
Abstract
The liver is essential for metabolic homeostasis. The onset of liver cancer is often accompanied by dysregulated liver function, leading to metabolic rearrangements. Overwhelming evidence has illustrated that dysregulated cellular metabolism can, in turn, promote anabolic growth and tumor propagation in a hostile microenvironment. In addition to supporting continuous tumor growth and survival, disrupted metabolic process also creates obstacles for the anticancer immune response and restrains durable clinical remission following immunotherapy. In this review, we elucidate the metabolic communication between liver cancer cells and their surrounding immune cells and discuss how metabolic reprogramming of liver cancer impacts the immune microenvironment and the efficacy of anticancer immunotherapy. We also describe the crucial role of the gut–liver axis in remodeling the metabolic crosstalk of immune surveillance and escape, highlighting novel therapeutic opportunities.
Similar content being viewed by others

Background
Primary liver cancer (PLC) is one of the most common malignancies worldwide, with hepatocellular carcinoma (HCC) accounts for 75–85% and intrahepatic cholangiocarcinoma (iCCA) for 10–15% (Other rare PLCs are not discussed in this Review) [1]. The heterogeneous nature of PLC challenges the development of new treatment strategies, especially for patients at advanced stage. Systemic agents, such as multi-kinase inhibitors, have achieved great progress since 2007, and combined anti-vascular endothelial growth factor (VEGF) or tyrosine kinase inhibitors (TKIs) with immune checkpoint blockades (ICBs) has been endorsed as the new standard of care in first-line treatment for advanced HCC [2]. The ICB-based regimen has also obtained impressive survival benefits in two recent phase 3 trials for the treatment of advanced biliary tract cancer, including iCCA [3, 4]. However, most of these treatment options provide limited extensions of overall survival, often with miserable life qualities. A better understanding of the complex microenvironment within PLC may help in developing novel therapeutic interventions to augment immune-based therapies.
While the global incidence of hepatic virus B or C (HBV or HCV)-related PLCs is decreasing due to successful interventions of vaccinations or anti-viral treatments, obesity-related disorders and associated morbidity/mortality are continuously rising, especially in Western countries [2]. The liver is not only an immune-privileged organ with a high tolerance for gastrointestinal tract-derived antigens, but also the central hub for various metabolic processes and the maintenance of homeostasis. This underscores the importance of metabolic rearrangements during hepatic carcinogenesis [1]. Increasing evidence reveals that the liver receives gut bacterial metabolites through the blood supply from the intestine. Changes in the gut microbiome disturb immune cell infiltration and function in the tumor microenvironment (TME) of PLC, potentially affecting the efficacy of current immunotherapies [5].
Metabolism, a fundamental biological process of a living cell, converts nutrients to generate extensive energy (i.e., ATP), redox equivalents (i.e., NADPH), and macromolecules (i.e., lipids, proteins, and nucleic acids). Tumor cells often exhibit a high dependence on a reprogrammed metabolic state for stress adaptation and infinite proliferation, which can be leveraged for non-invasive cancer diagnosis [6,7,8]. Unlike normal cells, which usually acquire energy through oxidative phosphorylation (OXPHOS), tumor cells tend to choose glycolysis even in the presence of oxygen, also known as “Warburg effect” [9]. Although aerobic glycolysis is way more inefficient than OXPHOS (2 ATP vs. 36 ATP per glucose), the ATP production rate of aerobic glycolysis is much higher, which better satisfies the greedy reproduction of neoplastic cells [10]. On the other hand, excessive lactate generated by aerobic glycolysis also fuels neighboring oxygenated cells, leading to a metabolic symbiosis between glycolytic and oxidative metabolism [11,12,13]. Due to significant glucose uptake in PLC bulk tissues, 18F-2-deoxyglucose (18FDG) positron emission tomography (PET) imaging is widely used for PLC diagnosis and progression monitoring [14]. In addition to glucose metabolism, other central metabolic pathways are often reprogrammed, leading to dysregulated nutrient depletion, oncometabolite accumulation, and signaling pathway perturbations in the TME [15].
The metabolic changes during PLC progression contribute to identifying pathogenic mechanisms and therapeutic targets and developing novel prognostic and diagnostic biomarkers. Just like the TME, liver cancer metabolism is heterogeneous, encompassing metabolic signatures from tumor, stromal, and immune cells. Emerging evidence indicates that metabolic alterations in tumor cells affect the composition and function of surrounding cells [16, 17]. With the advent of immunometabolism in cancer treatment [18,19,20,21,22], greater attention should be given to the interplay of metabolism-related immune signaling. In this review, we focus on how metabolically rewired liver tumor cells cultivate an immunosuppressive microenvironment through tumor immune metabolic interactions. We also discuss the impact of the gut–liver axis on the liver microenvironment and ICB-based immunotherapies. The purpose of this review is to link recent findings on the crosstalk between liver cancer metabolism and immunometabolism, potentially revealing novel therapeutic opportunities.
General characteristics of liver cancer metabolism
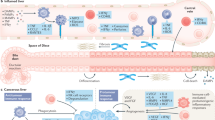
The liver is the largest internal organ for controlling metabolism, and metabolic disruption is closely associated with hepatocarcinogenesis. Common risk factors for PLC include HBV/HCV infection, alcohol abuse, obesity, metabolic dysfunction-associated steatohepatitis (MASH), and metabolic dysfunction-associated steatotic liver disease (MASLD). In addition, exposure to aflatoxin promotes the development of HCC, and liver fluke infection, biliary duct cysts, and primary sclerosing cholangitis (PSC) are established risk factors for iCCA [1]. These risk factors may promote the initiation and progression of PLC through metabolic reprogramming [1, 23]. Therefore, understanding metabolic alterations in liver cancer is vital for identifying pathogenic mechanisms and exploring therapeutic targets (Fig. 1).

Metabolic alterations in PLC. HBV: hepatitis B virus; HCV: hepatitis C virus; MASH: metabolic dysfunction-associated steatohepatitis; MASLD: metabolic dysfunction-associated steatotic liver disease; OE: overexpression; Mut: mutation; PPP: pentose phosphate pathway; HBP: hexosamine biosynthesis pathway; FASN: fatty acid synthase; and SCD: stearoyl-CoA desaturase
Metabolic classification aids in understanding the heterogeneous metabolic microenvironment and developing personalized interventions. Systemic analysis using multi-omics has been crucial in outlining the varied metabolic landscape of HCC. By integrating genomics, transcriptomics, and proteomics data retrieved from several public datasets, genome-wide metabolic models (GEMs) stratified HCC patients into three prognostic subgroups with significant differences in dysregulated kynurenine metabolism (iHCC1), WNT/β-catenin-related lipid metabolism (iHCC2), and PI3K/AKT/mTOR signaling (iHCC3) [24]. Consistent with the histological features [25], both iHCC1 and iHCC2 exhibit hepatocyte differentiation and maturation, whereas iHCC3 is associated with proliferation and immune activation [24]. Multi-omics studies have nominated metabolic pathways as the most dramatic alterations in HCC and iCCA, compared with normal adjacent tissues [26,27,28,29]. During liver cancer progression, typical hepatocyte metabolic functions, such as gluconeogenesis, bile acid (BA) metabolism, detoxication, and ureagenesis-ammonia, are diminished. This decline is accompanied by an increase in tumor malignancy [30, 31], most likely due to the de-differentiation from functional hepatocytes to HCC cells. Recently, we identified two metabolic subtypes in 65 human liver cancer organoids through multi-omics profiling, which complements our understanding of HCC tissue metabolism. Glucose-6-phosphate dehydrogenase (G6PD) was identified as a potential target in the subtype with an enriched drug metabolism pathway, consistent with the previous results [32,33,34,35]. Meanwhile, classifying patients based on a single metabolic pathway can guide personalized therapy. For instance, HCC patients were categorized into three metabolic subtypes (F1, F2, and F3) based on the expression pattern of 42 fatty acid degradation (FAD) genes, revealing distinct clinical/molecular characteristics and treatment vulnerabilities. Interestingly, the F1 subtype with the lowest expression levels of FAD genes shows a high degree of immune infiltration, designated “hot” tumor. Thus, HCC mouse models derived from the F1 subtype are responsive to anti-PD-1 (programmed cell death protein 1) therapy, in contrast with mouse models derived from the F2 and F3 subtypes [36]. Taken together, systems biological approaches in metabolic signature deconvolution can illuminate metabolic heterogeneity and identify potential metabolic targets for PLC (Fig. 1).
Systemic analysis focusing on metabolic gene expressions and non-targeted metabolic profiling has shown that aerobic glycolysis, lipid metabolism, and amino acid metabolism are the main metabolic alterations in HCC tissues [31, 37]. Cancer cells often face hypoxic and hypo-nutrient environments, necessitating metabolic rearrangement to satisfy energy demands and biomass synthesis. It has been demonstrated that liver cancer cells typically utilize glycolysis under hypoxic conditions to produce lactate via lactic dehydrogenase (LDH) [38]. In addition to creating an acidic microenvironment that promotes tumor progression, lactate accumulation also causes lactylation of adenylate kinase 2 (AK2) at K28, compromising its kinase activity and disrupting energy homeostasis in HCC cells, thereby facilitating tumor proliferation, invasion, and metastasis as shown in several xenograft mouse models [39]. Based on several retrospective analyses [40,41,42], high serum LDH levels are associated with poor prognosis after curative resection or standard therapies in both HCC and iCCA. Hypoxia also induces micropinocytosis for nutrient scavenging via the hypoxia-inducible factor (HIF)/EH domain-containing protein 2 (EHD2) pathways in several HCC cell lines and mouse models [43]. Other glucose metabolic pathways, such as the pentose phosphate pathway (PPP) and the hexosamine biosynthetic pathway (HBP), are also more active in HCC tissue compared to normal adjacent tissues [44,45,46,47] (Fig. 1).
In high-fat diet (HFD)-induced HCC, or steatohepatitic HCC, the fatty acid oxidation (FAO) pathway tends to be downregulated to protect HCC cells from lipotoxicity [48]. Concordantly, de novo lipogenesis gradually increases from normal liver tissue to liver tumors and is generally associated with advanced HCC and worse patient prognoses [49,50,51,52]. However, an independent study in a diethylnitrosamine (DEN)-induced mouse model showed that liver-specific knockout of acetyl-CoA carboxylase (ACC) inhibiting de novo lipogenesis accelerates HCC progression by activating antioxidant defense. This discrepancy may be attributed to differences between clinical samples and preclinical models of HCC, highlighting the need for a thorough exploration of tumor-driving events and metabolic plasticity [53]. Lipid metabolic pathways, including fatty acid synthase (FASN) and stearoyl-COA desaturase (SCD) signaling, also sustain cancer stem cells in HCC, contributing to metastasis and drug resistance [54]. High-cholesterol diets induce HCC in mice, partly through dysregulation of metabolism and calcium signaling [55,56,57]. Integrated proteomics and phosphoproteomics have revealed that targeting sterol O-acyltransferase 1 (SOAT1) to reduce cholesterol content in plasma membranes presents effective treatment options for early-stage HCC patients, which was further verified in a patient-derived tumor xenograft mouse model [58]. Conversely, high serum cholesterol levels are linked with better patient outcomes by inhibiting tumor metastasis [59], implying that cholesterol distribution and homeostasis significantly influence HCC tumorigenesis (Fig. 1).
