
Abstract
In one decade, immunotherapy based on immune checkpoint blockades (ICBs) has become a new pillar of cancer treatment following surgery, radiation, chemotherapy, and targeted therapies. However, not all cancer patients benefit from single or combination therapy with anti-CTLA-4 and anti-PD-1/PD-L1 monoclonal antibodies. Thus, an increasing number of immune checkpoint proteins (ICPs) have been screened and their effectiveness evaluated in preclinical and clinical trials. Lymphocyte activation gene-3 (LAG-3), T cell immunoglobulin and mucin-domain-containing-3 (TIM-3), and T cell immunoreceptor with immunoglobulin and tyrosine-based inhibitory motif (ITIM) domain (TIGIT) constitute the second wave of immunotherapy targets that show great promise for use in the treatment of solid tumors and leukemia. To promote the research and clinical application of ICBs directed at these targets, we summarize their discovery, immunotherapy mechanism, preclinical efficiency, and clinical trial results in this review.
Similar content being viewed by others
Introduction
Immune suppression resulting from cancer cell immune escape is closely related to tumor development and progression, treatment resistance, and poor prognosis. The mechanisms underlying immune suppression include complex steps and factors, and a key aspect is cytotoxic immune cell (CD8+ T cells and natural killer (NK) cell) exhaustion. T/NK cell exhaustion is generally recognized by the increased expression of several immune checkpoint proteins (ICPs), such as programmed cell death protein 1 (PD-1), cytotoxic T lymphocyte antigen-4 (CTLA-4), lymphocyte activation gene-3 (LAG-3), T cell immunoglobulin and mucin-domain-containing-3 (TIM-3), T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT), and B and T lymphocyte attenuator. These ICPs then inhibit the tumor-killing capacity of T/NK cells by ligating with respective ligands expressed on antigen-presenting cells (APCs), tumor cells, and other cells in the tumor microenvironment (TME) [1,2,3]. These ICPs have been aptly called the “brakes” of T and NK cells. The preponderance of evidence has shown that blocking the binding of ICPs or their ligands reverses the antitumor immune response of immune cells, resulting in tumor regression [4, 5]. In addition to expression on immune cells, some ICPs are expressed in tumor cells, sometimes promoting the proliferation and survival of these cells. Thus, blocking ICPs may have a “one stone, two birds” effect in tumor treatment [6]. Since the approval of the first immune checkpoint inhibitor (ICI), ipilimumab (an anti-CTLA-4 monoclonal antibody (mAb)), for the treatment of unresectable and metastatic melanoma by the US Food and Drug Administration (FDA) in 2014, immunotherapy based on immune checkpoint blockers (ICBs) has been approved for the treatment of more tumor types in earlier disease stages. Currently, four types of ICIs (anti-PD-1, PD-L1, CTLA-4, and LAG-3 mAbs) have been approved by the FDA for tumor treatment [7, 8]. Other ICIs, such as TIM-3 and TIGIT inhibitors, have been extensively evaluated in clinical trials as treatments for different solid tumors and leukemia [9, 10].
Due to varying expression and coexpression levels of other ICPs in different tumors, the efficiency of ICBs varies greatly. For example, while PD-1/PD-L1 blockade monotherapy has achieved a satisfactory response in patients with different cancers, a meta-analysis demonstrated that approximately four-fifths of patients do not respond to PD-1/PD-L1 monotherapy in clinical trials [11]. The reasons for resistance to PD-1/PD-L1 blockade remain unclear, but related factors may include the lack of PD-L1 expression [11]; heterogeneity in the TME, including reduced immune cell diversity [12,13,14]; the lack of active immune cells [15]; the existence of specific TCR clones; and the coexpression of other ICPs [16,17,18]. Thus, numerous clinical trials are currently evaluating the efficiency of different ICI monotherapies or combined therapies in different tumors. PD-1/PD-L1 and CTLA-4 blockers are widely used and extensively studied in the clinic, but the characteristics and efficiency of LAG-3, TIM-3, and TIGIT blockers have not been completely described. In this review, we describe the gene and protein characteristics, biological functions, and abnormal expression profiles of LAG-3, TIM-3, and TIGIT in solid tumors and leukemia. Most importantly, we summarize the advancements shown by studies related to the development of three ICPs from the bench to bedside and discuss their advantages and limitations.
Structural characteristics and biological functions of LAG-3, TIM-3, and TIGIT
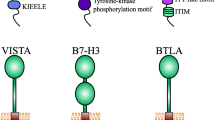
LAG-3, TIM-3, and TIGIT were identified in 1990, 2001, and 2009, respectively. The structures of these three ICPs are distinct, but their biological functions, namely inducing the exhaustion of immune cells and mediating immune suppression, are similar. The timeline of the discovery and antibody development for LAG-3, TIM-3, and TIGIT is shown in Fig. 1. The details of LAG-3, TIM-3, and TIGIT ligands are listed in Table 1.

Timeline of discovery and antibody development of LAG-3, TIM-3 and TIGIT. A. LAG-3, B. TIM-3, C. TIGIT. Notes: AML: Acute Myelocytic Leukemia; AST: Advanced Solid Tumors; BC: Breast Cancer; BLCA: Bladder Cancer; BLCA: Bladder Cancer; CAR-T: Chimeric Antigen Receptor T Cell Immunotherapy; CRC: Colorectal Cancer; DLBCL: Diffuse Large B cell Lymphoma; ESCC: Esophageal Squamous Cell Carcinoma; Fap2: Fibroblast activation protein 2; FGL-1: Fibrinogen-like protein 1; Gal-9: Galectin-9; GC: GC; HCC: Hepatocellular Carcinoma; HNSCC: Head and neck squamous cell carcinoma; LCA: Lung cancer; LSECtin: Liver and lymph node sinusoidal endothelial cell C-type lectin; MDS: Myelodysplastic Syndromes; MHC-II: MHC class II; MM: Multiple Myeloma; NPC: Nasopharyngeal Carcinoma; NSCLC: Non-small Cell Lung Cancer; OV: Ovarian Cancer; PAAD: Pancreatic Cancer; RCC: Renal Cell Carcinoma; SCC: Squamous Cell Carcinoma; SCLC: Small Cell Lung Carcinoma; TNBC: Triple-Negative Breast Cancer
LAG-3
LAG-3 (also named CD223 or FDC) was identified in 1990 by Triebel and colleagues while screening molecules that were selectively expressed in F5 cells, a CD3-negative interleukin (IL)-2-dependent NK cell line [19]. LAG-3 is located on the distal part of the short arm of chromosome 12 (12p13.31) in humans and chromosome 6 (6;6 F2) in mice. LAG-3 encodes a 525 amino acid protein that carries a signal peptide of 23 amino acids and an approximately 70 kDa mature Type I transmembrane glycoprotein in the immunoglobulin superfamily. To date, three isomers of LAG-3 have been identified; they range from LAG-3 protein isoform 1 precursor to LAG-3 protein isoform 3 precursor [20]. The structure of LAG-3 is different from that of CD3 and CD8, but it is highly homologous to that of CD4; LAG-3 consists of a transmembrane region, an extracellular region, and a cytoplasmic region. The extracellular structure consists of four IgSF domains, namely D1, D2, D3, and D4, which are critical for binding ligands. The D1 domain contains a loop domain rich in proline and an in-chain disulfide bond, which is species-specific and is in the V immunoglobulin superfamily. However, D2, D3, and D4 belong to the C2 family. The cytoplasmic region of LAG-3 consists of three parts: a serine phosphorylation site S454; a highly conserved “KIEELE” motif, which is known to be highly conserved in primates, mice, and rats; and a glutamate-proline dipeptide repeat motif (EP sequence). Soluble LAG-3 (sLAG-3) detaches from the cell membrane through the action of the metalloprotein (ADAM10/17) enriched in lipid rafts at the 20-aa connecting peptide between D4 and the transmembrane domain [20, 21].
At present, the LAG-3-related signal remains unclear. Louzalen et al. identified a novel protein called LAG-3-related protein (LAP) that binds to repetitive EP sequences in the LAG-3 intracellular region and may be involved in the downregulation of the CD3/T cell receptor (TCR) activation pathway (19). In addition, LAP may facilitate LAG-3 colocalization with CD3, CD4, and/or CD8 within glycosphingolipid-enriched microdomains (lipid rafts) to form immune synapses that regulate TCR signaling [22]. Studies have also shown that the KIEELE motif in the cytoplasmic domain is crucial for the activity of LAG-3. A single lysine residue (K468) in the conserved “KIEELE” sequence may recruit or mediate the activation of currently unknown signaling molecules, leading to downstream protein signaling [23].
LAG-3 is mainly expressed on activated T and B cells, NK cells, and dendritic cells (DCs) under physiological conditions, and it can negatively regulate T cell function [24]. Interestingly, LAG-3 was also found to be expressed on a proportion of malignant B cells from patients with diffuse large B cell lymphoma (DLBCL) [25]. To date, five LAG-3 ligands have been identified: MHC class II (MHC-II) [24], liver sinusoidal endothelial cell lectin (LSECtin) [26], galectin-3 [27], α-synuclein fibrils, and fibrinogen-like protein 1 (FGL-1) [28, 29].
LAG-3 can negatively regulate the function of T cells, exerting important effects on maintaining the homeostasis of the immune system under normal physiological conditions and promoting tumor cell immune escape in the TME [30]. LAG-3 also mediates bidirectional signaling in APCs. During Treg-DC interactions, engagement of LAG-3 on Tregs can enhance Treg activity to promote immune tolerance and indirectly inhibit DC function. Given its important biological role, LAG-3 is considered a promising target for cancer immunotherapy [31]. To date, more than twenty anti-LAG-3 antibodies have been used in clinical trials for cancer immunotherapy. Relatlimab, the first commercially developed anti-LAG-3 mAb, was entered into clinical trials in 2013, and it received FDA approval in March 2022, along with the PD-1 inhibitor nivolumab in the combination treatment Opdualag (Bristol Myers Squibb), which is used for the treatment of unresectable or metastatic melanoma.
TIM-3
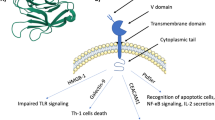
TIM-3 is a member of the TIM gene family, and it was identified in 2001 during a study of asthma susceptibility genes in congenic inbred mice. Murine TIM-3 is located on chromosome 11 within the Tapr region, and the human TIM-3 gene is located on chromosome 5 at q33.2. The full-length human TIM-3 cDNA is 906 bp and encodes a predicted membrane protein of 281 amino acid residues [32]. TIM-3 isoforms were identified in 2003, and an 800-bp amplicon was shown to encode an alternatively spliced soluble form of TIM-3 (sTIM-3) [33]. Initially, it was hypothesized that sTIM-3 competitively prevents TIM-3 from binding the TIM-3 ligand, which results in Th1 cells continuing to proliferate and perform effector functions [34]. In contrast, however, other studies suggested that sTIM-3 binds to ligands on T cells and suppresses antitumor immunity [33, 35]. TIM-3 is a single transmembrane molecule with an extracellular tail that carries an N-terminal immunoglobulin variable domain. This domain is successively followed by a mucin domain with glycosylation sites, a peptide linker with N-linked glycosylation sites, a transmembrane domain, and the C-terminus [36, 37]. TIM-3 contains a conserved region with five tyrosine residues. Two residues, Y265 and Y272 in humans, are assumed to be phosphorylated after the interaction of TIM-3 with its ligands. Itk, a Tec family tyrosine kinase, and Fyn and Lck, two kinases in the Src family, are involved in the TIM-3 signaling pathway. The activation of these tyrosine kinases leads to the accumulation of proteins with SH2 domains, such as the p85 subunit of phosphoinositide 3-kinase and phospholipase C-γ1, in the cytoplasmic tail of TIM-3. Furthermore, TIM-3 activation enhances nuclear factor of activated T cells and nuclear factor kB (NF-κB) activity through its interaction with zeta-chain-associated protein kinase 70 and SLP-76, which are components of the TCR signaling pathway. Importantly, the SH2 domain-binding motif is a trans-regulatory sequence that controls TIM-3-mediated signal transduction [38]. In addition, human leukocyte antigen-B-associated transcript 3 directly binds to the cytoplasmic tail of TIM-3 and prevents signaling in the absence of TIM-3 ligand(s) [38].
To date, TIM-3 has been found to be expressed on T cells (except for Th2 cells), and other immune cells, such as NK cells, macrophages, DCs, myeloid-derived suppressor cells, and mast cells. Moreover, TIM-3 is also expressed on certain malignant cells, such as melanoma [39, 40], myeloid leukemia [41], non-small cell lung cancer (NSCLC) [42], prostate cancer [43], osteosarcoma [44], colon carcinoma [45], and hepatocellular carcinoma (HCC) cells [46]. When TIM-3 binds to a ligand, immune cell or adaptive immune cell maturation and activation is attenuated, which is beneficial to tumor cell proliferation and survival. To date, four TIM-3 ligands have been identified. The first and most extensively characterized ligand is galectin-9 (Gal-9), followed by high-mobility group protein B1 (HMGB1), phosphatidylserine (PtdSer), and carcinoembryonic antigen cell adhesion molecule 1 (CEACAM-1) (Table 1). Different patterns of TIM-3 and ligand binding in various types of cells may result in different biological effects; e.g., HMGB1 binds to TIM-3 in different contexts that does not always lead to the same outcome [47]. In addition to the four aforementioned ligands, retinoic acid-inducible gene I (RIG-I), which is a member of the (RIG-I)-like receptor family, has also been reported to interact directly with Tim-3. Specifically, Tim-3 inhibits RIG-I expression in macrophages through the action of STAT1, promotes RIG-I ubiquitination and degradation through the action of the E3 ligase RNF-122, and subsequently inhibits type I interferon (IFN) production and antiviral activity [48,49,50]. The first anti-TIM-3 mAb, sabatolimab, was developed for use in solid tumor therapy, and it works by blocking the binding of TIM-3 to its ligands Gal-9 and PtdSer. To date, more than 33 TIM-3 mAbs have been evaluated in clinical trials as cancer immunotherapies.
TIGIT
TIGIT was identified in 2009 through the combination of two genome-wide search strategies used in studies to determine whether activated human T cells express costimulatory or inhibitory molecules, particularly genes expressed in T cells and NK cells [51]. The mouse TIGIT gene is located at the B4 position of chromosome 16, while the human TIGIT gene is located at q13.31 on chromosome 3. Human TIGIT cDNA is 2926 bp in length and encodes 244 amino acids. Six variants encode TIGIT isoforms [51, 52]. TIGIT is expressed on lymphocytes, including Tregs, memory T cell subsets, and NK cells [53], and its expression can be upregulated when these cells are activated [51]. In addition, TIGIT has been reported to be expressed on tumor cells in mice [54].
It is now believed that there are five TIGIT ligands, namely CD155 (also known as PVR), CD112, CD113, Nectin4 [55], and Fab2 [56]. TIGIT encodes a protein carrying an immunoglobulin variable domain, a transmembrane domain, and an immunoreceptor tyrosine-based inhibitory motif (ITIM). Human TIGIT shares 58% sequence identity with mouse TIGIT, and the ITIM-containing sequence is identical in the cytoplasmic tails of mouse and human TIGIT [51, 52]. The binding of TIGIT to its ligands triggers the activation of a series of signaling pathways that affect the function of immune cells and the immune response, thereby causing an overall immune suppressive response in cells. Notably, in addition to binding with ligands, TIGIT can carry out its the immune suppression function via interference of the co-stimulation signaling in T cells mediated by CD226 or CD96 [51, 57].
More than 45 types of TIGIT inhibitors have been developed, and most of them are used in clinical practice for solid tumors and leukemia; however, only a few anti-TIGIT mAbs have been entered into Phase III clinical trials. IBI939 is the first anti-TIGIT mAb approved for clinical use in China, and it is currently in Phase I trials for patients with leukemia and solid tumors [58]. Another mAb, tiragolumab, is a fully human anti-TIGIT IgG1/kappa mAb developed by Roche that carries a complete Fc region that blocks the binding of TIGIT to its receptor CD155 [59].
Upregulating the expression of LAG-3, TIM-3 and TIGIT in solid tumors and leukemia
In solid tumors and leukemia, ICPs are generally increased expression on immune cells and bind to ligands on malignant cells or APCs, leading to the depletion of T/NK cells or the disruption of their antitumor function [39]. ICPs can also be expressed on malignant cells and may promote their proliferation [60,61,62]. For example, TIM-3 is expressed in patients with solid tumors and leukemia [42, 62, 63]. The expression of LAG-3, TIM-3 and TIGIT in solid tumors and leukemia and their correlation with clinical outcomes are summarized in Table 2.
LAG-3
Overexpression of LAG-3 has been identified on tumor-infiltrating lymphocytes (TILs) in a number of solid tumors, including melanoma, glioma, NSCLC, head and neck squamous cell carcinoma (HNSCC), breast cancer (BC), gastric cancer (GC), and lymphoma, as well as in leukemia [64,65,66,67,68,69,70,71,72,73,74,75]. Early studies have found that in the melanoma context, metastatic lymph nodes were infiltrated not only by T cells (CD4+, CD8+, and Tregs) but also by a substantial percentage of NKT and NK cells that express LAG-3. LAG-3 expression mediated immune escape in melanoma cells by impairing immune cell function while also protecting against Fas- and drug-induced apoptosis via its interaction with MHC-II [76]. LAG-3 is expressed on a subset of human pDCs, and LAG-3+ pDCs are enriched in the tumor sites of melanoma patients. These LAG-3+ pDCs interact with MHC-II to induce Toll-like receptor-independent pDC activation, which may contribute to the formation of an immunosuppressive microenvironment by increasing IL-6 production [77, 78]. On the other hand, LAG-3 is expressed on a certain proportion of malignant B cells in patients with DLBCL and chronic lymphocytic leukemia (CLL), and digital protein spatial analysis showed that LAG-3 is strongly associated with macrophages in the TME. In contrast to TIM-3, no studies have reported the mechanism by which LAG-3 expressed on tumor cells promotes the proliferation of these cells [25].
