
Abstract
Background
Alternative polyadenylation (APA) is an important mechanism of gene expression regulation through generation of RNA isoforms with distinct 3′ termini. Increasing evidence has revealed that APA is actively involved in development and disease, including hepatocellular carcinoma (HCC). However, how APA functions in tumor formation and progression remains elusive. In this study, we investigated the role of cleavage factor I (CFIm) subunit CPSF6 in human hepatocellular carcinoma (HCC).
Methods
Expression levels of CPSF6 in clinical tissues and cell lines were determined by qRT-PCR and western blot. Functional assays, including the cell number, MTT, colony formation and transwell, were used to determine the oncogenic role of CPSF6 in HCC. Animal experiments were used to determine the role of CPSF6 in HCC tumorigenicity in vivo. Deep sequencing-based 3 T-seq was used to profile the transcriptome-wide APA sites in both HCC cells and CPSF6 knockdown HCC cells. The function of CPSF6-affected target NQO1 with distinct 3′UTRs was characterized by metabolism assays.
Results
We observed CPSF6 was upregulated in HCC and the high expression of CPSF6 was associated with poor prognosis in patients. Overexpression of CPSF6 promoted proliferation, migration and invasion of HCC cells in vitro and in vivo. Transcriptome-wide APA profiling analysis indicated that high expression of CPSF6 promoted the favorable usage of the proximal poly(A) site in the 3′UTR of NQO1. We demonstrated CPSF6-induced tumorigenic activities were mediated by the NQO1 isoform with short 3′UTR. Furthermore, we found that CPSF6 induced metabolic alterations in liver cells through NQO1.
Conclusion
CPSF6 plays a critical role in HCC progression by upregulating NQO1 expression through APA. These findings provide evidence to demonstrate that APA of NQO1 contributes to HCC progression and may have implications for developing new therapeutic strategy against this disease.
Similar content being viewed by others


Background
Hepatocellular carcinoma (HCC), the main form of liver cancer, is one of the most common cancer types and the third leading cause of cancer-related mortality worldwide [1, 2]. Many risk factors, such as hepatitis B virus (HBV) and hepatitis C virus (HCV) infections, autoimmune hepatitis, cirrhosis, alcohol abuse and toxic chemical exposures, may play a role in HCC progression [3, 4]. Moreover, genetic alterations in the TERT, TP53, and CTNNB1 loci have been detected in early stage or advanced HCC [5]. Despite significant progress, the overall survival rate of HCC patients is still unsatisfactory, mainly owing to their high rates of recurrence [6]. Therefore, the molecular mechanisms of HCC remain to be investigated and new therapeutic targets need to be identified.
Alternative polyadenylation (APA) is a post-transcriptional mechanism to generate distinct 3′-untranslated regions (UTRs) of a given gene, increasing transcriptome complexity [7]. The 3′UTRs contain regulatory elements for miRNA and RNA-binding protein binding sites, which allows for regulation of those gene products and provides an important layer of gene expression regulation [8,9,10,11]. APA potentially affects mRNA stability, translation efficiency and subcellular localization of the transcript isoforms. About 70% of human genes have multiple poly(A) sites. The selective usage of APA sites has been characterized in various biological contexts, including cellular proliferation, differentiation, neuron activation and cancer [7, 12,13,14,15,16].
An interesting topic of cancer biology is that APA contributes to proto-oncogene activation and thereby promotes oncogenic transformation [17]. The preferential usage of APA sites has been observed in various types of cancer, such as breast, kidney, colon, liver and lung tumors [18,19,20]. Moreover, APA patterns could be used as biomarkers for cancer diagnosis and prognosis [21]. These observations suggest APA is actively associated with the initiation and progression of cancer. However, the mechanistic links between APA and tumorigenicity remain elusive.
A number of factors have been reported to regulate APA in global or gene-specific manner, among which are RNA 3′-end-processing factors [7]. Some APA regulatory factors are functionally involved in tumor formation. For example, upregulation of CFIm25 in glioblastoma cells suppresses the tumorigenic properties and inhibits tumor growth [22]. In contrast, activation of CSTF2 was observed to enhance the oncogenic activities in urothelial carcinoma of the bladder [23]. These studies suggest the complicated biological consequence of APA factors on tumorigenicity.
We have developed a deep sequencing-based approach 3 T-seq for APA site profiling [24]. Recently, we have reported that CFIm25 (encoded by Nudt21) exerted suppressive effect on liver cancer cells [25]. The polyadenylation complex cleavage factor I (CFIm), one of four core components of APA machinery, is composed of two small subunits CFIm25 and two large subunits CFIm68 (also known as CPSF6) and CFIm59 (also known as CPSF7) [26, 27]. CFIm plays a key regulator of cleavage and polyadenylation site choice during APA through its binding to 5′-UGUA-3′ elements localized in the 3’UTR for a huge number of pre-mRNAs [28, 29]. CPSF6 is a component of the CFIm complex that functions as an activator of the pre-mRNA cleavage and polyadenylation processing required for the maturation of pre-mRNA into functional mRNAs [26, 29,30,31]. Although CPSF6 was reported to be involved in tumor progression in cancer [32], the underlying mechanisms are yet to be completely characterized. In this study, we investigated the role of CPSF6 in HCC. We showed that CPSF6 expression was significantly increased in HCC tissues. CPSF6 knockdown obviously inhibits the proliferation, migration and invasion ability of liver cancer cells Huh-7 and HepG2 in vitro and in vivo. Transcriptome-wide APA profiling analysis indicated that high expression of CPSF6 promoted the favorable usage of the proximal poly(A) site in the 3′UTR of a number of genes, including NQO1. We demonstrated CPSF6-induced tumorigenic activities were mediated by the NQO1 isoform with short 3′UTR. Furthermore, we found that CPSF6 induced metabolic alterations in liver cells through NQO1.
Methods
Cells and culture conditions
Human immortalized liver cell line (HL-7702) and liver cancer cell lines (Huh-7, HepG2, SK-HEP-1, PLC/PRF/5, Hep3B) were purchased from Shanghai Cell Bank (Shanghai, China). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, Carlsbad, CA, USA), 1% penicillin–streptomycin (Sangon, Shanghai, China), and 2 mM glutamine (Invitrogen, Carlsbad, CA, USA) at 37 °C in a 5% CO2 humidified atmosphere.