Moreover, numerous studies indicate enhanced amino acid metabolism in liver tumors compared to non-tumor tissues [1, 31, 37, 60, 61]. Sustained urea cycle repression in liver cancer shifts metabolism from arginine production to pyrimidine biosynthesis. HCC cells depend on external arginine sources, with arginine restriction inducing a general control nonderepessible 2 (GCN2) kinase-related stress response. GCN2 suppression leads to cell senescence and increases sensitivity to senolytic treatment both in vitro and in vivo. Thus, combining GCN2 inhibition with senolytic agents could be an effective treatment strategy in arginine-deprived HCC cells [62]. In an mTOR-driven HCC mouse model, tumor cells also increased arginine import and reduced its conversion to polyamines, driving oncogenic metabolism via the arginine-binding factor RNA-binding motif protein 39 (RBM39). Targeting RBM39 instead of circulating arginine may offer a way to reverse the oncogenic pathway triggered by high arginine pools in HCC cells, thereby avoiding the adverse side effects of circulating arginine-depleting therapy [63]. Glutamine, the most abundant amino acid in human blood, is a key carbon source for de novo lipogenesis in mitochondrial dysfunctional HCC [64]. Glutamine addiction in glutamine synthetase (GS)-overexpressing HCC supports mTOR-dependent cell proliferation and survival in clinically relevant HCC models [65]. Additionally, the glutamate-to-proline biosynthetic flux is elevated in tumor tissues, promoting HCC cell proliferation and tumor growth in both tumor models and regenerating tissues [66]. Folate-mediated one-carbon (1C) metabolism contributes to the availability of various building blocks for tumor cell proliferation [67,68,69,70], and the expression of central enzymes involved in 1C metabolism is largely dysregulated in PLC [71, 72]. Serine, glycine, and methionine metabolism is tightly linked to the generation of 1C units. In HCC cells, glycine-derived 1C units support purine and pyrimidine biosynthesis and tumor progression through glycine cleavage system (GCS) flux [73]. Recent findings have also highlighted the promotion of tumor development by dietary folate supplementation through the integration of methionine and 1C metabolism in the HCC mouse model induced by DEN/HFD [74].
We and others have identified numerous putative driver genes that reshape PLC metabolism [29, 30, 75,76,77,78,79] (Table 1). The proto-oncogene Myc is overexpressed in nearly 70% of viral and alcohol-related HCC. Studies in HCC cell lines show that MYC overexpression upregulates glucose transporters GLUT1 and GLUT2, hexokinase HK2, and pyruvate kinase isoforms PKL/PKM, thereby enhancing tumor glycolysis. High levels of GLUT1 expression are also associated with poor prognosis in both HCC and iCCA [80,81,82,83,84]. Under glucose/glutamine-deprived conditions, overexpressed cMYC also activates the serine biosynthesis pathway to adopt metabolic switch through transcriptionally upregulated the final rate-limiting enzyme phosphoserine phosphatase in both HCC cell lines and xenograft mouse models [85]. Furthermore, the “Warburg effect” is promoted by TP53 mutations and PI3K/AKT/mTOR pathway activation in PLCs through upregulating related glycolytic enzymes [86, 87]. Wnt-β-catenin signaling is often hyperactivated, promoting PLC growth and dissemination [88]. β-catenin (encoded by CTNNB1) oncogenic activation in HCC cells induces FAO through the transcription factor peroxisome proliferator-activated receptor α (PPARα) [89]. Additionally, CTNNB1-mutated HCC cells rely on glutamine synthetase-dominated mTORC1 signaling for metabolism [65]. In an iCCA patient cohort, KRAS alterations lead to GLUT1-mediated glycolysis and poor patient outcomes [90]. Significant alterations in metabolic genes, including ALB, APOB, and IDH1/2, also induce metabolic changes in PLC [77, 91]. Leveraging genetically engineered mouse models of iCCA, IDH mutations were shown to increase the production of D-2-hydroxyglutarate (D-2-HG), affecting α-ketoglutarate (αKG)-dependent dioxygenases involved in DNA repair and epigenetic remodeling [92]. Thus, oncogenic alterations can also drive metabolic rearrangements in PLC (Fig. 1).
Metabolic interactions between tumor cells and the TME
The cancer-immunity cycle (CIC) describes the consecutive anti-tumor responses of the immune system, including the release of cancer cell antigens, the presentation of cancer-associated antigens, the priming and activation of T-cell, and their trafficking to the tumor site, followed by infiltration into the tumor and stroma, recognition of the tumor cells, and, ultimately, killing of the tumor cells [93]. High metabolic turnover is the typical feature of rapidly proliferating and differentiating cells. Metabolic derangements within the TME are increasingly recognized as one of the most important factors halting the CIC [94]. Tumor cells orchestrate metabolism to meet their prodigious anabolic demands, creating a microenvironment featured by hypoxia, acidification, and essential nutrient depletion for adjacent immune cells. Generally, intense nutrient competition occurs between tumor cells and anti-tumor immune cells [18]. Glucose, amino acids, and fatty acids are critical energy sources not only for tumors, but also for proliferative cells, particularly anti-tumor immune cells [95]. Tumor cells often extract these nutrients from the tumor interstitial fluid (TIF) to hinder tumorolytic activities [10, 16, 96,97,98,99,100,101]. Meanwhile, the aberrant consumption of macromolecules and metabolic substances leads to the production of numerous byproducts, and some of them could be harmful to immunosurveillance [10]. Oncometabolites, such as lactic acid [102,103,104,105,106,107,108,109,110,111,112,113,114], kynurenine [115,116,117], adenosine [117], 2-HG [118, 119], and prostaglandin E2 (PGE2) [120], typically antagonize the anti-tumor immune response and/or promote the immunosuppressive activities of TME components, ultimately leading to immune evasion. Currently, the monocarboxylate transporter MCT1 inhibitor AZD3965 (for lactate symporter) [121], the indoleamine 2,3-dioxygenase IDO1 inhibitor Indoximod/Epacadostat (for kynurenine synthesis) [122], the CD73 inhibitor Oleclumab (for adenosine conversion) [123], the isocitrate dehydrogenase IDH1/2 inhibitor Enasidenib/Ivosidenib (for 2-HG production) [124], and the cyclooxygenase COX-2 inhibitor Celecoxib (for PGE2 accumulation) [125, 126] are being evaluated or approved to target cancer metabolism, aiming to enhance the efficacies of current therapies [127].
Metabolically reprogrammed tumors can foster an immune-suppressed TME by modulating the expression of signaling molecules such as immune checkpoints, chemokines, cytokines, etc. On the other hand, the pro-tumor attributes trigger dysregulated signaling or metabolic pathways, leading to the metabolic reprogramming of immune cells. Notably, anti-tumor immune cells often exhibit metabolic profiles complementary to their pro-tumor counterparts. For instance, immune-activated cells, including effector T (Teff) cells, nature killer (NK) cells, dendritic cells (DCs), and inflammatory tumor-associated macrophages (TAMs), primarily exhibit high glycolysis activity. In contrast, immunosuppressive cells such as regulatory T (Treg) cells, TAMs, and myeloid-derived suppressor cells (MDSCs) typically rely on OXPHO or FAO to sustain their function [18] (Table 2). This metabolic heterogeneity underpins the immunosuppressive TME and supports the unrestrained growth of tumor cells.
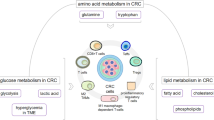
Nutrient competition in liver cancer
Glutamine metabolism is essential for both proliferative cancer cells and activated CD8 + T cells. In the TCGA HCC cohort, glutamine metabolism-related gene expression scores inversely correlate with patient prognoses. In the glutamine-dominant HCC subgroup, CD8 + Teff cells shift to metabolizing exogenous lipids due to limited access of glutamine, reducing their quantity and cytolytic function [128]. In vitro co-culture assays have indicated that glutamine deprivation in the TME induces mitochondrial damage and CD8 T-cell apoptosis, impairing their tumorolytic function [129]. Apart from glutamine, glucose is critical for the metabolic fitness of tumor-infiltrating cytotoxic CD8 + T cells [18]. The results of a recent clinical trial demonstrated the promising efficacy of combined IFNα and ICB therapy in patients with advanced melanoma [130]. Our group further demonstrated that IFNα therapy could strongly enhance the efficacy of ICB in both patients with HCC and preclinical HCC mouse models. Mechanistically, IFNα therapy inhibits HIF1α signaling to reduce glucose consumption in tumor cells. The consequent accumulation of glucose in the TME stimulates the expression of the costimulatory molecule CD27 via mTOR–FOXM1 signaling in CD8 + T cells, thereby reinforcing the functions of cytotoxic T cells in both immunocompetent orthotopic and spontaneous HCC models [131].
Nutrient depletion in liver cancer also influences the shift from anti-tumor M1-like macrophages (M1φ) to pro-tumor M2-like macrophages (M2φ). Compared to M1φ, M2φ tends to polarize under low ferrous iron levels [132]. The hypoxic HCC microenvironment prompts tumor cells to compete with macrophages for iron through increased transferrin receptor (TFRC) expression, the primary receptor for transferrin‑mediated iron uptake. This iron competition culminates in an M2-like TAM polarization in vitro [133]. Taken together, nutrient competition between liver cancer cells and immune cells can either weaken anti-tumor immunity or enhance pro-tumor activities, contributing to the initiation and progression of liver cancer (Fig. 2). However, given the diversity of metabolites and nutrients, the precise impact of their depletion on immune cells in PLC requires further elucidation.

Immune regulation by nutrient competition and oncometabolite production. SAM: S-adenosylmethionine and MTA: 5-methylthioadenosine
The effect of liver cancer metabolites on immune cells
Lactate
The well-known “Warburg effect” rearranges glucose metabolism to produce excessive lactic acid, which has emerged as an important regulator in promoting immune evasion in PLC [134, 135]. For instance, upregulated MCT4 expression in HCC cells contributes to lactate exportation and subsequent TME acidification, ultimately leading to CD8 + T-cell exhaustion and M2φ polarization [136,137,138]. Therefore, targeting MCT4 can reinvigorate anti-tumor immunity in HCC and may be a promising therapeutic strategy to improve the efficacy of ICB-based therapy. In vitro, lactate also strengthens Treg cell functionality through Lys72 lactylation on MOESIN. This contributes to the activation of TGF-β signaling and the expression of key transcription factor FOXP3. Considering the critical role of Treg cells in immunotherapy, lactylation of MOESIN in Treg cells may predict the response of anti-PD-1 therapy in HCC [139]. Mitochondria are involved in cancer energy metabolism, and mitoribosome defects in HCC have been associated with an aggressive phenotype. Mechanistically, hepatic mitoribosomal defects induce elevated reactive oxygen species (ROS) and lactate production to circumvent a hostile environment for cytolytic T cells [140]. Lenvatinib, a multi-kinase inhibitor approved for systemic first-line treatment of HCC [141], promotes neutrophil recruitment by inducing CXCL2 and CXCL5 secretion in the TME. Simultaneously, tumor-derived lactate induces programmed cell death ligand 1 (PD-L1) expression in infiltrated neutrophils through the MCT1/NF-κB/COX-2 pathway, thus counteracting the efficacy of lenvatinib monotherapy in HCC mouse models [142].