Most data from different clinical investigations have indicated that higher LAG-3 expression is related to poor clinical outcomes. For example, increased LAG-3 expression is used to stratify patients with HNSCC into high-risk groups [79], and a high level of LAG-3 expression in soft tissue sarcoma tissues has been found to be significantly correlated with high pathological grade and late stage [69]. High LAG-3 expression levels have been associated with poorer prognosis for renal cell carcinoma (RCC) [80], poor overall survival (OS) of patients with either high- or low-grade glioma [81], and low disease-free survival in patients with pancreatic cancer [82]. In patients with NSCLC, positive LAG-3 expression has been associated with early postoperative relapse and worsened prognosis [72]. In addition, high expression of the LAG-3 gene in patients with DLBCL is associated with poor survival and prognosis. Moreover, compared with LAG-3low/PD-L1high-expressing patients with DLBCL, those who expressed LAG-3high/PD-L1high showed lower progression-free survival (PFS) and OS rates [25].
In contrast, an increase in the density of LAG-3 TILs indicated a trend toward higher OS times in patients with triple-negative BC and Her2-positive BC [83]. Interestingly, similar findings have been reported in which LAG-3 expression on TILs at the tumoral front predicted better treatment outcomes for all Stage II patients and for a subgroup of patients with Stage II microsatellite-stable colon cancer (CC) [84]. Whether these results are related to targeted therapy or immunotherapy is unclear, and further analysis is needed.
Here we summarize the immunosuppression mechanisms underlying LAG-3 expression in the TME as follows: 1) The interaction of LAG-3 and MHC-II between CD4 and tumor cells inhibits the proliferation of CD4 T cells and cytokine secretion from these cells; moreover, the signaling downstream of MHC-II supports the survival of tumor cells [85], (2) The interaction of LAG-3 and MHC-II between Treg and tumor cells/DCs may enhance the stability and immunosuppression capacity of the Tregs; however, the maturation and immunostimulatory capacity of DCs may be impaired by MHC-II downstream signaling [26, 28, 86], (3) The interaction of LAG-3 and MHC-II between Treg and tumor cells/DCs may enhance the stability and immunosuppression capacity of the Tregs; however, the maturation and immunostimulatory capacity of DCs may be impaired by MHC-II downstream signaling [31], (4) sLAG-3 in the TME impaired the antigen-presentation function of monocyte-derived DCs (mDCs) in the TME or even inhibit the differentiation of mDCs [87] (Fig. 2). Therefore, blocking LAG-3 may be an effective strategy to enhance antitumor T cell responses. The effects of targeting LAG-3 in melanoma were first reported in an early clinical trial in 2013.

The immunosuppression mechanisms underlying LAG-3 action in the TME. ① The interaction of LAG-3 and MHC-II between CD4 and tumor cells inhibits the proliferation and cytokine secretion of CD4 T cells, and the downstream MHC-II signal may support the survival of tumor cells. ② The interaction of LAG-3 and Galectin-3/LSECtin/FGL-1 between CTL/NK cells and the TME compartment inhibits the proliferation and cytotoxicity of CTL/NK cells. ③ The interaction of LAG-3 and MHC-II between Tregs and tumor cells/DCs enhances the stability and immunosuppression capacity of Tregs. On the other hand, the maturation and immunostimulatory capacity of DCs are impaired by MHC-II downstream signaling. ④ The TME contains soluble LAG-3 (sLAG-3), which can impair the antigen-presenting function of monocyte-derived DCs (mDCs) or even inhibit the differentiation of monocytes into DCs
TIM-3
In cancer patients, TIM-3 overexpression can be detected on most immune cells, particularly antigen-specific CD8+ T cells, CD4+ T cells, and NK cells [46, 88]. TIM-3 coexpression with PD-1 is frequently found on immune cells of peripheral blood and bone marrow and on TILs from tumor patients, and this expression is correlated with decreased T cell proliferation and cytokine production, resulting in immune cell dysfunction and tumor immune escape [89,90,91,92].
In addition to expression on immune cells, TIM-3 is expressed on a number of tumor cells [42, 62, 93, 94]. In 2010, Kikushige Y et al. first reported TIM-3 on the surface of leukemia stem cells (LSCs) but not on hematopoietic stem cells (HSCs) [41]; Since this report, TIM-3 has been thought of as a biomarker for acute myeloid leukemia (AML) stem cells as well as a target for treatments directed against myeloid leukemia stem cells for patients with AML and myelodysplastic syndromes (MDS) [95, 96]. Further study has demonstrated that TIM-3 is expressed on endothelial cells, but in this context, it does not function as a Gal-9 receptor but rather interacts with melanoma cells to trigger the NF-κB signaling pathway, promoting cell proliferation and reducing the apoptosis rate [62]. Furthermore, TIM-3 was also reported to be expressed on tumor cells from patients with NSCLC, HCC, and GC [42, 46, 88].
Most studies indicated that overexpression of TIM-3 either on immune cells or tumor cells is associated with poor OS for patients, such as patients with GC [88], and high-risk groups, showing poor prognosis, and lower complete response (CR) rates following induction chemotherapy in patients with AML as well as high-risk patients with B cell acute lymphoblastic lymphoma (B-ALL) relapse after allogeneic hematopoietic stem cell transplantation (allo-HSCT) [97,98,99,100]. Moreover, a Phase I trial (NCT02573363) demonstrated that higher TIM-3/Gal-9 expression was associated with chemotherapy resistance in patients with AML [101].
Several studies have demonstrated that TIM-3 expression in tumors may contribute to cancer cell immune escape via different mechanisms, including (1) inhibiting CD4+ T cell activation via the IL-6-STAT3 pathway, thereby preventing Th1 polarization and promoting tumor occurrence, growth, and metastasis [42, 63], (2) reducing the adhesion of tumor cells and promoting the survival of melanoma cells [39, 60]. (3) regulating the epithelial-mesenchymal transition by reducing E-cadherin and upregulating N-cadherin expression, which increases HCC cell migration and invasion rates [46], (4) by autocrine signaling mediated through TIM-3 binding ligands, including Gal-9, thereby enabling these cells to avoid recognition and clearance by immune cells to sustain their survival and self-renewal [102,103,104,105,106]. Blocking TIM-3 has been explored as a therapeutic strategy in a number of clinical trials since 2015. The immunosuppressive mechanisms of TIM-3 in the TME are depicted in Fig. 3.

The immunosuppression mechanisms underlying TIM-3 action in the TME. ① Inhibition of CD4 + T cell activation via the IL-6-STAT3 pathway, preventing Th1 polarization and promoting tumor occurrence, growth, and metastasis. ② Expression of TIM-3 on melanoma cells can reduce the adhesion ability of tumor cells and promote their survival. ③ Expression of TIM-3 on HCC cells regulate the epithelial-mesenchymal transition (EMT) by reducing E-cadherin and upregulating N-cadherin expression, which increases the migration and invasion of HCC cells. ④ TIM-3+ tumor cells sustain their own survival and self-renewal via the autocrine action a variety of TIM-3 ligands and avoiding recognition and clearance by immune cells
TIGIT
TIGIT upregulation was identified in several kinds of solid tumors and leukemia, including melanoma, NSCLC, GC, AML, and multiple myeloma [8, 57, 107,108,109,110,111]. In melanoma patients, CD8+ TILs highly expressed TIGIT together with PD-1, this high-TIGIT expression was consistent with that observed in NSCLC patients. TIGIT+ CD8+ T cells from patients with AML, GC, or multiple myeloma showed reduced cytokine production, high susceptibility to apoptosis, and significantly reduced proliferation and killing ability. In addition, CD8+ T cells in patients with AML or multiple myeloma with high-TIGIT expression expressed lower levels of CD226. Moreover, the increased expression of TIGIT on CD8+ T cells has been related to poor prognosis during leukemia relapse after allo-HSCT and in advanced GC patients [111, 112].
In addition to its expression on TILs, TIGIT is expressed on NK cells. In human CC NK cells, the expression of TIGIT in intratumoral areas was significantly higher than that in the peritumoral area, and the expression of TIGIT on CD8+ T cells in the intratumoral area was not significantly different from that in the peritumoral area. In a variety of tumor bearing mice models (B16 melanoma, CT26 CC, 4T1 BC lung metastasis model mice, among others), TIGIT was more specifically associated with tumor progression in NK cells than in other cells, and TIGIT+ NK cells acquired an exhaustion phenotype, with reduced effector function and antitumor potential. Lack of TIGIT expression in NK cells in vivo retarded tumor growth, and blockade of TIGIT action via mAb reversed antitumor NK cell exhaustion in multiple tumor models, resulting in increased overall host survival [113]. TIGIT is commonly expressed on Tregs. TIGIT+ Tregs acquired both highly activated and suppressed phenotypes in tumor tissues [114]. For example, in bladder cancer, TIGIT + Tregs accumulated around cancer tissues, promoted cancer cell metastasis and suppressed the antitumor immune response by promoting IL-32 expression in Tregs [115].
Recently, a study showed that TIGIT is expressed on human memory B cells and controls the immune response by directly acting on T cells and blocking the proinflammatory function of dendritic cells, thereby inhibiting Th1-, Th2-, Th17- and CXCR5+ICOS+ T cell responses while promoting the immune regulatory function of the T cells [116]. All these studies suggest that the increased expression of TIGIT in the TME leads to immune escape of tumor cells, thereby affecting the development and progression of tumors and the prognosis of patients.
TIGIT has been shown to potentially suppress innate and adaptive immunity through the following mechanisms: (1) TIGIT acts on DCs by binding to its ligand CD155, promoting the formation of immune tolerogenic DCs and indirectly hindering the function of T cells [51]. (2) TIGIT can act directly on the T cells by attenuating TCR driven activation signals, which is independent of the APC directed inhibition of T cell responses [117]. (3) TIGIT directly inhibits T cell function by competing with CD226 to bind their common ligand CD155, reducing IL-2 and IFN-γ production and increasing the production of IL-10, thereby exerting immunosuppressive effects [118]. (4) TIGIT can directly bind to CD226 in cis, which disrupts the binding of CD226 to CD155 [119]. (5) TIGIT is enriched in Tregs and enhances the immunosuppressive function of Tregs through its effects on an exogenous pathway. TIGIT interferes with selective Treg-mediated suppression of proinflammatory Th1 and Th17 cells, but not Th2 cells, by inducing the secretion of the soluble effector molecule fibrin-like protein-like protein 2 [114, 120]. (6) Furthermore, TIGIT binds to its ligands and transmits inhibitory signals directly to T cells and NK cells through its cytoplasmic tail [121] (Fig. 4). Hence, blocking TIGIT activity is an effective approach to resolve the exhaustion of immune cells and restore their antitumor function. Therapies blocking TIGIT have been explored in a number of clinical trials since approximately 2016.

The immunosuppression mechanisms underlying TIGIT action in the TME. ① TIGIT binds to D155 expressed on DCs, making DCs tolerant, resulting in increased IL-10 secretion and decreased IL-12 production. ② TIGIT acts directly on T cells independent of APCs by weakening the activation signal mediated by TCR, thereby inhibiting the T cell response ③ TIGIT and CD226 are coexpressed on T cells, and TIGIT competes with CD226 for binding their common ligand CD155, thereby directly inhibiting the T cell response and increasing the secretion rate of IL-10. ④ TIGIT directly binds to CD226 and prevents the dimerization of CD226, thereby preventing the binding of CD155 to CD226. ⑤ TIGIT expressed on Tregs binds to CD155 expressed on tumor cells or APCs, resulting in increased secretion of the Fgl2 protein, thereby inhibiting Th1/Th17 cell differentiation. ⑥ TIGIT binds to ligands and transmits inhibitory signals directly to T cells and NK cells through its cytoplasmic tail
Preclinical studies evaluating therapies targeting LAG-3, TIM-3, and TIGIT in solid tumors and leukemia
Several preclinical studies have demonstrated that targeting LAG-3, TIM-3, or TIGIT restores T cell function and inhibits tumor progression, and most studies indicated that coinhibition of different ICPs may effectively enhance antitumor activity.
LAG-3
Increasing the expression of LAG-3 and LAG-3+ immune cells in patients with solid tumors or leukemia has been associated with tumor progression, poor prognosis, and unfavorable clinical outcomes, strongly indicating that LAG-3 contributes to immune escape by tumor cells. These findings are similar to those reported for PD-1, but different mechanisms may be involved. Therefore, LAG-3 has been proposed as a promising therapeutic target for cancer immunotherapy based on results from in vitro and in vivo animal model studies.
Recently, strategies targeting LAG-3 have mainly includes sLAG-3-Ig and anti-LAG-3 antagonistic antibodies. LAG-3 antagonistic Abs can directly bind to LAG-3 molecules, block the interaction between ligands and LAG-3, and down-regulate the inhibitory effect of LAG-3 on the immune system. sLAG-3-Ig (such as IMP321) is composed of the Fc part of the human antibody and the four extracellular domains of LAG-3, which can target the MHC-II molecules on APCs and activate APCs to activate other immune cells, including T cells [30].
Shortly after it was generated for use in biochemical and functional studies, a sLAG-3-Ig fusion protein was studied in vivo in murine tumor models. For example, in an HNSCC mouse model with overexpressed LAG-3 on CD4+ and CD8+ T cells and Tregs, administration of LAG-3-Ig retarded tumor growth in a manner associated with an enhanced systemic antitumor response; specifically, LAG-3-Ig potentiated the cytotoxicity of CD8+ T cells and reduced the population of immunosuppressive cells [73]. sLAG-3-Ig mediated tumor control and regression in mice bearing RCC, sarcoma, or BC. sLAG-3-Ig upregulates the expression of costimulatory molecules and increases IL-12 expression in DCs [122]. These phenotypic changes result in an enhanced sLAG-3-Ig-induced DC maturation, which leads to Th1 cell responses and increases the production of IFN-γ in responding T cells. Based on these findings, sLAG-3-Ig has been proposed to function as an adjuvant that likely can potentiate a response to a vaccine. This proposal was realized, as sLAG-3-Ig has been shown to markedly enhance the CD8+ T cell response to a soluble antigen vaccine (ovalbumin) as well as the humoral response to a particulate antigen (hepatitis B surface antigen) in mice [123]. This adjuvant effect has been extended to cancer vaccines as well.
In contrast to the effects of sLAG-3-Ig, anti-LAG-3 mAbs mainly block the LAG-3/MHC-II interaction to restore immune cell function. High expression of LAG-3 in patients with CLL has been associated with poor cytogenetics and poor prognosis. After anti-LAG-3 mAb treatment in vitro, peripheral blood mononuclear cells from CLL patients eliminated leukemic cells and exhibited restored NK and T cell-mediated responses [124]. In soft tissue sarcoma model mice, LAG-3 blockade decreased tumor growth and enhanced the secretion of IFN-γ by CD8+ and CD4+ T cells [69]. However, due to the limited efficacy of the anti-LAG-3 mAb administered alone, it is generally used in combination with other ICIs, including CTLA-4 inhibitors or PD-1 inhibitors, increasing their efficacy synergistically.
It has been shown that LAG-3 functions in concert with PD-1 to suppress antitumor immunity. Coexpression of LAG-3 and PD-1 on tumor-infiltrating CD4+ and CD8+ T cells and the profound therapeutic effects of coblockers or the genetic deletion of LAG-3 and PD-1 have been observed in various model mice of tumors, including B16 melanoma, MC38 colon adenocarcinoma, Sa1N fibrosarcoma, IE9mp1 ovarian cancer (OC), Em-TCL1 CLL, and recurrent melanoma [75, 125,126,127]. The blockade of both LAG-3 and PD-1 augmented the proliferation and cytokine production of tumor-infiltrating CD8+ T cells after ex vivo stimulation with the tumor-associated antigen NY-ESO-1 in OC cells [128]. Therefore, targeting multiple ICPs simultaneously has become a promising therapeutic strategy.
In 2019, Fianlimab (REGN3767), a fully human IgG4 mAb that targets LAG-3, was developed. This mAb binds human and monkey LAG-3 with high affinity, and specificity blocks the interaction between LAG-3 and MHC-II. In an engineered T/APC bioassay, fianlimab, alone or in combination with cemiplimab (REGN2810, a human anti-PD-1 mAb), blocked inhibitory signaling to T cells mediated by hLAG-3/MHC-II in the presence of PD-1/PD-L1. In humanized PD-1xLAG-3 knock-in mice, treatment with cemiplimab and fianlimab showed increased efficacy and enhanced the amount of proinflammatory cytokines secreted by tumor-specific T cells compared with the effect of fianlimab alone [129]. Another study described the profound effects of combined inhibition of LAG-3 (BI 754111) and PD-1 (ezabenlimab) in an in vitro model of antigen-exposed memory T cells expressing PD-1 and LAG-3. IFN-γ secretion was increased as high as 13.2-fold compared to that of the isotype control in the BI 754111 plus ezabenlimab group and was increased 1.8-fold and 6.9-fold in the BI 754111 and ezabenlimab monotherapy groups, respectively [130]. These results supported the clinical investigation of a combination treatment to inhibit PD-1 and LAG-3. Clinical trials involving anti-LAG-3 mAb treatment have been conducted since 2006.
