectin
- GBR:
-
Glycan-binding receptors
- HSC:
-
Human hematopoietic stem cells
- hu-PBL:
-
Human peripheral blood leukocytes model
- ICI:
-
Immune checkpoint inhibitor
- IL:
-
Interleukin
- ITAM:
-
Immunoreceptor tyrosine-based activation motif
- ITIM:
-
Immunoreceptor tyrosine-based inhibitory motif
- LIBRA-seq:
-
Linking B cell receptor to antigen specificity through sequencing
- MHC:
-
Major histocompatibility complex
- MS:
-
Mass spectrometry
- PBMC:
-
Peripheral blood mononuclear cells
- PD-L1:
-
Programmed cell death ligand 1
- PDO:
-
Patient derived cancer organoids
- PDX:
-
Patient-derived cancer xenograft
- RNA-seq:
-
RNA-sequencing
- SCID:
-
Severe combined immune deficiency
- SFC:
-
Full spectrum flow cytometry
- SRC:
-
Scid-repopulating cell
- TAM:
-
Tumour-associated macrophage
- TCR:
-
T-cell receptor
- TIM3:
-
T cell immunoglobulin and mucin domain 3
- TME:
-
Tumour microenvironment
References
Xin YuJ, Hubbard-Lucey VM, Tang J. Immuno-oncology drug development goes global. Nat Rev Drug Discov. 2019;18(12):899–900.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509–20.
Osipov A, Lim SJ, Popovic A, Azad NS, Laheru DA, Zheng L, et al. Tumor Mutational Burden, Toxicity, and Response of Immune Checkpoint Inhibitors Targeting PD(L)1, CTLA-4, and Combination: A Meta-regression Analysis. Clin Cancer Res. 2020;26(18):4842–51.
Haslam A, Prasad V. Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Network Open. 2019;2(5):e192535-e.
Wang DY, Salem JE, Cohen JV, Chandra S, Menzer C, Ye F, et al. Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol. 2018;4(12):1721–8.
Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol. 2020;20(1):25–39.
Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Frontiers in Immunology. 2019;10:168.
Labani-Motlagh A, Ashja-Mahdavi M, Loskog A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front Immunol. 2020;11:940.
Peixoto A, Relvas-Santos M, Azevedo R, Santos LL, Ferreira JA. Protein Glycosylation and Tumor Microenvironment Alterations Driving Cancer Hallmarks. Front Oncol. 2019;9:380.
Cotton S, Azevedo R, Gaiteiro C, Ferreira D, Lima L, Peixoto A, et al. Targeted O-glycoproteomics explored increased sialylation and identified MUC16 as a poor prognosis biomarker in advanced-stage bladder tumours. Mol Oncol. 2017;11(8):895–912.
Peixoto A, Fernandes E, Gaiteiro C, Lima L, Azevedo R, Soares J, et al. Hypoxia enhances the malignant nature of bladder cancer cells and concomitantly antagonizes protein O-glycosylation extension. Oncotarget. 2016;7(39):63138–57.
Cotton S, Ferreira D, Soares J, Peixoto A, Relvas-Santos M, Azevedo R, et al. Target Score—A Proteomics Data Selection Tool Applied to Esophageal Cancer Identifies GLUT1-Sialyl Tn Glycoforms as Biomarkers of Cancer Aggressiveness. Int J Mol Sci. 2021;22(4):1664.
Fernandes E, Freitas R, Ferreira D, Soares J, Azevedo R, Gaiteiro C, et al. Nucleolin-Sle A Glycoforms as E-Selectin Ligands and Potentially Targetable Biomarkers at the Cell Surface of Gastric Cancer Cells. Cancers (Basel). 2020;12(4):861.
Fernandes E, Sores J, Cotton S, Peixoto A, Ferreira D, Freitas R, et al. Esophageal, gastric and colorectal cancers: Looking beyond classical serological biomarkers towards glycoproteomics-assisted precision oncology. Theranostics. 2020;10(11):4903–28.
RodrÍguez E, Schetters STT, van Kooyk Y. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat Rev Immunol. 2018;18(3):204–11.
Wolfert MA, Boons G-J. Adaptive immune activation: glycosylation does matter. Nat Chem Biol. 2013;9(12):776–84.
Duan S, Paulson JC. Siglecs as Immune Cell Checkpoints in Disease. Annu Rev Immunol. 2020;38(1):365–95.
Peixoto A, Relvas-Santos M, Azevedo R, Santos LL, Ferreira JA. Protein Glycosylation and Tumor Microenvironment Alterations Driving Cancer Hallmarks. Frontiers in oncology. 2019;9:380.
Ferreira JA, Magalhães A, Gomes J, Peixoto A, Gaiteiro C, Fernandes E, et al. Protein glycosylation in gastric and colorectal cancers: Toward cancer detection and targeted therapeutics. Cancer Lett. 2017;387:32–45.
Ferreira JA, Peixoto A, Neves M, Gaiteiro C, Reis CA, Assaraf YG, et al. Mechanisms of cisplatin resistance and targeting of cancer stem cells: Adding glycosylation to the equation. Drug Resist Updat. 2016;24:34–54.
Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015;15(9):540–55.
Gill DJ, Tham KM, Chia J, Wang SC, Steentoft C, Clausen H, et al. Initiation of GalNAc-type O-glycosylation in the endoplasmic reticulum promotes cancer cell invasiveness. Proc Natl Acad Sci U S A. 2013;110(34):E3152–61.
Ju T, Cummings RD. Chaperone mutation in Tn syndrome. Nature. 2005;437(7063):1252.
Peixoto A, Freitas R, Ferreira D, Relvas-Santos M, Paulo P, Cardoso M, et al. Metabolomics, Transcriptomics and Functional Glycomics Reveals Bladder Cancer Cells Plasticity and Enhanced Aggressiveness Facing Hypoxia and Glucose Deprivation. bioRxiv. 2021:2021.02.14.431133. https://doi.org/10.1101/2021.02.14.431133.
Dewald JH, Colomb F, Bobowski-Gerard M, Groux-Degroote S, Delannoy P. Role of Cytokine-Induced Glycosylation Changes in Regulating Cell Interactions and Cell Signaling in Inflammatory Diseases and Cancer. Cells. 2016;5(4):43.
Cotton S, Ferreira D, Soares J, Peixoto A, Relvas-Santos M, Azevedo R, et al. Target Score-A Proteomics Data Selection Tool Applied to Esophageal Cancer Identifies GLUT1-Sialyl Tn Glycoforms as Biomarkers of Cancer Aggressiveness. Int J Mol Sci. 2021;22(4):1664.
Peixoto A, Ferreira D, Azevedo R, Freitas R, Fernandes E, Relvas-Santos M, et al. Glycoproteomics identifies HOMER3 as a potentially targetable biomarker triggered by hypoxia and glucose deprivation in bladder cancer. J Exp Clin Cancer Res. 2021;40(1):191.
Lima L, Neves M, Oliveira MI, Dieguez L, Freitas R, Azevedo R, et al. Sialyl-Tn identifies muscle-invasive bladder cancer basal and luminal subtypes facing decreased survival, being expressed by circulating tumor cells and metastases. Urol Oncol. 2017;35(12):675.e1-675.e8.
Azevedo R, Peixoto A, Gaiteiro C, Fernandes E, Neves M, Lima L, et al. Over forty years of bladder cancer glycobiology: Where do glycans stand facing precision oncology? Oncotarget. 2017;8(53):91734–64.
Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015;15(9):540–55.
Chia J, Goh G, Bard F. Short O-GalNAc glycans: regulation and role in tumor development and clinical perspectives. Biochim Biophys Acta. 2016;1860(8):1623–39.
Zlocowski N, Grupe V, Garay YC, Nores GA, Lardone RD, Irazoqui FJ. Purified human anti-Tn and anti-T antibodies specifically recognize carcinoma tissues. Sci Rep. 2019;9(1):8097.
Ju T, Aryal RP, Kudelka MR, Wang Y, Cummings RD. The Cosmc connection to the Tn antigen in cancer. Cancer Biomark. 2014;14(1):63–81.
Kölbl AC, Jeschke U, Friese K, Andergassen U. The role of TF- and Tn-antigens in breast cancer metastasis. Histol Histopathol. 2016;31(6):613–21.
Cornelissen LAM, Blanas A, Zaal A, van der Horst JC, Kruijssen LJW, O’Toole T, et al. Tn Antigen Expression Contributes to an Immune Suppressive Microenvironment and Drives Tumor Growth in Colorectal Cancer. Frontiers in Oncology. 2020;10:1622.
Matsumoto T, Okayama H, Nakajima S, Saito K, Nakano H, Endo E, et al. Tn Antigen Expression Defines an Immune Cold Subset of Mismatch-Repair Deficient Colorectal Cancer. Int J Mol Sci. 2020;21(23):9081.