Histopathological analysis
Human liver tissues were obtained from the First Affiliated Hospital of Anhui Medical University (Hefei, China). The study was approved by the Biomedical Ethics Committee of Anhui Medical University and informed consent was obtained from each patient. For IHC staining of clinical samples and xenograft tumors, tissue samples were first fixed in formalin and performed following the manufacturer’s instruction. IHC labeling intensity was reviewed and scored by two independent pathologists. The mouse lung tissues were fixed with paraformaldehyde (PFA, 4%) for 12 h, then dehydrated with ethanol and embedded in paraffin. Paraffin-embedded mouse lung tissue was sectioned and then stained with hematoxylin and eosin. Tissue samples for RNA extraction were quickly frozen in liquid nitrogen after resection and kept in RNAlater (CAT #AM7021, Invitrogen, Carlsbad, CA, USA). Total RNA was extracted using Trizol (CAT #15596026, Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instruction.
Protein extraction and western blot analysis
Cells or tissues were lysed in ice-cold RIPA buffer (50 mM Tris, 150 mM NaCl, 1.0% Triton, 1 mM EDTA, 0.1% SDS) supplemented with protease (CAT #11697498001, Roche, Basel, Switzerland) and phosphatase inhibitor cocktail (CAT #4693132001, Roche, Basel, Switzerland). The protein concentrations were determined by the BCA Assay using BCA Assay protein Kit (Sangon, Shanghai, China). Soluble protein was separated by 10% SDS-PAGE and transfer onto PVDF membranes (Millipore, Billerica, MA, USA). The membrane was incubated in blocking buffer (1xPBST with 1% BSA) for 1 h at room temperature. The membrane was then blotted with the diluted primary antibodies: anti-CPSF6 (1:1000, CAT #ab175237, Abcam), anti-NQO1 (1:1000, CAT #11451–1-AP, Proteintech) and anti-β-actin (1:5000, CAT #A5316, Sigma). Immunoreactive protein was visualized using ECL start Western Blotting Substrate (GE Healthcare Life Sciences, USA) and image quant LAS 4000 mini (GE Healthcare Life Sciences, USA).
Reverse transcription and quantitative PCR analysis
Total RNA was extracted using TRIzol (CAT #15596026, Invitrogen, Carlsbad, CA, USA) and reversely transcribed using RevertAid™ First Strand cDNA Synthesis Kit (CAT #K1621, ThermoFisher, USA) according to the manufacturer’s instructions. The qRT-PCR reactions were performed using SYBR-GREEN (Applied Biosystems) according to the manufacturer’s instructions. Gene-specific primers for qRT-PCR analysis were as follows: CPSF6 (F: GGAGCAGCACCAAATGTTGTC, R: CTCCCAAAGAATGAACTGCTTC); NQO1 (F: CTGATCGTACTGGCTCACTCAG, R: GCAGGATACTGAAAGTTCGCAG); GAPDH (F: CACAGTCCATGCCATCACTG, R: CTTGGCAGCGCCAGTAGAG). GAPDH was used as an internal control.
Generation of stable cell lines
The lentiviral vectors pSIN-GFP-puro-CPSF6 and pSIN-GFP-puro-NQO1 were constructed for CPSF6 and NQO1 expression, respectively. The pSIN-GFP-puro was purchased from Genecopoeia. The lentiviral vectors pLKO.1-puro-shRNA-CPSF6 and pLKO.1-puro-shRNA-NQO1 were constructed for CPSF6 and NQO1 knockdown, respectively. The pLKO.1-puro was obtained from Genecopoeia. The 293 T cells were used for the lentiviral production and supernatant was collected at 48 h and 72 h after transfection. The viruses were filtered using 0.45 μm filters and appropriate amounts of viruses were used to infect liver cells. Finally, cells were maintained in the presence of puromycin (2 μg/ml) for 1–2 weeks.
Luciferase assays
NQO1-luciferase reporter construct (psiCHECK2, Promega, USA) contained the short or the long 3′UTR of the human NQO1 gene. Liver cells were transfected with NQO1-luciferase reporter vector in 24-well plates (100 ng/well) using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). After transfection for 48 h, cells were harvested and assayed using dual luciferase assay according to the manufacturer’s protocol (Promega, USA). Firefly luciferase read-outs were normalized to Renilla luciferase read-outs. To ensure the reporter with long 3′UTR was expressed, the proximal PAS signals were mutated (AATAAA was mutated to ACAAAC) for these reporters, which contains the long 3′UTR of NQO1.
MTT assay
Liver cells were plated in 96-well plates (1000–3000 cells/well). 3–5 days after seeding, methyl thiazol tetrazolium (MTT) was added into each well (5 mg/mL) for 1–2 h. Then, the media was removed and each well was washed using PBS. DMSO was added into each well and the absorbance was measured at 570 nm.
Colony formation assays
Liver cells were seeded into 6-well plates (100–300 cells/well) and cultured for 2–3 weeks. At the end points of the experiments, cells were fixed with 4% methanol, stained with crystal violet (0.1%), and photographed.
Migration and invasion assays
Liver cells were resuspended in serum-free medium. For the migration assays, cells (5–10 × 104 /100 μl) were seeded onto the chamber without Matrigel. For the invasion assays, 10–20 × 104 cells (5–10 × 104 /100 μl) were seeded onto the chamber with 100 μl of Matrigel. The bottom chamber was prepared using 800 μl of DMEM medium (supplemented with 10% FBS) as a chemoattractant. After 24–48 h of incubation, the cells on the outer surface were washed by PBS and fixed with 4% paraformaldehyde, stained in a dye solution containing 0.1% crystal violet for visualization.
Glucose uptake and lactate measurements assay
The glucose uptake in HL-7702 or Huh-7 cells (2 × 104) was measured using the commercially available kit Glucose Uptake-Glo™ Assay (CAT #ab136955, Abcam, UK) according to the manufacturer’s instructions. HL-7702 or Huh-7 cells (2 × 104) were plated in 96-well plates and lactate production was measured using the commercially available kit (CAT #600450, Cayman, USA) according to the manufacturer’s instructions.
Xenograft model in vivo
The design and protocol of in vivo experiments were approved by the Institutional Animal Care and Use Committee, Shanghai Jiao Tong University. For tumorigenicity assay, liver cells were resuspended in PBS/Matrigel Matrix mix at 1:1 ratio. 5 × 106 cells in 100 μL solution were injected into the flank of 4–6 weeks old athymic male nude mice. Tumor volumes were calculated as follows: Volume = ½ (L × W2), where L is the length and W is the width. For the lung colonization assay, cells were resuspended in PBS and injected into the lateral tail vein of 4–6 weeks old NOD/SCID mice (5 × 105 cells/animal).
3 T-seq analysis
3 T-seq analysis was performed as we described previously [24]. Briefly, M280 beads were coated with Bio-oligo dT (20) then incubated with 50 μg total RNA. The cDNA was synthesized and released by Gsu I digestion. The 3′UTR fragments were ligated to the Illumina sequencing adaptors and subjected to deep sequencing. The qualified reads were mapped to UCSC human reference genome (hg19) with bowtie2 (version 2.3.3.1) [33]. The 3′UTR switching for each gene in the cell lines was detected by linear trend test (the FDR-adjusted p-value < 0.05) [34]. The accession number of raw data is E-MTAB-9458, and available in the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress).