TIM-3
Similar to the effects of PD-1 blockade, the effects of blocking TIM-3 in vitro and ex vivo experiments demonstrated that it can improve cytotoxicity and IFN-γ release by both TILs and NK cells in RCC, melanoma, lung adenocarcinoma, and OC contexts [131].
The first study demonstrating that blocking the TIM-3/Gal-9 pathway by an anti-TIM-3 mAb increases the activation and numbers of macrophages in a mouse model of autoimmune disease was reported in 2002 [132]. Further study using a TIM-3 fusion protein confirmed that the TIM-3–TIM-3 ligand pathway may inhibit the expansion and effector functions of Th1 cell populations and may be essential for tolerance induction in Th1 cells [133]. Later, lower immune tolerance mediated via a reduction in the expansion of myeloid-derived suppressor cells was demonstrated in mice with TIM-3−/−4T1 mammary adenocarcinoma after TIM-3 Ig fusion protein treatment [134]. In 2010, anti-TIM-3 mAbs were first used in both CT26 CC mice and mice bearing B16 melanoma, but they demonstrated little effect. However, significant antitumor effects were demonstrated when TIM-3-Ig was administered in combination with an anti-PD-L1 mAb [135]. Similarly, studies demonstrated that anti-TIM-3 (5D12 clone) alone was not effective in reducing tumor growth in CC (CT26 and MC38) model mice, while blockade of both CEACAM-1 and PtdSer via TIM-3 showed greater efficacy [102, 136]. In contrast, other experiments have shown that anti-TIM-3 mAb alone exerted an antitumor effect on WT3 sarcoma, a MC38 tumor, and B16 melanoma models [41, 131, 137].
As previously described, TIM-3 is expressed on myeloid LSCs; thus, Kikushige Y et al. successfully reconstructed an AML model using TIM-3+ AML cells in immunodeficient mice and established the first anti-human TIM-3 mouse IgG2a mAb that did not disrupt the reconstitution of normal human HSCs but blocked LSCs [41]. TIM-3+ AML LSCs secrete the ligand Gal-9 in an autocrine manner, activating the NF-κB and β-catenin pathways to increase survival and self-renewal [106]. In addition, anti-TIM-3 alone can inhibit leukemia cell proliferation in AML model mice [41, 131, 137]. However, additional findings indicate the necessity of targeting multiple coinhibitory ICPs rather than targeting TIM-3 alone to maximize therapeutic efficacy in the context of AML and solid tumors in mice [131, 137].
The antitumor effects by targeting TIM-3 may be required under certain conditions; for example, in the cases of IFN-γ-producing CD8+ and CD4+ T cells and when the ratio of tumor-infiltrating CD8+:CD4+ T cells is high [131]. Moreover, in a CT26 subcutaneous tumor model, blocking TIM-3 was effective only before the appearance and accumulation of a significant number of TIM-3+PD-1+ T cells [131]. In contrast, many preclinical studies have revealed that TIM-3 is upregulated in immunotherapy resistance, and high expression of TIM-3 on T cells may be related to adaptive resistance to anti-PD-1 or anti-CTLA-4 treatment. Blocking TIM-3 can increase the antitumor effects of anti-PD-1 or anti-CTLA-4 immunotherapy. TIM-3 and PD-1 coblockers increased antitumor immune responses and tumor growth-reducing efficacy in melanoma and GC mouse models [60, 135, 138,139,140,141]. The anti-TIM-3 mAb has been shown to increase the resistance of anti-PD-1 therapy in mouse models of lung adenocarcinoma with genetically engineered EGFR and extended the median survival from 5 to 11.9 weeks. The effects not only included enhanced T cell function following anti-PD-1 mAb failure but also decreases in the levels of tumor-promoting cytokines, such as IL-6 and progranulin [138]. Overall, targeting TIM-3 can be considered a strategy to overcome resistance to anti-PD-1 therapy. In addition, several attempts have been made to investigate the synergistic efficacy of TIM-3 inhibitor combined with chemotherapy or radiotherapy and anti-PD-1/PD-L1 therapy in tumor model mice [139, 142, 143]. For example, adding anti-TIM-3 mAbs to an anti-PD-1 mAb therapy regimen prolonged median survival from 33 to 100 days and increased OS from 27.8 to 57.9% in model mice with glioblastoma [142]. TIM-3 blocker has been evaluated in several clinical trials since 2015.
TIGIT
Because TIGIT hinders multiple stages of antitumor immunity, it is abnormally expressed in several cancer types and associated with poor clinical outcome. Numerous preclinical studies have evaluated TIGIT blockade immunotherapy in the contexts of various solid tumors and leukemia.
TIGIT blockade was first observed in a study that showed that deletion of the TIGIT gene in mice significantly enhanced the cytotoxic effects of NK cells and CD8+ T cells against tumor cells [144]. In addition, TIGIT modulate the suppressive activity of Tregs, thereby promoting tumor growth in B16F10 melanoma model mice, and these findings were further demonstrated in TIGIT knockout mice. In OC model mice, blocking TIGIT significantly reduced tumor growth and the proportion of CD4+ Tregs and increased the survival rate [145, 146]. Overall, TIGIT blockade can enhance NK cell cytotoxicity and CD4+ and CD8+ T cell activation, inhibit Treg activity, and improve antitumor effects in vitro and in vivo in mouse models.
TIGIT expression is closely associated with PD-1 on T cells in patients with solid tumors or leukemia [147,148,149,150]. Anti-TIGIT mAbs alone or in combination with anti-PD-L1 mAbs synergistically exerted their effects in a CT26 colorectal cancer model mice [57]. Treatment with a combination of anti-PD-1 and anti-TIGIT mAbs more effectively controlled tumor growth [57]. In ex vivo experiments, combination treatment including atezolizumab (anti-PD-L1 mAb) and tiragolumab (anti-TIGIT mAb) restored the functionality of TILs from colorectal cancer patients [151].
In contrast, the immunosuppressive effects of TIGIT can be leveraged for acute graft-versus-host disease (GVHD) therapy after allo-HSCT. A study using a TIGIT-Fc fusion protein, which exerted immunosuppressive effects by binding to CD155 on DCs, demonstrated that TIGIT-Fc delayed the onset of GVHD symptoms and increased survival in model mice with acute GVHD. TIGIT inhibition can also be utilized in transplant immunotherapy because it enhances the activity and function of graft immune cells with disease relapse after allo-HSCT [152]. Therefore, targeting TIGIT holds clear clinical potential as a cancer treatment, and various TIGIT-targeting mAbs have been evaluated in clinical trials for the treatment of solid tumors since 2016.
Clinical trials of anti-LAG-3, anti-TIM-3, and anti-TIGIT mAbs
Several clinical trials have been established to evaluate anti-LAG-3, anti-TIM-3, or anti-TIGIT mAbs as different tumor therapies. Most of these trials are in Phase I/II for patients with advanced and metastatic cancers; the final results have not been reported. Multiple Phase III trials have shown positive results, and on 2022, both of the FDA and the European Medical Agency (EMA) approved Opdualag (a fixed-dose combination of the anti-LAG-3-blocking mAb relatlimab and the anti-PD-1-blocking mAb nivolumab) for the treatment of adults and children 12 years of age or older with unresectable or metastatic melanoma [7].
Anti-LAG-3 mAbs
A number of anti-LAG-3 mAbs have been developed in the past year, and some are currently being evaluated in clinical trials as cancer immunotherapies (Table 3). These trials have been completed, are underway, or are recruiting participants (ClinicalTrials.gov). Two types of inhibitors have been developed for LAG-3-targeting therapies: anti-LAG-3 mAbs and LAG-3-bispecific antibodies (BsAbs). In this section, we describe the clinical trials in which anti-LAG-3 mAbs are being evaluated.
Relatlimab (BMS-986016) is an anti-LAG-3 fully human IgG4-κ mAb and the first LAG-3 blocker to be clinically developed. Relatlimab binds LAG-3 with high affinity and inhibits its binding to MHC-II. Currently, relatlimab is being evaluated alone or in combination with anti-PD-1/PD-L1 mAbs in Phase I to III trials for patients with solid tumors or leukemia, and it presented good tolerance profile and clinical activity [153]. One important trial for relatlimab evaluation is RELATIVITY-047, a Phase III trial that evaluated the effect of inhibiting both LAG-3 and PD-1 by the combination of relatlimab and nivolumab compared with the effect of nivolumab alone for patients with untreated metastatic or unresectable melanoma (NCT03470922). Blinded independent assessment of the primary end point showed that patients taking relatlimab–nivolumab dual checkpoint inhibitors experienced a PFS that was twice the median PFS and a 25% lower risk of disease progression or death than patients receiving nivolumab alone. The relatlimab–nivolumab cohort showed a slightly greater incidence of adverse events than the nivolumab cohort, but the quality of life measurements was similar. The benefit of relatlimab–nivolumab compared to that of nivolumab was also observed among BRAF-mutant melanoma patients and wild-type individuals. Overall, the trial provided solid data supporting relatlimab–nivolumab as a potential new treatment option for patients with previously untreated metastatic or unresectable melanoma [7]. In June 2022, Bristol Myers Squibb announced the first-in-class dual immunotherapy, relatlimab–nivolumab fixed-dose combination Opdualag, which received accelerated FDA approval for the treatment of metastatic melanoma. This is the first FDA-approved anti-LAG-3 mAb combination therapy, making LAG-3 the third clinical ICI target in the clinic, following PD-1/PD-L1 and CTLA-4.
To date, 65 clinical trials have been established to evaluate relatlimab. In a clinical trial with CLL patients (NCT02061761), relatlimab induced the depletion of leukemia cells in vitro, restored T and NK cell-mediated antitumor responses and promoted T cell production of cytokines such as IL-2. These results provide new insights into the anti-leukemia potential of relatlimab, which may be related to reduced anti-apoptotic signaling in malignant cells and enhanced responses mediated by NK and T cells [124].
Favezelimab (MK-4280) is a humanized IgG4 anti-LAG-3 mAb developed by Merck. Favezelimab treatment increases the production of cytokines, such as IFN-γ, IL-2, IL-8, and TNF-α, and chemokines (CCL4, CXCL10, and CCL22) in Jurkat Clone G10-PD-1 cells (a group of cell lines obtained by coculturing Jurkat and Raji B cell lymphoma lines that express high levels of LAG-3 and PD-1). The expression of CD69, CD44, and CD25 was also upregulated [154]. To date, 15 clinical trials have been established to evaluate favezelimab alone or in combination with other ICIs in different types of tumors. The preliminary results have demonstrated that this drug shows good safety and efficacy and controllable tolerance when administered alone or in combination with other ICIs [155]. The first Phase I/II trial confirmed the safety and effectiveness of pembrolizumab (anti-PD-1) for treating solid tumors [156]. In addition, the combination treatment consisting of favezelimab with pembrolizumab was entered into three Phase III trials for evaluation as a colorectal cancer and Hodgkin lymphoma (HL) treatment (NCT05600309, NCT05064059, and NCT05508867).
Fianlimab (REGN3767) is a fully human, hinge-stabilized, high-affinity IgG4 mAb developed by Regeneron Pharmaceuticals [157, 158]. Fianlimab blocks the interaction between LAG-3 and MHC-II to activate T cells and enhance tumor cell damage mediated by cytotoxic T cells [129]. To date, six clinical trials evaluating fianlimab as a monotherapy or in combination with anti-PD-1 inhibitors in patients with melanoma, NSCLC, lymphoma, and HNSCC have been established.
89Zr-DFO-REGN3767 (a fianlimab tracer) is composed of the anti-LAG-3 mAb fianlimab labeled with a radioactive isotope, the positron-emitter zirconium-89 (89Zr), through a chelator linker [159]. 89Zr-DFO-REGN3767 is currently under evaluation in two clinical trials established to monitor patient response to anti-LAG-3 therapy (NCT04566978 and NCT04706715). The main objective of the clinical trials is to better understand how the body absorbs, distributes, and disposes of 89Zr-DFO-REGN3767 and to identify the best dose and best time to perform a PET scan after injection.
Sym022 is a recombinant, Fc-inert, and fully human mAb developed by Symphogen that blocks LAG-3/MHC-II binding. Similar to the above mentioned mAbs, Sym022 induces T cells to produce cytokines in vitro and inhibits tumor growth in vivo [157]. Three clinical trials have been established to investigate Sym022 alone or in combination with Sym021 (an anti-PD-1 mAb) and Sym023 (an anti-TIM-3 mAb) (NCT03489369, NCT03489369, and NCT03311412). The preliminary data have shown that Sym021 monotherapy is well tolerated and exhibits both immune modulation and antitumor activity, and in combination with Sym022 and Sym023, a synergistic antitumor effect was reported [160]. The data from clinical trials of Sym022 used for patients with advanced solid tumors or lymphomas demonstrated no serious adverse drug reactions after the first and second doses. The third dose caused chest pain in one of three patients, and the fourth dose caused gastrointestinal hemorrhaging, increased lipase levels, and tumor pain in one of the six patients in the trial [161].
INCAGN02385 is an Fc-engineered IgG1κ mAb with the ability to potently block LAG-3 binding with MHC-II. INCAGN02385 increases T cell reactivity to TCR stimulation during monotherapy and in the presence of anti-PD-1/PD-L1 mAbs. INCAGN02385 treatment in cynomolgus monkeys was well tolerated, and a safe pharmacokinetic profile was reported [162]. Clinical studies on the safety and tolerability of INCAGN02385 in patients with certain advanced malignancies have been completed (NCT03538028), and four clinical trials with patients with HNSCC, melanoma, urothelial carcinoma (UC), or endometrial cancer are recruiting (NCT05287113, NCT04370704, NCT04586244, and NCT04463771).
Two other anti-LAG-3 mAbs include Ieramilimab (LAG525) and TSR-033. Leramilimab is a humanized IgG4 mAb that blocks LAG-3 binding to MHC-II. Five clinical trials in different phases have been conducted to evaluate Ieramilimab effectiveness. Four of these trials were completed, and one was terminated. Leramilimab in combination with spartalizumab (anti-PD-1 mAb) was well tolerated and showed initial antitumor activity in patients with mesothelioma and triple-negative BC, neuroendocrine tumors, small cell lung cancer (SCLC), and DLBCL. A clinical benefit rate of 86% was reported for gastrointestinal and pancreatic neuroendocrine tumor cohorts [163, 164]. TSR-033 is a humanized IgG4 mAb that shows high binding affinity for LAG-3 and is a functional antagonist. A preclinical study showed that double blockade of LAG-3 and PD-1 with TSR-033 and TSR-042 increased the total amount and proliferation rate of T cells in model mice harboring humanized NSCLC tumors compared to the effects of TSR-042 monotherapy; these results are consistent with increased antitumor efficacy [165]. TSR-033 is currently being evaluated in two clinical trials in the recruiting phase for the treatment of advanced solid tumors (NCT03250832 and NCT02817633).
Anti-TIM-3 mAbs
To date, 33 mAbs have been designed to target TIM-3 and for use alone or in combination with other ICIs, chemotherapy agents, targeted therapy drugs, or radiotherapy in clinical trials to evaluate their antitumor activity. The clinical trial details for these drugs are summarized in Table 4.
Clinical studies for three anti-TIM-3 mAbs, including Sym023 (NCT03489343 and NCT03311412), INCAGN02390 (NCT03652077), and sabatolimab (MBG453) (NCT04812548, NCT05020912), have been completed. The safety of these anti-TIM-3 mAbs alone in treating patients with advanced solid tumors or leukemia has been preliminarily demonstrated by the completed studies. In a Phase I/Ib clinical trial for patients with advanced and metastatic solid tumors, sabatolimab treatment alone led to no response; however, five patients who received combination treatment showed partial responses (6%; lasting 12–27 months) [166].
Similarly, different anti-TIM-3 mAbs combined with anti-PD-1 mAbs in the treatment of advanced lymphomas or NSCLC displayed higher efficacy than TIM-3 blockers alone (objective response rate (ORR): 42.9% vs. 0%; disease control rate (DCR): 42.9% vs. 11.1%) [167]. At the ASCO-SITC Clinical Immuno-Oncology Symposium of 2019, it was shown that in a Phase I a/b trial, the efficacy of LY3321367, an anti-TIM-3 mAb, in patients with NSCLC varied depending on the anti-PD-1/L1 efficacy; that is, the efficacy in anti-PD-1/L1 refractory patients (N = 23, ORR: 0%, DCR: 35%, PFS: 1.9 months) was compared to that in anti-PD-1/L1 responders (N = 14, ORR: 7%, DCR: 50%, PFS: 7.3 months). For patients receiving the combination anti-PD-L1 treatment, the ORR and DCR were 4% and 42%, respectively [168]. In addition, ICIs show higher response rates and durable clinical benefit in microsatellite instability-high/mismatch repair-deficient tumors. A Phase I trial (NCT02791334) demonstrated that combining a TIM-3 inhibitor (LY3321367) with anti-PD-L1 (LY3300054) therapy did not compromise the safety or tolerability of either treatment, and the results suggested numerically higher response rates in the anti–PD-1/PD-L1 inhibitor-naïve microsatellite instability-high/mismatch repair-deficient tumor group (ORR: 45%; DCR: 70%; 1-year OS: 64%, vs. ORR: 33%; DCR: 60%; 1-year OS: 71%) [169]. Clinical trials focused on anti-TIM-3 mAbs combined with inhibitors of the anti-PD-1/PD-L1 axis for the treatment of solid tumors included NCT05400876, NCT04139902, and NCT05645315. Together, the aforementioned data show that although anti-TIM-3 mAbs alone are safe for tumor treatment, the combination treatment blocking the PD-1/PD-L1 axis demonstrates significantly greater effects, and the microsatellite status and ICB treatment history were closely related to efficacy.