Lipid
In addition to glucose metabolism and lactate, anti-liver cancer immunity is also largely dismantled by lipid metabolism and its products. Aberrant lipid metabolism in MASLD promotes hepatocarcinogenesis partly through intrahepatic CD4 + T-cell deprivation. Mechanistically, MASLD-associated linoleic acid production predominantly causes the accumulation of mitochondrial-derived ROS in mouse models and human samples. This mediates selective loss of intrahepatic CD4 + T lymphocytes due to their great mitochondrial mass [143]. Sirtuin 5 (SIRT5) is a metabolic regulator that removes succinyl, malonyl, and glutaryl groups from the lysine residues of mitochondrial and peroxisomal metabolic enzymes. Sun et al. reported that SIRT5 expression is repressed in tumor cells, which leads to aberrant BA biosynthesis in the peroxisome and subsequent M2φ-induced immunosuppression in oncogene-induced HCC mouse models [144]. PGE2, a bioactive lipid generated from arachidonic acid, has recently been implicated in immune evasion through multiple mechanisms [145]. In HCC cells, higher expression of COX-2, the rate-limiting enzyme of the PGE2 production, induces M2φ polarization to inhibit CD8 T-cell function in a multi-cellular co-culture system [146]. Recently, we also found that KRAS mutations upregulate COX-2 expression to promote PGE2 production in vitro. This results in an immunosuppressive TME dominated by excessive neutrophil infiltration and contributes to poor prognosis in iCCA [147].
Amino acids and adenosine
Other metabolites, such as amino acids and adenosine, also play pivotal roles in investigating the immune-suppressed TME. Intracellular arginine concentrations directly determine the metabolic fitness and functionality of activated T cells [100]. Chronic viral infection activates hepatocyte-intrinsic type I interferon (IFN-I) responses to break the urea cycle, leading to decreased arginine/ornithine ratios in the circulation and subsequently suppressed virus-specific CD8 + T-cell responses in chronic lymphocytic choriomeningitis virus (LCMV)-infected mice [148]. Other amino acid metabolisms, such as the methionine recycling pathway, have also been reported in HCC immune modulation. S-adenosylmethionine (SAM) and 5-methylthioadenosine (MTA) are two critical metabolites for methionine salvage. They promote T-cell exhaustion and exert significant impacts on HCC progression in both human samples and mouse models [6]. In HCC cells, hypoxia contributes to adenosine accumulation and extracellular secretion. This results in adenosine-mediated immunosuppressive roles on T cells and myeloid cells [149, 150]. Tumor cell-derived adenosine also synergizes with autocrine granulocyte–macrophage colony-stimulating factor (GM-CSF) secreted from activated TAM to promote their proliferation, thus maintaining the Mφ pools in HCC [151]. Collectively, the metabolic by-products produced by dysregulated cancer cells could directly play a profound role in immune cells within the TME (Fig. 2).
Dysregulated PLC metabolism acts as signaling molecules regulating the TME
Hypoxia
Metabolic reprogramming in PLC cells can also impact the TME through signaling molecules. Chen et al. proposed that sorafenib treatment increases intra-tumoral hypoxia, promoting immunosuppression through the stromal cell-derived factor 1α (SDF1α)-CXCR4 axis-induced Treg cells/M2φ accumulation in orthotopic HCC mouse models [152]. In hypoxia-high HCC regions of patient samples, the upregulation of tumor-derived chemokines, such as CCL20 and CCL5, leads to excessive Treg cell and cDC2 infiltration. Subsequently, in vitro assays demonstrated that the direct interaction of infiltrated Treg cells with cDC2 mediates HLA-DR loss, a critical antigen presentation molecule required for anti-tumor T-cell activation [153]. Additionally, in HCC patient samples, hypoxia-inducible gene 2 (HIG2), a HIF-1 target gene, fosters IL-10 secretion by HCC cells. This suppresses NK cell cytotoxicity through the activation of the STAT3 signaling pathway in co-culture assays [154]. Another study showed that hypoxic TME drives NK cell loss and dysfunction via mTOR-GTPase dynamin-related protein 1 (Drp1) mitochondrial fragmentation, leading to HCC immune evasion in human liver cancer and mouse liver models [106]. On the other hand, hypoxia often promotes tumor cell phagocytosis via CD103 + DCs, further recruiting and activating anti-tumor NK cells. However, tumor cells upregulate the innate immune checkpoint CD47 under hypoxic conditions, counteracting NK cell-mediated cytotoxicity with the “do not eat me” signal. Thus, blocking CD47 on the cell surface enhances NK cell-mediated anti-tumor immunity in the hypoxic microenvironment of HCC [155]. In iCCA, hypoxic surroundings can induce HIF1α and its downstream PD-1/PD-L1 pathway, creating an immunosuppressive TME [156]. Due to the crucial role of hypoxia in regulating intratumor immune components, targeting hypoxia depletion synergistically with current therapies (like TKIs plus ICBs) is warranted to advance the systemic treatment of unresectable HCC (Fig. 3).

Metabolic signaling-mediated immune escape in tumor cells and immune cells. HA: hyaluronan; OSM: oncostatin M; NET: neutrophil extracellular traps; FN1: fibronectin 1; αKG: α-ketoglutarate; and D-2-HG: D-2-hydroxyglutarate
Lipid
In addition to hypoxia, lipid metabolism is closely involved in the signaling molecules between PLC cells and the TME. Compared with HBV-HCC, MASLD-HCC is associated with a high incidence of mutations in CTNNB1 over TP53. This shift in the driver mutations results in immune exclusion through the repression of the TNF receptor superfamily member 19 (TNFRSF19)-mediated senescence-associated secretory phenotype (SASP), as shown in a syngeneic immunocompetent mouse model [157]. Apart from MASLD, HCC patients with etiologies of HCV infections or alcohol abuse also harbor unique genetic variations, which may impact the metabolic reprogramming and the TME [75, 158]. Multi-omics study showed lipid accumulation in HCC elevates PD-L1 expression, inducing an immunosuppressive TME [28]. Due to excessive lipid accumulation, obesity heightens the risk of HCC, particularly in men, though the underlying molecular mechanism remains unclear [159, 160]. In MASH-related HCC mouse models, the androgen receptor (AR)-driven oncogene, cell cycle-related kinase (CCRK), combined with obesity-induced IL-6/STAT3 signaling, induces lipid metabolic reprogramming and MDSC-dominated immunosuppression [161]. Tumor cells increase Piwi-like RNA-Mediated Gene Silencing 1 (PIWIL1) to boost oxygen consumption and energy production through fatty acid metabolism, advancing HCC progression. Meanwhile, PIWIL1 regulates the secretion of Complement C3 to mediate interaction between HCC cells and MDSC, promoting the immunosuppressive cytokine IL10 in the TME [162]. Concerning the crucial role of lipid metabolism in the TME, serum lipids can predict the efficacy of anti-PD-1 therapy in iCCA patients, with apolipoproteinA-1 (APOA1) and triglycerides (TG) as notable independent predictors [163] (Fig. 3).
Epigenetics
Metabolic alterations accompanying hepatocarcinogenesis may also prompt epigenetic reprogramming of immune cells through the accumulation of epigenetically regulated metabolites. For instance, NAD + metabolism triggers PD-L1 expression on tumor cells, impairing the cytolytic activity of PD-1 + T cells through αKG-mediated epigenetic modifications [164]. In iCCA, IDH mutations produce the oncometabolite D-2-HG, suppressing the TET2-dependent epigenetic response to CD8 T-cell-derived IFN-γ in tumor cells. Consequently, the IDH1/2 inhibitor AG120 synergizes with ICBs for advancing immunotherapy in the treatment of mIDH1-driven genetically engineered mouse models [165]. In a phase 3 clinical trial for mIDH1 iCCA, Ivosidenib/AG120 was found to significantly improve the progression-free survival (PFS) of patients compared with placebo, although the absolute improvement in the median PFS appeared modest [166, 167] (Table 3). These findings underscore that metabolic reprogramming of tumor cells acts as signaling molecules, contributing to immunosuppressive pathway deregulation in PLC (Fig. 3). Therefore, targeting these molecules is a viable treatment option for related metabolic diseases.
Metabolic reprogramming of immune cells diminishes anti-tumor immunity
Recent advances in cancer immunology have highlighted metabolic fuels/nutrients as a fourth signal beyond the three-signal model for effective T-cell priming and differentiation [179]. In the context of tumor-dysregulated metabolism, tumor-infiltrating immune cells inevitably experience metabolic stress. Consequently, they adapt metabolic characteristics to fulfill their duties [19]. In MASH-associated HCC mouse models, neutrophil extracellular traps (NETs) interact with naïve CD4 + T cells to facilitate its mitochondrial OXPHOS through TLR4 expression, contributing to their differentiation into immunosuppressive Treg cells [180]. Following anti-PD1 treatment, MASH also promoted aberrant activation of PD-1 + CD8 + T cells, leading to tissue damage, immune anergy, and reduced response to immunotherapy in preclinical HCC models [181]. Another study using multiple murine MASH models elucidated that MASH also impaired the mitochondrial fitness and motility of tumor-infiltrating CD8 + T cells. This impairment diminishes the efficacy of ICB therapy, which could be salvaged by metformin [182]. There are currently several clinical trials assessing the efficacy of metformin in treating HCC (Table 3). Meanwhile, gastrointestinal IgA+ metabolically activated B cells can license auto-aggressive T cells, promoting HCC development in an antigen-independent manner in MASH-induced HCC mouse models [183]. Of note, high serum cholesterol levels drive NK cell accumulation and subsequent lipid raft formation, further enhancing the anti-tumor activity in both DEN-induced HCC mouse models and Hepa1-6 (mouse hepatoma) subcutaneous models [184]. Due to the differences between the mouse models, the impact of lipid metabolism reprogramming on the TME remains controversial and requires further exploration.