Lo-Man R, Vichier-Guerre S, Perraut R, Dériaud E, Huteau V, BenMohamed L, et al. A fully synthetic therapeutic vaccine candidate targeting carcinoma-associated Tn carbohydrate antigen induces tumor-specific antibodies in nonhuman primates. Cancer Res. 2004;64(14):4987–94.
Freire T, Zhang X, Dériaud E, Ganneau C, Vichier-Guerre S, Azria E, et al. Glycosidic Tn-based vaccines targeting dermal dendritic cells favor germinal center B-cell development and potent antibody response in the absence of adjuvant. Blood. 2010;116(18):3526–36.
Scheid E, Major P, Bergeron A, Finn OJ, Salter RD, Eady R, et al. Tn-MUC1 DC Vaccination of Rhesus Macaques and a Phase I/II Trial in Patients with Nonmetastatic Castrate-Resistant Prostate Cancer. Cancer Immunol Res. 2016;4(10):881–92.
Zhai X, You F, Xiang S, Jiang L, Chen D, Li Y, et al. MUC1-Tn-targeting chimeric antigen receptor-modified Vγ9Vδ2 T cells with enhanced antigen-specific anti-tumor activity. Am J Cancer Res. 2021;11(1):79–91.
Sharma P, Marada V, Cai Q, Kizerwetter M, He Y, Wolf SP, et al. Structure-guided engineering of the affinity and specificity of CARs against Tn-glycopeptides. Proc Natl Acad Sci U S A. 2020;117(26):15148–59.
Welinder C, Baldetorp B, Borrebaeck C, Fredlund BM, Jansson B. A new murine IgG1 anti-Tn monoclonal antibody with in vivo anti-tumor activity. Glycobiology. 2011;21(8):1097–107.
Julien S, Videira PA, Delannoy P. Sialyl-tn in cancer: (how) did we miss the target? Biomolecules. 2012;2(4):435–66.
Munkley J, Oltean S, Vodák D, Wilson BT, Livermore KE, Zhou Y, et al. The androgen receptor controls expression of the cancer-associated sTn antigen and cell adhesion through induction of ST6GalNAc1 in prostate cancer. Oncotarget. 2015;6(33):34358–74.
Ferreira JA, Videira PA, Lima L, Pereira S, Silva M, Carrascal M, et al. Overexpression of tumour-associated carbohydrate antigen sialyl-Tn in advanced bladder tumours. Mol Oncol. 2013;7(3):719–31.
Santos SN, Junqueira MS, Francisco G, Vilanova M, Magalhães A, Dias Baruffi M, et al. O-glycan sialylation alters galectin-3 subcellular localization and decreases chemotherapy sensitivity in gastric cancer. Oncotarget. 2016;7(50):83570–87.
Carrascal MA, Severino PF, Guadalupe Cabral M, Silva M, Ferreira JA, Calais F, et al. Sialyl Tn-expressing bladder cancer cells induce a tolerogenic phenotype in innate and adaptive immune cells. Mol Oncol. 2014;8(3):753–65.
Takamiya R, Ohtsubo K, Takamatsu S, Taniguchi N, Angata T. The interaction between Siglec-15 and tumor-associated sialyl-Tn antigen enhances TGF-β secretion from monocytes/macrophages through the DAP12-Syk pathway. Glycobiology. 2013;23(2):178–87.
Eavarone DA, Al-Alem L, Lugovskoy A, Prendergast JM, Nazer RI, Stein JN, et al. Humanized anti-Sialyl-Tn antibodies for the treatment of ovarian carcinoma. PloS One. 2018;13(7):e0201314-e.
Prendergast JM, Galvao da Silva AP, Eavarone DA, Ghaderi D, Zhang M, Brady D, et al. Novel anti-Sialyl-Tn monoclonal antibodies and antibody-drug conjugates demonstrate tumor specificity and anti-tumor activity. Mabs. 2017;9(4):615–27.
Loureiro LR, Sousa DP, Ferreira D, Chai W, Lima L, Pereira C, et al. Novel monoclonal antibody L2A5 specifically targeting sialyl-Tn and short glycans terminated by alpha-2–6 sialic acids. Sci Rep. 2018;8(1):12196.
Trabbic KR, Kleski KA, Shi M, Bourgault JP, Prendergast JM, Dransfield DT, et al. Production of a mouse monoclonal IgM antibody that targets the carbohydrate Thomsen-nouveau cancer antigen resulting in in vivo and in vitro tumor killing. Cancer Immunol Immunother. 2018;67(9):1437–47.
Shi M, Kleski KA, Trabbic KR, Bourgault JP, Andreana PR. Sialyl-Tn Polysaccharide A1 as an Entirely Carbohydrate Immunogen: Synthesis and Immunological Evaluation. J Am Chem Soc. 2016;138(43):14264–72.
Zhou Z, Mondal M, Liao G, Guo Z. Synthesis and evaluation of monophosphoryl lipid A derivatives as fully synthetic self-adjuvanting glycoconjugate cancer vaccine carriers. Org Biomol Chem. 2014;12(20):3238–45.
Song C, Zheng XJ, Liu CC, Zhou Y, Ye XS. A cancer vaccine based on fluorine-modified sialyl-Tn induces robust immune responses in a murine model. Oncotarget. 2017;8(29):47330–43.
Huo CX, Zheng XJ, Xiao A, Liu CC, Sun S, Lv Z, et al. Synthetic and immunological studies of N-acyl modified S-linked STn derivatives as anticancer vaccine candidates. Org Biomol Chem. 2015;13(12):3677–90.
Fu C, Zhao H, Wang Y, Cai H, Xiao Y, Zeng Y, et al. Tumor-associated antigens: Tn antigen, sTn antigen, and T antigen. Hla. 2016;88(6):275–86.
Andergassen U, Zebisch M, Kölbl AC, Schindlbeck C, Ilmer M, Hutter S, et al. Detection of breast cancer cells in blood samples by immunostaining of the Thomsen-Friedenreich antigen. Future Oncol. 2013;9(5):747–52.
Glinsky VV, Huflejt ME, Glinsky GV, Deutscher SL, Quinn TP. Effects of Thomsen-Friedenreich antigen-specific peptide P-30 on beta-galactoside-mediated homotypic aggregation and adhesion to the endothelium of MDA-MB-435 human breast carcinoma cells. Cancer Res. 2000;60(10):2584–8.
Khaldoyanidi SK, Glinsky VV, Sikora L, Glinskii AB, Mossine VV, Quinn TP, et al. MDA-MB-435 human breast carcinoma cell homo- and heterotypic adhesion under flow conditions is mediated in part by Thomsen-Friedenreich antigen-galectin-3 interactions. J Biol Chem. 2003;278(6):4127–34.
Yu LG, Andrews N, Zhao Q, McKean D, Williams JF, Connor LJ, et al. Galectin-3 interaction with Thomsen-Friedenreich disaccharide on cancer-associated MUC1 causes increased cancer cell endothelial adhesion. J Biol Chem. 2007;282(1):773–81.
Newton-Northup JR, Dickerson MT, Ma L, Besch-Williford CL, Deutscher SL. Inhibition of metastatic tumor formation in vivo by a bacteriophage display-derived galectin-3 targeting peptide. Clin Exp Metastasis. 2013;30(2):119–32.
Hung JS, Huang J, Lin YC, Huang MJ, Lee PH, Lai HS, et al. C1GALT1 overexpression promotes the invasive behavior of colon cancer cells through modifying O-glycosylation of FGFR2. Oncotarget. 2014;5(8):2096–106.
Yu L-G. The oncofetal Thomsen-Friedenreich carbohydrate antigen in cancer progression. Glycoconj J. 2007;24(8):411–20.
Chaturvedi R, Heimburg J, Yan J, Koury S, Sajjad M, Abdel-Nabi HH, et al. Tumor immunolocalization using 124 I-iodine-labeled JAA-F11 antibody to Thomsen-Friedenreich alpha-linked antigen. Appl Radiat Isot. 2008;66(3):278–87.
Ferguson K, Yadav A, Morey S, Abdullah J, Hrysenko G, Eng JY, et al. Preclinical studies with JAA-F11 anti-Thomsen-Friedenreich monoclonal antibody for human breast cancer. Future Oncol. 2014;10(3):385–99.
Ulsemer P, Henderson G, Toutounian K, Löffler A, Schmidt J, Karsten U, et al. Specific humoral immune response to the Thomsen-Friedenreich tumor antigen (CD176) in mice after vaccination with the commensal bacterium Bacteroides ovatus D-6. Cancer Immunol Immunother. 2013;62(5):875–87.
Mereiter S, Balmaña M, Campos D, Gomes J, Reis CA. Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell. 2019;36(1):6–16.
Cotton S, Azevedo R, Gaiteiro C, Ferreira D, Lima L, Peixoto A, et al. Targeted O-glycoproteomics explored increased sialylation and identified MUC16 as a poor prognosis biomarker in advanced-stage bladder tumours. Mol Oncol. 2017;11(8):895–912.