In addition, anti-TIM-3 mAbs combined with conventional chemotherapy or demethylation therapy showed increased antitumor effects and are novel options that showed initial efficacy, particularly in the treatment of MDS and AML. The STIMULUS trial (NCT03066648) (data cutoff date June 15, 2021) is another important trial in which 53 patients with very high/high-risk myelodysplastic syndrome (vHR/HR-MDS) and 48 with newly diagnosed AML (ND-AML) were treated with sabatolimab plus a hypomethylating agent (HMA). The incidence of common adverse events (AEs) with a Grade ≥ 3 in both groups was similar to that after HMA administered alone. Higher efficacy was shown for 51 patients with vHR/HR-MDS compared with 40 patients with ND-AML (ORR: 56.9% vs. 40.0%, PFS: 51.9% vs. 27.9%). Moreover, 24.5% of the vHR/HR-MDS patients showed disease attenuation, allowing them to undergo HSCT. Importantly, durable responses were observed in patients with adverse-risk mutations, such as TP53 mutations in vHR/HR-MDS patients (ORR: 71.4%; median duration of response (mDOR): 21.5 months). The AML group presented with a higher mDOR but a higher immune-mediated AE (im-AE) rate than the MDS group (mDOR: 23 vs. 21.5 months, im-AE rate: 25% vs. 11.7%, respectively). Patients with vHR/HR-MDS did not demonstrate excessive GVHD toxicity after subsequent allo-HSCT [96]. Another Phase II clinical study (NCT04150029) reported preliminarily data on the three-drug combination of sabatolimab + venetoclax + azacytidine in 18 ND-AML patients. The addition of 400 and 800 mg sabatolimab led to safety and tolerability comparable to that of the venetoclax + azacytidine combination [170]. A retrospective study of 28 patients with relapsed/refractory (R/R) AML and HR-MDS who received sabatolimab + HMA and subsequently underwent HSCT reported high 2-year PFS (64%) and OS (69%). The data also suggested that treatment with MGB-453 plus MHA before HSCT did not increase posttransplant GVHD and led to improved clinical outcomes for a RAS mutation subgroup, but it did not alter the adverse outcomes of TP53 patients [171]. An additional clinical study (NCT02608268) tentatively demonstrated the safety of TIM-3 inhibitors in the treatment of cancer [96]. In addition to studies focusing on HR-MDS, the program includes a doublet or triplet sabatolimab study for patients with low-risk MDS, which is in the recruiting stage (NCT04810611), for patients with AML that makes them unfit for intensive chemotherapy (STIMULUS-AML1, NCT04150029), and for AML posttransplant patients with measurable residual disease, which is also in the recruiting stage (STIMULUS-AML2, NCT04623216) [172].
In addition, radiotherapy elicits a potent antitumor immune response driven by the activation of T cells infiltrating tumors and an increase in cross-presentation by APCs; these outcomes are considered effective for transforming “cold” tumors into “hot” tumors [173]. Clinical studies analyzing the expression of TIM-3, PD-1, and CTLA-4 on T cells after radiotherapy given to patients with prostate cancer have been conducted to evaluate changes in immune status after radiotherapy (NCT04624828). Similarly, clinical studies on photodynamic therapy are used for basal cell carcinoma (NCT05020912). There are even Phase I clinical trials that use anti-TIM-3 and anti-PD-1 mAbs and stereotactic radiosurgery in combination for the treatment of recurrent glioblastoma multiforme to verify the effectiveness of this strategy (NCT03961971).
Anti-TIGIT mAbs
Several TIGIT mAbs are being evaluated in clinical trials, with Vibostolimab, Tiragolumab, Ociperlimab, and Domvanalimab being the most advanced candidates in Phase III clinical trials (Table 5).
IBI939 is the first anti-TIGIT mAb approved for clinical trials for leukemia and solid tumor therapy in China [58]. This treatment is a fully human mAb that directly binds to TIGIT to relieve the inhibition and depletion of T cells and NK cells and thus promotes the antitumor effects of these cells. IBI939 is expected to synergistically enhance the antitumor activity of anti-PD-1/PD-L1 mAbs and delay the acquisition of drug resistance. A Phase I clinical trial (NCT04353830) of IBI939 for advanced malignancies established to evaluate its safety, tolerability, pharmacokinetics, and efficacy has been completed.
Vibostolimab (MK-7684) is a humanized immunoglobulin G1 mAb that binds to TIGIT and blocks its interaction with its ligands CD112 and CD155, thereby activating T cells to help kill tumor cells. Vibostolimab plus pembrolizumab was entered into a Phase I clinical trial in 2016 (NCT02964013), and it was well tolerated and showed antitumor activity in patients with advanced solid tumors, including advanced NSCLC [174].
Tiragolumab (RG6058, MTIG7192A) is a fully humanized anti-TIGIT IgG1/kappa mAb developed by Roche with an intact Fc region that blocks the binding of TIGIT to its receptor CD155 [59]. The first Phase I clinical trial of this drug, designed to evaluate tiragolumab alone and in combination with atezolizumab, was conducted in 2016 for patients with advanced/metastatic tumors, including NSCLC and HNSCC (NCT02794571). In this study, 73 patients with multiple tumor types were treated in dose-escalation studies (24 patients were treated with tiragolumab in Phase Ia, and 49 patients were treated with tiragolumab plus atezolizumab in Phase Ib). There was no objective response among the patients in Phase Ia, but stable disease of > 4 months duration was observed (n = 4). In the Phase Ib cohort, 3 patients showed responses, with all responses related to PD-L1-positive tumors (2 NSCLC patients: 1 patient with a CR and 1 patient with a PR, and 1 HNSCC patient, who showed a PR), with two patients not receiving prior immunotherapy. Therefore, expansion cohorts were generated for a Phase Ib trial. In the metastatic NSCLC expansion cohort (N = 14), the ORR was 50%. Dose-limiting toxicities were not observed in Phase Ia or Ib, and the most common adverse effects were fatigue among the patients in Phase Ia (38%) and anemia among those in Phase Ib (31%) [175]. Tiragolumab combined with other targeted therapy drugs, such as bevacizumab (a vascular endothelial growth factor inhibitor), has also been entered in Phase II clinical trials for patients with NSCLC.
Ociperlimab (BGB-A1217) is a humanized anti-TIGIT IgG1 mAb. This drug binds to the extracellular domain of human TIGIT with high affinity and blocks the interaction between TIGIT and its ligand CD155 or CD112 while preserving intact Fc segment function, which is essential for its antitumor activity; it exerted synergistic antitumor effects when combined with anti-PD-1/PD-L1 mAbs [176]. The results of Phase I clinical trials designed to evaluate ociperlimab combined with tislelizumab (anti-PD-1 mAb) were first reported at ASCO in 2021, and they confirmed that ociperlimab combined with tislelizumab was well tolerated; moreover, antitumor efficacy was observed in patients with advanced solid tumors (NCT04693234) [177]. In June 2023, a Phase III clinical trial evaluating the efficacy and safety of ociperlimab in combination with tislelizumab and platinum-based doublet chemotherapy as first-line treatment for patients with locally advanced or metastatic NSCLC without an operational driver mutation was initiated (NCT05791097).
Domvanalimab (AB154) is another humanized anti-TIGIT mAb that binds human TIGIT and blocks the TIGIT-CD155 interaction, reducing the inhibition of T cells and NK cells, thereby promoting antitumor effects. Moreover, domvanalimab abolished the function of the Fc end of the antibody, which blocked the activity of TIGIT at the nanomolecular level, thereby blocking immunosuppression and increasing immune activity. A Phase III clinical trial evaluating the efficacy and safety of durvalumab (anti-PD-L1 mAb) and domvanalimab compared to that of durvalumab plus placebo in adult patients with locally advanced (Stage III), unresectable NSCLC is currently underway worldwide (NCT05211895) [178].
HLX53, a TIGIT-targeting nano-mAb, is composed of the variable region (VHH) of a heavy chain antibody and the Fc terminus of wild-type IgG1. This drug was approved for a clinical trial of advanced solid tumors or lymphomas in June 2022 by the Center for Drug Evaluation. Preclinical studies have shown that HLX53 exhibited excellent tumor suppression and showed a favorable safety profile [179]. A Phase I clinical trial to evaluate the safety, tolerability, kinetics, and preliminary antitumor efficacy of HLX53 is currently ongoing for patients with advanced/metastatic solid tumors or lymphomas (NCT05394168).
Other approaches to targeting LAG-3, TIM-3, and TIGIT for immunotherapy
In addition to using mAbs to block ICPs expression and restore tumor-infiltrating immune cell function, several approaches for targeting LAG-3, TIM-3, and TIGIT via immunotherapy, such as the development of BsAbs, have been reported. Since 2009, when the CD3 and CD19 BsAb (a bispecific T cell engager, BiTE) blinatumomab was approved by the FDA for the treatment of Philadelphia chromosome-negative R/R B-ALL [180], increasing numbers of BsAbs have been developed. The clinical therapeutic effects of BsAbs are better than those of mAbs, and BsAbs have a wide range of applications to the treatment of tumors and other diseases [181]. BsAbs that were developed for LAG-3, TIM-3, and TIGIT are summarized in Table 6.
LAG-3-PD-1/PD-L1/CTLA-4/TIGIT BsAbs
Four types of BsAbs have been developed for LAG-3: LAG-3-PD-1, LAG-3-PD-L1, LAG-3-CTLA-4, and LAG-3-TIGIT. The first LAG-3-PD-1 BsAb developed was tebotelimab (MGD013), which specifically binds to PD-1 and LAG-3 with high affinity and targets cell lines expressing these proteins and chronically activated T cells. Functional characterization of tebotelimab revealed enhanced cytokine secretion in response to antigen rechallenge of previously stimulated T cells treated with tebotelimab compared to that after PD-1 or LAG-3 pathway blockade alone. In a Phase I clinical trial for relapsed or refractory DLBCL patients, after tebotelimab treatment, serum IFN-γ levels were found to be significantly elevated, and CD8+ T cell function was restored with an increase in cytolytic marker (i.e., perforin, granzyme B) levels. Encouraging early evidence suggests that tebotelimab exhibits good pharmacodynamic, safety and antitumor activity, regardless of whether patients with R/R DLBCL had previously received chimeric antigen receptor T cell (CAR-T) therapy [182]. Tebotelimab was used in combination with margetuximab, a HER2−targeting mAb, for patients with HER2+ BC therapy [183]. There are currently seven clinical trials evaluating tebotelimab monotherapy or combination therapy. In addition, tebotelimab monotherapy has shown antitumor activity in multiple tumor types, such as melanoma and advanced HCC [184]. There is a newly launched LAG-3-PD-1 BsAb, CB213, and its structure is composed of a human nanobody (recombinant variable domains of a heavy-chain-only antibody) with an asymmetric 2:1 binding format involving bivalent human LAG-3 and monovalent human PD-1. The antitumor efficacy of CB213 was characterized by the potent inhibition of tumor growth and an increase in the number of CD8+ T cells with tumor antigen specificity [185]. Another LAG-3-PD-1 BsAb, RO7247669, targets and binds PD-1/LAG-3+ T cells and leads to CTL-induced immune responses against tumor cells [186]. The safety and efficacy of RO7247669 are still being evaluated in the contexts of metastatic melanoma, NSCLC, esophageal squamous cell carcinoma (ESCC), and advanced HCC. Additionally, EMB-02 is a LAG-3-PD-1 BsAb that has been shown to restore effector T cell function and enhance antitumor activity (NCT04618393).
Two LAG-3-PD-L1 BsAbs have been developed, FS118 and IBI323. FS118 targets LAG-3 and PD-L1 and shows the potential to activate exhausted immune cells and to target and overcome resistance to PD-L1 blockade. FS118 binds both LAG-3 and PD-L1, blocking PD-1/PD-L1, CD80/PD-L1, and LAG-3/MHC-II interactions, thereby reversing T cell suppression and promoting the production of cytokines by CD4+ and CD8+ T cells [187]. A Phase I clinical trial (NCT03440437) for patients with advanced cancers and PD-L1 drug resistance initially showed that FS118 was well tolerated, but further studies are needed to determine the clinical benefit for patients refractory to anti-PD-(L)1 therapy [188]. IBI323 demonstrated similar potency in blocking the interactions of PD-1/PD-L1, CD80/PD-L1, and LAG-3/MHC-II. In PD-L1/LAG-3 double knock-in mice bearing human PD-L1 knock-in MC3 tumors, IBI323 exhibited stronger antitumor activity than each parental antibody, and these antitumor responses were associated with an increase in the number of tumor-specific CD8+ and CD4+ T cells [189].
XmAb22841 is a CTLA-4-LAG-3 BsAb developed by Xencor that enhances T cell activation. The structure of XmAb22841 consists of a bispecific Fc domain that functions as a scaffold between the two binding domains, conferring stability and making purification and fabrication easy. This structure promotes heterodimer formation and leads to a long half-life in the circulatory system. XmAb22841 enhances allogeneic antitumor activity and facilitates triple checkpoint blockade in combination with anti-PD-1 blockade [190]. There are two ongoing clinical trials for evaluating XmAb22841 as a melanoma treatment, either alone or in combination with pembrolizumab or XmAb23104 (PD-1 × ICOS) (NCT03849469, NCT05695898).
ZGGS15 is a humanized anti-LAG-3 and anti-TIGIT BsAb that not only blocks the signaling pathways activated by LAG-3 and its ligand MHC-II but also activates the TCR signaling pathway, thereby promoting the activation and proliferation of T and NK cells and cytokine production, synergistically enhancing the ability of the immune system to kill tumor cells. The combination of ZGGS15 and anti-PD-1 mAb showed high efficacy than anti-LAG-3 mAbs or anti-TIGIT mAbs combined with anti-PD-1 mAbs [190]. In June 2023, the FDA approved ZGGS15 injection for the treatment of patients with advanced solid tumors in a clinical trial (NCT05864573).
TIM-3-PD-1/PD-L1 BsAbs
In preclinical studies, a PD-1-TIM-3 BsAb was shown to increase the abundance of activated (HLA-DR+CD25+GranzymeB+) and proliferating CD8+ T cells and NK cells [191].
Compared with anti-PD-1 antibodies alone, TIM-3-PD-1 BsAbs significantly increased the proportion of proliferating NK cells [191]. In addition, 45% (5/11) of samples from patients with solid tumors demonstrated an effective response, namely a twofold in IFN-γ secretion after 96 h of coculture with PD-1-TIM-3 BsAbs in vitro [191]. Moreover, in responsive tumor samples, TIM-3-PD-1 BsAbs indirectly promoted B cell activation by inhibiting the production of PD-1+CXCL13+CD4+ T cells [191]. Mechanistically, the chemokine CXCL13 in the TME may play a crucial role as a B cell attractant [192]. Another PD-1-TIM-3 BsAb, MCLA-134, is currently being evaluated in preclinical studies [193].
To date, two types of BsAbs, TIM-3-PD-1 and TIM-3-PD-L1, have been entered into clinical trials. The TIM-3-PD-L1 BsAb, known as LY3415244, was entered into a Phase Ia/Ib study for the treatment of advanced solid tumors in November 2018, and it was terminated early in October 2019 due to an unpredictable immune response (NCT03752177). Two patients (16.7%) developed clinically significant infusion-related anaphylactic reactions, and all 12 patients developed treatment-related antidrug antibodies [194]. Although the study was stopped early, one patient with NSCLC who was resistant to PD-1 blockade achieved a near partial response with tumor regression by 29.6% [194].
The first TIM-3-PD-1 BsAb, R07121661, was used in a Phase I study to treat advanced and/or metastatic solid tumors, including ESCC, melanoma, SCLC, and NSCLC (NCT03708328), and in a Phase II study, it was used to treat advanced or metastatic ESCC (NCT04785820) [195]. The second TIM-3-PD-1 BsAb, AZD7789, was used in a Phase I/II open-label, multicenter study designed to assess its safety, tolerability, pharmacokinetics, and preliminary efficacy in patients with R/R classical HL (NCT05216835) and advanced solid tumors (NCT04931654). These studies are currently in the recruitment stage. Based on adjustments to both the heavy and light chain complementarity-determining regions, differences in the TIM-3-PD-1 BsAbs are described in detail in patents EP33564A1 and WO2019009727 [196].
TIGIT-PD-1/CTLA-4 BsAbs
A few clinical trials have shown that anti-TIGIT mAbs administered in combination with various other ICIs are much more effective than monotherapy, but determination of the optimal combinations with different ICIs remains a challenge. Therefore, many dual-target antibodies targeting TIGIT and other ICPs have been developed. BsAbs compensate for a deficiency of single drug therapy and generally show greater safety and efficacy.
IBI321 was the first dual-targeting IC (TIGIT and PD-1) BsAb to enter clinical practice. This molecule inhibits both the PD-1 and TIGIT signaling pathways. On July 26, 2021, Cinda announced completion of the first Phase I clinical trial of IBI321 with Chinese patients. The study was established mainly to evaluate the safety, tolerability, and antitumor activity of the dual-target antibody IBI321 in 16 patients with advanced malignant solid tumors who failed to respond to standard treatment (NCT04911894). The trial was completed on February 17, 2023, but no data on the drug have been reported.