In addition to immune-activated cells, immunosuppressive cells in liver cancer also undergo metabolic reprogramming. Chen et al. proposed that tumor-derived hyaluronan (HA) fragments induce aerobic glycolysis in monocytes and upregulate PD-L1 expression through the PFKFB3-NF-κB pathway [185]. Notably, the same group further illustrated that glycolytic monocytes in HCC produce large amounts of chemokines, such as CXCL2 and CXCL8. These chemokines induce neutrophil infiltration, while glycolytic monocytes secrete TNF-α to seduce oncostatin M (OSM) production from accumulated neutrophils. These processes ultimately lead to the metastasis of HCC, as shown by both ex vivo and in vitro experiments [186]. The acidic TME also induces a metabolic switch in monocytes to produce tremendous amounts of CCL8. This production promotes epithelial-mesenchymal transition (EMT) and HCC metastasis [187]. On the other hand, macrophage polarization is tightly correlated with metabolic rearrangement [188]. In mouse and human HCC, the serine/threonine kinase RIPK3 is downregulated in macrophages, leading to FAO-dominated M2φ polarization via the ROS/Caspase1/PPAR pathway [189]. M2-like TAMs upregulate the glycolysis pathway under hypoxic TME and produce excessive IL1β to facilitate EMT and subsequent metastasis in HCC [190]. Furthermore, the PKM2/HIF-1α axis in human HCC samples and syngeneic mouse models drives fibronectin 1 (FN1) production to instigate pluripotent polarization of macrophages, concurrent with anti-tumorigenic IL-12p70 production from glycolytic macrophages [191].
In this section, we discussed the metabolic interactions between liver cancer cells and immune cells (Fig. 3), highlighting a promising field for liver cancer treatment in the future. Some clinical trials have been conducted to assess the targeting of metabolic changes in the treatment of PLC (Table 3); however, combined targeting of these metabolic alterations using ICB should be specifically considered as this would significantly reinvigorate the anti-tumor immune response and thus achieve better therapeutic effects.
Impact of gut microbiota-derived metabolites on the liver cancer microenvironment
The gut and the liver are physiologically connected due to their unique anatomical location. This gut–liver axis executes critical functions in nutrient metabolism and bacterial metabolite clearance, mainly through the portal vein [192]. Emerging studies have addressed the pivotal roles of the gut microbiota in PLC pathogenesis and anti-tumor therapy [5, 193,194,195,196]. The gut microbiota is closely associated with immunity and metabolism, underscoring their vital role in health and disease. The interaction between intestinal microbiota-secreted metabolites, including short-chain fatty acids, BA, indoles, and ethanol, and the PLC microenvironment requires further exploration [197]. In mice exposed to chemical carcinogens, obesity increases the levels of deoxycholic acid (DCA), a secondary bile acid produced by gut bacteria such as Clostridium cluster XI and XIVa strains. Through the gut–liver axis, DCA induces SASP in hepatic stellate cells (HSC) to trigger the secretion of various inflammatory factors, thus facilitating HCC progression [198]. Using 16S rRNA sequencing and serum metabolomic analysis, another independent group found that the development of MASLD-HCC induced by a high-cholesterol diet in mice was closely correlated to dysbiosis of the gut microbiota and the resultant alterations in metabolites [199]. The gut microbiota has also been implicated in HCC induced by high dietary intake of fructose. Investigation of the underlying mechanism showed that microbiota-derived acetate enhanced UDP-GlcNAc biosynthesis and O-GlcNAcylation, and hyper-O-GlcNAcylation of eukaryotic elongation factor 1A1 (eEF1A1) at T279 promoted the proliferation of tumor cells and HCC progression in the DEN + CCl4-induced HCC mouse model [200]. Butyrate is mainly produced by the gut microbiota during fermentation and is subsequently absorbed by the liver via the gut–liver axis [201]. Butyrate accumulation disrupts intracellular calcium homeostasis and induces the production of ROS, thus improving the efficacy of TKI therapy in HCC mouse models [202]. PSC and colitis, two well-known risk factors for iCCA, promote the exposure of gut-derived bacteria and lipopolysaccharide to the liver. This induces TLR4-dependent CXCL1 expression in hepatocytes, leading to the recruitment of CXCR2 + PMN-MDSC, and ultimately promoting an immunosuppressive TME [203]. Multi-omics studies incorporating 16S rRNA MiSeq sequencing in different cohorts from various regions of China have also identified gut microbes as non-invasive biomarkers for the early diagnosis of PLC [204,205,206].
Increasing evidence suggests that the gut microbiota modulates immune responses in the TME of PLC [5, 194, 203, 207, 208]. The liver immune system precludes gut-derived microbes and corresponding metabolites without evoking a systemic immune response, thus demonstrating immune privilege [209, 210]. Within intestinal microbiota, gram-positive bacteria predominantly convert immunostimulatory/primary into immunosuppressive/secondary BA. These recirculate to the liver through enterohepatic circulation. Secondary BA suppresses the recruitment of CXCR6 + tumorolytic NKT cells into the liver by reducing CXCL16 expression in liver sinusoidal endothelial cells (LSECs) [211,212,213]. Considering the impact of primary and secondary BA balance on the TME and PLC therapy, Ji et al. developed a strategy targeting BA receptors via nanoparticle-based delivery of modulators. This approach effectively reverses the immune privilege of HCC [214]. Bile acid metabolites such as isoalloLCA and isoDCA promote Treg cell differentiation through FOXP3 induction, while 3-oxoLCA and isoLCA inhibit Th17 cell differentiation by targeting RORγt in vitro and in vivo [215,216,217]. Thus, the role of the bile acid metabolic pathway in Treg/Th17 cell balance and anti-tumor immunity in the liver cancer microenvironment remains to be explored.
The gut microbiome strongly influences the tumor immune microenvironment and immunotherapy response [218,219,220,221,222,223]. Lipoteichoic acid (LTA), an obesity-induced gram-positive gut microbial metabolite, elevates COX-2 expression in the DCA-induced senescent HSCs. This leads to an immunosuppressive TME through PGE2 production in obesity-associated HCC mouse models [198, 224], potentially causing ICB resistance [225, 226]. Innate lymphoid cells, including ILC1, ILC2, and ILC3 subsets, are now recognized as crucial in tumor regulation by releasing specific cytokines. Hu et al. found a significant reduction of Lactobacillus reuteri in gut microbiota of mice with HCC. This leads to decreased short-chain fatty acid secretion, particularly acetate. The lack of acetate in TME weakens ILC3 anti-tumor functionality by increasing IL17A production. Thus, combining acetate supplementation with PD-1 blockades significantly boosts anti-tumor immunity [227]. Additionally, gut microbiome-derived metabolite D-lactate can shift TAMs from the M2 to M1 phenotype, remodeling the immunosuppressive TME in HCC mice [111, 228]. Lee et al. suggested that Lachnoclostridium, along with ursodeoxycholic and ursocholic acids, produce better responses to ICB treatment in patients with unresectable HCC [229].
People often consume dietary supplements for health benifits [230]. However, highly refined fermentable fibers may promote cholestasis and the development of HCC. In wild-type mice, a diet rich in inulin-enriched soluble fiber leads to microbiota-dependent cholestasis, hepatocyte death, and subsequent neutrophilic inflammation in the liver, culminating in HCC [231]. Ex vivo studies showed that gut bacteria convert dietary fiber into short-chain fatty acids in MASLD-HCC, leading to an immunosuppressive TME with an elevated CD4 + Treg cell/CD8 + T-cell ratio [208]. In conclusion, we discuss mechanisms by which the gut microbiota-related metabolites affect liver cancer TME (Table 4). Harnessing the gut microbiome could offer novel therapeutic strategies that target liver metabolism for PLC treatment.
Conclusions
The cancer type and location influence nutrient availability and subsequent metabolism within the TME. Since the liver is the largest metabolic organ, the initiation and progression of liver cancer disrupt the metabolic homeostasis of the TME. In recent decades, there has been limited progress in the use of compounds targeting specific metabolic alterations in the treatment of PLC, likely due to metabolic plasticity, drug specificity, and the heterogeneous metabolic microenvironment [127]. The term “tumor metabolism” is used to indicate a common set of metabolic alterations accompanying malignancies. However, tumors are metabolically heterogeneous, mainly due to the complex cellular composition of the TME, including tumor, immune, and stromal cells. As both tumor cells and their surrounding anti-tumor immune cells share certain metabolic activities, agents designed to block these metabolic pathways in tumor cells may impair the proliferation, activation, and function of cytolytic immune cells, ultimately leading to unfavorable side effects. Thus, investigation of the mechanism involved in tumor immune metabolic crosstalk may help identify novel therapeutic targets. These less toxic and more specific metabolic targets could improve the unsatisfactory response rates of current therapies.
The role of the gut microbiome in health and disease has seen increasing research attention recently. Gut microbial dysbiosis promotes hepatocarcinogenesis by altering metabolic programs within TME. Exploring the metabolic connections among liver tumor cells, the gut microbiome, and immune cells may shed light on how to harness the gut microbiome to enhance current treatment strategies, particularly immunotherapies.
Many current studies have focused on animal models, while data derived from multi-omics analysis of clinical samples are generally descriptive and lack further validation. There is also a need for in-depth mechanistic studies to establish causalities between metabolites in the TME and liver cancer outcomes. Given that perturbations of metabolic processes and the gut microbiome are generally influenced by confounding factors, such as diet, environment, and host genetics, study of diverse cohorts from patients of different demographics, ethnicities, and geographical regions will be needed to determine the broader implications of the gut microbiome in PLC pathogenesis.
Availability of data and materials
Not applicable.
References
Satriano L, Lewinska M, Rodrigues PM, Banales JM, Andersen JB. Metabolic rearrangements in primary liver cancers: cause and consequences. Nat Rev Gastroenterol Hepatol. 2019;16:748–66. https://doi.org/10.1038/s41575-019-0217-8.
Vogel A, Meyer T, Sapisochin G, Salem R, Saborowski A. Hepatocellular carcinoma. Lancet. 2022;400:1345–62. https://doi.org/10.1016/S0140-6736(22)01200-4.
Oh D-Y, et al. Durvalumab plus gemcitabine and cisplatin in advanced biliary tract cancer. NEJM Evidence. 2022;1:EVIDoa2200015. https://doi.org/10.1056/EVIDoa2200015.
Kelley RK, et al. Pembrolizumab in combination with gemcitabine and cisplatin compared with gemcitabine and cisplatin alone for patients with advanced biliary tract cancer (KEYNOTE-966): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2023;401:1853–65. https://doi.org/10.1016/S0140-6736(23)00727-4.
Schwabe RF, Greten TF. Gut microbiome in HCC: mechanisms, diagnosis and therapy. J Hepatol. 2020;72:230–8. https://doi.org/10.1016/j.jhep.2019.08.016.
Hung MH, et al. Tumor methionine metabolism drives T-cell exhaustion in hepatocellular carcinoma. Nat Commun. 2021;12:1455. https://doi.org/10.1038/s41467-021-21804-1.
Miallot R, Galland F, Millet V, Blay JY, Naquet P. Metabolic landscapes in sarcomas. J Hematol Oncol. 2021;14:114. https://doi.org/10.1186/s13045-021-01125-y.
Schmidt DR, et al. Metabolomics in cancer research and emerging applications in clinical oncology. CA Cancer J Clin. 2021;71:333–58. https://doi.org/10.3322/caac.21670.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. https://doi.org/10.1126/science.1160809.
Xia L, et al. The cancer metabolic reprogramming and immune response. Mol Cancer. 2021;20:28. https://doi.org/10.1186/s12943-021-01316-8.
Sonveaux P, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–42. https://doi.org/10.1172/JCI36843.
Pavlides S, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. https://doi.org/10.4161/cc.8.23.10238.