Bai R, Luan X, Zhang Y, Robbe-Masselot C, Brockhausen I, Gao Y. The expression and functional analysis of the sialyl-T antigen in prostate cancer. Glycoconj J. 2020;37(4):423–33.
Chen Q, Tan Z, Guan F, Ren Y. The Essential Functions and Detection of Bisecting GlcNAc in Cell Biology. Front Chem. 2020;8:511.
Kizuka Y, Taniguchi N. Enzymes for N-Glycan Branching and Their Genetic and Nongenetic Regulation in Cancer. Biomolecules. 2016;6(2):25.
Silva MC, Fernandes Â, Oliveira M, Resende C, Correia A, de-Freitas-Junior JC, et al. Glycans as Immune Checkpoints: Removal of Branched N-glycans Enhances Immune Recognition Preventing Cancer Progression. Cancer Immunol Res. 2020;8(11):1407–25.
Nagae M, Kizuka Y, Mihara E, Kitago Y, Hanashima S, Ito Y, et al. Structure and mechanism of cancer-associated N-acetylglucosaminyltransferase-V. Nat Commun. 2018;9(1):3380.
Greco B, Paolella K, Camisa B, Malacarne V, Falcone L, Graziani A, et al. Combining De-Glycosylating Agents with CAR-T Cells for Targeting Solid Tumors and Reducing Toxicity. Blood. 2018;132(Supplement 1):4544.
Trinchera M, Aronica A, Dall’Olio F. Selectin Ligands Sialyl-Lewis a and Sialyl-Lewis x in Gastrointestinal Cancers. Biology (Basel). 2017;6(1):16.
Blanas A, Sahasrabudhe NM, Rodríguez E, van Kooyk Y, van Vliet SJ. Fucosylated Antigens in Cancer: An Alliance toward Tumor Progression, Metastasis, and Resistance to Chemotherapy. Frontiers in Oncology. 2018;8:39.
Terraneo L, Avagliano L, Caretti A, Bianciardi P, Tosi D, Bulfamante GP, et al. Expression of carbohydrate-antigen sialyl-Lewis a on colon cancer cells promotes xenograft growth and angiogenesis in nude mice. Int J Biochem Cell Biol. 2013;45(12):2796–800.
Tei K, Kawakami-Kimura N, Taguchi O, Kumamoto K, Higashiyama S, Taniguchi N, et al. Roles of cell adhesion molecules in tumor angiogenesis induced by cotransplantation of cancer and endothelial cells to nude rats. Cancer Res. 2002;62(21):6289–96.
Mathieu S, Gerolami R, Luis J, Carmona S, Kol O, Crescence L, et al. Introducing alpha(1,2)-linked fucose into hepatocarcinoma cells inhibits vasculogenesis and tumor growth. Int J Cancer. 2007;121(8):1680–9.
Ohyama C, Tsuboi S, Fukuda M. Dual roles of sialyl Lewis X oligosaccharides in tumor metastasis and rejection by natural killer cells. Embo j. 1999;18(6):1516–25.
Ohyama C, Kanto S, Kato K, Nakano O, Arai Y, Kato T, et al. Natural killer cells attack tumor cells expressing high levels of sialyl Lewis x oligosaccharides. Proc Natl Acad Sci U S A. 2002;99(21):13789–94.
Fabricius H-Å, Starzonek S, Lange T. The Role of Platelet Cell Surface P-Selectin for the Direct Platelet-Tumor Cell Contact During Metastasis Formation in Human Tumors. Front Oncol. 2021;11:642761.
Weitzenfeld P, Bournazos S, Ravetch JV. Antibodies targeting sialyl Lewis A mediate tumor clearance through distinct effector pathways. J Clin Investig. 2019;129(9):3952–62.
Tivadar ST, McIntosh RS, Chua JX, Moss R, Parsons T, Zaitoun AM, et al. Monoclonal Antibody Targeting Sialyl-di-Lewis(a)-Containing Internalizing and Noninternalizing Glycoproteins with Cancer Immunotherapy Development Potential. Mol Cancer Ther. 2020;19(3):790–801.
Kawamura YI, Adachi Y, Curiel DT, Kawashima R, Kannagi R, Nishimoto N, et al. Therapeutic adenoviral gene transfer of a glycosyltransferase for prevention of peritoneal dissemination and metastasis of gastric cancer. Cancer Gene Ther. 2014;21(10):427–33.
Mondal N, Silva M, Castano AP, Maus MV, Sackstein R. Glycoengineering of chimeric antigen receptor (CAR) T-cells to enforce E-selectin binding. J Biol Chem. 2019;294(48):18465–74.
Kawahara R, Chernykh A, Alagesan K, Bern M, Cao W, Chalkley RJ, et al. Community evaluation of glycoproteomics informatics solutions reveals high-performance search strategies for serum glycopeptide analysis. Nat Methods. 2021;18(11):1304–16.
Ferreira JA, Relvas-Santos M, Peixoto A, Silva M N, Lara Santos L. Glycoproteogenomics: Setting the Course for Next-generation Cancer Neoantigen Discovery for Cancer Vaccines. Genomics Proteomics Bioinformatics. 2021;19(1):25–43.
van Kooyk Y, Rabinovich GA. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat Immunol. 2008;9(6):593–601.
van Kooyk Y, Rabinovich GA. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat Immunol. 2008;9(6):593–601.
Compagno D, Tiraboschi C, Garcia JD, Rondón Y, Corapi E, Velazquez C, et al. Galectins as Checkpoints of the Immune System in Cancers, Their Clinical Relevance, and Implication in Clinical Trials. Biomolecules. 2020;10(5):750.
Gianchecchi E, Arena A, Fierabracci A. Sialic Acid-Siglec Axis in Human Immune Regulation, Involvement in Autoimmunity and Cancer and Potential Therapeutic Treatments. Int J Mol Sci. 2021;22(11):5774.
Geijtenbeek TBH, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol. 2009;9(7):465–79.
Popa SJ, Stewart SE, Moreau K. Unconventional secretion of annexins and galectins. Semin Cell Dev Biol. 2018;83:42–50.
Modenutti CP, Capurro JIB, Di Lella S, Martí MA. The Structural Biology of Galectin-Ligand Recognition: Current Advances in Modeling Tools, Protein Engineering, and Inhibitor Design. Front Chem. 2019;7:823.
Dimitroff CJ. Galectin-Binding O-Glycosylations as Regulators of Malignancy. Can Res. 2015;75(16):3195–202.
Pace KE, Lee C, Stewart PL, Baum LG. Restricted receptor segregation into membrane microdomains occurs on human T cells during apoptosis induced by galectin-1. J Immunol. 1999;163(7):3801–11.
Toscano MA, Bianco GA, Ilarregui JM, Croci DO, Correale J, Hernandez JD, et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat Immunol. 2007;8(8):825–34.
Motran CC, Molinder KM, Liu SD, Poirier F, Miceli MC. Galectin-1 functions as a Th2 cytokine that selectively induces Th1 apoptosis and promotes Th2 function. Eur J Immunol. 2008;38(11):3015–27.
Yang R, Sun L, Li C-F, Wang Y-H, Yao J, Li H, et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat Commun. 2021;12(1):832.
Kandel S, Adhikary P, Li G, Cheng K. The TIM3/Gal9 signaling pathway: An emerging target for cancer immunotherapy. Cancer Lett. 2021;510:67–78.
Wang F, Wan L, Zhang C, Zheng X, Li J, Chen ZK. Tim-3-Galectin-9 pathway involves the suppression induced by CD4+CD25+ regulatory T cells. Immunobiology. 2009;214(5):342–9.
Ilarregui JM, Croci DO, Bianco GA, Toscano MA, Salatino M, Vermeulen ME, et al. Tolerogenic signals delivered by dendritic cells to T cells through a galectin-1-driven immunoregulatory circuit involving interleukin 27 and interleukin 10. Nat Immunol. 2009;10(9):981–91.
Robinson BS, Arthur CM, Evavold B, Roback E, Kamili NA, Stowell CS, et al. The Sweet-Side of Leukocytes: Galectins as Master Regulators of Neutrophil Function. Front Immunol. 2019;10:1762.
Barrionuevo P, Beigier-Bompadre M, Ilarregui JM, Toscano MA, Bianco GA, Isturiz MA, et al. A novel function for galectin-1 at the crossroad of innate and adaptive immunity: galectin-1 regulates monocyte/macrophage physiology through a nonapoptotic ERK-dependent pathway. J Immunol. 2007;178(1):436–45.
Golden-Mason L, McMahan RH, Strong M, Reisdorph R, Mahaffey S, Palmer BE, et al. Galectin-9 functionally impairs natural killer cells in humans and mice. J Virol. 2013;87(9):4835–45.
Tsuboi S, Sutoh M, Hatakeyama S, Hiraoka N, Habuchi T, Horikawa Y, et al. A novel strategy for evasion of NK cell immunity by tumours expressing core2 O-glycans. Embo j. 2011;30(15):3173–85.