The second PD-1-TIGIT BsAb is ZG005, which was declared for clinical application in China and is mainly suitable for advanced malignant tumors. As a dual blockade of the PD-1/TIGIT ICPs, ZG005 showed sustained occupancy of both targets and was able to specifically inhibit their pathways simultaneously, resulting in synergistic effects and enhanced ability of the immune system to kill tumor cells. A Phase I clinical trial (CTR20220021) of ZG005 in advanced cancer is ongoing [197].
Another PD-1-TIGIT BsAb treatment is BC008-1A injection. This BsAb can enhance the role of immune surveillance by recognizing and killing tumor cells and blocking the potential synergistic effects of PD-1 and TIGIT to enhance antitumor effects. This treatment is suitable mainly for patients with advanced solid tumors and was approved for clinical trials in September 2022. A Phase I clinical trial (CTR20230047) is still in the recruitment phase, and 36 patients are expected to be enrolled. The aim of this trial is primarily to evaluate the safety and tolerability of BC008-1A injection in subjects with advanced solid tumors and determine the dose-limiting toxicities and maximum tolerated dose.
In addition, Yang et al. developed an anti-TIGIT mAb that exerts strong antitumor effects through mechanisms including a CD8+ T immune response and Fc-mediated effector functions that cause a significant reduction in the number of intratumoral Tregs. This result suggests that TIGIT-Fc treatment alone or in combination with other checkpoint receptor blockers is a promising anticancer therapeutic strategy [198]. In 2021, Bristol Myers Squibb and Agenus Inc. announced a license for a BsAb, AGEN1777, which blocks TIGIT and a second undisclosed target. The enhanced Fc region shows higher binding affinity for T and NK cells, thus increasing the activation of these cells [199]. This BsAb is being evaluated to determine its safety, tolerability, pharmacokinetics, and pharmacodynamics as a single agent and in combination with PD-1 inhibitors in patients with advanced solid tumors (NCT05025085).
In contrast to dual ICP BsAbs, AK130 is a TIGIT/human transforming growth factor-β (TGF-β) dual target antibody fusion protein independently developed by Kangfang Biological. This BsAb is composed of an anti-TIGIT mAb fused to the extracellular domain of TGF-β receptor II, and it is the first and only TIGIT/TGFβ dual target fusion protein antibody developed to date. In preclinical studies, AK130 blocked the TIGIT-CD155 and TGFβ-TGFβR signaling pathways, and it showed a profound ability to increase IL-2 secretion. Moreover, AK130 exhibited significant antitumor activity without inducing antibody-dependent cell-mediated cytotoxicity or complement-dependent cytotoxic effects in HCC model mice. Overall, AK130 simultaneously targets TIGIT and TGF-β to relieve immunosuppression, activate antitumor immune responses, and inhibit tumors at the same time [200]. It will be evaluated for its safety, tolerability, pharmacokinetics, and antitumor activity in patients with advanced malignancies in a Phase I trial (NCT05653284). Recruitment has not yet begun, and the trial is expected to be completed in 2025.
sLAG-3-Ig fusion protein induces APC activation
Eftilagimod alpha (IMP321), developed by IMMUTEP S.A.S., is an original, first-in-class LAG-3 regulator, and it is the only soluble recombinant LAG-3 used in clinical research. Eftilagimod alpha is a fusion protein consisting of the LAG-3 extracellular domains fused to a human Ig Fc region, and it was developed by replacing the Fab Ig domains of IgG1 with four Ig-like domains from the extracellular region of LAG-3. Eftilagimod alpha activates APCs through its interaction with MHC-II. Eftilagimod alpha interaction with MHC-II on human immature DCs induces the secretion of IL-12 and TNF-α, and it promotes morphological changes, such as the formation of dendritic projections. Hence, eftilagimod alpha functions differently from antagonistic LAG-3 mAbs that block the LAG-3/MHC-II interaction and thus the LAG-3-mediated reducing in T cells [201, 202]. Five clinical trials of eftilagimod alpha have been completed (NCT02676869, NCT02614833, NCT00349934, NCT00351949, and NCT00324623), and other trials are ongoing. The design of most of these trials is based on combination treatments. Combining eftilagimod alpha1 and PD-1 mAbs for treating patients with metastatic melanoma demonstrated that eftilagimod alpha was well tolerated and showed encouraging antitumor activity. An ORR of 33% was observed in patients refractory to pembrolizumab treatment in the dose-escalation part of the study, and an ORR of 50% was observed for PD-1-naïve patients in the extension part of the study [203].
Small molecules targeting TIM-3
The small-molecule LPX-TI641 is an orally bioavailable TIM family agonist (TIM-3 and TIM-4) designed to promote immune tolerance restoration/induction. As an orally administered therapeutic, LPX-TI641 represents a new approach that unleashes the power of immune tolerance while being unrestrained by the limitations of previous antigen-specific immune tolerance approaches. LAPIX Therapeutics initially designed immune system restoration therapies for both autoimmune disease and oncology applications, but only LPX-TI641 has been entered into a Phase I clinical trial, and its safety and efficacy in neuro-autoimmune indications such as multiple sclerosis are being evaluated (NCT05853835).
Combination of targeting LAG-3, TIM-3, and TIGIT mAbs with other immune therapy strategies
CAR-T cell therapy has been widely used to treat B cell malignancies [204]. Current challenges to the use of CAR-T cell therapy include failure to persist and produce prolong antitumor responses in the immunosuppressive TME [204, 205]. The response rate of CAR-T cell therapy for B cell malignancies is different. To understand why only 26% of CLL patients benefited from CD19 CAR-T therapy while more than 90% of CD19+ B-ALL patients experienced CR, a detailed transcriptomic analysis was performed to compare CLL responses after CD19 CAR-T therapy using CAR-T cells from responders and non-responders. CAR-T cells from non-responders upregulated pathways involved in exhaustion and apoptosis [206, 207]. The expression levels of ICPs such as PD-1, TIM-3, and LAG-3 were upregulated on CAR-T cells after infusion, which may be related to CAR-T cell dysfunction [208, 209]. Moreover, CAR-T cells deficient in PD-1 or LAG-3 demonstrated better antitumor efficacy both in vitro and in vivo [210]. In mesothelin-CAR-T cells, a lower expression level of the exhausted phenotype, including PD-1, LAG-3, and TIM-3 expression, has been shown to increase the strength and prolong clinical responses in the treatment of OC models [211].
Significantly, T cell exhaustion induced by coinhibitory pathways has been thought to contribute to the low persistence and highly dysfunctional activity of CAR-T cells. Thus, several studies have explored selective blockers of these inhibitory receptors in CAR-T cells. To date, at least two TIM-3-CD28 fusion proteins have been designed to increase the proliferation, activation, and cytotoxic capacity of conventional anti-CD19 CAR-T cells [212]. In addition, TIGIT has been identified is a marker for CD19 CAR-T cell dysfunction in experiments involving single-cell RNA sequencing and in analysis of surface protein marker levels before and after CAR-T cell infusion in NHL patients. Simultaneous downregulation of PD-1 and TIGIT enhances the in vivo function of CD19 CAR-T cells, resulting in coordinated antitumor effects [213]. TIGIT inhibition alone has also been shown to increase the efficacy of CAR-T cells [214]. Moreover, evidence suggests that TIGIT is highly expressed in mantle cell lymphoma cells in patients after relapse, and cotargeting TIGIT prevented CAR-T cell relapses, thereby promoting the long-term PFS of mantle cell lymphoma patients [215]. In addition, a group of investigators constructed anti-MLSN-CAR-T cells combined with anti-α-TIGIT for the treatment of solid tumors, e.g., pancreatic cancer, BC, and OC. Blocking TIGIT significantly promoted the release of cytokines, thereby enhancing the tumor-killing effects of the anti-MLSN-CAR-T cells. Moreover, anti-α-TIGIT scFv expression and secretion interrupted the interaction between TIGIT and its ligand CD155, enhancing the infiltration and activation of CAR-T cells in the TME to achieve increased tumor regression in vivo [216]. Additionally, TIM-3 was used to develop a second-generation 41BB-CD19-CAR linked with a switch receptor T3/28 chimera, known as T3/28 CAR-T cells, which significantly prolonged the persistence of CAR-T cells and showed potent antitumor activity both in vitro and in MM model mice [217]. Moreover, TIM-3 is expressed on most LSCs of AML but not on normal HSCs [41, 106]. The first anti-TIM-3 CAR-T cell was designed and demonstrated effective anti-myeloid leukemia effects both in vitro and in AML model mice [218].
Similar to CAR-T cells, BiTEs target tumor-specific antigens but depend on the normal function of T cells [219]. In the multicohort, open-label, phase 1/2 MajesTEC-1 study, in which the safety/efficacy of teclistamab (a B cell maturation antigen (BCMA)-CD3 BsAb IgG4) were evaluated in patients with RR-MM, encouraging efficacy was demonstrated and indicated that a higher frequency of T cells expressed IC markers, including TIM-3, and this may be an underlying reason for non-responders observed with unfavorable immune characteristics at baseline [220]. Thus, a combination of BiTEs with ICIs may be increase the effect of the BiTEs.
Conclusion and perspectives
LAG-3, TIM-3, and TIGIT are the next wave of targets for ICBs, and their efficacy has been extensively evaluated in clinical trials, following the evaluation of PD-1/PD-L1 and CTLA-4 blockers [1, 10]. In addition to single-target blockade, dual inhibition of PD-1/PD-L1 and other ICPs, such as CTLA-4, LAG-3, TIM-3, and TIGIT, has been tested in patients with different types of cancer in recent years. Encouragingly, the FDA approved the first dual inhibitor of LAG-3 and PD-1 drug (Opdualag) for treating adult and pediatric patients with unresectable or metastatic melanoma [7]. In addition to the combined application of two ICIs, BsAbs targeting two ICPs is a strategy to overcome cell resistance to a single ICB [221, 222]. This year, the FDA approved injection of an anti-LAG-3/TIGIT BsAb for the treatment of patients with advanced solid tumors in clinical trials. However, whether the BsAbs targeting two ICPs exhibit a “1 + 1 > 2” effect compared with the combined use of two types of ICP mAbs and induce fewer side effects remains to be seen in the future. Moreover, it is known that ICP-mediated exhaustion significantly influences the function of CAR-T cells, and ICB can increase CAR-T cell function. However, the determination of which ICI is the best choice for reversing CAR-T cell function requires further investigation. Finally, ICBs combined with CAR-T cells, BiTEs, and tumor vaccines and the development of BsAbs targeting one ICP and one immune suppressive cytokine are promising strategies to overcome immune escape by different types of cancer cells. The development of these new ICP-based treatment strategies offers new hope for cancer patients; however, to develop a strategy for choosing combination therapy targets for specific types of tumors and specific individuals, further exploration is needed. The identification of the most efficacious combination might depend on additional exploration into ICP expression profiles in different types of tumors and a deeper understanding of the molecular mechanisms underlying the effect of each ICP. Undoubtably, only a set of biomarkers that can predict the efficacy of ICBs will enable guided clinical drug administration.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Change history
29 September 2023
A Correction to this paper has been published: https://doi.org/10.1186/s13045-023-01503-8
Abbreviations
- AE:
-
Adverse events
- AML:
-
Acute myeloid leukemia
- APCs:
-
Antigen-presenting cells
- BC:
-
Breast cancer
- B-ALL:
-
B cell acute lymphoblastic lymphoma
- BsAbs:
-
Bispecific antibodies
- BiTE:
-
Bispecific T cell engager
- CEACAM-1:
-
Carcinoembryonic antigen cell adhesion molecule 1
- CC:
-
Colon cancer
- CR:
-
Complete response
- CTL:
-
Cytotoxic T lymphocyte
- CTLA-4:
-
Cytotoxic T lymphocyte antigen-4
- CLL:
-
Chronic lymphocytic leukemia
- CAR-T cell:
-
Chimeric antigen receptor T cell
- DCR:
-
Disease control rate
- DCs:
-
Dendritic cells
- DLBCL:
-
Diffuse large B cell lymphoma
- ESCC:
-
Esophageal squamous cell carcinoma
- FGL-1:
-
Fibrinogen-like protein 1
- FDA:
-
Food and Drug Administration
- Gal-9:
-
Galectin-9
- GC:
-
Gastric cancer
- GVHD:
-
Graft-versus-host disease
- IFN:
-
Interferon
- ICPs:
-
Immune checkpoint proteins
- ICB:
-
Immune checkpoint blocker
- ICI:
-
Immune checkpoint inhibitor
- IL:
-
Interleukin
- ITIM:
-
Immunoreceptor tyrosine-based inhibitory motif
- LAG-3:
-
Lymphocyte activation gene-3
- LAP:
-
LAG-3-related protein
- LSECtin:
-
Liver sinusoidal endothelial cell lectin
- LSCs:
-
Leukemia stem cells
- mAb:
-
Monoclonal antibody
- MHC-II:
-
MHC class II
- MDS:
-
Myelodysplastic syndromes
- mDOR:
-
Median duration of response
- mDCs:
-
Monocyte-derived DCs
- ND-AML:
-
Newly diagnosed AML
- HCC:
-
Hepatocellular carcinoma
- HL:
-
Hodgkin lymphoma
- HMGB1:
-
High-mobility group protein B1
- HNSCC:
-
Head and neck squamous cell carcinoma
- NHL:
-
Non-Hodgkin lymphoma
- HR:
-
High risk
- NK:
-
Natural killer cells
- HMA:
-
Hypomethylating agent
- NSCLC:
-
Non-small cell lung cancer
- ORR:
-
Objective response rate
- OS:
-
Overall survival
- OC:
-
Ovarian cancer
- allo-HSCT:
-
Allogeneic hematopoietic stem cell transplantation
- PD-1:
-
Programmed cell death protein 1
- PFS:
-
Progression-free survival
- PR:
-
Partial response
- PtdSer:
-
Phosphatidylserine
- RCC:
-
Renal cell carcinoma
- RIG-I:
-
Retinoic acid-inducible gene I
- R/R:
-
Relapsed/refractory
- sTIM-3:
-
Soluble form of TIM-3
- TME:
-
Tumor microenvironment
- TCR:
-
T cell receptor
- TIGIT:
-
T cell immunoreceptor with immunoglobulin and ITIM domain
- TIM-3:
-
T cell immunoglobulin and mucin-domain-containing-3
- Tregs:
-
Regulatory T cells
- TILs:
-
Tumor-infiltrating lymphocytes
- TGF-β:
-
Transforming growth factor-β
- SCLC:
-
Small cell lung cancer
- vHR:
-
Very high risk
References
Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. 2016;44(5):989–1004.
Korman AJ, Garrett-Thomson SC, Lonberg N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat Rev Drug Discov. 2022;21(7):509–28.
Tian C, Chen Z. Immune therapy: a new therapy for acute myeloid leukemia. Blood Sci. 2023;5(1):15–24.
Littman DR. Releasing the brakes on cancer immunotherapy. Cell. 2015;162(6):1186–90.
Ma W, Xue R, Zhu Z, Farrukh H, Song W, Li T, Zheng L. Pan C-x: increasing cure rates of solid tumors by immune checkpoint inhibitors. Exp Hematol Oncol. 2023;12(1):10.
Wang Z, Chen J, Wang M, Zhang L, Yu L. One stone, two birds: the roles of Tim-3 in acute myeloid leukemia. Front Immunol. 2021;12: 618710.
Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutierrez E, Rutkowski P, Gogas HJ, Lao CD, De Menezes JJ, Dalle S, Arance A, Grob JJ, Srivastava S, Abaskharoun M, Hamilton M, Keidel S, Simonsen KL, Sobiesk AM, Li B, Hodi FS, Long GV. Investigators R-: relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med. 2022;386(1):24–34.
Wang Y, Zhang H, Liu C, Wang Z, Wu W, Zhang N, Zhang L, Hu J, Luo P, Zhang J, Liu Z, Peng Y, Liu Z, Tang L, Cheng Q. Immune checkpoint modulators in cancer immunotherapy: Recent advances and emerging concepts. J Hematol Oncol. 2022;15(1):111.
Marin-Acevedo JA, Kimbrough EO, Lou Y. Next generation of immune checkpoint inhibitors and beyond. J Hematol Oncol. 2021;14(1):45.
Liu S, Sun Q, Ren X. Novel strategies for cancer immunotherapy: Counter-immunoediting therapy. J Hematol Oncol. 2023;16(1):38.
Zhao B, Zhao H, Zhao J. Efficacy of PD-1/PD-L1 blockade monotherapy in clinical trials. Ther Adv Med Oncol. 2020;12:1758835920937612.
Wang X, Chen Y, Li Z, Huang B, Xu L, Lai J, Lu Y, Zha X, Liu B, Lan Y, Li Y. Single-Cell RNA-Seq of T cells in B-ALL patients reveals an exhausted subset with remarkable heterogeneity. Adv Sci (Weinh). 2021;8(19): e2101447.
Jia Q, Wang A, Yuan Y, Zhu B, Long H. Heterogeneity of the tumor immune microenvironment and its clinical relevance. Exp Hematol Oncol. 2022;11(1):24.
Niu M, Yi M, Wu Y, Lyu L, He Q, Yang R, Zeng L, Shi J, Zhang J, Zhou P, Zhang T, Mei Q, Chu Q, Wu K. Synergistic efficacy of simultaneous anti-TGF-β/VEGF bispecific antibody and PD-1 blockade in cancer therapy. J Hematol Oncol. 2023;16(1):94.
Li X, Song W, Shao C, Shi Y, Han W. Emerging predictors of the response to the blockade of immune checkpoints in cancer therapy. Cell Mol Immunol. 2019;16(1):28–39.