Locasale JW, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869–74. https://doi.org/10.1038/ng.890.
Schwenck J, et al. Advances in PET imaging of cancer. Nat Rev Cancer. 2023;23:474–90. https://doi.org/10.1038/s41568-023-00576-4.
Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17:351–9. https://doi.org/10.1038/ncb3124.
Chang CH, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229–41. https://doi.org/10.1016/j.cell.2015.08.016.
Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–54. https://doi.org/10.1172/JCI31178.
Leone RD, Powell JD. Metabolism of immune cells in cancer. Nat Rev Cancer. 2020;20:516–31. https://doi.org/10.1038/s41568-020-0273-y.
Li X, et al. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat Rev Clin Oncol. 2019;16:425–41. https://doi.org/10.1038/s41571-019-0203-7.
Wang T, et al. Inosine is an alternative carbon source for CD8(+)-T-cell function under glucose restriction. Nat Metab. 2020;2:635–47. https://doi.org/10.1038/s42255-020-0219-4.
Liu L, et al. Proinflammatory signal suppresses proliferation and shifts macrophage metabolism from Myc-dependent to HIF1alpha-dependent. Proc Natl Acad Sci U S A. 2016;113:1564–9. https://doi.org/10.1073/pnas.1518000113.
Miller A, et al. Exploring metabolic configurations of single cells within complex tissue microenvironments. Cell Metab. 2017;26:788–800. https://doi.org/10.1016/j.cmet.2017.08.014.
Hepatocellular carcinoma. Nat Rev Dis Primers 7, 7, https://doi.org/10.1038/s41572-021-00245-6 (2021).
Bidkhori G, et al. Metabolic network-based stratification of hepatocellular carcinoma reveals three distinct tumor subtypes. Proc Natl Acad Sci U S A. 2018;115:E11874–83. https://doi.org/10.1073/pnas.1807305115.
Calderaro J, Ziol M, Paradis V, Zucman-Rossi J. Molecular and histological correlations in liver cancer. J Hepatol. 2019;71:616–30. https://doi.org/10.1016/j.jhep.2019.06.001.
Xing X, et al. Integrated omics landscape of hepatocellular carcinoma suggests proteomic subtypes for precision therapy. Cell Rep Med. 2023;4: 101315. https://doi.org/10.1016/j.xcrm.2023.101315.
Deng M, et al. Proteogenomic characterization of cholangiocarcinoma. Hepatology. 2023;77:411–29. https://doi.org/10.1002/hep.32624.
Murai H, et al. Multiomics identifies the link between intratumor steatosis and the exhausted tumor immune microenvironment in hepatocellular carcinoma. Hepatology. 2023;77:77–91. https://doi.org/10.1002/hep.32573.
Dong L, et al. Proteogenomic characterization identifies clinically relevant subgroups of intrahepatic cholangiocarcinoma. Cancer Cell. 2022;40:70–87. https://doi.org/10.1016/j.ccell.2021.12.006.
Gao Q, et al. Integrated proteogenomic characterization of HBV-related hepatocellular carcinoma. Cell. 2019;179:561–77. https://doi.org/10.1016/j.cell.2019.08.052.
Nwosu ZC, et al. Identification of the consistently altered metabolic targets in human hepatocellular carcinoma. Cell Mol Gastroenterol Hepatol. 2017;4:303–23. https://doi.org/10.1016/j.jcmgh.2017.05.004.
Ji S, et al. Pharmaco-proteogenomic characterization of liver cancer organoids for precision oncology. Sci Transl Med. 2023;15:eadg3358. https://doi.org/10.1126/scitranslmed.adg3358.
Yang HC, Stern A, Chiu DT. G6PD: a hub for metabolic reprogramming and redox signaling in cancer. Biomed J. 2021;44:285–92. https://doi.org/10.1016/j.bj.2020.08.001.
Hong X, et al. PTEN antagonises Tcl1/hnRNPK-mediated G6PD pre-mRNA splicing which contributes to hepatocarcinogenesis. Gut. 2014;63:1635–47. https://doi.org/10.1136/gutjnl-2013-305302.
Gao LP, et al. Ineffective GSH regeneration enhances G6PD-knockdown Hep G2 cell sensitivity to diamide-induced oxidative damage. Free Radic Biol Med. 2009;47:529–35. https://doi.org/10.1016/j.freeradbiomed.2009.05.029.
Li B, et al. Multiomics identifies metabolic subtypes based on fatty acid degradation allocating personalized treatment in hepatocellular carcinoma. Hepatology. 2023. https://doi.org/10.1097/HEP.0000000000000553.
Huang Q, et al. Metabolic characterization of hepatocellular carcinoma using nontargeted tissue metabolomics. Cancer Res. 2013;73:4992–5002. https://doi.org/10.1158/0008-5472.CAN-13-0308.
Yang F, et al. Metabolic reprogramming and its clinical implication for liver cancer. Hepatology. 2023;78:1602–24. https://doi.org/10.1097/HEP.0000000000000005.
Yang Z, et al. Lactylome analysis suggests lactylation-dependent mechanisms of metabolic adaptation in hepatocellular carcinoma. Nat Metab. 2023;5:61–79. https://doi.org/10.1038/s42255-022-00710-w.
Wang ZX, et al. Preoperative serum liver enzyme markers for predicting early recurrence after curative resection of hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int. 2015;14:178–85. https://doi.org/10.1016/s1499-3872(15)60353-8.
Faloppi L, et al. The role of LDH serum levels in predicting global outcome in HCC patients treated with sorafenib: implications for clinical management. BMC Cancer. 2014;14:110. https://doi.org/10.1186/1471-2407-14-110.
Faloppi L, et al. The correlation between LDH serum levels and clinical outcome in advanced biliary tract cancer patients treated with first line chemotherapy. Sci Rep. 2016;6:24136. https://doi.org/10.1038/srep24136.
Zhang MS, et al. Hypoxia-induced macropinocytosis represents a metabolic route for liver cancer. Nat Commun. 2022;13:954. https://doi.org/10.1038/s41467-022-28618-9.
Lu M, et al. Elevated G6PD expression contributes to migration and invasion of hepatocellular carcinoma cells by inducing epithelial-mesenchymal transition. Acta Biochim Biophys Sin (Shanghai). 2018;50:370–80. https://doi.org/10.1093/abbs/gmy009.
Kowalik MA, et al. Metabolic reprogramming identifies the most aggressive lesions at early phases of hepatic carcinogenesis. Oncotarget. 2016;7:32375–93. https://doi.org/10.18632/oncotarget.8632.
Zhu Q, et al. O-GlcNAcylation plays a role in tumor recurrence of hepatocellular carcinoma following liver transplantation. Med Oncol. 2012;29:985–93. https://doi.org/10.1007/s12032-011-9912-1.
Phoomak C, et al. High glucose levels boost the aggressiveness of highly metastatic cholangiocarcinoma cells via O-GlcNAcylation. Sci Rep. 2017;7:43842. https://doi.org/10.1038/srep43842.
Lin M, et al. Downregulation of CPT2 promotes tumorigenesis and chemoresistance to cisplatin in hepatocellular carcinoma. Onco Targets Ther. 2018;11:3101–10. https://doi.org/10.2147/OTT.S163266.
Calvisi DF, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology. 2011;140:1071–83. https://doi.org/10.1053/j.gastro.2010.12.006.
Wang MD, et al. Acetyl-coenzyme A carboxylase alpha promotion of glucose-mediated fatty acid synthesis enhances survival of hepatocellular carcinoma in mice and patients. Hepatology. 2016;63:1272–86. https://doi.org/10.1002/hep.28415.
Tomacha J, et al. Targeting fatty acid synthase modulates metabolic pathways and inhibits cholangiocarcinoma cell progression. Front Pharmacol. 2021;12: 696961. https://doi.org/10.3389/fphar.2021.696961.
Xu D, et al. The gluconeogenic enzyme PCK1 phosphorylates INSIG1/2 for lipogenesis. Nature. 2020;580:530–5. https://doi.org/10.1038/s41586-020-2183-2.
Nelson ME, et al. Inhibition of hepatic lipogenesis enhances liver tumorigenesis by increasing antioxidant defence and promoting cell survival. Nat Commun. 2017;8:14689. https://doi.org/10.1038/ncomms14689.
Nakagawa H, et al. Lipid metabolic reprogramming in hepatocellular carcinoma. Cancers Basel. 2018. https://doi.org/10.3390/cancers10110447.
Paul B, Lewinska M, Andersen JB. Lipid alterations in chronic liver disease and liver cancer. JHEP Rep. 2022;4: 100479. https://doi.org/10.1016/j.jhepr.2022.100479.
Liang JQ, et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat Commun. 2018;9:4490. https://doi.org/10.1038/s41467-018-06931-6.
Hall Z, et al. Lipid remodeling in hepatocyte proliferation and hepatocellular carcinoma. Hepatology. 2021;73:1028–44. https://doi.org/10.1002/hep.31391.
Jiang Y, et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature. 2019;567:257–61. https://doi.org/10.1038/s41586-019-0987-8.
Yang Z, et al. Cholesterol inhibits hepatocellular carcinoma invasion and metastasis by promoting CD44 localization in lipid rafts. Cancer Lett. 2018;429:66–77. https://doi.org/10.1016/j.canlet.2018.04.038.
Budhu A, et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology. 2013;144:1066–75. https://doi.org/10.1053/j.gastro.2013.01.054.
Murakami Y, et al. Comprehensive analysis of transcriptome and metabolome analysis in Intrahepatic Cholangiocarcinoma and Hepatocellular Carcinoma. Sci Rep. 2015;5:16294. https://doi.org/10.1038/srep16294.
Missiaen R, et al. GCN2 inhibition sensitizes arginine-deprived hepatocellular carcinoma cells to senolytic treatment. Cell Metab. 2022;34:1151–67. https://doi.org/10.1016/j.cmet.2022.06.010.
Mossmann D, et al. Arginine reprograms metabolism in liver cancer via RBM39. Cell. 2023;186:5068–83. https://doi.org/10.1016/j.cell.2023.09.011.
Dai W, et al. OGDHL silencing promotes hepatocellular carcinoma by reprogramming glutamine metabolism. J Hepatol. 2020;72:909–23. https://doi.org/10.1016/j.jhep.2019.12.015.
Adebayo Michael AO, et al. Inhibiting glutamine-dependent mTORC1 activation ameliorates liver cancers driven by beta-catenin mutations. Cell Metab. 2019;29:1135–50. https://doi.org/10.1016/j.cmet.2019.01.002.
Ding Z, et al. Metabolic pathway analyses identify proline biosynthesis pathway as a promoter of liver tumorigenesis. J Hepatol. 2020;72:725–35. https://doi.org/10.1016/j.jhep.2019.10.026.
Ducker GS, Rabinowitz JD. One-carbon metabolism in health and disease. Cell Metab. 2017;25:27–42. https://doi.org/10.1016/j.cmet.2016.08.009.
Newman AC, Maddocks ODK. One-carbon metabolism in cancer. Br J Cancer. 2017;116:1499–504. https://doi.org/10.1038/bjc.2017.118.