Results
CPSF6 is upregulated in HCC and high expression of CPSF6 is associated with poor prognosis in HCC patients
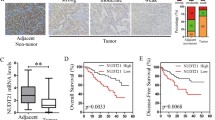
We firstly examined the CPSF6 expression in HCC patients. We collected HCC tissues (n = 159) and the paired non-tumor tissues (n = 159) and performed immunohistochemical (IHC) assay. CPSF6 staining was scored as a strong or moderate expression in 31 and 55% of tumor tissues, as compared with 9 and 33% in corresponding adjacent non-tumor liver tissues (Fig. 1a). The elevated expression of CPSF6 was also observed in 33 out of 36 HCC tissues by western blot assay when compared with the adjacent non-tumor counterparts (Fig. 1b and Supplementary Fig. S1). Consistent with the detected protein abundance, we observed the elevated mRNA level of CPSF6 in HCC tissues by real-time quantitative PCR (Fig. 1c). These data indicated that CPSF6 was upregulated in HCC. The patients with high CPSF6 expression (n = 135) had shorter overall survival (OS) (p = 0.0091) and disease-free survival (DFS) (p = 0.0038) than the patients with low CPSF6 expression (n = 121) (Fig. 1d, e). To understand the clinical implications of the increased expression, we examined the correlation of CPSF6 with the clinicopathologic features of HCC. The higher CPSF6 expression was observed to be associated with tumor size (p < 0.001), advanced tumor-node metastasis (TNM) stage (p < 0.001), venous invasion (p < 0.001) and distant metastasis (p < 0.001) of the HCC patients (Supplementary Table S1).

CPSF6 is upregulated and predicts poor prognosis in HCC. a IHC staining of CPSF6 in surrounding non-tumor and HCC tissues. Percentage of CPSF6 IHC was shown in bar graph. Scale bar, 50 μm. b Western blot assay for detecting CPSF6 expression in 16 paired surrounding non-tumor (N) and HCC tumor (T) tissues. β-actin was used as internal control. c qRT-PCR analysis of CPSF6 mRNA in 36 paired surrounding non-tumor (N) and HCC tumor (T) tissues. GAPDH was used as internal control. d, e Kaplan–Meier analysis of the correlation of CPSF6 protein expression with OS (d) and DFS (e). The expression level of CPSF6 protein was detected by immunohistochemistry. f qRT-PCR analysis of CPSF6 mRNA in TCGA liver cancer samples. g,h Kaplan–Meier analysis of OS (g) and DFS (h) data from TCGA liver cancer data containing 364 patients. The data of g and f can be obtained through online website (http://kmplot.com/analysis/).**p < 0.001, Student’s t-test
We extended our analysis by examining the expression of CPSF6 with publicly available human clinical data in The Cancer Genome Atlas (TCGA) database. We found that the expression level of CPSF6 was significantly increased in HCC patients (Fig. 1f). Similarly, we observed that the patients with high CPSF6 expression also had shorter OS (P = 0.00042) and DFS (P = 0.000043) (Fig. 1g, h). Together, these observations suggest that CPSF6 is upregulated in HCC and high CPSF6 expression is associated with poor clinical outcomes in the HCC patients.
CPSF6 promotes HCC cell growth in vitro and in vivo
We next examined the protein expression of CPSF6 in non-tumor liver cell line HL-7702 and human HCC cell lines (Huh-7, HepG2, SK-HEP-1, PLC/PRF/5 and Hep3B). Compared with HL-7702, higher level of CPSF6 was observed in four HCC cell lines (Huh-7, HepG2, SK-HEP-1 and PLC/PRF/5) (Fig. 2a). To explore the function of CPSF6 in HCC, CPSF6 was ectopically expressed in HL-7702 and repressed with short hairpin RNAs (shRNAs) in Huh-7 and HepG2, respectively (Fig. 2b, c and Supplementary Fig. S2A). Compared with HL-7702 cells (the control), overexpression of CPSF6 in HL-7702 significantly increased cell proliferation (Fig. 2d), viability (Fig. 2f), and generated more and larger colonies in vitro (Fig. 2h). In contrast, CPSF6 knockdown in Huh-7 and HepG2 significantly reduced cell proliferation (Fig. 2e and Supplementary Fig. S2B), viability (Fig. 2g and Supplementary Fig. S2C) and clonogenicity in vitro (Fig. 2i and Supplementary Fig. S2D). To understand CPSF6 role in vivo, HL-7702 cells with overexpression of CPSF6 were injected subcutaneously into nude mice. We observed a significant increase in both tumor growth and weight (Fig. 2j, k); whereas a significant reduction in both tumor growth and weight was observed in CPSF6 knockdown tumors (Fig. 2l, m). Similarly, the alterations in abundance of Ki67 protein were found in CPSF6-overexpressing tumors (Fig. 2n) and CPSF6 knockdown tumors (Fig. 2o). Collectively, our results demonstrated that enforced expression of CPSF6 significantly promoted tumorgenicity in vitro and in vivo.

CPSF6 accelerates the growth of HCC cells in vitro and in vivo. a The protein level of CPSF6 in different cell lines was assessed by western blotting. β-actin was used as internal control. b The protein level of CPSF6 in the CPSF6-overexpressing (CPSF6-OE) HL-7702 was assessed by western blotting. β-actin was used as internal control. c CPSF6 knockdown efficiency in Huh-7 with two CPSF6 specific shRNAs (shCPSF6–1 and shCPSF6–2) was examined by western blotting. β-actin was used as internal control. d Proliferation curves of the CPSF6-overexpressing HL-7702 cells and the control (Ctrl) were shown. e Proliferation curves of the CPSF6 knockdown Huh-7 cells and the control (shCtrl) were shown. f The cell viability of HL-7702 cells with CPSF6 overexpression and the control was examined by MTT assay. g The cell viability of CPSF6 knockdown Huh-7 cells and Huh-7 cells (the control) was examined by MTT assay. h,i Colony formation analysis of the indicated cell lines. j HL-7702 xenograft tumor growth with or without overexpression of CPSF6. k The weight of HL-7702 tumors at the end point was shown. l Huh-7 xenograft tumor growth with or without CPSF6 knockdown. m The weight of Huh-7 tumors at the end point was shown. n, o IHC analysis of CPSF6 and Ki67 expression in tumors. Scale bars, 100 μm. **p < 0.001, Student’s t test
CPSF6 promotes HCC cell migration and invasion in vitro and metastasis in vivo
Considering the clinical relevance between CPSF6 and metastasis, we examined whether CPSF6 affects the migration and invasion of HCC cells. As shown in Fig. 3a, forced expression of CPSF6 significantly promoted cell migration capacity in HL-7702 cells, while CPSF6 knockdown significantly inhibited migration of Huh-7 and HepG2 cells (Fig. 3b and Supplementary Fig. S3A). The similar phenotype of cell invasion was observed in the CPSF6-overexpressing HL-7702 cells (Fig. 3c) or the CPSF6 knockdown Huh-7 and HepG2 cells (Fig. 3d and Supplementary Fig. S3B). These results indicated that CPSF6 promoted migration and invasion of HCC cells in vitro. We next injected hepatic cells (HL-7702-Ctrl, HL-7702-CPSF6-OE, Huh-7-shCtrl and Huh-7-shCPSF6) into the tail vein of nude mice (n = 6 for each group) and examined the metastatic nodules in lungs. The frequency of lung metastasis from the lungs in HL-7702-CPSF6-OE group was significantly higher when compared with the HL-7702-Ctrl group at one month after injection (Fig. 3e). Meanwhile, we observed that the frequency of lung metastasis was decreased in Huh-7-shCPSF6 group as compared with Huh-7-shCtrl group (Fig. 3f). These observations demonstrated that CPSF6 knockdown suppresses metastasis of HCC cells in vivo.