Chen C, Liu SM, Chen Y, Han M, Ou Q, Bao H, Xu L, Zhang Y, Zhang JT, Zhong W, Zhou Q, Yang XN, Shao Y, Wu YL, Liu SY, Li Y. Poor prognosis of intra-tumoural TRBV6-6 variants in EGFR-mutant NSCLC: Results from the ADJUVANT-CTONG1104 trial. Clin Transl Med. 2022;12(4): e775.
Han J, Duan J, Bai H, Wang Y, Wan R, Wang X, Chen S, Tian Y, Wang D, Fei K, Yao Z, Wang S, Lu Z, Wang Z, Wang J. TCR repertoire diversity of peripheral PD-1(+)CD8(+) T Cells predicts clinical outcomes after immunotherapy in patients with non-small cell lung cancer. Cancer Immunol Res. 2020;8(1):146–54.
Li CCY. Predictive value of co-expression patterns of immune checkpoint molecules for clinical outcomes of hematological malignancies. Chin J Cancer Res. 2023;35(3):245–51.
Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, Hercend T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. 1990;171(5):1393–405.
Huard B, Mastrangeli R, Prigent P, Bruniquel D, Donini S, El-Tayar N, Maigret B, Dreano M, Triebel F. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc Natl Acad Sci. 1997;94(11):5744–9.
Wang JH, Meijers R, Xiong Y, Liu JH, Sakihama T, Zhang R, Joachimiak A, Reinherz EL. Crystal structure of the human CD4 N-terminal two-domain fragment complexed to a class II MHC molecule. Proc Natl Acad Sci. 2001;98(19):10799–804.
Iouzalen N, Andreae S, Hannier S, Triebel F. LAP, a lymphocyte activation gene-3 (LAG-3)-associated protein that binds to a repeated EP motif in the intracellular region of LAG-3, may participate in the down-regulation of the CD3/TCR activation pathway. Eur J Immunol. 2001;31(10):2885–91.
Workman CJ, Dugger KJ, Vignali DA. Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J Immunol. 2002;169(10):5392–5.
Long L, Zhang X, Chen F, Pan Q, Phiphatwatchara P, Zeng Y, Chen H. The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes Cancer. 2018;9(5–6):176–89.
Keane C, Law SC, Gould C, Birch S, Sabdia MB, Merida Long L, Thillaiyampalam G, Abro E, Tobin JW, Tan X, Xu-Monette ZY, Young KH, Gifford G, Gabreilli S, Stevenson WS, Gill A, Talaulikar D, Jain S, Hernandez A, Halliday SJ, Bird R, Cross D, Hertzberg M, Gandhi MK. LAG3: a novel immune checkpoint expressed by multiple lymphocyte subsets in diffuse large B-cell lymphoma. Blood Adv. 2020;4(7):1367–77.
Xu F, Liu J, Liu D, Liu B, Wang M, Hu Z, Du X, Tang L, He F. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. 2014;74(13):3418–28.
Dumic J, Dabelic S, Flogel M. Galectin-3: an open-ended story. Biochim Biophys Acta. 2006;1760(4):616–35.
Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, Chen L, Chen Y, Zhu G, Yin W, Zheng L, Zhou T, Badri T, Yao S, Zhu S, Boto A, Sznol M, Melero I, Vignali DAA, Schalper K, Chen L. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell. 2019;176(1–2):334-347 e312.
Qian W, Zhao M, Wang R, Li H. Fibrinogen-like protein 1 (FGL1): the next immune checkpoint target. J Hematol Oncol. 2021;14(1):147.
Prigent P, El Mir S, Dreano M, Triebel F. Lymphocyte activation gene-3 induces tumor regression and antitumor immune responses. Eur J Immunol. 1999;29(12):3867–76.
Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, Flores M, Li N, Schweighoffer E, Greenberg S, Tybulewicz V, Vignali D, Clynes R. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol. 2008;180(9):5916–26.
McIntire JJ, Umetsu SE, Akbari O, Potter M, Kuchroo VK, Barsh GS, Freeman GJ, Umetsu DT, DeKruyff RH. Identification of Tapr (an airway hyperreactivity regulatory locus) and the linked Tim gene family. Nat Immunol. 2001;2(12):1109–16.
Sabatos CA, Chakravarti S, Cha E, Schubart A, Sanchez-Fueyo A, Zheng XX, Coyle AJ, Strom TB, Freeman GJ, Kuchroo VK. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol. 2003;4(11):1102–10.
Meyers JH, Sabatos CA, Chakravarti S, Kuchroo VK. The Tim gene family regulates autoimmune and allergic diseases. Trends Mol Med. 2005;11(8):362–9.
Geng H, Zhang GM, Li D, Zhang H, Yuan Y, Zhu HG, Xiao H, Han LF, Feng ZH. Soluble form of T cell Ig mucin 3 is an inhibitory molecule in T cell-mediated immune response. J Immunol. 2006;176(3):1411–20.
Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, Chandwaskar R, Karman J, Su EW, Hirashima M, Bruce JN, Kane LP, Kuchroo VK, Hafler DA. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science. 2007;318(5853):1141–3.
Ferris RL, Lu B, Kane LP. Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion. J Immunol. 2014;193(4):1525–30.
Rangachari M, Zhu C, Sakuishi K, Xiao S, Karman J, Chen A, Angin M, Wakeham A, Greenfield EA, Sobel RA, Okada H, McKinnon PJ, Mak TW, Addo MM, Anderson AC, Kuchroo VK. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3-mediated cell death and exhaustion. Nat Med. 2012;18(9):1394–400.
Wiener Z, Kohalmi B, Pocza P, Jeager J, Tolgyesi G, Toth S, Gorbe E, Papp Z, Falus A. TIM-3 is expressed in melanoma cells and is upregulated in TGF-beta stimulated mast cells. J Invest Dermatol. 2007;127(4):906–14.
Cazzato G, Cascardi E, Colagrande A, Lettini T, Filosa A, Arezzo F, Lupo C, Casatta N, Loizzi V, Pellegrini C, Fargnoli MC, Maiorano E, Cicco G, Tamma R, Ingravallo G. T cell immunoglobulin and mucin domain 3 (TIM-3) in cutaneous melanoma: a narrative review. Cancers (Basel). 2023;15(6):1697.
Kikushige Y, Shima T, Takayanagi S, Urata S, Miyamoto T, Iwasaki H, Takenaka K, Teshima T, Tanaka T, Inagaki Y, Akashi K. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7(6):708–17.
Zhuang X, Zhang X, Xia X, Zhang C, Liang X, Gao L, Zhang X, Ma C. etcopic expression of TIM-3 in lung cancers: a potential independent prognostic factor for patients with NSCLC. Am J Clin Pathol. 2012;137(6):978–85.
Piao YR, Piao LZ, Zhu LH, Jin ZH, Dong XZ. Prognostic value of T cell immunoglobulin mucin-3 in prostate cancer. Asian Pac J Cancer Prev. 2013;14(6):3897–901.
Shang Y, Li Z, Li H, Xia H, Lin Z. TIM-3 expression in human osteosarcoma: correlation with the expression of epithelial-mesenchymal transition-specific biomarkers. Oncol Lett. 2013;6(2):490–4.
Zhou E, Huang Q, Wang J, Fang C, Yang L, Zhu M, Chen J, Chen L, Dong M. Up-regulation of Tim-3 is associated with poor prognosis of patients with colon cancer. Int J Clin Exp Pathol. 2015;8(7):8018–27.
Lin H, Yang B, Teng M. T-cell immunoglobulin mucin-3 as a potential inducer of the epithelial-mesenchymal transition in hepatocellular carcinoma. Oncol Lett. 2017;14(5):5899–905.
Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD, Hirashima M, Uede T, Takaoka A, Yagita H, Jinushi M. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. 2012;13(9):832–42.
Shi Q, Li G, Dou S, Tang L, Hou C, Wang Z, Gao Y, Gao Z, Hao Y, Mo R, Shen B, Wang R, Li Y, Han G. Negative regulation of RIG-I by Tim-3 promotes H1N1 infection. Immunol Invest. 2022;52(1):1–19.
Jiang Y, Zhang H, Wang J, Chen J, Guo Z, Liu Y, Hua H. Exploiting RIG-I-like receptor pathway for cancer immunotherapy. J Hematol Oncol. 2023;16(1):8.
Ma B, Akosman B, Kamle S, Lee C-M, He CH, Koo JS, Lee CG, Elias JA. CHI3L1 regulates PD-L1 and anti–CHI3L1–PD-1 antibody elicits synergistic antitumor responses. J Clin Invest. 2021;131(21): e137750.
Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, Tom I, Ivelja S, Refino CJ, Clark H, Eaton D, Grogan JL. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10(1):48–57.
Stanietsky N, Rovis TL, Glasner A, Seidel E, Tsukerman P, Yamin R, Enk J, Jonjic S, Mandelboim O. Mouse TIGIT inhibits NK-cell cytotoxicity upon interaction with PVR. Eur J Immunol. 2013;43(8):2138–50.
Ge Z, Peppelenbosch MP, Sprengers D, Kwekkeboom J. TIGIT, the next step towards successful combination immune checkpoint therapy in cancer. Front Immunol. 2021;12: 699895.
Zhou XM, Li WQ, Wu YH, Han L, Cao XG, Yang XM, Wang HF, Zhao WS, Zhai WJ, Qi YM, Gao YF. Intrinsic expression of immune checkpoint molecule TIGIT could help tumor growth in vivo by suppressing the function of NK and CD8(+) T cells. Front Immunol. 2018;9:2821.
Reches A, Ophir Y, Stein N, Kol I, Isaacson B, Charpak Amikam Y, Elnekave A, Tsukerman P, Kucan Brlic P, Lenac T, Seliger B, Jonjic S, Mandelboim O. Nectin4 is a novel TIGIT ligand which combines checkpoint inhibition and tumor specificity. J Immunother Cancer. 2020;8(1): e000266.
Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, Enk J, Bar-On Y, Stanietsky-Kaynan N, Coppenhagen-Glazer S, Shussman N, Almogy G, Cuapio A, Hofer E, Mevorach D, Tabib A, Ortenberg R, Markel G, Miklic K, Jonjic S, Brennan CA, Garrett WS, Bachrach G, Mandelboim O. Binding of the Fap2 protein of fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. 2015;42(2):344–55.
Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, Park S, Javinal V, Chiu H, Irving B, Eaton DL, Grogan JL. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. 2014;26(6):923–37.
Qiu D, Liu X, Wang W, Jiang X, Wu X, Zheng J, Zhou K, Kong X, Wu X, Jin Z. TIGIT axis: novel immune checkpoints in anti-leukemia immunity. Clin Exp Med. 2023;23(2):165–74.
Houssaini MS, Damou M, Ismaili N. Advances in the management of non-small cell lung cancer (NSCLC): a new practice changing data from asco 2020 annual meeting. Cancer Treat Res Commun. 2020;25: 100239.
Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Kuchroo V, Zarour HM. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207(10):2175–86.
da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, Osman I, Bhardwaj N. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res. 2014;2(5):410–22.
Wu FH, Yuan Y, Li D, Lei Z, Song CW, Liu YY, Li B, Huang B, Feng ZH, Zhang GM. Endothelial cell-expressed Tim-3 facilitates metastasis of melanoma cells by activating the NF-kappaB pathway. Oncol Rep. 2010;24(3):693–9.
Huang X, Bai X, Cao Y, Wu J, Huang M, Tang D, Tao S, Zhu T, Liu Y, Yang Y, Zhou X, Zhao Y, Wu M, Wei J, Wang D, Xu G, Wang S, Ma D, Zhou J. Correction: lymphoma endothelium preferentially expresses Tim-3 and facilitates the progression of lymphoma by mediating immune evasion. J Exp Med. 2021;218(4):505–20.
Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61.
Mair MJ, Kiesel B, Feldmann K, Widhalm G, Dieckmann K, Wohrer A, Mullauer L, Preusser M, Berghoff AS. LAG-3 expression in the inflammatory microenvironment of glioma. J Neurooncol. 2021;152(3):533–9.
Radwan SM, Elleboudy NS, Nabih NA, Kamal AM. The immune checkpoints cytotoxic T lymphocyte antigen-4 and lymphocyte activation gene-3 expression is up-regulated in acute myeloid leukemia. HLA. 2020;96(1):3–12.
Ohmura H, Yamaguchi K, Hanamura F, Ito M, Makiyama A, Uchino K, Shimokawa H, Tamura S, Esaki T, Mitsugi K, Shibata Y, Oda H, Tsuchihashi K, Ariyama H, Kusaba H, Oda Y, Akashi K, Baba E. OX40 and LAG3 are associated with better prognosis in advanced gastric cancer patients treated with anti-programmed death-1 antibody. Br J Cancer. 2020;122(10):1507–17.
Murga-Zamalloa CA, Brown NA, Wilcox RA. Expression of the checkpoint receptors LAG-3, TIM-3 and VISTA in peripheral T cell lymphomas. J Clin Pathol. 2020;73(4):197–203.
Que Y, Fang Z, Guan Y, Xiao W, Xu B, Zhao J, Chen H, Zhang X, Zeng M, Liang Y, Zhang X. LAG-3 expression on tumor-infiltrating T cells in soft tissue sarcoma correlates with poor survival. Cancer Biol Med. 2019;16(2):331–40.
Chen BJ, Dashnamoorthy R, Galera P, Makarenko V, Chang H, Ghosh S, Evens AM. The immune checkpoint molecules PD-1, PD-L1, TIM-3 and LAG-3 in diffuse large B-cell lymphoma. Oncotarget. 2019;10(21):2030–40.
Yarchoan M, Xing D, Luan L, Xu H, Sharma RB, Popovic A, Pawlik TM, Kim AK, Zhu Q, Jaffee EM, Taube JM, Anders RA. Characterization of the immune microenvironment in hepatocellular carcinoma. Clin Cancer Res. 2017;23(23):7333–9.
He Y, Yu H, Rozeboom L, Rivard CJ, Ellison K, Dziadziuszko R, Suda K, Ren S, Wu C, Hou L, Zhou C, Hirsch FR. LAG-3 protein expression in non-small cell lung cancer and its relationship with PD-1/PD-L1 and tumor-infiltrating lymphocytes. J Thorac Oncol. 2017;12(5):814–23.
Deng WW, Mao L, Yu GT, Bu LL, Ma SR, Liu B, Gutkind JS, Kulkarni AB, Zhang WF, Sun ZJ. LAG-3 confers poor prognosis and its blockade reshapes antitumor response in head and neck squamous cell carcinoma. Oncoimmunology. 2016;5(11): e1239005.
Yang ZZ, Grote DM, Ziesmer SC, Niki T, Hirashima M, Novak AJ, Witzig TE, Ansell SM. IL-12 upregulates TIM-3 expression and induces T cell exhaustion in patients with follicular B cell Non-Hodgkin Lymphoma. J Clin Invest. 2012;122(4):1271–82.
Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, Eppolito C, Qian F, Lele S, Shrikant P, Old LJ, Odunsi K. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci. 2010;107(17):7875–80.
Hemon P, Jean-Louis F, Ramgolam K, Brignone C, Viguier M, Bachelez H, Triebel F, Charron D, Aoudjit F, Al-Daccak R, Michel L. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J Immunol. 2011;186(9):5173–83.
Kwiatkowska D, Kluska P, Reich A. Beyond PD-1 immunotherapy in malignant melanoma. Dermatol Ther (Heidelb). 2019;9(2):243–57.
Camisaschi C, De Filippo A, Beretta V, Vergani B, Villa A, Vergani E, Santinami M, Cabras AD, Arienti F, Triebel F, Rodolfo M, Rivoltini L, Castelli C. Alternative activation of human plasmacytoid DCs in vitro and in melanoma lesions: involvement of LAG-3. J Invest Dermatol. 2014;134(7):1893–902.
Botticelli A, Zizzari IG, Scagnoli S, Pomati G, Strigari L, Cirillo A, Cerbelli B, Di Filippo A, Napoletano C, Scirocchi F, Rughetti A, Nuti M, Mezi S, Marchetti P. The role of soluble LAG3 and soluble immune checkpoints profile in advanced head and neck cancer: a pilot study. J Pers Med. 2021;11(7):651.
Giraldo NA, Becht E, Pages F, Skliris G, Verkarre V, Vano Y, Mejean A, Saint-Aubert N, Lacroix L, Natario I, Lupo A, Alifano M, Damotte D, Cazes A, Triebel F, Freeman GJ, Dieu-Nosjean MC, Oudard S, Fridman WH, Sautes-Fridman C. Orchestration and prognostic significance of immune checkpoints in the microenvironment of primary and metastatic renal cell cancer. Clin Cancer Res. 2015;21(13):3031–40.
Wang M, Du Q, Jin J, Wei Y, Lu Y, Li Q. LAG3 and its emerging role in cancer immunotherapy. Clin Transl Med. 2021;11(3): e365.
Seifert L, Plesca I, Muller L, Sommer U, Heiduk M, von Renesse J, Digomann D, Gluck J, Klimova A, Weitz J, Schmitz M, Seifert AM. LAG-3-expressing tumor-infiltrating T cells are associated with reduced disease-free survival in pancreatic cancer. Cancers (Basel). 2021;13(6):1297.
Hu G, Wang S, Wang S, Ding Q, Huang L. LAG-3(+) tumor-infiltrating lymphocytes ameliorates overall survival in triple-negative breast cancer patients. Front Oncol. 2022;12: 986903.
Rhyner Agocs G, Assarzadegan N, Kirsch R, Dawson H, Galvan JA, Lugli A, Zlobec I, Berger MD. LAG-3 expression predicts outcome in stage II colon cancer. J Pers Med. 2021;11(8):749.