Giovannone N, Liang J, Antonopoulos A, Geddes Sweeney J, King SL, Pochebit SM, et al. Galectin-9 suppresses B cell receptor signaling and is regulated by I-branching of N-glycans. Nat Commun. 2018;9(1):3287.
Cao A, Alluqmani N, Buhari FHM, Wasim L, Smith LK, Quaile AT, et al. Galectin-9 binds IgM-BCR to regulate B cell signaling. Nat Commun. 2018;9(1):3288.
Drouin M, Saenz J, Chiffoleau E. C-Type Lectin-Like Receptors: Head or Tail in Cell Death Immunity. Front Immuno. 2020;11:251.
Drickamer K, Taylor ME. Recent insights into structures and functions of C-type lectins in the immune system. Curr Opin Struct Biol. 2015;34:26–34.
Gringhuis SI, Kaptein TM, Wevers BA, Mesman AW, Geijtenbeek TB. Fucose-specific DC-SIGN signalling directs T helper cell type-2 responses via IKKε- and CYLD-dependent Bcl3 activation. Nat Commun. 2014;5:3898.
Gringhuis SI, Kaptein TM, Wevers BA, van der Vlist M, Klaver EJ, van Die I, et al. Fucose-based PAMPs prime dendritic cells for follicular T helper cell polarization via DC-SIGN-dependent IL-27 production. Nat Commun. 2014;5(1):5074.
Rodriguez E, Boelaars K, Brown K, Madunić K, van Ee T, Dijk F, et al. Analysis of the glyco-code in pancreatic ductal adenocarcinoma identifies glycan-mediated immune regulatory circuits. Commun Biol. 2022;5(1):41.
García-Vallejo JJ, Ilarregui JM, Kalay H, Chamorro S, Koning N, Unger WW, et al. CNS myelin induces regulatory functions of DC-SIGN–expressing, antigen-presenting cells via cognate interaction with MOG. J Exp Med. 2014;211(7):1465–83.
Freire T, Lo-Man R, Bay S, Leclerc C. Tn glycosylation of the MUC6 protein modulates its immunogenicity and promotes the induction of Th17-biased T cell responses. J Biol Chem. 2011;286(10):7797–811.
van Vliet SJ, Bay S, Vuist IM, Kalay H, García-Vallejo JJ, Leclerc C, et al. MGL signaling augments TLR2-mediated responses for enhanced IL-10 and TNF-α secretion. J Leukoc Biol. 2013;94(2):315–23.
van Vliet SJ, Gringhuis SI, Geijtenbeek TBH, van Kooyk Y. Regulation of effector T cells by antigen-presenting cells via interaction of the C-type lectin MGL with CD45. Nat Immunol. 2006;7(11):1200–8.
Beatson R, Maurstad G, Picco G, Arulappu A, Coleman J, Wandell HH, et al. The Breast Cancer-Associated Glycoforms of MUC1, MUC1-Tn and sialyl-Tn, Are Expressed in COSMC Wild-Type Cells and Bind the C-Type Lectin MGL. PLoS One. 2015;10(5):e0125994.
Mortezai N, Behnken HN, Kurze AK, Ludewig P, Buck F, Meyer B, et al. Tumor-associated Neu5Ac-Tn and Neu5Gc-Tn antigens bind to C-type lectin CLEC10A (CD301, MGL). Glycobiology. 2013;23(7):844–52.
Julien S, Videira PA, Delannoy P. Sialyl-Tn in Cancer: (How) Did We Miss the Target? Biomolecules. 2012;2(4):435–66.
Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7(4):255–66.
Ravetch JV, Lanier LL. Immune inhibitory receptors. Science. 2000;290(5489):84–9.
Perdicchio M, Ilarregui JM, Verstege MI, Cornelissen LA, Schetters ST, Engels S, et al. Sialic acid-modified antigens impose tolerance via inhibition of T-cell proliferation and de novo induction of regulatory T cells. Proc Natl Acad Sci U S A. 2016;113(12):3329–34.
Beatson R, Tajadura-Ortega V, Achkova D, Picco G, Tsourouktsoglou TD, Klausing S, et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec-9. Nat Immunol. 2016;17(11):1273–81.
Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7(10):790–802.
Freitas R, Relvas-Santos M, Azevedo R, Soares J, Fernandes E, Teixeira B, et al. Single-pot enzymatic synthesis of cancer-associated MUC16 O-glycopeptide libraries and multivalent protein glycoconjugates: a step towards cancer glycovaccines. New J Chem. 2021;45(20):9197–211.
Daniels MA, Hogquist KA, Jameson SC. Sweet “n” sour: the impact of differential glycosylation on T cell responses. Nat Immunol. 2002;3(10):903–10.
Bax M, García-Vallejo JJ, Jang-Lee J, North SJ, Gilmartin TJ, Hernández G, et al. Dendritic Cell Maturation Results in Pronounced Changes in Glycan Expression Affecting Recognition by Siglecs and Galectins. J Immunol. 2007;179(12):8216.
Comelli EM, Sutton-Smith M, Yan Q, Amado M, Panico M, Gilmartin T, et al. Activation of murine CD4+ and CD8+ T lymphocytes leads to dramatic remodeling of N-linked glycans. J Immunol. 2006;177(4):2431–40.
Morgan R, Gao G, Pawling J, Dennis JW, Demetriou M, Li B. N-acetylglucosaminyltransferase V (Mgat5)-mediated N-glycosylation negatively regulates Th1 cytokine production by T cells. J Immunol. 2004;173(12):7200–8.
Méndez-Huergo SP, Blidner AG, Rabinovich GA. Galectins: emerging regulatory checkpoints linking tumor immunity and angiogenesis. Curr Opin Immunol. 2017;45:8–15.
Rubinstein N, Alvarez M, Zwirner NW, Toscano MA, Ilarregui JM, Bravo A, et al. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection; A potential mechanism of tumor-immune privilege. Cancer Cell. 2004;5(3):241–51.
Toscano MA, Bianco GA, Ilarregui JM, Croci DO, Correale J, Hernandez JD, et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat Immunol. 2007;8(8):825–34.
Narimatsu Y, Büll C, Chen YH, Wandall HH, Yang Z, Clausen H. Genetic glycoengineering in mammalian cells. J Biol Chem. 2021;296:100448.
Steentoft C, Vakhrushev SY, Joshi HJ, Kong Y, Vester-Christensen MB, Schjoldager KT, et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. Embo j. 2013;32(10):1478–88.
Petersen BL, Möller SR, Mravec J, Jørgensen B, Christensen M, Liu Y, et al. Improved CRISPR/Cas9 gene editing by fluorescence activated cell sorting of green fluorescence protein tagged protoplasts. BMC Biotechnol. 2019;19(1):36.
Steentoft C, Vakhrushev SY, Vester-Christensen MB, Schjoldager KT, Kong Y, Bennett EP, et al. Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat Methods. 2011;8(11):977–82.
Lonowski LA, Narimatsu Y, Riaz A, Delay CE, Yang Z, Niola F, et al. Genome editing using FACS enrichment of nuclease-expressing cells and indel detection by amplicon analysis. Nat Protoc. 2017;12(3):581–603.
König S, Yang Z, Wandall HH, Mussolino C, Bennett EP. Fast and Quantitative Identification of Ex Vivo Precise Genome Targeting-Induced Indel Events by IDAA. Methods Mol Biol. 2019;1961:45–66.
Narimatsu Y, Joshi HJ, Yang Z, Gomes C, Chen YH, Lorenzetti FC, et al. A validated gRNA library for CRISPR/Cas9 targeting of the human glycosyltransferase genome. Glycobiology. 2018;28(5):295–305.
Freitas D, Campos D, Gomes J, Pinto F, Macedo JA, Matos R, et al. O-glycans truncation modulates gastric cancer cell signaling and transcription leading to a more aggressive phenotype. EBioMedicine. 2019;40:349–62.
Stolfa G, Mondal N, Zhu Y, Yu X, Buffone A, Neelamegham S. Using CRISPR-Cas9 to quantify the contributions of O-glycans, N-glycans and Glycosphingolipids to human leukocyte-endothelium adhesion. Sci Rep. 2016;6(1):30392.
Dabelsteen S, Pallesen EMH, Marinova IN, Nielsen MI, Adamopoulou M, Rømer TB, et al. Essential Functions of Glycans in Human Epithelia Dissected by a CRISPR-Cas9-Engineered Human Organotypic Skin Model. Dev Cell. 2020;54(5):669-84.e7.
Bagdonaite I, Pallesen EMH, Nielsen MI, Bennett EP, Wandall HH. Mucin-Type O-GalNAc Glycosylation in Health and Disease. Adv Exp Med Biol. 2021;1325:25–60.
Walsh G. Biopharmaceutical benchmarks 2014. Nat Biotechnol. 2014;32(10):992–1000.
Yang Z, Wang S, Halim A, Schulz MA, Frodin M, Rahman SH, et al. Engineered CHO cells for production of diverse, homogeneous glycoproteins. Nat Biotechnol. 2015;33(8):842–4.