CPSF6 promotes migration, invasion and metastasis of HCC cells. a Effect of CPSF6 overexpression on HL-7702 cell migration. b Effect of CPSF6 knockdown on Huh-7 cell migration. c Effect of CPSF6 overexpression on HL-7702 cell invasion. d Effect of CPSF6 knockdown on Huh-7 cell invasion. Representative pictures of H&E staining of lungs and incidence of lung metastasis from mice inoculated with (e) HL-7702 and (f) Huh-7 cells. Red arrows indicated the lung metastases. Results represented mean ± SD using bar graph. Scale bars, 100 μm. **p < 0.001, Student’s t test
CPSF6 modulates the widespread 3′UTR alteration in HCC cells
As a member of APA machinery, CPSF6 causes the oncogenic activities described above. We thus hypothesized that CPSF6 functions as an oncogene, at least in part, by influencing APA and 3′UTR. To test this hypothesis, we performed APA site profiling analysis in CPSF6-overexpressing HL-7702 cells, CPSF6 knockdown Huh-7 cells and the corresponding control cells with 3 T-seq we reported previously [24]. A total of 73.1 million reads were obtained, of which 87.7% were uniquely mapped to the reference genome (Supplementary Table S2). After filtering internal priming events, we found most of the qualified reads were mapped to the annotated transcription termination sites (TTSs) or 3′UTRs (Fig. 4a, b), yielding 26,946 poly(A) sites (Supplementary Table S2).

Characteristics of 3 T-seq data. a, b Genomic locations of qualified 3 T-seq reads mapped to the reference genome. c, d Genomic distribution of the poly(A) sites. e, f The statistics of genes with various number of detected poly(A) sites. g, h Scatterplot of CULI for measuring the 3′UTR alteration in CPSF6-overexpressing or CPSF6 knockdown cells when compared with the corresponding control cells (false discovery rate (FDR) = 0.05). i CPSF6-modulated 7 candidate genes. These genes had the shortened 3′UTR in CPSF6-overexpressing HL-7702 cells and the lengthened 3′UTR in CPSF6 knockdown Huh-7 cells. j CPSF6-induced APA shift of NQO1. Left, Integrative Genomics Viewer (IGV) genome browser exhibited the poly(A) site usage of NQO1 3′UTR. Right, histogram showed the relative expression of the isoform with distal polyadenylation site (dPAS) relative to the one with proximal PAS (pPAS). k, l The protein levels of NQO1 in the CPSF6-overexpressing HL-7702 cells (k) and the CPSF6 knockdown Huh-7 cells (l) were assessed by western blotting. β-actin was used as internal control
Using the snowball clustering method [34], we found the majority of identified APA sites in each 3 T-seq library were located at the 3′ terminus. In particular, 34–59% of these poly(A) sites were mapped to TTSs and 24–47% to the 3′UTR regions in polyA-DB database [35], respectively. About 30% of these poly(A) sites have been annotated in the polyA-DB database, and thus the remaining sites are putatively novel (Fig. 4c, d). In addition, 28% of genes harbor three or more poly(A) sites (Fig. 4e, f). We adopted the cancer 3′UTR length index (CULI) [34] to quantitatively measure the 3′UTR alteration. Comparing with HL-7702, we identified 433 genes with altered 3′UTR in CPSF6-overexpressing HL-7702 cells, 51.3% of which showed a shift from distal to proximal poly(A) site usage and thus possessed shortened 3′UTRs (Fig. 4g and Supplementary Table S2). Meanwhile, we also identified 74 genes with lengthened 3′UTR in CPSF6 knockdown Huh-7 cells (Fig. 4h and Supplementary Table S3).
To explore how CPSF6-modulated APA contributes to tumorigenicity, we examined the genes with 3′UTR alteration simultaneously influenced under both CPSF6-overexpressing and knockdown conditions, yielding seven candidates (Fig. 4i and Supplementary Table S4). The association between HCC and the metabolic alterations has been recently reported [36, 37]. Interestingly, among seven CPSF6 responsive genes we found the metabolism regulator NQO1 (Fig. 4i). Furthermore, overexpression of CPSF6 induced the preferential usage of the proximal poly(A) site in NQO1 3’UTR; while CPSF6 knockdown caused the shift of polyadenylation from the proximal to the distal (Fig. 4j). Furthermore, western blotting results indicated that CPSF6 positively regulates NQO1, i.e., NQO1 expression increased under the condition of CPSF6 overexpression and decreased in the case of CPSF6 knockdown. (Fig. 4k, l).
NQO1 short 3’UTR isoform enhances the oncogenic and metastatic capacities of liver cells both in vitro and in vivo
We performed the prediction analysis of miRNA binding site with online tools TargetSan (http://www.targetscan.org/vert_72/) and starBase V2.0 (http://starbase.sysu.edu.cn/). Compared with the transcript isoform containing short 3′UTR, NQO1 transcript isoform with long 3′UTR contains multiple potential miRNA binding sites (Fig. 5a). We selected several predicted miRNAs, all of which were reported to have cancer suppression function [38,39,40,41]. The effect of the selected miRNAs on translation activity of NQO1 3’UTRs was examined by luciferase activity assay. We observed that the let-7a-5p and other three miRNAs had a significant inhibitory effect on the translation activity of the NQO1 isoform with long 3’UTR, and no significant effect on the isoform with short 3’UTR was observed (Fig. 5b, c). The translation activity of the short 3’UTR is 5.6- to 10.1- fold higher than that of the long 3’UTR (Fig. 5d). As expected, the NOQ1 protein level of the mRNA isoform with short 3′UTR was higher than that with long 3′UTR (Fig. 5e).