Huard B, Prigent P, Pages F, Bruniquel D, Triebel F. T cell major histocompatibility complex class II molecules down-regulate CD4+ T cell clone responses following LAG-3 binding. Eur J Immunol. 1996;26(5):1180–6.
Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, Jaffee E. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol Res. 2015;3(4):412–23.
Buisson S, Triebel F. LAG-3 (CD223) reduces macrophage and dendritic cell differentiation from monocyte precursors. Immunology. 2005;114(3):369–74.
Wang Y, Zhao E, Zhang Z, Zhao G, Cao H. Association between Tim-3 and Gal-9 expression and gastric cancer prognosis. Oncol Rep. 2018;40(4):2115–26.
Lu X, Yang L, Yao D, Wu X, Li J, Liu X, Deng L, Huang C, Wang Y, Li D, Liu J. Tumor antigen-specific CD8(+) T cells are negatively regulated by PD-1 and Tim-3 in human gastric cancer. Cell Immunol. 2017;313:43–51.
Piao Y, Jin X. Analysis of Tim-3 as a therapeutic target in prostate cancer. Tumour Biol. 2017;39(7):1010428317716628.
Wang Z, Yin N, Zhang Z, Zhang Y, Zhang G, Chen W. Upregulation of T-cell immunoglobulin and mucin-domain containing-3 (Tim-3) in monocytes/macrophages associates with gastric cancer progression. Immunol Invest. 2017;46(2):134–48.
Shapoorian H, Zalpoor H, Ganjalikhani-Hakemi M. The correlation between Flt3-ITD mutation in dendritic cells with TIM-3 expression in acute myeloid leukemia. Blood Sci. 2021;3(4):132–5.
Li H, Wu K, Tao K, Chen L, Zheng Q, Lu X, Liu J, Shi L, Liu C, Wang G, Zou W. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology. 2012;56(4):1342–51.
Li Z, Li N, Li F, Zhou Z, Sang J, Chen Y, Han Q, Lv Y, Liu Z. Immune checkpoint proteins PD-1 and TIM-3 are both highly expressed in liver tissues and correlate with their gene polymorphisms in patients with HBV-related hepatocellular carcinoma. Medicine (Baltimore). 2016;95(52): e5749.
Kikushige Y: Clinical roles of TIM-3 in myeloid malignancies and its importance in cellular therapy. Blood Cell Ther 2022, 5(Spec Edition):S1-S5.
Brunner AM, Esteve J, Porkka K, Knapper S, Vey N, Scholl S, Garcia-Manero G, Wermke M, Janssen J, Traer E, Loo S, Narayan R, Tovar N, Kontro M, Ottmann O, Naidu P, Kurtulus S, Makofske J, Liao S, Mohammed A, Sabatos-Peyton CA, Rinne ML, Borate U, Wei AH. Efficacy and safety of sabatolimab (MBG453) in combination with hypomethylating agents (HMAs) in patients with acute myeloid leukemia (AML) and high-risk myelodysplastic syndrome (HR-MDS): updated results from a phase 1b study. Blood. 2020;136(Supplement 1):1–2.
Xu L, Xu J, Ma S, Li X, Zhu M, Chen S, Han Y, Tang X, Fu Z, Qiu H, Yu J, Wu D, Wu X. High Tim-3 expression on AML blasts could enhance chemotherapy sensitivity. Oncotarget. 2017;8(60):102088–96.
Tan J, Yu Z, Huang J, Chen Y, Huang S, Yao D, Xu L, Lu Y, Chen S, Li Y. Increased PD-1+Tim-3+ exhausted T cells in bone marrow may influence the clinical outcome of patients with AML. Biomark Res. 2020;8:6.
Liu L, Chang YJ, Xu LP, Zhang XH, Wang Y, Liu KY, Huang XJ. T cell exhaustion characterized by compromised MHC class I and II restricted cytotoxic activity associates with acute B lymphoblastic leukemia relapse after allogeneic hematopoietic stem cell transplantation. Clin Immunol. 2018;190:32–40.
Yegin ZA, Can F, Aydin Kaynar L, Gokcen S, Eren Sadioglu R, Ozkurt ZN, Karacaoglu O. Pre-transplant sTIM-3 levels may have a predictive impact on transplant outcome in acute leukemia patients. Hematology. 2020;25(1):125–33.
Dama P, Tang M, Fulton N, Kline J, Liu H. Gal9/Tim-3 expression level is higher in AML patients who fail chemotherapy. J Immunother Cancer. 2019;7(1):175.
Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, Dougan SK, Petersen BS, Melum E, Pertel T, Clayton KL, Raab M, Chen Q, Beauchemin N, Yazaki PJ, Pyzik M, Ostrowski MA, Glickman JN, Rudd CE, Ploegh HL, Franke A, Petsko GA, Kuchroo VK, Blumberg RS. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. 2015;517(7534):386–90.
Du W, Yang M, Turner A, Xu C, Ferris RL, Huang J, Kane LP, Lu B. TIM-3 as a target for cancer immunotherapy and mechanisms of action. Int J Mol Sci. 2017;18(3):645.
Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, Yagita H, Okumura K. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. 2009;113(16):3821–30.
Wang J, Yu C, Zhuang J, Qi W, Jiang J, Liu X, Zhao W, Cao Y, Wu H, Qi J, Zhao RC. The role of phosphatidylserine on the membrane in immunity and blood coagulation. Biomark Res. 2022;10(1):4.
Kikushige Y, Miyamoto T, Yuda J, Jabbarzadeh-Tabrizi S, Shima T, Takayanagi S, Niiro H, Yurino A, Miyawaki K, Takenaka K, Iwasaki H, Akashi K. A TIM-3/Gal-9 autocrine stimulatory loop drives self-renewal of human myeloid leukemia stem cells and leukemic progression. Cell Stem Cell. 2015;17(3):341–52.
Swamydas M, Murphy EV, Ignatz-Hoover JJ, Malek E, Driscoll JJ. Deciphering mechanisms of immune escape to inform immunotherapeutic strategies in multiple myeloma. J Hematol Oncol. 2022;15(1):17.
Tian Y, Zhai X, Han A, Zhu H, Yu J. Potential immune escape mechanisms underlying the distinct clinical outcome of immune checkpoint blockades in small cell lung cancer. J Hematol Oncol. 2019;12(1):67.
Chauvin J-M, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, Kirkwood JM, Chen T-HT, Maurer M, Korman AJ, Zarour HM. TIGIT and PD-1 impair tumor antigen–specific CD8+ T cells in melanoma patients. J Clin Investig. 2015;125(5):2046–58.
Hu F, Wang W, Fang C, Bai C. TIGIT presents earlier expression dynamic than PD-1 in activated CD8+ T cells and is upregulated in non-small cell lung cancer patients. Exp Cell Res. 2020;396(1): 112260.
Kong Y, Zhu L, Schell TD, Zhang J, Claxton DF, Ehmann WC, Rybka WB, George MR, Zeng H, Zheng H. T-cell immunoglobulin and ITIM domain (TIGIT) associates with CD8+ T-cell exhaustion and poor clinical outcome in AML patients. Clin Cancer Res. 2016;22(12):3057–66.
He W, Zhang H, Han F, Chen X, Lin R, Wang W, Qiu H, Zhuang Z, Liao Q, Zhang W, Cai Q, Cui Y, Jiang W, Wang H, Ke Z. CD155T/TIGIT signaling regulates CD8(+) T-cell metabolism and promotes tumor progression in human gastric cancer. Cancer Res. 2017;77(22):6375–88.
Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, Wang Z, Wu Q, Peng H, Wei H, Sun R, Tian Z. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19(7):723–32.
Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, Xia J, Tan TG, Sefik E, Yajnik V, Sharpe AH, Quintana FJ, Mathis D, Benoist C, Hafler DA, Kuchroo VK. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. 2014;40(4):569–81.
Wu K, Zeng J, Shi X, Xie J, Li Y, Zheng H, Peng G, Zhu G, Tang D, Wu S. Targeting TIGIT inhibits bladder cancer metastasis through suppressing IL-32. Front Pharmacol. 2022;12: 801493.
Hasan MM, Nair SS, O’Leary JG, Thompson-Snipes L, Nyarige V, Wang J, Park W, Stegall M, Heilman R, Klintmalm GB, Joo H, Oh S. Implication of TIGIT+ human memory B cells in immune regulation. Nat Commun. 2021;12(1):1534.
Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, Sharpe AH, Kuchroo VK. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol. 2011;186(3):1338–42.
Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 axis regulates human T cell function. J Immunol. 2012;188(8):3869–75.
Chauvin JM, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer. 2020;8(2): e000957.
Fuhrman CA, Yeh WI, Seay HR, Saikumar Lakshmi P, Chopra G, Zhang L, Perry DJ, McClymont SA, Yadav M, Lopez MC, Baker HV, Zhang Y, Li Y, Whitley M, von Schack D, Atkinson MA, Bluestone JA, Brusko TM. Divergent phenotypes of human regulatory T cells expressing the receptors TIGIT and CD226. J Immunol. 2015;195(1):145–55.
Harjunpaa H, Guillerey C. TIGIT as an emerging immune checkpoint. Clin Exp Immunol. 2020;200(2):108–19.
Andreae S, Piras F, Burdin N, Triebel F. Maturation and activation of dendritic cells induced by lymphocyte activation gene-3 (CD223). J Immunol. 2002;168(8):3874–80.
El Mir S, Triebel F. A soluble lymphocyte activation gene-3 molecule used as a vaccine adjuvant elicits greater humoral and cellular immune responses to both particulate and soluble antigens. J Immunol. 2000;164(11):5583–9.
Sordo-Bahamonde C, Lorenzo-Herrero S, Gonzalez-Rodriguez AP, Payer AR, Gonzalez-Garcia E, Lopez-Soto A, Gonzalez S. LAG-3 blockade with relatlimab (BMS-986016) restores anti-leukemic responses in chronic lymphocytic leukemia. Cancers (Basel). 2021;13(9):2112.
Goding SR, Wilson KA, Xie Y, Harris KM, Baxi A, Akpinarli A, Fulton A, Tamada K, Strome SE, Antony PA. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J Immunol. 2013;190(9):4899–909.
Huang RY, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget. 2015;6(29):27359–77.
Wierz M, Pierson S, Guyonnet L, Viry E, Lequeux A, Oudin A, Niclou SP, Ollert M, Berchem G, Janji B, Guerin C, Paggetti J, Moussay E. Dual PD1/LAG3 immune checkpoint blockade limits tumor development in a murine model of chronic lymphocytic leukemia. Blood. 2018;131(14):1617–21.
Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, Tangsombatvisit S, Grosso JF, Netto G, Smeltzer MP, Chaux A, Utz PJ, Workman CJ, Pardoll DM, Korman AJ, Drake CG, Vignali DA. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–27.
Burova E, Hermann A, Dai J, Ullman E, Halasz G, Potocky T, Hong S, Liu M, Allbritton O, Woodruff A, Pei J, Rafique A, Poueymirou W, Martin J, MacDonald D, Olson WC, Murphy A, Ioffe E, Thurston G, Mohrs M. Preclinical development of the anti-LAG-3 antibody REGN3767: characterization and activity in combination with the anti-PD-1 antibody cemiplimab in human PD-1xLAG-3-knockin mice. Mol Cancer Ther. 2019;18(11):2051–62.
Zettl M, Wurm M, Schaaf O, Mostbock S, Tirapu I, Apfler I, Lorenz IC, Frego L, Kenny C, Thibodeau M, Oquendo Cifuentes E, Reschke M, Moll J, Kraut N, Vogt A, Sedgwick JD, Waizenegger IC. Combination of two novel blocking antibodies, anti-PD-1 antibody ezabenlimab (BI 754091) and anti-LAG-3 antibody BI 754111, leads to increased immune cell responses. Oncoimmunology. 2022;11(1):2080328.
Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-Tim3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011;71(10):3540–51.
Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, Manning S, Greenfield EA, Coyle AJ, Sobel RA, Freeman GJ, Kuchroo VK. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415(6871):536–41.
Sanchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos CA, Manlongat N, Bender O, Kamradt T, Kuchroo VK, Gutierrez-Ramos JC, Coyle AJ, Strom TB. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol. 2003;4(11):1093–101.
Dardalhon V, Anderson AC, Karman J, Apetoh L, Chandwaskar R, Lee DH, Cornejo M, Nishi N, Yamauchi A, Quintana FJ, Sobel RA, Hirashima M, Kuchroo VK. Tim-3/galectin-9 pathway: regulation of Th1 immunity through promotion of CD11b+Ly-6G+ myeloid cells. J Immunol. 2010;185(3):1383–92.
Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–94.
Zhang D, Jiang F, Zaynagetdinov R, Huang H, Sood VD, Wang H, Zhao X, Jenkins MH, Ji Q, Wang Y, Nannemann DP, Musil D, Wesolowski J, Paoletti A, Bartholomew T, Derner MG, An Q, Iffland C, Halle JP. Identification and characterization of M6903, an antagonistic anti-TIM-3 monoclonal antibody. Oncoimmunology. 2020;9(1):1744921.
Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, Murphy WJ, Azuma M, Anderson AC, Kuchroo VK, Blazar BR. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011;117(17):4501–10.
Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, Jones RE, Kulkarni MM, Kuraguchi M, Palakurthi S, Fecci PE, Johnson BE, Janne PA, Engelman JA, Gangadharan SP, Costa DB, Freeman GJ, Bueno R, Hodi FS, Dranoff G, Wong KK, Hammerman PS. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501.
Oweida A, Hararah MK, Phan A, Binder D, Bhatia S, Lennon S, Bukkapatnam S, Van Court B, Uyanga N, Darragh L, Kim HM, Raben D, Tan AC, Heasley L, Clambey E, Nemenoff R, Karam SD. Resistance to radiotherapy and PD-L1 blockade is mediated by TIM-3 upregulation and regulatory T-cell infiltration. Clin Cancer Res. 2018;24(21):5368–80.
Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, Nyman J, Marjanovic ND, Kowalczyk MS, Wang C, Kurtulus S, Law T, Etminan Y, Nevin J, Buckley CD, Burkett PR, Buenrostro JD, Rozenblatt-Rosen O, Anderson AC, Regev A, Kuchroo VK. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature. 2018;558(7710):454–9.
Cheng L, Ruan Z. Tim-3 and Tim-4 as the potential targets for antitumor therapy. Hum Vaccin Immunother. 2015;11(10):2458–62.
Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, Liu A, Sankey EW, Tam A, Xu H, Mathios D, Jackson CM, Harris-Bookman S, Garzon-Muvdi T, Sheu M, Martin AM, Tyler BM, Tran PT, Ye X, Olivi A, Taube JM, Burger PC, Drake CG, Brem H, Pardoll DM, Lim M. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res. 2017;23(1):124–36.
Kang CW, Dutta A, Chang LY, Mahalingam J, Lin YC, Chiang JM, Hsu CY, Huang CT, Su WT, Chu YY, Lin CY. Apoptosis of tumor infiltrating effector TIM-3+CD8+ T cells in colon cancer. Sci Rep. 2015;5:15659.
Wu L, Mao L, Liu JF, Chen L, Yu GT, Yang LL, Wu H, Bu LL, Kulkarni AB, Zhang WF, Sun ZJ. Blockade of TIGIT/CD155 signaling reverses T-cell exhaustion and enhances antitumor capability in head and neck squamous cell carcinoma. Cancer Immunol Res. 2019;7(10):1700–13.
Xu J, Fang Y, Chen K, Li S, Tang S, Ren Y, Cen Y, Fei W, Zhang B, Shen Y, Lu W. Single-cell RNA sequencing reveals the tissue architecture in human high-grade serous ovarian cancer. Clin Cancer Res. 2022;28(16):3590–602.
Chen F, Xu Y, Chen Y, Shan S. TIGIT enhances CD4(+) regulatory T-cell response and mediates immune suppression in a murine ovarian cancer model. Cancer Med. 2020;9(10):3584–91.
Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, Smyth MJ, Kuchroo VK, Anderson AC. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. 2015;125(11):4053–62.
Xu L, Liu L, Yao D, Zeng X, Zhang Y, Lai J, Zhong J, Zha X, Zheng R, Lu Y, Li M, Jin Z, Hebbar Subramanyam S, Chen S, Huang X, Li Y. PD-1 and TIGIT are highly co-expressed on CD8(+) T cells in AML patient bone marrow. Front Oncol. 2021;11: 686156.
Brauneck F, Weimer P, Schulze Zur Wiesch J, Weisel K, Leypoldt L, Vohwinkel G, Fritzsche B, Bokemeyer C, Wellbrock J, Fiedler W. Bone marrow-resident vdelta1 T cells co-express TIGIT with PD-1, TIM-3 or CD39 in AML and myeloma. Front Med (Lausanne). 2021;8: 763773.
Godfrey J, Chen X, Sunseri N, Cooper A, Yu J, Varlamova A, Zarubin D, Popov Y, Jacobson C, Postovalova E, Xiang Z, Nomie K, Bagaev A, Venkataraman G, Zha Y, Tumuluru S, Smith SM, Kline JP. TIGIT is a key inhibitory checkpoint receptor in lymphoma. J Immunother Cancer. 2023;11(6): e006582.
Thibaudin M, Limagne E, Hampe L, Ballot E, Truntzer C, Ghiringhelli F. Targeting PD-L1 and TIGIT could restore intratumoral CD8 T cell function in human colorectal cancer. Cancer Immunol Immunother. 2022;71(10):2549–63.