Tedeschi PM, et al. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis. 2013;4: e877. https://doi.org/10.1038/cddis.2013.393.
Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13:572–83. https://doi.org/10.1038/nrc3557.
Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol. 2013;10:656–65. https://doi.org/10.1038/nrgastro.2013.183.
Ren X, et al. The Protein Kinase Activity of NME7 Activates Wnt/beta-Catenin Signaling to Promote One-Carbon Metabolism in Hepatocellular Carcinoma. Cancer Res. 2022;82:60–74. https://doi.org/10.1158/0008-5472.CAN-21-1020.
Mukha D, et al. Glycine decarboxylase maintains mitochondrial protein lipoylation to support tumor growth. Cell Metab. 2022;34:775–82. https://doi.org/10.1016/j.cmet.2022.04.006.
Li JT, et al. Dietary folate drives methionine metabolism to promote cancer development by stabilizing MAT IIA. Signal Transduct Target Ther. 2022;7:192. https://doi.org/10.1038/s41392-022-01017-8.
Schulze K, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505–11. https://doi.org/10.1038/ng.3252.
Nakamura H, et al. Genomic spectra of biliary tract cancer. Nat Genet. 2015;47:1003–10. https://doi.org/10.1038/ng.3375.
Cancer Genome Atlas Research Network. Electronic address, w. b. e. & Cancer Genome Atlas Research, N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 169, 1327–1341, https://doi.org/10.1016/j.cell.2017.05.046 (2017).
Fernandez-Banet J, et al. Decoding complex patterns of genomic rearrangement in hepatocellular carcinoma. Genomics. 2014;103:189–203. https://doi.org/10.1016/j.ygeno.2014.01.003.
Baughman JM, et al. NeuCode proteomics reveals bap1 regulation of metabolism. Cell Rep. 2016;16:583–95. https://doi.org/10.1016/j.celrep.2016.05.096.
Amann T, et al. GLUT1 expression is increased in hepatocellular carcinoma and promotes tumorigenesis. Am J Pathol. 2009;174:1544–52. https://doi.org/10.2353/ajpath.2009.080596.
Kim YH, et al. SLC2A2 (GLUT2) as a novel prognostic factor for hepatocellular carcinoma. Oncotarget. 2017;8:68381–92. https://doi.org/10.18632/oncotarget.20266.
Sun HW, et al. GLUT1 and ASCT2 as Predictors for Prognosis of Hepatocellular Carcinoma. PLoS ONE. 2016;11: e0168907. https://doi.org/10.1371/journal.pone.0168907.
Kubo Y, et al. Different expression of glucose transporters in the progression of intrahepatic cholangiocarcinoma. Hum Pathol. 2014;45:1610–7. https://doi.org/10.1016/j.humpath.2014.03.008.
Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007;27:7381–93. https://doi.org/10.1128/MCB.00440-07.
Sun L, et al. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 2015;25:429–44. https://doi.org/10.1038/cr.2015.33.
Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004;64:2627–33. https://doi.org/10.1158/0008-5472.can-03-0846.
Mossmann D, Park S, Hall MN. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer. 2018;18:744–57. https://doi.org/10.1038/s41568-018-0074-8.
Perugorria MJ, et al. Wnt-beta-catenin signalling in liver development, health and disease. Nat Rev Gastroenterol Hepatol. 2019;16:121–36. https://doi.org/10.1038/s41575-018-0075-9.
Senni N, et al. beta-catenin-activated hepatocellular carcinomas are addicted to fatty acids. Gut. 2019;68:322–34. https://doi.org/10.1136/gutjnl-2017-315448.
Ikeno Y, et al. Preoperative metabolic tumor volume of intrahepatic cholangiocarcinoma measured by (18)F-FDG-PET is associated with the KRAS mutation status and prognosis. J Transl Med. 2018;16:95. https://doi.org/10.1186/s12967-018-1475-x.
Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell. 2017;168:657–69. https://doi.org/10.1016/j.cell.2016.12.039.
Raggi C, Taddei ML, Rae C, Braconi C, Marra F. Metabolic reprogramming in cholangiocarcinoma. J Hepatol. 2022;77:849–64. https://doi.org/10.1016/j.jhep.2022.04.038.
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. https://doi.org/10.1016/j.immuni.2013.07.012.
Mellman I, Chen DS, Powles T, Turley SJ. The cancer-immunity cycle: Indication, genotype, and immunotype. Immunity. 2023;56:2188–205. https://doi.org/10.1016/j.immuni.2023.09.011.
Arner EN, Rathmell JC. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell. 2023;41:421–33. https://doi.org/10.1016/j.ccell.2023.01.009.
Ho PC, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. 2015;162:1217–28. https://doi.org/10.1016/j.cell.2015.08.012.
Yu YR, et al. Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat Immunol. 2020;21:1540–51. https://doi.org/10.1038/s41590-020-0793-3.
Sullivan MR, et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife. 2019. https://doi.org/10.7554/eLife.44235.
Chantranupong L, et al. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell. 2016;165:153–64. https://doi.org/10.1016/j.cell.2016.02.035.
Geiger R, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167:829–42. https://doi.org/10.1016/j.cell.2016.09.031.
Wang W, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–4. https://doi.org/10.1038/s41586-019-1170-y.
Fischer K, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–9. https://doi.org/10.1182/blood-2006-07-035972.
Elia I, et al. Tumor cells dictate anti-tumor immune responses by altering pyruvate utilization and succinate signaling in CD8(+) T cells. Cell Metab. 2022;34:1137–50. https://doi.org/10.1016/j.cmet.2022.06.008.
Brand A, et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016;24:657–71. https://doi.org/10.1016/j.cmet.2016.08.011.
Dodard G, et al. Inflammation-Induced Lactate Leads to Rapid Loss of Hepatic Tissue-Resident NK Cells. Cell Rep. 2020;32: 107855. https://doi.org/10.1016/j.celrep.2020.107855.
Zheng X, et al. Mitochondrial fragmentation limits NK cell-based tumor immunosurveillance. Nat Immunol. 2019;20:1656–67. https://doi.org/10.1038/s41590-019-0511-1.
Watson MJ, et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature. 2021;591:645–51. https://doi.org/10.1038/s41586-020-03045-2.
Kumagai S, et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell. 2022;40:201–18. https://doi.org/10.1016/j.ccell.2022.01.001.
Colegio OR, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–63. https://doi.org/10.1038/nature13490.
Chaudagar K, et al. Reversal of Lactate and PD-1-mediated Macrophage Immunosuppression Controls Growth of PTEN/p53-deficient Prostate Cancer. Clin Cancer Res. 2023;29:1952–68. https://doi.org/10.1158/1078-0432.CCR-22-3350.
Han S, et al. d-lactate modulates M2 tumor-associated macrophages and remodels immunosuppressive tumor microenvironment for hepatocellular carcinoma. Sci Adv. 2023;9:eadg2697. https://doi.org/10.1126/sciadv.adg2697.
Zhao JL, et al. Notch-mediated lactate metabolism regulates MDSC development through the Hes1/MCT2/c-Jun axis. Cell Rep. 2022;38: 110451. https://doi.org/10.1016/j.celrep.2022.110451.
Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. 2013;191:1486–95. https://doi.org/10.4049/jimmunol.1202702.
Yang X, et al. Lactate-modulated immunosuppression of myeloid-derived suppressor cells contributes to the radioresistance of pancreatic cancer. Cancer Immunol Res. 2020;8:1440–51. https://doi.org/10.1158/2326-6066.CIR-20-0111.
Fallarino F, et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002;9:1069–77. https://doi.org/10.1038/sj.cdd.4401073.
Mezrich JD, et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185:3190–8. https://doi.org/10.4049/jimmunol.0903670.
Holmgaard RB, et al. Tumor-expressed IDO recruits and activates MDSCs in a Treg-dependent manner. Cell Rep. 2015;13:412–24. https://doi.org/10.1016/j.celrep.2015.08.077.
Bunse L, et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med. 2018;24:1192–203. https://doi.org/10.1038/s41591-018-0095-6.
Notarangelo G, et al. Oncometabolite d-2HG alters T cell metabolism to impair CD8(+) T cell function. Science. 2022;377:1519–29. https://doi.org/10.1126/science.abj5104.
Zelenay S, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. 2015;162:1257–70. https://doi.org/10.1016/j.cell.2015.08.015.
Halford SER, et al. A first-in-human first-in-class (FIC) trial of the monocarboxylate transporter 1 (MCT1) inhibitor AZD3965 in patients with advanced solid tumours. J Clin Oncol. 2017;35:2516–2516. https://doi.org/10.1200/JCO.2017.35.15_suppl.2516.
Girithar HN, et al. Involvement of the kynurenine pathway in breast cancer: updates on clinical research and trials. Br J Cancer. 2023;129:185–203. https://doi.org/10.1038/s41416-023-02245-7.
Bendell JC, et al. Safety and efficacy of the anti-CD73 monoclonal antibody (mAb) oleclumab ± durvalumab in patients (pts) with advanced colorectal cancer (CRC), pancreatic ductal adenocarcinoma (PDAC), or EGFR-mutant non-small cell lung cancer (EGFRm NSCLC). J Clin Oncol. 2021;39:9047–9047. https://doi.org/10.1200/JCO.2021.39.15_suppl.9047.
DiNardo CD, et al. Mutant isocitrate dehydrogenase 1 inhibitor ivosidenib in combination with azacitidine for newly diagnosed acute myeloid leukemia. J Clin Oncol. 2021;39:57–65. https://doi.org/10.1200/jco.20.01632.
Antonarakis ES, et al. Phase II, randomized, placebo-controlled trial of neoadjuvant celecoxib in men with clinically localized prostate cancer: evaluation of drug-specific biomarkers. J Clin Oncol. 2009;27:4986–93. https://doi.org/10.1200/jco.2009.21.9410.
Altorki NK, et al. Celecoxib, a selective cyclo-oxygenase-2 inhibitor, enhances the response to preoperative paclitaxel and carboplatin in early-stage non–small-cell lung cancer. J Clin Oncol. 2003;21:2645–50. https://doi.org/10.1200/jco.2003.07.127.
Stine ZE, Schug ZT, Salvino JM, Dang CV. Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discov. 2022;21:141–62. https://doi.org/10.1038/s41573-021-00339-6.
Chen J, et al. Unbalanced glutamine partitioning between CD8T cells and cancer cells accompanied by immune cell dysfunction in hepatocellular carcinoma. Cells. 2022. https://doi.org/10.3390/cells11233924.
Wang W, Guo MN, Li N, Pang DQ, Wu JH. Glutamine deprivation impairs function of infiltrating CD8(+) T cells in hepatocellular carcinoma by inducing mitochondrial damage and apoptosis. World J Gastrointest Oncol. 2022;14:1124–40. https://doi.org/10.4251/wjgo.v14.i6.1124.
Davar D, et al. Phase Ib/II study of pembrolizumab and pegylated-interferon Alfa-2b in advanced melanoma. J Clin Oncol. 2018;36:JCO1800632. https://doi.org/10.1200/JCO.18.00632.