The NQO1 short 3’UTR isoform has oncogenic function and increases aggressiveness in liver cells. a Schematic illustration of the NQO1 isoform with long or short 3’UTR. Positions of the binding sites of miRNA were indicated by yellow horizontal lines. The activity of short 3’UTR (3’UTR-S) and long 3’UTR (3’UTR-L) of NQO1 after enforced expression of the specific miRNAs was examined using luciferase reporter assay in HL-7702 (b) and Huh-7 (c) cells. d Luciferase expression from a reporter containing the short 3’UTR of NQO1, as compared to that from the reporter containing the long 3’UTR of NQO1 in 4 liver cell lines. e The protein levels of NQO1 in HL-7702 cells stably transfected with NQO1-overexpressing plasmids were assessed by western blotting. β-actin was used as internal control. f Proliferation curves of HL-7702 cells stably transfected with NQO1-overexpressing plasmids or control (Ctrl) were shown. g The cell viability of HL-7702-NQO1-OE and HL-7702-Ctrl cells was examined by MTT assay. h Colony formation analysis of HL-7702-NQO1-OE and HL-7702-Ctrl cells. i Transwell analysis of the migration ability of HL-7702 cells. j Transwell analysis of the invasion ability of HL-7702 cells. k HL-7702 xenograft tumor growth with or without overexpression of different NQO1 isoforms. l The weight of HL-7702 tumors at the end point was shown. m IHC analysis of NQO1 and Ki67 expression in tumors. Scale bars, 100 μm. n Representative pictures of H&E staining of lungs and incidence of lung metastasis from mice inoculated with HL-7702 cells. Black arrows indicate the lung metastases. **p < 0.001, Student’s t test
To understand the function of distinct NQO1 transcript isoforms in liver cells, the NQO1 isoforms with short and long 3′UTRs were transfected into HL-7702 cells, respectively. The results showed that enforced expression of the isoform with short 3′UTR dramatically increased cell growth (Fig. 5f, g), colony formation (Fig. 5h), migration (Fig. 5i) and invasion (Fig. 5j), when compared with that of vector control and ectopic expression of isoform with long 3′UTR. Furthermore, we observed that enforced expression of the short isoform substantially enhanced the activities of subcutaneous tumor formation (Fig. 5k) and increased the tumor weight (Fig. 5l) in mice. The expression of NQO1 and Ki67 was increased in tumors of mice injected with NQO1 short isoform-expressing HL-7702 cells; while no significant change was observed in the tumors injected with NQO1 long isoform-expressing HL-7702 cells (Fig. 5m). We next injected HL-7702 cells with NOQ1 isoforms into the tail vein of nude mice (n = 6 for each group) and examined the metastatic nodules in lungs. The results showed that enforced expression of the short 3’UTR isoform of NQO1 enhanced lung metastasis when compared with that of either ectopic expression of long 3’UTR isoform or the control cells (Fig. 5n).
The oncogenic effect of CPSF6 is mediated by NQO1
To ask whether NQO1 was required for the oncogenic effect of CPSF6, we knocked down NQO1 expression in HL-7702 (the control) and CPSF6-overexpressing HL-7702 cells by shRNA, respectively (Fig. 6a). Although CPSF6 overexpression promoted cell proliferation, migration and invasion in HL-7702, this effect was almost depleted in NQO1 knockdown cells (Fig. 6b-f), suggesting that CPSF6 function is NQO1-dependent. Furthermore, we ectopically expressed NQO1 in CPSF6 knockdown Huh-7 cells (Fig. 6g), and found that although knockdown of CPSF6 inhibited cell proliferation, migration and invasion in Huh-7, restoring the expression of NQO1 obviously reversed these tumorigenic activities (Fig. 6h-l).

Function of CPSF6 is NQO1-dependent. a Western blot analysis of the efficiency of NQO1 knockdown and overexpression of CPSF6 in HL-7702 cells. b Proliferation curves of HL-7702 cells were shown. c The cell viability of HL-7702 cells was examined by MTT assay. d Colony formation analysis of HL-7702 cells. e Transwell analysis of cell migration of HL-7702. f Transwell analysis of the cell invasion of HL-7702. g Western blot analysis of the efficiency of NQO1 overexpression (NQO1-OE) and CPSF6 knockdown in Huh-7. h Proliferation curves of Huh-7 cells were shown. i The cell viability of Huh-7 cells was examined by MTT assay. j Colony formation analysis of Huh-7 cells. k Transwell analysis of the cell migration of Huh-7. l Transwell analysis of the cell invasion of Huh-7. **p < 0.001, Student’s t test
CPSF6 regulates liver cell metabolism through NQO1
It has been reported that NQO1 can regulate cell metabolism [42]. It is unclear whether the NQO1 isoforms are involved in metabolism regulation in HCC. To address this issue, we tested the role of distinct NQO1 isoforms in HL-7702 cells. We found the enforced expression of the NQO1 isoform with short 3′UTR dramatically increased glucose uptake (Fig. 7a) and lactate production (Fig. 7b), when compared with that of vector control and ectopic expression of long 3′UTR isoform. In addition, the enforced expression of the short isoform dramatically decreased basal as well as maximum oxygen consumption rate (OCR) (Fig. 7c) but increased extracellular acidification rate (ECAR) (Fig. 7d), when compared with that of vector control and ectopic expression of long 3′UTR isoform.

CPSF6 upregulates glycolysis and promotes aerobic glycolysis through NQO1. a Glucose uptake in control and NQO1-overexpressing HL-7702 cells. b Cellular lactate levels in control and NQO1-overexpressing HL-7702 cells. OCR (c) and ECAR (d) of control and NQO1- overexpressing HL-7702 cells measured by the Seahorse Bioscience XF96 analyzer. e Glucose uptake in control and CPSF6-overexpressing HL-7702 cells with or without NQO1 knockdown. f Cellular lactate levels in control and CPSF6-overexpressing HL-7702 cells with or without NQO1 knockdown. OCR (g) and ECAR (h) of control and CPSF6-overexpressing HL-7702 cells with or without NQO1 knockdown measured by the Seahorse Bioscience XF96 analyzer. i Glucose uptake in control and CPSF6 knockdown Huh-7 cells with or without overexpression of NQO1. j Cellular lactate levels in control and CPSF6 knockdown Huh-7 cells with or without overexpression of NQO1. OCR (k) and ECAR (l) of control and CPSF6 knockdown Huh-7 cells with or without overexpression of NQO1 measured by the Seahorse Bioscience XF96 analyzer. **p < 0.001, Student’s t test
Although overexpression of CPSF6 in HL-7702 dramatically increased glucose uptake, lactate production, ECAR, and decreased OCR (Fig. 7e-h), we found this trend was obviously reversed after NQO1 knockdown (Fig. 7e-h). Similar to NQO1 knockdown, CPSF6 knockdown cells also exhibited reduced glucose uptake and lactate production (Fig. 7i, j), increased basal as well as maximum OCR (Fig. 7k), and decreased ECAR (Fig. 7l). These observations suggest that CPSF6 promotes aerobic glycolysis and suppresses oxidative phosphorylation in liver cells through NQO1.