Mathiesen CBK, Carlsson MC, Brand S, Möller SR, Idorn M, Thor Straten P, et al. Genetically engineered cell factories produce glycoengineered vaccines that target antigen-presenting cells and reduce antigen-specific T-cell reactivity. J Allergy Clin Immunol. 2018;142(6):1983–7.
Kitowski A, Corzana F, Bernardes GJL. Precise protein conjugation technology for the construction of homogenous glycovaccines. Drug Discov Today Technol. 2021;38:69–75.
Anderluh M, Berti F, Bzducha-Wróbel A, Chiodo F, Colombo C, Compostella F, et al. Recent advances on smart glycoconjugate vaccines in infections and cancer. FEBS J. 2022. https://doi.org/10.1111/febs.15909.
Samaras JJ, Mauri M, Kay EJ, Wren BW, Micheletti M. Development of an automated platform for the optimal production of glycoconjugate vaccines expressed in Escherichia coli. Microb Cell Fact. 2021;20(1):104.
Marinova IN, Wandall HH, Dabelsteen S. Protocol for CRISPR-Cas9 modification of glycosylation in 3D organotypic skin models. STAR Protoc. 2021;2(3):100668.
Balmaña M, Mereiter S, Diniz F, Feijão T, Barrias CC, Reis CA. Multicellular Human Gastric-Cancer Spheroids Mimic the Glycosylation Phenotype of Gastric Carcinomas. Molecules. 2018;23(11):2815.
Balmaña M, Diniz F, Feijão T, Barrias CC, Mereiter S, Reis CA. Analysis of the Effect of Increased α2,3-Sialylation on RTK Activation in MKN45 Gastric Cancer Spheroids Treated with Crizotinib. Int J Mol Sci. 2020;21(3):722.
Spahn PN, Hansen AH, Kol S, Voldborg BG, Lewis NE. Predictive glycoengineering of biosimilars using a Markov chain glycosylation model. Biotechnol J. 2017;12(2). https://doi.org/10.1002/biot.201600489.
Spahn PN, Hansen AH, Hansen HG, Arnsdorf J, Kildegaard HF, Lewis NE. A Markov chain model for N-linked protein glycosylation–towards a low-parameter tool for model-driven glycoengineering. Metab Eng. 2016;33:52–66.
Sommer F, Adam N, Johansson MEV, Xia L, Hansson GC, Bäckhed F. Altered mucus glycosylation in core 1 O-glycan-deficient mice affects microbiota composition and intestinal architecture. PloS One. 2014;9(1):e85254-e.
Ghosh SK, Uchida M, Yoo B, Ross AW, Gendler SJ, Gong J, et al. Targeted imaging of breast tumor progression and therapeutic response in a human uMUC-1 expressing transgenic mouse model. Int J Cancer. 2013;132(8):1860–7.
Dias AM, Correia A, Pereira MS, Almeida CR, Alves I, Pinto V, et al. Metabolic control of T cell immune response through glycans in inflammatory bowel disease. Proc Natl Acad Sci U S A. 2018;115(20):E4651–60.
Pereira MS, Alves I, Vicente M, Campar A, Silva MC, Padrão NA, et al. Glycans as Key Checkpoints of T Cell Activity and Function. Front Immunol. 2018;9:2754.
Freeze HH, Sharma V. Metabolic manipulation of glycosylation disorders in humans and animal models. Semin Cell Dev Biol. 2010;21(6):655–62.
Wang Y, Tan J, Sutton-Smith M, Ditto D, Panico M, Campbell RM, et al. Modeling human congenital disorder of glycosylation type IIa in the mouse: conservation of asparagine-linked glycan-dependent functions in mammalian physiology and insights into disease pathogenesis. Glycobiology. 2001;11(12):1051–70.
Sommer F, Adam N, Johansson ME, Xia L, Hansson GC, Bäckhed F. Altered mucus glycosylation in core 1 O-glycan-deficient mice affects microbiota composition and intestinal architecture. PLoS One. 2014;9(1):e85254.
Reily C, Stewart TJ, Renfrow MB, Novak J. Glycosylation in health and disease. Nat Rev Nephrol. 2019;15(6):346–66.
Buchlis G, Odorizzi P, Soto PC, Pearce OM, Hui DJ, Jordan MS, et al. Enhanced T cell function in a mouse model of human glycosylation. J Immunol. 2013;191(1):228–37.
Pereira MS, Alves I, Vicente M, Campar A, Silva MC, Padrão NA, et al. Glycans as Key Checkpoints of T Cell Activity and Function. Front Immunol. 2018;9:2754.
Gupta R, Leon F, Rauth S, Batra SK, Ponnusamy MP. A Systematic Review on the Implications of O-linked Glycan Branching and Truncating Enzymes on Cancer Progression and Metastasis. Cells. 2020;9(2):446.
Bernardo C, Costa C, Amaro T, Gonçalves M, Lopes P, Freitas R, et al. Patient-derived sialyl-Tn-positive invasive bladder cancer xenografts in nude mice: an exploratory model study. Anticancer Res. 2014;34(2):735–44.
Sinha A, Hussain A, Ignatchenko V, Ignatchenko A, Tang KH, Ho VWH, et al. N-Glycoproteomics of Patient-Derived Xenografts: A Strategy to Discover Tumor-Associated Proteins in High-Grade Serous Ovarian Cancer. Cell Syst. 2019;8(4):345-51.e4.
Ponath P, Menezes D, Pan C, Chen B, Oyasu M, Strachan D, et al. A Novel, Fully Human Anti–fucosyl-GM1 Antibody Demonstrates Potent <em>In Vitro</em> and <em>In Vivo</em> Antitumor Activity in Preclinical Models of Small Cell Lung Cancer. Clin Cancer Res. 2018;24(20):5178.
Li C-W, Lim S-O, Xia W, Lee H-H, Chan L-C, Kuo C-W, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7(1):12632.
Costa C, Pereira S, Lima L, Peixoto A, Fernandes E, Neves D, et al. Abnormal Protein Glycosylation and Activated PI3K/Akt/mTOR Pathway: Role in Bladder Cancer Prognosis and Targeted Therapeutics. PLoS One. 2015;10(11):e0141253.
Silva MC, Fernandes Â, Oliveira M, Resende C, Correia A, de-Freitas-Junior JC, et al. Glycans as Immune Checkpoints: Removal of Branched N-glycans Enhances Immune Recognition Preventing Cancer Progression. Cancer Immunol Res. 2020;8(11):1407.
Macauley MS, Crocker PR, Paulson JC. Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol. 2014;14(10):653–66.
Garcia-Vallejo JJ, van Kooyk Y. The physiological role of DC-SIGN: a tale of mice and men. Trends Immunol. 2013;34(10):482–6.
Costache M, Apoil PA, Cailleau A, Elmgren A, Larson G, Henry S, et al. Evolution of fucosyltransferase genes in vertebrates. J Biol Chem. 1997;272(47):29721–8.
Gonzalez-Exposito R, Semiannikova M, Griffiths B, Khan K, Barber LJ, Woolston A, et al. CEA expression heterogeneity and plasticity confer resistance to the CEA-targeting bispecific immunotherapy antibody cibisatamab (CEA-TCB) in patient-derived colorectal cancer organoids. J Immunother Cancer. 2019;7(1):101.
Dias AM, Correia A, Pereira MS, Almeida CR, Alves I, Pinto V, et al. Metabolic control of T cell immune response through glycans in inflammatory bowel disease. Proc Natl Acad Sci. 2018;115(20):E4651–60.
Demetriou M, Granovsky M, Quaggin S, Dennis JW. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature. 2001;409(6821):733–9.
Soleimani L, Roder JC, Dennis JW, Lipina T. Beta N-acetylglucosaminyltransferase V (Mgat5) deficiency reduces the depression-like phenotype in mice. Genes Brain Behav. 2008;7(3):334–43.
Mak IW, Evaniew N, Ghert M. Lost in translation: animal models and clinical trials in cancer treatment. Am J Transl Res. 2014;6(2):114–8.
Ostrand-Rosenberg S. Animal models of tumor immunity, immunotherapy and cancer vaccines. Curr Opin Immunol. 2004;16(2):143–50.
Perrin S. Preclinical research: Make mouse studies work. Nature. 2014;507(7493):423–5.
Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110(9):3507–12.
von Herrath MG, Nepom GT. Lost in translation: barriers to implementing clinical immunotherapeutics for autoimmunity. J Exp Med. 2005;202(9):1159–62.
Warren HS, Tompkins RG, Moldawer LL, Seok J, Xu W, Mindrinos MN, et al. Mice are not men. Proc Natl Acad Sci U S A. 2015;112(4):E345.
Grimm D, Staeheli P, Hufbauer M, Koerner I, Martínez-Sobrido L, Solórzano A, et al. Replication fitness determines high virulence of influenza A virus in mice carrying functional Mx1 resistance gene. Proc Natl Acad Sci U S A. 2007;104(16):6806–11.