Hu B, et al. IFNalpha Potentiates Anti-PD-1 Efficacy by Remodeling Glucose Metabolism in the Hepatocellular Carcinoma Microenvironment. Cancer Discov. 2022;12:1718–41. https://doi.org/10.1158/2159-8290.CD-21-1022.
Handa P, et al. Iron alters macrophage polarization status and leads to steatohepatitis and fibrogenesis. J Leukoc Biol. 2019;105:1015–26. https://doi.org/10.1002/JLB.3A0318-108R.
Sun JL, et al. Tumor cell-imposed iron restriction drives immunosuppressive polarization of tumor-associated macrophages. J Transl Med. 2021;19:347. https://doi.org/10.1186/s12967-021-03034-7.
Li Y, Mo H, Wu S, Liu X, Tu K. A novel lactate metabolism-related gene signature for predicting clinical outcome and tumor microenvironment in hepatocellular carcinoma. Front Cell Dev Biol. 2021;9: 801959. https://doi.org/10.3389/fcell.2021.801959.
Certo M, Tsai CH, Pucino V, Ho PC, Mauro C. Lactate modulation of immune responses in inflammatory versus tumour microenvironments. Nat Rev Immunol. 2021;21:151–61. https://doi.org/10.1038/s41577-020-0406-2.
Contreras-Baeza Y, et al. Monocarboxylate transporter 4 (MCT4) is a high affinity transporter capable of exporting lactate in high-lactate microenvironments. J Biol Chem. 2019;294:20135–47. https://doi.org/10.1074/jbc.RA119.009093.
Fang Y, et al. Monocarboxylate transporter 4 inhibition potentiates hepatocellular carcinoma immunotherapy through enhancing T cell infiltration and immune attack. Hepatology. 2023;77:109–23. https://doi.org/10.1002/hep.32348.
Xu M, et al. Tumor associated macrophages-derived exosomes facilitate hepatocellular carcinoma malignance by transferring lncMMPA to tumor cells and activating glycolysis pathway. J Exp Clin Cancer Res. 2022;41:253. https://doi.org/10.1186/s13046-022-02458-3.
Gu J, et al. Tumor metabolite lactate promotes tumorigenesis by modulating MOESIN lactylation and enhancing TGF-beta signaling in regulatory T cells. Cell Rep. 2022;39: 110986. https://doi.org/10.1016/j.celrep.2022.110986.
Song BS, et al. Mitoribosomal defects aggravate liver cancer via aberrant glycolytic flux and T cell exhaustion. J Immunother Cancer. 2022. https://doi.org/10.1136/jitc-2021-004337.
Llovet JM, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7:6. https://doi.org/10.1038/s41572-020-00240-3.
Deng H, et al. Tumor-derived lactate inhibit the efficacy of lenvatinib through regulating PD-L1 expression on neutrophil in hepatocellular carcinoma. J Immunother Cancer. 2021. https://doi.org/10.1136/jitc-2020-002305.
Ma C, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531:253–7. https://doi.org/10.1038/nature16969.
Sun R, et al. Loss of SIRT5 promotes bile acid-induced immunosuppressive microenvironment and hepatocarcinogenesis. J Hepatol. 2022;77:453–66. https://doi.org/10.1016/j.jhep.2022.02.030.
Bayerl F, et al. Tumor-derived prostaglandin E2 programs cDC1 dysfunction to impair intratumoral orchestration of anti-cancer T cell responses. Immunity. 2023;56:1341–58. https://doi.org/10.1016/j.immuni.2023.05.011.
Xun X, et al. Cyclooxygenase-2 expressed hepatocellular carcinoma induces cytotoxic T lymphocytes exhaustion through M2 macrophage polarization. Am J Transl Res. 2021;13:4360–75.
Lin J, et al. Multimodule characterization of immune subgroups in intrahepatic cholangiocarcinoma reveals distinct therapeutic vulnerabilities. J Immunother Cancer. 2022. https://doi.org/10.1136/jitc-2022-004892.
Lercher A, et al. Type I interferon signaling disrupts the hepatic urea cycle and alters systemic metabolism to suppress T cell function. Immunity. 2019;51:1074–87. https://doi.org/10.1016/j.immuni.2019.10.014.
Cheu JW, et al. Hypoxia-inducible factor orchestrates adenosine metabolism to promote liver cancer development. Sci Adv. 2023;9:eade5111. https://doi.org/10.1126/sciadv.ade5111.
Fu Y, et al. MicroRNA-223 attenuates hepatocarcinogenesis by blocking hypoxia-driven angiogenesis and immunosuppression. Gut. 2023;72:1942–58. https://doi.org/10.1136/gutjnl-2022-327924.
Wang J, et al. Tumor-derived adenosine promotes macrophage proliferation in human hepatocellular carcinoma. J Hepatol. 2021;74:627–37. https://doi.org/10.1016/j.jhep.2020.10.021.
Chen Y, et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology. 2015;61:1591–602. https://doi.org/10.1002/hep.27665.
Suthen S, et al. Hypoxia-driven immunosuppression by Treg and type-2 conventional dendritic cells in HCC. Hepatology. 2022;76:1329–44. https://doi.org/10.1002/hep.32419.
Cui C, et al. Hypoxia-inducible gene 2 promotes the immune escape of hepatocellular carcinoma from nature killer cells through the interleukin-10-STAT3 signaling pathway. J Exp Clin Cancer Res. 2019;38:229. https://doi.org/10.1186/s13046-019-1233-9.
Wang S, et al. Blocking CD47 promotes antitumour immunity through CD103(+) dendritic cell-NK cell axis in murine hepatocellular carcinoma model. J Hepatol. 2022;77:467–78. https://doi.org/10.1016/j.jhep.2022.03.011.
Wang J, et al. Three-in-one oncolytic adenovirus system initiates a synergetic photodynamic immunotherapy in immune-suppressive cholangiocarcinoma. Small. 2023;19: e2207668. https://doi.org/10.1002/smll.202207668.
Wong AM, et al. Unique molecular characteristics of NAFLD-associated liver cancer accentuate beta-catenin/TNFRSF19-mediated immune evasion. J Hepatol. 2022;77:410–23. https://doi.org/10.1016/j.jhep.2022.03.015.
Totoki Y, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46:1267–73. https://doi.org/10.1038/ng.3126.
El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264–73. https://doi.org/10.1053/j.gastro.2011.12.061.
Le Duc D, et al. Reduced lipolysis in lipoma phenocopies lipid accumulation in obesity. Int J Obes (Lond). 2021;45:565–76. https://doi.org/10.1038/s41366-020-00716-y.
Sun H, et al. An inflammatory-CCRK circuitry drives mTORC1-dependent metabolic and immunosuppressive reprogramming in obesity-associated hepatocellular carcinoma. Nat Commun. 2018;9:5214. https://doi.org/10.1038/s41467-018-07402-8.
Wang N, et al. PIWIL1 governs the crosstalk of cancer cell metabolism and immunosuppressive microenvironment in hepatocellular carcinoma. Signal Transduct Target Ther. 2021;6:86. https://doi.org/10.1038/s41392-021-00485-8.
Yang Z, et al. Levels of pretreatment serum lipids predict responses to PD-1 inhibitor treatment in advanced intrahepatic cholangiocarcinoma. Int Immunopharmacol. 2023;115: 109687. https://doi.org/10.1016/j.intimp.2023.109687.
Lv H, et al. NAD(+) metabolism maintains inducible PD-L1 expression to drive tumor immune evasion. Cell Metab. 2021;33:110–27. https://doi.org/10.1016/j.cmet.2020.10.021.
Wu MJ, et al. Mutant IDH Inhibits IFNgamma-TET2 Signaling to Promote Immunoevasion and Tumor Maintenance in Cholangiocarcinoma. Cancer Discov. 2022;12:812–35. https://doi.org/10.1158/2159-8290.CD-21-1077.
Abou-Alfa GK, et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020;21:796–807. https://doi.org/10.1016/S1470-2045(20)30157-1.
Zhu AX, et al. Final results from ClarIDHy, a global, phase III, randomized, double-blind study of ivosidenib (IVO) versus placebo (PBO) in patients (pts) with previously treated cholangiocarcinoma (CCA) and an isocitrate dehydrogenase 1 (IDH1) mutation. J Clin Oncol. 2021;39:266–266. https://doi.org/10.1200/JCO.2021.39.3_suppl.266.
Halford S, et al. A Phase I Dose-escalation Study of AZD3965, an Oral Monocarboxylate Transporter 1 Inhibitor, in Patients with Advanced Cancer. Clin Cancer Res. 2023;29:1429–39. https://doi.org/10.1158/1078-0432.CCR-22-2263.
Chu QS, et al. A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Invest New Drugs. 2015;33:603–10. https://doi.org/10.1007/s10637-015-0221-y.
Sehdev A, et al. A pharmacodynamic study of sirolimus and metformin in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2018;82:309–17. https://doi.org/10.1007/s00280-018-3619-3.
Molenaar RJ, et al. Study protocol of a phase IB/II clinical trial of metformin and chloroquine in patients with IDH1-mutated or IDH2-mutated solid tumours. BMJ Open. 2017;7: e014961. https://doi.org/10.1136/bmjopen-2016-014961.
Zhu AX, et al. Final Overall Survival Efficacy Results of Ivosidenib for Patients With Advanced Cholangiocarcinoma With IDH1 Mutation: The Phase 3 Randomized Clinical ClarIDHy Trial. JAMA Oncol. 2021;7:1669–77. https://doi.org/10.1001/jamaoncol.2021.3836.
Lowery MA, et al. Safety and activity of ivosidenib in patients with IDH1-mutant advanced cholangiocarcinoma: a phase 1 study. Lancet Gastroenterol Hepatol. 2019;4:711–20. https://doi.org/10.1016/S2468-1253(19)30189-X.
Tap WD, et al. Phase I study of the mutant IDH1 inhibitor ivosidenib: safety and clinical activity in patients with advanced chondrosarcoma. J Clin Oncol. 2020;38:1693–701. https://doi.org/10.1200/JCO.19.02492.
Jouve JL, et al. Pravastatin combination with sorafenib does not improve survival in advanced hepatocellular carcinoma. J Hepatol. 2019;71:516–22. https://doi.org/10.1016/j.jhep.2019.04.021.
Blanc JF, et al. Phase 2 trial comparing sorafenib, pravastatin, their combination or supportive care in HCC with Child-Pugh B cirrhosis. Hepatol Int. 2021;15:93–104. https://doi.org/10.1007/s12072-020-10120-3.
Riano I, et al. Efficacy and Safety of the Combination of Pravastatin and Sorafenib for the Treatment of Advanced Hepatocellular Carcinoma (ESTAHEP Clinical Trial). Cancers Basel. 2020. https://doi.org/10.3390/cancers12071900.
Britten CD, et al. A Phase I Study of ABC294640, a First-in-Class Sphingosine Kinase-2 Inhibitor, in Patients with Advanced Solid Tumors. Clin Cancer Res. 2017;23:4642–50. https://doi.org/10.1158/1078-0432.CCR-16-2363.