NQO1 is upregulated in HCC and positively correlates with CPSF6 expression
We lastly examined the NQO1 expression in both HCC and the paired non-tumor tissues. A total of 159 cases of HCC and paired non-tumor samples were collected for IHC assay. We observed the upregulated expression of NQO1 in tumor tissues when comparing with the paired non-tumor samples (Fig. 8a). This observation was confirmed by western blotting assay of another 36 patients (Fig. 8b and Supplementary Fig. S4). We next examined the expression relationship of NOQ1 and CPSF6. We observed a moderately positive correlation between CPSF6 and NQO1 in the 124 patients with stage III and IV (r = 0.501, Fig. 8c; Supplementary Table S1). These findings suggest NQO1 is upregulated in HCC and positively correlates with CPSF6 expression.

Expression of NQO1 in human HCC tissues. a IHC staining of NQO1 in surrounding non-tumor and HCC tissues. Scale bar, 50 μm. b Western blot analysis of NQO1 expression in 16 paired surrounding non-tumor (N) and HCC tumor (T) tissues. β-actin was used as internal control. c The correlation analysis between NQO1 expression and CPSF6 level in 124 HCC tissues (TNM, III/IV) with linear regression and pearson’s correlation significance (P < 0.0001, ANOVA test). d The proposed mechanism of CPSF6 in regulation of HCC progression
Discussion
As an important mechanism in regulation of gene expression, APA is widespread across human genome. Dysregulation of APA is increasingly recognized as tumorigenesis driver [15]. APA is specified by a number of regulatory factors [7], among which polyadenylation factors have been intensively characterized. For example, CFIm25 causes 3′UTR shortening of a number of genes in glioblastoma cells and promotes tumor suppression [22]; however, CSTF2 in urothelial carcinoma of the bladder elicits 3′UTR shortening of RAC1 and contributes to UCB pathogenesis [23]. These studies underline the necessity of APA factor characterization at different cellular context. CPSF6 has been recently reported to be of clinical relevance in breast cancer [32]. However, the role of CPSF6 in HCC is still largely unknown.
Some recent studies have reported the altered expression pattern of APA factors in HCC. For example, Ji and colleagues found CPSF7 is activated and regulates liver cancer growth and metastasis by targeting the WWP2/PTEN/AKT signaling pathway [43]. In contrast, we reported that the expression of NUDT21 is repressed, which contributes to hepatocellular carcinoma suppression [25]. In the present study we observed that CPSF6 is highly expressed in HCC tissues and associated with poor prognosis. We then performed a combination of in vitro and in vivo assays to understand the function of CPSF6. Our findings clearly showed that overexpression of CPSF6 significantly promoted the tumorigenic and metastatic activities either in vitro or in vivo. Our results suggest that CPSF6 exerts oncogenic effect in HCC.
Given CPSF6 as an APA factor per se, one of intriguing question was thus raised up, i.e., what mechanisms underlie the oncogenic effect of this CFIm subunit? We then investigated APA profiles under the conditions of high and low levels of CPSF6. Seven genes with altered 3’UTR were simultaneously found under both conditions, including NQO1.
NQO1 is a cytosolic flavoprotein that catalyzes the reduction of quinones and protects cells from oxidative stress through preventing the generation of free radicals [42]. Upregulation of NQO1 was frequently observed in various types of cancer, such as breast, prostate, lung and liver cancer [44,45,46,47]. Nevertheless, the regulatory mechanism of NQO1 expression has not well characterized. Our results showed that overexpression of CPSF6 caused the preferential proximal poly(A) site usage in the 3′UTR of NQO1 and increased the level of NQO1 protein. The activation of CPSF6-induced NQO1 isoform with short 3′UTR, at least partially due to the escape from miRNA repression, was confirmed by luciferase activity assays. In addition to CPSF6, it should be noted that another APA factor CstF-64 also modulates APA pattern of NQO1 in HEK 293 cells [48], suggesting that the selective poly(A) site usage of NQO1 is regulated in cellular context-dependent manner.
The Warburg effect is a general feature of glucose metabolism in cancer cells, which is intimately linked to the uncontrolled and continuing proliferation characteristic of cancer cells [49,50,51]. Numerous studies have documented a strong association between HCC and the metabolic alterations [36, 37, 52, 53]. Very recently, NQO1 has been reported to be involved in metabolic adaptation and affect glycolysis and glutaminolysis in HCC [54]. Our results showed that enforced expression of the NQO1 isoform with short 3′UTR dramatically increased glucose uptake and lactate production. We further observed that such enforced expression dramatically decreased basal as well as maximum OCR, but increased ECAR. In HCC cells overexpression of NQO1 activated the PI3K/Akt and MAPK/ERK pathways and promoted metabolic adaptation [54]. Whether the CPSF6-induced high expression of NQO1 contributes to the activation of these pathways is unknown. In addition, the finding of CPSF6-associated metabolic adaptation was observed in the cell culture system, it remains unclear whether the similar scenario happens in vivo and requires further investigation.
Conclusions
In summary, our study described that CPSF6 exerts oncogenic effect on HCC cells by promoting 3′UTR shortening of NQO1 (Fig. 8d). To the best of our knowledge, this study, for the first time, suggested a link between APA and metabolism adaption in cancer. The functional and mechanistic results generated in our study might offer potential therapeutic targets for the treatment options against HCC.
Availability of data and materials
All data generated or analysed during this study are included in this published article [and its supplementary information files].
Abbreviations
- HCC:
-
Hepatocellular carcinoma
- IHC:
-
Immunological histological chemistry
- TCGA:
-
The cancer genome atlas
- OS:
-
Overall survival
- PFS:
-
Progress-free survival
- OE:
-
Overexpression
- KD:
-
Knockdown
- miRNA:
-
MicroRNA
- NS:
-
No significant difference
- HE staining:
-
Hematoxylin-eosin staining
- ECAR:
-
extracellular acidification rate
- OCR:
-
Oxygen consumption rate
- TNM:
-
Tumor node metastasis
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, Gores G. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018.
Marengo A, Rosso C, Bugianesi E. Liver Cancer: connections with obesity, fatty liver, and cirrhosis. Annu Rev Med. 2016;67:103–17.
Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16(10):589–604.
Craig AJ, von Felden J, Garcia-Lezana T, Sarcognato S, Villanueva A. Tumour evolution in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2020;17(3):139–52.
El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118–27.
Tian B, Manley JL. Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol. 2017;18(1):18–30.
Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11(9):597–610.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–33.
Hentze MW, Castello A, Schwarzl T, Preiss T. A brave new world of RNA-binding proteins. Nat Rev Mol Cell Biol. 2018;19(5):327–41.
Corley M, Burns MC, Yeo GW. How RNA-binding proteins interact with RNA: molecules and mechanisms. Mol Cell. 2020;78(1):9–29.
Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet. 2013;14(7):496–506.
Gruber AJ, Zavolan M. Alternative cleavage and polyadenylation in health and disease. Nat Rev Genet. 2019;20(10):599–614.
Mayr C. Evolution and biological roles of alternative 3'UTRs. Trends Cell Biol. 2016;26(3):227–37.
Masamha CP, Wagner EJ. The contribution of alternative polyadenylation to the cancer phenotype. Carcinogenesis. 2018;39(1):2–10.
Neve J, Patel R, Wang Z, Louey A, Furger AM. Cleavage and polyadenylation: ending the message expands gene regulation. RNA Biol. 2017;14(7):865–90.
Mayr C, Bartel DP. Widespread shortening of 3'UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138(4):673–84.
Lin Y, Li Z, Ozsolak F, Kim SW, Arango-Argoty G, Liu TT, Tenenbaum SA, Bailey T, Monaghan AP, Milos PM, et al. An in-depth map of polyadenylation sites in cancer. Nucleic Acids Res. 2012;40(17):8460–71.
Xiang Y, Ye Y, Lou Y, Yang Y, Cai C, Zhang Z, Mills T, Chen NY, Kim Y, Muge Ozguc F, et al. Comprehensive characterization of alternative Polyadenylation in human Cancer. J Natl Cancer Inst. 2018;110(4):379–89.
Xia Z, Donehower LA, Cooper TA, Neilson JR, Wheeler DA, Wagner EJ, Li W. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat Commun. 2014;5:5274.
Lembo A, Di Cunto F, Provero P. Shortening of 3'UTRs correlates with poor prognosis in breast and lung cancer. PLoS One. 2012;7(2):e31129.
Masamha CP, Xia Z, Yang J, Albrecht TR, Li M, Shyu AB, Li W, Wagner EJ. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature. 2014;510(7505):412–6.
Chen X, Zhang JX, Luo JH, Wu S, Yuan GJ, Ma NF, Feng Y, Cai MY, Chen RX, Lu J, et al. CSTF2-induced shortening of the RAC1 3'UTR promotes the pathogenesis of Urothelial carcinoma of the bladder. Cancer Res. 2018;78(20):5848–62.
Lai DP, Tan S, Kang YN, Wu J, Ooi HS, Chen J, Shen TT, Qi Y, Zhang X, Guo Y, et al. Genome-wide profiling of polyadenylation sites reveals a link between selective polyadenylation and cancer metastasis. Hum Mol Genet. 2015;24(12):3410–7.
Tan S, Li H, Zhang W, Shao Y, Liu Y, Guan H, Wu J, Kang Y, Zhao J, Yu Q, et al. NUDT21 negatively regulates PSMB2 and CXXC5 by alternative polyadenylation and contributes to hepatocellular carcinoma suppression. Oncogene. 2018;37(35):4887–900.
Ruegsegger U, Beyer K, Keller W. Purification and characterization of human cleavage factor Im involved in the 3′ end processing of messenger RNA precursors. J Biol Chem. 1996;271(11):6107–13.
Yang Q, Gilmartin GM, Doublie S. Structural basis of UGUA recognition by the Nudix protein CFI(m)25 and implications for a regulatory role in mRNA 3′ processing. Proc Natl Acad Sci U S A. 2010;107(22):10062–7.
Kim S, Yamamoto J, Chen Y, Aida M, Wada T, Handa H, Yamaguchi Y. Evidence that cleavage factor Im is a heterotetrameric protein complex controlling alternative polyadenylation. Genes Cells. 2010;15(9):1003–13.
Zhu Y, Wang X, Forouzmand E, Jeong J, Qiao F, Sowd GA, Engelman AN, Xie X, Hertel KJ, Shi Y. Molecular mechanisms for CFIm-mediated regulation of mRNA alternative Polyadenylation. Mol Cell. 2018;69(1):62–74 e64.
Ruegsegger U, Blank D, Keller W. Human pre-mRNA cleavage factor Im is related to spliceosomal SR proteins and can be reconstituted in vitro from recombinant subunits. Mol Cell. 1998;1(2):243–53.
Brown KM, Gilmartin GM. A mechanism for the regulation of pre-mRNA 3′ processing by human cleavage factor Im. Mol Cell. 2003;12(6):1467–76.
Binothman N, Hachim IY, Lebrun JJ, Ali S. CPSF6 is a clinically relevant breast Cancer vulnerability target: role of CPSF6 in breast Cancer. EBioMedicine. 2017;21:65–78.
Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9(4):357–9.
Fu Y, Sun Y, Li Y, Li J, Rao X, Chen C, Xu A. Differential genome-wide profiling of tandem 3′ UTRs among human breast cancer and normal cells by high-throughput sequencing. Genome Res. 2011;21(5):741–7.
Wang R, Nambiar R, Zheng D, Tian B. PolyA_DB 3 catalogs cleavage and polyadenylation sites identified by deep sequencing in multiple genomes. Nucleic Acids Res. 2018;46(D1):D315–9.
Rahman R, Hammoud GM, Almashhrawi AA, Ahmed KT, Ibdah JA. Primary hepatocellular carcinoma and metabolic syndrome: an update. World J Gastrointest Oncol. 2013;5(9):186–94.
Satriano L, Lewinska M, Rodrigues PM, Banales JM, Andersen JB. Metabolic rearrangements in primary liver cancers: cause and consequences. Nat Rev Gastroenterol Hepatol. 2019;16(12):748–66.
Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J, et al. Let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131(6):1109–23.
Fang Y, Xue JL, Shen Q, Chen J, Tian L. MicroRNA-7 inhibits tumor growth and metastasis by targeting the phosphoinositide 3-kinase/Akt pathway in hepatocellular carcinoma. Hepatology. 2012;55(6):1852–62.
Sun X, Dai G, Yu L, Hu Q, Chen J, Guo W. miR-143-3p inhibits the proliferation, migration and invasion in osteosarcoma by targeting FOSL2. Sci Rep. 2018;8(1):606.
Li N, Fu H, Tie Y, Hu Z, Kong W, Wu Y, Zheng X. miR-34a inhibits migration and invasion by down-regulation of c-met expression in human hepatocellular carcinoma cells. Cancer Lett. 2009;275(1):44–53.