Du Y, Deng W, Wang Z, Ning M, Zhang W, Zhou Y, et al. Differential subnetwork of chemokines/cytokines in human, mouse, and rat brain cells after oxygen-glucose deprivation. J Cereb Blood Flow Metab. 2017;37(4):1425–34.
High KP, Akbar AN, Nikolich-Zugich J. Translational research in immune senescence: assessing the relevance of current models. Semin Immunol. 2012;24(5):373–82.
Chebib J, Jackson BC, López-Cortegano E, Tautz D, Keightley PD. Inbred lab mice are not isogenic: genetic variation within inbred strains used to infer the mutation rate per nucleotide site. Heredity. 2021;126(1):107–16.
Abolins S, King EC, Lazarou L, Weldon L, Hughes L, Drescher P, et al. The comparative immunology of wild and laboratory mice, Mus musculus domesticus. Nat Commun. 2017;8(1):14811.
Tian H, Lyu Y, Yang Y-G, Hu Z. Humanized Rodent Models for Cancer Research. Front Oncol. 2020;10:1696.
Walsh NC, Kenney LL, Jangalwe S, Aryee KE, Greiner DL, Brehm MA, et al. Humanized Mouse Models of Clinical Disease. Annu Rev Pathol. 2017;12:187–215.
Lan P, Tonomura N, Shimizu A, Wang S, Yang YG. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood. 2006;108(2):487–92.
Traggiai E, Chicha L, Mazzucchelli L, Bronz L, Piffaretti JC, Lanzavecchia A, et al. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science. 2004;304(5667):104–7.
Abarrategi A, Mian SA, Passaro D, Rouault-Pierre K, Grey W, Bonnet D. Modeling the human bone marrow niche in mice: From host bone marrow engraftment to bioengineering approaches. J Exp Med. 2018;215(3):729–43.
Laudanski K, Stentz M, DiMeglio M, Furey W, Steinberg T, Patel A. Potential Pitfalls of the Humanized Mice in Modeling Sepsis. Int J Inflamm. 2018;2018:6563454.
Douam F, Ploss A. The use of humanized mice for studies of viral pathogenesis and immunity. Curr Opin Virol. 2018;29:62–71.
Finak G, Langweiler M, Jaimes M, Malek M, Taghiyar J, Korin Y, et al. Standardizing Flow Cytometry Immunophenotyping Analysis from the Human ImmunoPhenotyping Consortium. Sci Rep. 2016;6:20686.
Maecker HT, McCoy JP Jr, Amos M, Elliott J, Gaigalas A, Wang L, et al. A model for harmonizing flow cytometry in clinical trials. Nat Immunol. 2010;11(11):975–8.
Nicholas KJ, Greenplate AR, Flaherty DK, Matlock BK, Juan JS, Smith RM, et al. Multiparameter analysis of stimulated human peripheral blood mononuclear cells: A comparison of mass and fluorescence cytometry. Cytometry A. 2016;89(3):271–80.
Alcántara-Hernández M, Leylek R, Wagar LE, Engleman EG, Keler T, Marinkovich MP, et al. High-Dimensional Phenotypic Mapping of Human Dendritic Cells Reveals Interindividual Variation and Tissue Specialization. Immunity. 2017;47(6):1037-50.e6.
Heather JM, Ismail M, Oakes T, Chain B. High-throughput sequencing of the T-cell receptor repertoire: pitfalls and opportunities. Brief Bioinform. 2018;19(4):554–65.
Glanville J, Huang H, Nau A, Hatton O, Wagar LE, Rubelt F, et al. Identifying specificity groups in the T cell receptor repertoire. Nature. 2017;547(7661):94–8.
Dash P, Fiore-Gartland AJ, Hertz T, Wang GC, Sharma S, Souquette A, et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature. 2017;547(7661):89–93.
Trück J, Ramasamy MN, Galson JD, Rance R, Parkhill J, Lunter G, et al. Identification of antigen-specific B cell receptor sequences using public repertoire analysis. J Immunol. 2015;194(1):252–61.
Galson JD, Trück J, Fowler A, Clutterbuck EA, Münz M, Cerundolo V, et al. Analysis of B Cell Repertoire Dynamics Following Hepatitis B Vaccination in Humans, and Enrichment of Vaccine-specific Antibody Sequences. EBioMedicine. 2015;2(12):2070–9.
Qi Q, Cavanagh MM, Le Saux S, NamKoong H, Kim C, Turgano E, et al. Diversification of the antigen-specific T cell receptor repertoire after varicella zoster vaccination. Sci Transl Med. 2016;8(332):332ra46.
Schmidt D, Wilson MD, Spyrou C, Brown GD, Hadfield J, Odom DT. ChIP-seq: using high-throughput sequencing to discover protein-DNA interactions. Methods. 2009;48(3):240–8.
Chaussabel D, Pascual V, Banchereau J. Assessing the human immune system through blood transcriptomics. BMC Biol. 2010;8:84.
Qu K, Zaba LC, Giresi PG, Li R, Longmire M, Kim YH, et al. Individuality and variation of personal regulomes in primary human T cells. Cell Syst. 2015;1(1):51–61.
Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63.
Satpathy AT, Saligrama N, Buenrostro JD, Wei Y, Wu B, Rubin AJ, et al. Transcript-indexed ATAC-seq for precision immune profiling. Nat Med. 2018;24(5):580–90.
Porichis F, Hart MG, Griesbeck M, Everett HL, Hassan M, Baxter AE, et al. High-throughput detection of miRNAs and gene-specific mRNA at the single-cell level by flow cytometry. Nat Commun. 2014;5:5641.
Frei AP, Bava FA, Zunder ER, Hsieh EW, Chen SY, Nolan GP, et al. Highly multiplexed simultaneous detection of RNAs and proteins in single cells. Nat Methods. 2016;13(3):269–75.
Chowdhury F, Williams A, Johnson P. Validation and comparison of two multiplex technologies, Luminex and Mesoscale Discovery, for human cytokine profiling. J Immunol Methods. 2009;340(1):55–64.
Pai JA, Satpathy AT. High-throughput and single-cell T cell receptor sequencing technologies. Nat Methods. 2021;18(8):881–92.
Lakshmikanth T, Olin A, Chen Y, Mikes J, Fredlund E, Remberger M, et al. Mass Cytometry and Topological Data Analysis Reveal Immune Parameters Associated with Complications after Allogeneic Stem Cell Transplantation. Cell Rep. 2017;20(9):2238–50.
Shukla GS, Olson WC, Pero SC, Sun YJ, Carman CL, Slingluff CL Jr, et al. Vaccine-draining lymph nodes of cancer patients for generating anti-cancer antibodies. J Transl Med. 2017;15(1):180.
Wendel BS, Del Alcazar D, He C, Del Río-Estrada PM, Aiamkitsumrit B, Ablanedo-Terrazas Y, et al. The receptor repertoire and functional profile of follicular T cells in HIV-infected lymph nodes. Sci Immunol. 2018;3(22):eaan8884.
Linterman MA, Hill DL. Can follicular helper T cells be targeted to improve vaccine efficacy? F1000Res. 2016;5:F1000 Faculty Rev-88.
Havenar-Daughton C, Carnathan DG, Torrents de la Peña A, Pauthner M, Briney B, Reiss SM, et al. Direct Probing of Germinal Center Responses Reveals Immunological Features and Bottlenecks for Neutralizing Antibody Responses to HIV Env Trimer. Cell Rep. 2016;17(9):2195–209.
Tatovic D, Young P, Kochba E, Levin Y, Wong FS, Dayan CM. Fine-Needle Aspiration Biopsy of the Lymph Node: A Novel Tool for the Monitoring of Immune Responses after Skin Antigen Delivery. J Immunol. 2015;195(1):386–92.
Roskell DE, Buley ID. Fine needle aspiration cytology in cancer diagnosis. BMJ. 2004;329(7460):244–5.
Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. 2016;6(8):827–37.
Goode EF, Roussos Torres ET, Irshad S. Lymph Node Immune Profiles as Predictive Biomarkers for Immune Checkpoint Inhibitor Response. Front Mol Biosci. 2021;8:674558. https://doi.org/10.3389/fmolb.2021.674558.
Durand M, Segura E. Dendritic Cell Subset Purification from Human Tonsils and Lymph Nodes. Methods Mol Biol. 2016;1423:89–99.
Pinto ML, Rios E, Durães C, Ribeiro R, Machado JC, Mantovani A, et al. The Two Faces of Tumor-Associated Macrophages and Their Clinical Significance in Colorectal Cancer. Front Immunol. 2019;10:1875.
Valpione S, Galvani E, Tweedy J, Mundra PA, Banyard A, Middlehurst P, et al. Immune-awakening revealed by peripheral T cell dynamics after one cycle of immunotherapy. Nat Cancer. 2020;1(2):210–21.
Huse M. The T-cell-receptor signaling network. J Cell Sci. 2009;122(Pt 9):1269–73.