Giles JR, Globig AM, Kaech SM, Wherry EJ. CD8(+) T cells in the cancer-immunity cycle. Immunity. 2023;56:2231–53. https://doi.org/10.1016/j.immuni.2023.09.005.
Wang H, et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J Hepatol. 2021;75:1271–83. https://doi.org/10.1016/j.jhep.2021.07.032.
Pfister D, et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. 2021;592:450–6. https://doi.org/10.1038/s41586-021-03362-0.
Wabitsch S, et al. Metformin treatment rescues CD8(+) T-cell response to immune checkpoint inhibitor therapy in mice with NAFLD. J Hepatol. 2022;77:748–60. https://doi.org/10.1016/j.jhep.2022.03.010.
Kotsiliti E, et al. Intestinal B cells license metabolic T-cell activation in NASH microbiota/antigen-independently and contribute to fibrosis by IgA-FcR signalling. J Hepatol. 2023;79:296–313. https://doi.org/10.1016/j.jhep.2023.04.037.
Qin WH, et al. High serum levels of cholesterol increase antitumor functions of nature killer cells and reduce growth of liver tumors in mice. Gastroenterology. 2020;158:1713–27. https://doi.org/10.1053/j.gastro.2020.01.028.
Chen DP, et al. Glycolytic activation of peritumoral monocytes fosters immune privilege via the PFKFB3-PD-L1 axis in human hepatocellular carcinoma. J Hepatol. 2019;71:333–43. https://doi.org/10.1016/j.jhep.2019.04.007.
Peng ZP, et al. Glycolytic activation of monocytes regulates the accumulation and function of neutrophils in human hepatocellular carcinoma. J Hepatol. 2020;73:906–17. https://doi.org/10.1016/j.jhep.2020.05.004.
Ning WR, et al. Carbonic anhydrase XII mediates the survival and prometastatic functions of macrophages in human hepatocellular carcinoma. J Clin Invest. 2022. https://doi.org/10.1172/JCI153110.
Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol. 2011;6:275–97. https://doi.org/10.1146/annurev-pathol-011110-130138.
Wu L, et al. RIPK3 orchestrates fatty acid metabolism in tumor-associated macrophages and hepatocarcinogenesis. Cancer Immunol Res. 2020;8:710–21. https://doi.org/10.1158/2326-6066.CIR-19-0261.
Zhang J, et al. Hypoxia-inducible factor-1alpha/interleukin-1beta signaling enhances hepatoma epithelial-mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatology. 2018;67:1872–89. https://doi.org/10.1002/hep.29681.
Lu LG, et al. PD-L1 blockade liberates intrinsic antitumourigenic properties of glycolytic macrophages in hepatocellular carcinoma. Gut. 2022;71:2551–60. https://doi.org/10.1136/gutjnl-2021-326350.
Liu J, et al. Integrative metabolomic characterisation identifies altered portal vein serum metabolome contributing to human hepatocellular carcinoma. Gut. 2022;71:1203–13. https://doi.org/10.1136/gutjnl-2021-325189.
Daniel N, et al. The role of the gut microbiome in the development of hepatobiliary cancers. Hepatology. 2023. https://doi.org/10.1097/HEP.0000000000000406.
Silveira MAD, Bilodeau S, Greten TF, Wang XW, Trinchieri G. The gut-liver axis: host microbiota interactions shape hepatocarcinogenesis. Trends Cancer. 2022;8:583–97. https://doi.org/10.1016/j.trecan.2022.02.009.
Bauer KC, Littlejohn PT, Ayala V, Creus-Cuadros A, Finlay BB. Nonalcoholic Fatty liver disease and the gut-liver axis: exploring an undernutrition perspective. Gastroenterology. 2022;162:1858–75. https://doi.org/10.1053/j.gastro.2022.01.058.
Yu LX, Schwabe RF. The gut microbiome and liver cancer: mechanisms and clinical translation. Nat Rev Gastroenterol Hepatol. 2017;14:527–39. https://doi.org/10.1038/nrgastro.2017.72.
Bi C, et al. Molecular Immune Mechanism of Intestinal Microbiota and Their Metabolites in the Occurrence and Development of Liver Cancer. Front Cell Dev Biol. 2021;9: 702414. https://doi.org/10.3389/fcell.2021.702414.
Yoshimoto S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. https://doi.org/10.1038/nature12347.
Zhang X, et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut. 2021;70:761–74. https://doi.org/10.1136/gutjnl-2019-319664.
Zhou P, et al. High dietary fructose promotes hepatocellular carcinoma progression by enhancing O-GlcNAcylation via microbiota-derived acetate. Cell Metab. 2023;35:1961–75. https://doi.org/10.1016/j.cmet.2023.09.009.
Blaak EE, et al. Short chain fatty acids in human gut and metabolic health. Benef Microbes. 2020;11:411–55. https://doi.org/10.3920/BM2020.0057.
Che Y, et al. Gut microbial metabolite butyrate improves anticancer therapy by regulating intracellular calcium homeostasis. Hepatology. 2023;78:88–102. https://doi.org/10.1097/HEP.0000000000000047.
Zhang Q, et al. Gut Microbiome Directs Hepatocytes to Recruit MDSCs and Promote Cholangiocarcinoma. Cancer Discov. 2021;11:1248–67. https://doi.org/10.1158/2159-8290.CD-20-0304.
Ren Z, et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut. 2019;68:1014–23. https://doi.org/10.1136/gutjnl-2017-315084.
Jia X, et al. Characterization of Gut Microbiota, Bile Acid Metabolism, and Cytokines in Intrahepatic Cholangiocarcinoma. Hepatology. 2020;71:893–906. https://doi.org/10.1002/hep.30852.
Huang H, et al. Integrated analysis of microbiome and host transcriptome reveals correlations between gut microbiota and clinical outcomes in HBV-related hepatocellular carcinoma. Genome Med. 2020;12:102. https://doi.org/10.1186/s13073-020-00796-5.
Schneider KM, et al. Imbalanced gut microbiota fuels hepatocellular carcinoma development by shaping the hepatic inflammatory microenvironment. Nat Commun. 2022;13:3964. https://doi.org/10.1038/s41467-022-31312-5.
Behary J, et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat Commun. 2021;12:187. https://doi.org/10.1038/s41467-020-20422-7.
Protzer U, Maini MK, Knolle PA. Living in the liver: hepatic infections. Nat Rev Immunol. 2012;12:201–13. https://doi.org/10.1038/nri3169.
Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. 2013;14:996–1006. https://doi.org/10.1038/ni.2691.
Ma C, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018. https://doi.org/10.1126/science.aan5931.
Schramm C. Bile Acids, the Microbiome, Immunity, and Liver Tumors. N Engl J Med. 2018;379:888–90. https://doi.org/10.1056/NEJMcibr1807106.
Dart A. Gut microbiota bile acid metabolism controls cancer immunosurveillance. Nat Rev Microbiol. 2018;16:453. https://doi.org/10.1038/s41579-018-0053-9.
Ji G, et al. Manipulating Liver Bile Acid Signaling by Nanodelivery of Bile Acid Receptor Modulators for Liver Cancer Immunotherapy. Nano Lett. 2021;21:6781–91. https://doi.org/10.1021/acs.nanolett.1c01360.
Hang S, et al. Bile acid metabolites control T(H)17 and T(reg) cell differentiation. Nature. 2019;576:143–8. https://doi.org/10.1038/s41586-019-1785-z.
Campbell C, et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature. 2020;581:475–9. https://doi.org/10.1038/s41586-020-2193-0.
Paik D, et al. Human gut bacteria produce Tau(Eta)17-modulating bile acid metabolites. Nature. 2022;603:907–12. https://doi.org/10.1038/s41586-022-04480-z.
Gopalakrishnan V, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103. https://doi.org/10.1126/science.aan4236.
Routy B, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–7. https://doi.org/10.1126/science.aan3706.
Zheng Y, et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J Immunother Cancer. 2019;7:193. https://doi.org/10.1186/s40425-019-0650-9.
Mao J, et al. Gut microbiome is associated with the clinical response to anti-PD-1 based immunotherapy in hepatobiliary cancers. J Immunother Cancer. 2021. https://doi.org/10.1136/jitc-2021-003334.
Loeuillard E, et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J Clin Invest. 2020;130:5380–96. https://doi.org/10.1172/JCI137110.
Pan B, et al. 2,5-dimethylcelecoxib alleviated NK and T-cell exhaustion in hepatocellular carcinoma via the gastrointestinal microbiota-AMPK-mTOR axis. J Immunother Cancer. 2023. https://doi.org/10.1136/jitc-2023-006817.
Loo TM, et al. Gut microbiota promotes obesity-associated liver cancer through PGE(2)-Mediated Suppression of Antitumor immunity. Cancer Discov. 2017;7:522–38. https://doi.org/10.1158/2159-8290.CD-16-0932.
Bonavita E, et al. Antagonistic Inflammatory phenotypes dictate tumor fate and response to immune checkpoint blockade. Immunity. 2020;53:1215–29. https://doi.org/10.1016/j.immuni.2020.10.020.
Pelly VS, et al. Anti-inflammatory drugs remodel the tumor immune environment to enhance immune checkpoint blockade efficacy. Cancer Discov. 2021;11:2602–19. https://doi.org/10.1158/2159-8290.CD-20-1815.
Hu C, et al. Gut microbiota-derived short-chain fatty acids regulate group 3 innate lymphoid cells in HCC. Hepatology. 2023;77:48–64. https://doi.org/10.1002/hep.32449.
McDonald B, et al. Programing of an intravascular immune firewall by the gut microbiota protects against pathogen dissemination during infection. Cell Host Microbe. 2020;28:660–8. https://doi.org/10.1016/j.chom.2020.07.014.
Lee PC, et al. Gut microbiota and metabolites associate with outcomes of immune checkpoint inhibitor-treated unresectable hepatocellular carcinoma. J Immunother Cancer. 2022. https://doi.org/10.1136/jitc-2022-004779.
Rautiainen S, Manson JE, Lichtenstein AH, Sesso HD. Dietary supplements and disease prevention - a global overview. Nat Rev Endocrinol. 2016;12:407–20. https://doi.org/10.1038/nrendo.2016.54.
Singh V, et al. Dysregulated Microbial fermentation of soluble fiber induces cholestatic liver cancer. Cell. 2018;175:679–94. https://doi.org/10.1016/j.cell.2018.09.004.
Acknowledgements
We thank SCIER (www.scier.com.cn) for its linguistic assistance during the preparation of this manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grants 82130077 and 82103314) and Jinshan Hospital Flexible Mobile Talent Research Startup Fund (RXRC-2020-1).
Author information
Authors and Affiliations
Contributions
QG conceived and supervised the manuscript. JL designed and wrote the manuscript. DR and MZ prepared the figures and collected the references. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
All other authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lin, J., Rao, D., Zhang, M. et al. Metabolic reprogramming in the tumor microenvironment of liver cancer. J Hematol Oncol 17, 6 (2024). https://doi.org/10.1186/s13045-024-01527-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-024-01527-8