Ross D, Kepa JK, Winski SL, Beall HD, Anwar A, Siegel D. NAD(P)H:quinone oxidoreductase 1 (NQO1): chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem Biol Interact. 2000;129(1–2):77–97.
Fang S, Zhang D, Weng W, Lv X, Zheng L, Chen M, Fan X, Mao J, Mao C, Ye Y, et al. CPSF7 regulates liver cancer growth and metastasis by facilitating WWP2-FL and targeting the WWP2/PTEN/AKT signaling pathway. Biochim Biophys Acta Mol Cell Res. 1867;2020(2):118624.
Yang Y, Zhang Y, Wu Q, Cui X, Lin Z, Liu S, Chen L. Clinical implications of high NQO1 expression in breast cancers. J Exp Clin Cancer Res. 2014;33:14.
Thapa D, Meng P, Bedolla RG, Reddick RL, Kumar AP, Ghosh R. NQO1 suppresses NF-kappaB-p300 interaction to regulate inflammatory mediators associated with prostate tumorigenesis. Cancer Res. 2014;74(19):5644–55.
Li WY, Zhou HZ, Chen Y, Cai XF, Tang H, Ren JH, Wai Wong VK, Kwan Law BY, Chen Y, Cheng ST, et al. NAD(P)H: Quinone oxidoreductase 1 overexpression in hepatocellular carcinoma potentiates apoptosis evasion through regulating stabilization of X-linked inhibitor of apoptosis protein. Cancer Lett. 2019;451:156–67.
Li Z, Zhang Y, Jin T, Men J, Lin Z, Qi P, Piao Y, Yan G. NQO1 protein expression predicts poor prognosis of non-small cell lung cancers. BMC Cancer. 2015;15:207.
Kandala DT, Mohan N, A V, A PS, G R, Laishram RS. CstF-64 and 3′-UTR cis-element determine star-PAP specificity for target mRNA selection by excluding PAPalpha. Nucleic Acids Res. 2016;44(2):811–23.
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33.
Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85–95.
De Matteis S, Ragusa A, Marisi G, De Domenico S, Casadei Gardini A, Bonafe M, Giudetti AM. Aberrant metabolism in hepatocellular carcinoma provides diagnostic and therapeutic opportunities. Oxidative Med Cell Longev. 2018;2018:7512159.
Piccinin E, Villani G, Moschetta A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: the role of PGC1 coactivators. Nat Rev Gastroenterol Hepatol. 2019;16(3):160–74.
Dimri M, Humphries A, Laknaur A, Elattar S, Lee TJ, Sharma A, Kolhe R, Satyanarayana A. NAD(P) H Quinone dehydrogenase 1 ablation inhibits activation of the Phosphoinositide 3-kinase/Akt serine/threonine kinase and mitogen-activated protein kinase/extracellular signal-regulated kinase pathways and blocks metabolic adaptation in hepatocellular carcinoma. Hepatology. 2020;71(2):549–68.
Acknowledgments
We thank Dr. Jun Wu of Center for Bioinformatics and Computational Biology, East China Normal University, China and Ms. Pengxiao Li of Shanghai Center for Systems Biomedicine, Shanghai Jiao Tong University, China for the helpful discussion.
Funding
This work was supported by National Key R&D Program of China (2018YFC1003501); The Science and Technology Commission of Shanghai Municipality (17JC1400804); Interdisciplinary Program of Shanghai Jiao Tong University (YG2019ZDA25); Special Fund Project for Youth Scientific Research of the Key Laboratory of Systems Biomedicine (Ministry of Education) (KLSB2020QN-07) and the National Natural Science Foundation of China (81672609, 31671299 and 81720108017).
Author information
Authors and Affiliations
Contributions
ST, TZ, JS and XZ contributed to the conception and design. ST, MZ, and XS performed most of the experiments and interpretation of the data. KD, QZ and YK contributed to the analysis of the data. QG, HW, and ZW advised on the experimental design. ST and XS wrote the manuscript. MZ, KD and QZ helped to draft the manuscript. TZ, JS, and XZ critically revised the manuscript. All authors read and approve the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Human liver tissues were obtained from the First Affiliated Hospital of Anhui Medical University (Hefei, China). The study was approved by the Biomedical Ethics Committee of Anhui Medical University and informed consent was obtained from each patient. Humane care of animals was objected to the guideline for the Care and Use of Laboratory Animals published by the National Academy of Science.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1 Supplementary Fig. S1.
The expression of CPSF6 in HCC. Western blot assay for the CPSF6 expression in 20 paired surrounding non-tumor (N) and HCC tumor (T) tissues. β-actin was used as internal control.
Additional file 2 Supplementary Table S1.
Clinicopathologic correlation of CPSF6 expression in HCCs. The expression level of CPSF6 protein was detected by immunohistochemistry.
Additional file 3 Supplementary Fig. S2.
CPSF6 accelerates the growth of HepG2 cells. A, CPSF6 knockdown efficiency in HepG2 cells with two CPSF6 specific shRNAs (shCPSF6–1 and shCPSF6–2) was examined by western blotting. β-actin was used as internal control. B, Proliferation curves of HepG2 cells with stable expression of CPSF6 shRNA or the control (shCtrl) were shown. C, The cell viability of CPSF6-silencing HepG2 cells and HepG2 cells (the control) was examined by MTT assay. D, Colony formation analysis of HepG2 cells. **p < 0.001, Student’s t test.
Additional file 4 Supplementary Fig. S3.
CPSF6 promotes migration and invasion of HepG2 cells. A, Effects of CPSF6-silencing on HepG2 cell migration. B, Effects of CPSF6-silencing on HepG2 cell invasion. Scale bars, 100 μm. **p < 0.001, Student’s t test.
Additional file 5 Supplementary Table S2.
The sequencing statistics of 3 T-seq data.
Additional file 6 Supplementary Table S3.
The 3′UTR shortened genes in CPSF6-OE HL-7702 cells and the 3′UTR lengthened genes in CPSF6-KD Huh7 cells.
Additional file 7 Supplementary Table S4.
The list of genes with altered 3’UTR shared by HL-7702 and Huh7 cells.
Additional file 8 Supplementary Fig. S4.
The expression of NQO1 in HCC. Western blot assay for the NQO1 expression in 20 paired surrounding non-tumor (N) and HCC tumor (T) tissues. β-actin was used as internal control.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tan, S., Zhang, M., Shi, X. et al. CPSF6 links alternative polyadenylation to metabolism adaption in hepatocellular carcinoma progression. J Exp Clin Cancer Res 40, 85 (2021). https://doi.org/10.1186/s13046-021-01884-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-021-01884-z