Lin J, Weiss A. T cell receptor signalling. J Cell Sci. 2001;114(Pt 2):243–4.
Samelson LE. Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu Rev Immunol. 2002;20:371–94.
Schwartzberg PL. Genetic approaches to tyrosine kinase signaling pathways in the immune system. Immunol Res. 2003;27(2–3):481–8.
Balagopalan L, Sherman E, Barr VA, Samelson LE. Imaging techniques for assaying lymphocyte activation in action. Nat Rev Immunol. 2011;11(1):21–33.
Higbee RG, Byers AM, Dhir V, Drake D, Fahlenkamp HG, Gangur J, et al. An Immunologic Model for Rapid Vaccine Assessment — A Clinical Trial in a Test Tube. Altern Lab Anim. 2009;37(1_suppl):19–27.
Dauner A, Agrawal P, Salvatico J, Tapia T, Dhir V, Shaik SF, et al. The in vitro MIMIC® platform reflects age-associated changes in immunological responses after influenza vaccination. Vaccine. 2017;35(41):5487–94.
Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell. 2018;174(6):1586-98.e12.
Drost J, Clevers H. Organoids in cancer research. Nat Rev Cancer. 2018;18(7):407–18.
Aboulkheyr Es H, Montazeri L, Aref AR, Vosough M, Baharvand H. Personalized Cancer Medicine: An Organoid Approach. Trends Biotechnol. 2018;36(4):358–71.
Purwada A, Singh A. Immuno-engineered organoids for regulating the kinetics of B-cell development and antibody production. Nat Protoc. 2017;12(1):168–82.
Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. 2018;8(2):196–215.
Grönholm M, Feodoroff M, Antignani G, Martins B, Hamdan F, Cerullo V. Patient-Derived Organoids for Precision Cancer Immunotherapy. Can Res. 2021;81(12):3149.
Bar-Ephraim YE, Kretzschmar K, Clevers H. Organoids in immunological research. Nat Rev Immunol. 2020;20(5):279–93.
Ye W, Luo C, Li C, Huang J, Liu F. Organoids to study immune functions, immunological diseases and immunotherapy. Cancer Lett. 2020;477:31–40.
Mattei F, Andreone S, Mencattini A, De Ninno A, Businaro L, Martinelli E, et al. Oncoimmunology Meets Organs-on-Chip. Front Mol Biosci. 2021;8:627454.
Pavesi A, Tan AT, Koh S, Chia A, Colombo M, Antonecchia E, et al. A 3D microfluidic model for preclinical evaluation of TCR-engineered T cells against solid tumors. JCI Insight. 2017;2(12):e89762.
Reza KK, Sina AA, Wuethrich A, Grewal YS, Howard CB, Korbie D, et al. A SERS microfluidic platform for targeting multiple soluble immune checkpoints. Biosens Bioelectron. 2019;126:178–86.
Beckwith AL, Velásquez-García LF, Borenstein JT. Microfluidic Model for Evaluation of Immune Checkpoint Inhibitors in Human Tumors. Adv Healthc Mater. 2019;8(11):e1900289.
Gérard A, Woolfe A, Mottet G, Reichen M, Castrillon C, Menrath V, et al. High-throughput single-cell activity-based screening and sequencing of antibodies using droplet microfluidics. Nat Biotechnol. 2020;38(6):715–21.
Businaro L, De Ninno A, Schiavoni G, Lucarini V, Ciasca G, Gerardino A, et al. Cross talk between cancer and immune cells: exploring complex dynamics in a microfluidic environment. Lab Chip. 2013;13(2):229–39.
Molino D, Quignard S, Gruget C, Pincet F, Chen Y, Piel M, et al. On-Chip Quantitative Measurement of Mechanical Stresses During Cell Migration with Emulsion Droplets. Sci Rep. 2016;6:29113.
Agliari E, Biselli E, De Ninno A, Schiavoni G, Gabriele L, Gerardino A, et al. Cancer-driven dynamics of immune cells in a microfluidic environment. Sci Rep. 2014;4:6639.
Vacchelli E, Ma Y, Baracco EE, Sistigu A, Enot DP, Pietrocola F, et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science. 2015;350(6263):972–8.
Cheah R, Srivastava R, Stafford ND, Beavis AW, Green V, Greenman J. Measuring the response of human head and neck squamous cell carcinoma to irradiation in a microfluidic model allowing customized therapy. Int J Oncol. 2017;51(4):1227–38.
Patra B, Lafontaine J, Bavoux M, Zerouali K, Glory A, Ahanj M, et al. On-chip combined radiotherapy and chemotherapy testing on soft-tissue sarcoma spheroids to study cell death using flow cytometry and clonogenic assay. Sci Rep. 2019;9(1):2214.
Lei Y, Tang R, Xu J, Wang W, Zhang B, Liu J, et al. Applications of single-cell sequencing in cancer research: progress and perspectives. J Hematol Oncol. 2021;14(1):91.
Lim B, Lin Y, Navin N. Advancing Cancer Research and Medicine with Single-Cell Genomics. Cancer Cell. 2020;37(4):456–70.
Baccin C, Al-Sabah J, Velten L, Helbling PM, Grünschläger F, Hernández-Malmierca P, et al. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat Cell Biol. 2020;22(1):38–48.
Armand EJ, Li J, Xie F, Luo C, Mukamel EA. Single-Cell Sequencing of Brain Cell Transcriptomes and Epigenomes. Neuron. 2021;109(1):11–26.
Vistain LF, Tay S. Single-Cell Proteomics. Trends Biochem Sci. 2021;46(8):661–72.
Hartmann FJ, Mrdjen D, McCaffrey E, Glass DR, Greenwald NF, Bharadwaj A, et al. Single-cell metabolic profiling of human cytotoxic T cells. Nat Biotechnol. 2021;39(2):186–97.
Leelatian N, Diggins KE, Irish JM. Characterizing Phenotypes and Signaling Networks of Single Human Cells by Mass Cytometry. Methods in molecular biology (Clifton, NJ). 2015;1346:99–113.
Gadalla R, Noamani B, MacLeod BL, Dickson RJ, Guo M, Xu W, et al. Validation of CyTOF Against Flow Cytometry for Immunological Studies and Monitoring of Human Cancer Clinical Trials. Front Oncol. 2019;9:415.
Bandura DR, Baranov VI, Ornatsky OI, Antonov A, Kinach R, Lou X, et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal Chem. 2009;81(16):6813–22.
Chevrier S, Crowell HL, Zanotelli VRT, Engler S, Robinson MD, Bodenmiller B. Compensation of Signal Spillover in Suspension and Imaging Mass Cytometry. Cell Syst. 2018;6(5):612-20.e5.
Ferrer-Font L, Mayer JU, Old S, Hermans IF, Irish J, Price KM. High-Dimensional Data Analysis Algorithms Yield Comparable Results for Mass Cytometry and Spectral Flow Cytometry Data. Cytometry A. 2020;97(8):824–31.
Ferrer-Font L, Small SJ, Lewer B, Pilkington KR, Johnston LK, Park LM, et al. Panel Optimization for High-Dimensional Immunophenotyping Assays Using Full-Spectrum Flow Cytometry. Curr Protoc. 2021;1(9):e222.
den Braanker H, Bongenaar M, Lubberts E. How to Prepare Spectral Flow Cytometry Datasets for High Dimensional Data Analysis: A Practical Workflow. Front Immunol. 2021;12:768113.
Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14(9):865–8.
Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC, et al. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol. 2017;35(10):936–9.
Setliff I, Shiakolas AR, Pilewski KA, Murji AA, Mapengo RE, Janowska K, et al. High-Throughput Mapping of B Cell Receptor Sequences to Antigen Specificity. Cell. 2019;179(7):1636-46.e15.
Black S, Phillips D, Hickey JW, Kennedy-Darling J, Venkataraaman VG, Samusik N, et al. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat Protoc. 2021;16(8):3802–35.
Allam M, Cai S, Coskun AF. Multiplex bioimaging of single-cell spatial profiles for precision cancer diagnostics and therapeutics. NPJ Precis Oncol. 2020;4(1):11.
Taverna JA, Hung CN, DeArmond DT, Chen M, Lin CL, Osmulski PA, et al. Single-Cell Proteomic Profiling Identifies Combined AXL and JAK1 Inhibition as a Novel Therapeutic Strategy for Lung Cancer. Cancer Res. 2020;80(7):1551–63.
Kwok I, Becht E, Xia Y, Ng M, Teh YC, Tan L, et al. Combinatorial Single-Cell Analyses of Granulocyte-Monocyte Progenitor Heterogeneity Reveals an Early Uni-potent Neutrophil Progenitor. Immunity. 2020;53(2):303-18.e5.
Reza KK, Dey S, Wuethrich A, Jing W, Behren A, Antaw F, et al. In Situ Single Cell Proteomics Reveals Circulating Tumor Cell Heterogeneity during Treatment. ACS Nano. 2021;15(7):11231–43.
Schoof EM, Furtwängler B, Üresin N, Rapin N, Savickas S, Gentil C, et al. Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nat Commun. 2021;12(1):3341.
Gebreyesus ST, Siyal AA, Kitata RB, Chen ESW, Enkhbayar B, Angata T, et al. Streamlined single-cell proteomics by an integrated microfluidic chip and data-independent acquisition mass spectrometry. Nat Commun. 2022;13(1):37.
Liang Y, Truong T, Zhu Y, Kelly RT. In-Depth Mass Spectrometry-Based Single-Cell and Nanoscale Proteomics. Methods Mol Biol. 2021;2185:159–79.
Li L, Yan S, Lin B, Shi Q, Lu Y. Chapter Eight - Single-Cell Proteomics for Cancer Immunotherapy. In: Broome A-M, editor. Advances in Cancer Research. vol. 139: Academic Press; 2018. p. 185–207.
Vistain LF, Tay S. Single-Cell Proteomics. Trends Biochem Sci. 2021;46(8):661–72.
Gohil SH, Iorgulescu JB, Braun DA, Keskin DB, Livak KJ. Applying high-dimensional single-cell technologies to the analysis of cancer immunotherapy. Nat Rev Clin Oncol. 2021;18(4):244–56.
Dyring-Andersen B, Løvendorf MB, Coscia F, Santos A, Møller LBP, Colaço AR, et al. Spatially and cell-type resolved quantitative proteomic atlas of healthy human skin. Nat Commun. 2020;11(1):5587.
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics Tissue-based map of the human proteome. Sci. 2015;347(6220):1260419.
Taylor MJ, Lukowski JK, Anderton CR. Spatially Resolved Mass Spectrometry at the Single Cell: Recent Innovations in Proteomics and Metabolomics. J Am Soc Mass Spectrom. 2021;32(4):872–94.
Liu Y, Zeng R, Wang R, Weng Y, Wang R, Zou P, et al. Spatiotemporally resolved subcellular phosphoproteomics. Proc Natl Acad Sci U S A. 2021;118(25):e2025299118.
McDowell CT, Klamer Z, Hall J, West CA, Wisniewski L, Powers TW, et al. Imaging Mass Spectrometry and Lectin Analysis of N-Linked Glycans in Carbohydrate Antigen-Defined Pancreatic Cancer Tissues. Mol Cell Proteomics. 2021;20:100012.
Boyaval F, van Zeijl R, Dalebout H, Holst S, van Pelt G, Fariña-Sarasqueta A, et al. N-Glycomic Signature of Stage II Colorectal Cancer and Its Association With the Tumor Microenvironment. Mol Cell Proteomics. 2021;20:100057.
Drake RR, McDowell C, West C, David F, Powers TW, Nowling T, et al. Defining the human kidney N-glycome in normal and cancer tissues using MALDI imaging mass spectrometry. J Mass Spectrom. 2020;55(4):e4490.
Kearney CJ, Vervoort SJ, Ramsbottom KM, Todorovski I, Lelliott EJ, Zethoven M, et al. SUGAR-seq enables simultaneous detection of glycans, epitopes, and the transcriptome in single cells. Sci Adv. 2021;7(8):eabe3610.
Gaiteiro C, Soares J, Relvas-Santos M, Peixoto A, Ferreira D, Brandão A, et al. Glycoproteogenomics characterizes the CD44 splicing code driving bladder cancer invasion. Theranostics. 2022. https://doi.org/10.7150/thno.67409.
Vodovotz Y, Xia A, Read EL, Bassaganya-Riera J, Hafler DA, Sontag E, et al. Solving Immunology? Trends Immunol. 2017;38(2):116–27.
Comes MC, Filippi J, Mencattini A, Corsi F, Casti P, De Ninno A, et al. Accelerating the experimental responses on cell behaviors: a long-term prediction of cell trajectories using Social Generative Adversarial Network. Sci Rep. 2020;10(1):15635.
Hoops S, Sahle S, Gauges R, Lee C, Pahle J, Simus N, et al. COPASI—a COmplex PAthway SImulator. Bioinformatics. 2006;22(24):3067–74.
Mei Y, Abedi V, Carbo A, Zhang X, Lu P, Philipson C, et al. Multiscale modeling of mucosal immune responses. BMC Bioinformatics. 2015;16(Suppl 12):S2.
Angermann BR, Meier-Schellersheim M. Using Python for Spatially Resolved Modeling with Simmune. Methods Mol Biol. 2019;1945:161–77.
Czech J, Dittrich M, Stiles JR. Rapid creation, Monte Carlo simulation, and visualization of realistic 3D cell models. Methods Mol Biol. 2009;500:237–87.
Xu W, Smith AM, Faeder JR, Marai GE. RuleBender: a visual interface for rule-based modeling. Bioinformatics. 2011;27(12):1721–2.
Schaff JC, Vasilescu D, Moraru II, Loew LM, Blinov ML. Rule-based modeling with Virtual Cell. Bioinformatics. 2016;32(18):2880–2.
Shugay M, Bagaev DV, Zvyagin IV, Vroomans RM, Crawford JC, Dolton G, et al. VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res. 2018;46(D1):D419–27.
Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518(7539):337–43.
Sweeney TE, Braviak L, Tato CM, Khatri P. Genome-wide expression for diagnosis of pulmonary tuberculosis: a multicohort analysis. Lancet Respir Med. 2016;4(3):213–24.
Lofgren S, Hinchcliff M, Carns M, Wood T, Aren K, Arroyo E, et al. Integrated, multicohort analysis of systemic sclerosis identifies robust transcriptional signature of disease severity. JCI Insight. 2016;1(21):e89073.
Azad TD, Donato M, Heylen L, Liu AB, Shen-Orr SS, Sweeney TE, et al. Inflammatory macrophage-associated 3-gene signature predicts subclinical allograft injury and graft survival. JCI Insight. 2018;3(2):e95659. https://doi.org/10.1172/jci.insight.95659.
Sweeney TE, Perumal TM, Henao R, Nichols M, Howrylak JA, Choi AM, et al. A community approach to mortality prediction in sepsis via gene expression analysis. Nat Commun. 2018;9(1):694.
Gaujoux R, Starosvetsky E, Maimon N, Vallania F, Bar-Yoseph H, Pressman S, et al. Cell-centred meta-analysis reveals baseline predictors of anti-TNFα non-response in biopsy and blood of patients with IBD. Gut. 2019;68(4):604–14.
Sweeney TE, Haynes WA, Vallania F, Ioannidis JP, Khatri P. Methods to increase reproducibility in differential gene expression via meta-analysis. Nucleic Acids Res. 2017;45(1):e1.
Yu J, Peng J, Chi H. Systems immunology: Integrating multi-omics data to infer regulatory networks and hidden drivers of immunity. Curr Opin Syst Biol. 2019;15:19–29.
Liu J, Qu S, Zhang T, Gao Y, Shi H, Song K, et al. Applications of Single-Cell Omics in Tumor Immunology. Frontiers in immunology. 2021;12:697412.
Guo T, Li W, Cai X. Applications of Single-Cell Omics to Dissect Tumor Microenvironment. Front Genet. 2020;11:548719. https://doi.org/10.3389/fgene.2020.548719.
Kaminska B, Ochocka N, Segit P. Single-Cell Omics in Dissecting Immune Microenvironment of Malignant Gliomas—Challenges and Perspectives. Cells. 2021;10(9):2264.
Acknowledgements
The authors acknowledge the Portuguese Foundation for Science and Technology funding, IPO-Porto and i3s institutional support, and the MSc program in Medicine and Molecular Oncology of FMUP-University of Porto. All figures were created with BioRender.com.
Funding
The authors wish to acknowledge the Portuguese Foundation for Science and Technology (FCT) for the assistant researcher position CEECIND/03186/2017 (JAF). AP also acknowledges junior researcher position UIDP/00776/2020–5 funded through CI-IPOP Programmatic funding 2020–2023 (reference UIDP/00776/2020) from FCT. This work was also financed by national funds through FCT/MCTES within the scope of FCT projects RESOLVE (PTDC/MED-OUT/2512/2021) and IPOscore (DSAIPA/DS/0042/2018). FCT is co-financed by European Social Fund (ESF) under Human Potential Operation Programme (POPH) from National Strategic Reference Framework (NSRF). The authors also thank the financial support of the Portuguese Oncology Institute of Porto—Research Centre (CI-IPOP-29–2017-2020; CI-IPOP-58–2017-2021).
Author information
Authors and Affiliations
Contributions
AP, AM, and JAF conceptualized and wrote the manuscript, AM produced the artwork, LLS and JAF secured human resources funding, and all authors revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors do not have any financial and non-financial competing interests to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Peixoto, A., Miranda, A., Santos, L.L. et al. A roadmap for translational cancer glycoimmunology at single cell resolution. J Exp Clin Cancer Res 41, 143 (2022). https://doi.org/10.1186/s13046-022-02335-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-022-02335-z