Zhang D, Hu W, Xie J, Zhang Y, Zhou B, Liu X, Zhang Y, Su Y, Jin B, Guo S, Zhuang R. TIGIT-Fc alleviates acute graft-versus-host disease by suppressing CTL activation via promoting the generation of immunoregulatory dendritic cells. Biochim Biophys Acta Mol Basis Dis. 2018;1864(9):3085–98.
Chocarro L, Blanco E, Arasanz H, Fernandez-Rubio L, Bocanegra A, Echaide M, Garnica M, Ramos P, Fernandez-Hinojal G, Vera R, Kochan G, Escors D. Clinical landscape of LAG-3-targeted therapy. Immunooncol Technol. 2022;14: 100079.
Bhagwat B, Cherwinski H, Sathe M, Seghezzi W, McClanahan TK, de Waal MR, Willingham A. Establishment of engineered cell-based assays mediating LAG3 and PD1 immune suppression enables potency measurement of blocking antibodies and assessment of signal transduction. J Immunol Methods. 2018;456:7–14.
Berry S, Giraldo N, Nguyen P, Green B, Xu H, Ogurtsova A, Soni A, Succaria F, Wang D, Roberts C, Stein J, Engle E, Pardoll D, Anders R, Cottrell T, Taube JM, Tran B, Voskoboynik M, Kuo J, Bang YL, Chung HC, Ahn MJ, Kim SW, Perera A, Freeman D, Achour I, Faggioni R, Xiao F, Ferte C, Lemech C, et al. Correction to: 33rd annual meeting & pre-conference programs of the society for immunotherapy of cancer (SITC 2018). J Immunother Cancer. 2019;7(1):46.
Gregory GP, Zinzani PL, Palcza J, Healy JA, Orlowski RJ, Nahar A, Armand P. Abstract CT106: anti-LAG-3 antibody MK-4280 in combination with pembrolizumab for the treatment of hematologic malignancies: a phase I/II study. Cancer Res. 2019;79(13_Supplement):CT106.
Macdonald LE, Karow M, Stevens S, Auerbach W, Poueymirou WT, Yasenchak J, Frendewey D, Valenzuela DM, Giallourakis CC, Alt FW, Yancopoulos GD, Murphy AJ. Precise and in situ genetic humanization of 6 Mb of mouse immunoglobulin genes. Proc Natl Acad Sci. 2014;111(14):5147–52.
Murphy AJ, Macdonald LE, Stevens S, Karow M, Dore AT, Pobursky K, Huang TT, Poueymirou WT, Esau L, Meola M, Mikulka W, Krueger P, Fairhurst J, Valenzuela DM, Papadopoulos N, Yancopoulos GD. Mice with megabase humanization of their immunoglobulin genes generate antibodies as efficiently as normal mice. Proc Natl Acad Sci. 2014;111(14):5153–8.
Klingler S, Fay R, Holland JP. Light-induced radiosynthesis of (89)Zr-DFO-Azepin-Onartuzumab for imaging the hepatocyte growth factor receptor. J Nucl Med. 2020;61(7):1072–8.
Spreafico A, Janku F, Rodon JA, Tolcher AW, Chandana SR, Oliva M, Musalli S, Knauss L, Kragh M, Alifrangis L, Fröhlich C, Melander MC, Blondal T, Pedersen MW, Lantto J, Wood D, Nadler PI, Horak ID, Siu LL, Lakhani N. A phase I study of Sym021, an anti-PD-1 antibody (Ab), alone and in combination with Sym022 (anti-LAG-3) or Sym023 (anti-TIM-3). Ann Oncol. 2019;30:v488–9.
Sauer N, Szlasa W, Jonderko L, Oslizlo M, Kunachowicz D, Kulbacka J, Karlowicz-Bodalska K. LAG-3 as a potent target for novel anticancer therapies of a wide range of tumors. Int J Mol Sci. 2022;23(17):9958.
Savitsky D, Ward R, Riordan C, Mundt C, Jennings S, Connolly J, Findeis M, Sanicola M, Underwood D, Nastri H, Scherle P, Hollis G, Huber R, Stein R, Dijk MV, Wilson NS. Abstract 3819: INCAGN02385 is an antagonist antibody targeting the co-inhibitory receptor LAG-3 for the treatment of human malignancies. Cancer Res. 2018;78(13):3819–3819.
Hong DS, Schoffski P, Calvo A, Sarantopoulos J. Phase I/II study of LAG525 ± spartalizumab (PDR001) in patients (pts) with advanced malignancies. J Clin Oncol. 2018;36(15_suppl):3012–3012.
Uboha NV, Milhem MM, Kovacs C, Amin A, Magley A, Purkayastha DD, Piha-Paul SA. Phase II study of spartalizumab (PDR001) and LAG525 in advanced solid tumors and hematologic malignancies. J Clin Oncol. 2019;37(15_suppl):2553–2553.
Ghosh S, Sharma G, Travers J, Kumar S, Choi J, Jun HT, Kehry M, Ramaswamy S, Jenkins D. TSR-033, a novel therapeutic antibody targeting LAG-3, enhances T-cell function and the activity of PD-1 blockade in vitro and in vivo. Mol Cancer Ther. 2019;18(3):632–41.
Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM, Sarantopoulos J, Bedard PL, Lin CC, Hodi FS, Wilgenhof S, Santoro A, Sabatos-Peyton CA, Longmire TA, Xyrafas A, Sun H, Gutzwiller S, Manenti L, Naing A. Phase I/Ib clinical trial of sabatolimab, an anti-TIM-3 antibody, alone and in combination with spartalizumab, an anti-PD-1 antibody, in advanced solid tumors. Clin Cancer Res. 2021;27(13):3620–9.
Falchook GS, Ribas A, Davar D, Eroglu Z, Wang JS, Luke JJ, Hamilton EP, Di Pace B, Wang T, Ghosh S, Dhar A, Borgovan T, Waszak A, LoRusso P. Phase 1 trial of TIM-3 inhibitor cobolimab monotherapy and in combination with PD-1 inhibitors nivolumab or dostarlimab (AMBER). J Clin Oncol. 2022;40(16_suppl):2504–2504.
Harding JJ, Moreno V, Bang YJ, Hong MH, Patnaik A, Trigo J, Szpurka AM, Yamamoto N, Doi T, Fu S, Calderon B, Velez de Mendizabal N, Calvo E, Yu D, Gandhi L, Liu ZT, Galvao VR, Leow CC, de Miguel MJ. Blocking TIM-3 in treatment-refractory advanced solid tumors: a phase Ia/b study of LY3321367 with or without an anti-PD-L1 antibody. Clin Cancer Res. 2021;27(8):2168–78.
Hollebecque A, Chung HC, de Miguel MJ, Italiano A, Machiels JP, Lin CC, Dhani NC, Peeters M, Moreno V, Su WC, Chow KH, Galvao VR, Carlsen M, Yu D, Szpurka AM, Zhao Y, Schmidt SL, Gandhi L, Xu X, Bang YJ. Safety and antitumor activity of alpha-PD-L1 antibody as monotherapy or in combination with alpha-TIM-3 antibody in patients with microsatellite instability-high/mismatch repair-deficient tumors. Clin Cancer Res. 2021;27(23):6393–404.
Zeidan AM, Westermann J, Kovacsovics T, Assouline S, Schuh AC, Kim H-J, Rodriguez Macias G, Sanford D, Luskin MR, Stein EM, Malek K, Lyu J, Stegert M, Esteve J. P582: First results of a phase ii study (stimulus-aml1) investigating sabatolimab + azacitidine + venetoclax in patients with newly diagnosed acute myeloid leukemia. HemaSphere. 2022;6:481–2.
Brunner AM, Traer E, Vey N, Scholl S, Tovar N, Porkka K, Narayan R, Garcia-Manero G, Knapper S, Wermke M, Janssen JJ, Esteve J, Loo S, Kontro M, Defilipp Z, Wei AH, Borate U. Allogeneic hematopoietic cell transplantation outcomes of patients with R/R AML or higher-risk MDS treated with the TIM-3 inhibitor MBG453 (Sabatolimab) and hypomethylating agents. Blood. 2021;138(Supplement 1):3677–3677.
Zeidan AM, Giagounidis A, Sekeres MA, Xiao Z, Sanz GF, Hoef MV, Ma F, Hertle S, Santini V. STIMULUS-MDS2 design and rationale: a phase III trial with the anti-TIM-3 sabatolimab (MBG453) + azacitidine in higher risk MDS and CMML-2. Future Oncol. 2023;19(9):631–42.
Xu L, Zou C, Zhang S, Chu TSM, Zhang Y, Chen W, Zhao C, Yang L, Xu Z, Dong S, Yu H, Li B, Guan X, Hou Y, Kong FM. Reshaping the systemic tumor immune environment (STIE) and tumor immune microenvironment (TIME) to enhance immunotherapy efficacy in solid tumors. J Hematol Oncol. 2022;15(1):87.
Niu J, Maurice-Dror C, Lee DH, Kim DW, Nagrial A, Voskoboynik M, Chung HC, Mileham K, Vaishampayan U, Rasco D, Golan T, Bauer TM, Jimeno A, Chung V, Chartash E, Lala M, Chen Q, Healy JA, Ahn MJ. First-in-human phase 1 study of the anti-TIGIT antibody vibostolimab as monotherapy or with pembrolizumab for advanced solid tumors, including non-small-cell lung cancer. Ann Oncol. 2022;33(2):169–80.
Bendell JC, Bedard P, Bang Y-J, LoRusso P, Hodi S, Gordon M, D’Angelo S, D’Angelo S, Desai J, Garralda E, Italiano A, Ahn M-J, Cervantes A, Wainberg Z, Calvo E, Gil-Martin M, Martinez-Garcia M, Bahleda R, Cassier P, Delord J-P, Prawira A, Melero I, Emens L, Romano E, Miller K, Hsieh RW, Xue C, Morrissey K, Twomey P, Gash K, et al. Abstract CT302: phase Ia/Ib dose-escalation study of the anti-TIGIT antibody tiragolumab as a single agent and in combination with atezolizumab in patients with advanced solid tumors. Cancer Res. 2020;80(16_Supplement):CT302.
Chen X, Xue L, Ding X, Zhang J, Jiang L, Liu S, Hou H, Jiang B, Cheng L, Zhu Q, Zhang L, Zhou X, Ma J, Liu Q, Li Y, Ren Z, Jiang B, Song X, Song J, Jin W, Wei M, Shen Z, Liu X, Wang L, Li K, Zhang T. An Fc-competent anti-human TIGIT blocking antibody ociperlimab (BGB-A1217) elicits strong immune responses and potent anti-tumor efficacy in pre-clinical models. Front Immunol. 2022;13: 828319.
Wu L, Wang P-H, Hsiao S-Y, Chang C-L, Kim HS, Lee J-Y, Ryu S-Y, Zuo Y, Mu X, Gao Y, Yang S, Lee J-K. AdvanTIG-202: a phase 2 study investigating anti-TIGIT monoclonal antibody ociperlimab plus anti-PD-1 monoclonal antibody tislelizumab in patients with previously treated recurrent or metastatic cervical cancer. J Clin Oncol. 2021;39(15):TPS5595.
Ozguroglu M, Levy B, Horinouchi H, Yu J, Grainger E, Phuong P, Peterson DA, Newton M, Spira A. 971TiP - Phase III trial of durvalumab combined with domvanalimab following concurrent chemoradiotherapy (cCRT) in patients with unresectable stage III NSCLC (PACIFIC-8). Ann Oncol (2022). 2022;33:S438–47.
Hua B, Yang M, Xue J, Dong C, Mao Y-T, Li O, Cheung E, Issafras H, Xu W, Jiang W. Abstract 2451: a novel single domain antibody targeting TIGIT for cancer use in combination therapies. Cancer Res. 2021;81(13_Supplement):2451–2451.
Przepiorka D, Ko CW, Deisseroth A, Yancey CL, Candau-Chacon R, Chiu HJ, Gehrke BJ, Gomez-Broughton C, Kane RC, Kirshner S, Mehrotra N, Ricks TK, Schmiel D, Song P, Zhao P, Zhou Q, Farrell AT, Pazdur R. FDA approval: blinatumomab. Clin Cancer Res. 2015;21(18):4035–9.
Wu Y, Yi M, Zhu S, Wang H, Wu K. Recent advances and challenges of bispecific antibodies in solid tumors. Exp Hematol Oncol. 2021;10(1):56.
Wang J, Asch AS, Hamad N, Weickhardt A, Tomaszewska-Kiecana M, Dlugosz-Danecka M, Pylypenko H, Bahadur S, Ulahannan S, Koucheki J, Sun J, Li H, Chen F, Zhang X, Muth J, Kaminker P, Moore P, Sumrow BJ. A Phase 1, open-label study of MGD013, a bispecific DART® molecule binding PD-1 and LAG-3 in patients with relapsed or refractory diffuse large B-cell lymphoma. Blood. 2020;136(Supplement 1):21–2.
Luke JJ, Patel MR, Hamilton EP, Chmielowski B, Ulahannan SV, Kindler HL, Bahadur SW, Clingan PR, Mallesara G, Weickhardt AJ, Currence S, Xu L, Kaul S, Chen F, Moore PA, Bonvini E, Sumrow B, Blumenschein G. A phase I, first-in-human, open-label, dose-escalation study of MGD013, a bispecific DART molecule binding PD-1 and LAG-3, in patients with unresectable or metastatic neoplasms. J Clin Oncol. 2020;38(15):3004–3004.
Jenkins S, Wesolowski R, Gatti-Mays ME. Improving breast cancer responses to immunotherapy-a search for the achilles heel of the tumor microenvironment. Curr Oncol Rep. 2021;23(5):55.
Legg JW, McGuinness B, Arasanz H, Bocanegra A, Bartlett P, Benedetti G, Birkett N, Cox C, De Juan E, Enever C, Hames E, Kochan G, Garcia-Granda MJ, Sette A, Teng Y, Thompson L, Vera R, Williams R, Zuazo M, Escors D, Edwards C. Abstract 930: CB213: a half-life extended bispecific humabody VH delivering dual checkpoint blockade to reverse the dysfunction of LAG3+PD-1+ double-positive T cells. Cancer Res. 2020;80(16_Supplement):930–930.
Rohrberg KS, Garralda E, Calvo E, Moreno Garcia V, Guidi M, Kraus DG, McIntyre C, Kao H, Codarri Deak L, Michielin F, Liu T, Muecke M, Markert C, Melero I. 745P Clinical activity, safety, and PK/PD from the first in human study (NP41300) of RO7247669, a PD1-LAG3 bispecific antibody. Ann Oncol. 2022;33:S884–5.
Kraman M, Faroudi M, Allen NL, Kmiecik K, Gliddon D, Seal C, Koers A, Wydro MM, Batey S, Winnewisser J, Young L, Tuna M, Doody J, Morrow M, Brewis N. FS118, a bispecific antibody targeting LAG-3 and PD-L1, enhances T-cell activation resulting in potent antitumor activity. Clin Cancer Res. 2020;26(13):3333–44.
Yap TA, LoRusso PM, Wong DJ, Hu-Lieskovan S, Papadopoulos KP, Holz JB, Grabowska U, Gradinaru C, Leung KM, Marshall S, Reader CS, Russell R, Sainson RCA, Seal CJ, Shepherd CJ, Germaschewski F, Gliddon D, Stern O, Young L, Brewis N, Kayitalire L, Morrow M. A phase 1 first-in-human study of FS118, a tetravalent bispecific antibody targeting LAG-3 and PD-L1 in patients with advanced cancer and PD-L1 resistance. Clin Cancer Res. 2023;29(5):888–98.
Jiang H, Ni H, Zhang P, Guo X, Wu M, Shen H, Wang J, Wu W, Wu Z, Ding J, Tang R, Zhou S, Chen B, Yu M, Jing H, Liu J. PD-L1/LAG-3 bispecific antibody enhances tumor-specific immunity. Oncoimmunology. 2021;10(1):1943180.
Hedvat M, Bonzon C, Bernett MJ, Moore GL, Avery K, Rashid R, Nisthal A, Schubert S, Varma R, Lee S-H, Bogaert L, Leung IWL, Chu S, Muchhal U, Desjarlais J. Abstract 2784: simultaneous checkpoint-checkpoint or checkpoint-costimulatory receptor targeting with bispecific antibodies promotes enhanced human T cell activation. Cancer Res. 2018;78(13_Supplement):2784–2784.
Natoli M, Hatje K, Gulati P, Junker F, Herzig P, Jiang Z, Davydov II, Germann M, Trub M, Marbach D, Zwick A, Weber P, Seeber S, Wiese M, Lardinois D, Heinzelmann-Schwarz V, Rosenberg R, Tietze L, Mertz KD, Umana P, Klein C, Codarri-Deak L, Kao H, Zippelius A. Deciphering molecular and cellular ex vivo responses to bispecific antibodies PD1-TIM3 and PD1-LAG3 in human tumors. J Immunother Cancer. 2022;10(11): e005548.
Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, Gopalakrishnan V, Xi Y, Zhao H, Amaria RN, Tawbi HA, Cogdill AP, Liu W, LeBleu VS, Kugeratski FG, Patel S, Davies MA, Hwu P, Lee JE, Gershenwald JE, Lucci A, Arora R, Woodman S, Keung EZ, Gaudreau PO, Reuben A, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577(7791):549–55.
Dahlen E, Veitonmaki N, Norlen P. Bispecific antibodies in cancer immunotherapy. Ther Adv Vaccines Immunother. 2018;6(1):3–17.
Hellmann MD, Bivi N, Calderon B, Shimizu T, Delafontaine B, Liu ZT, Szpurka AM, Copeland V, Hodi FS, Rottey S, Aftimos P, Piao Y, Gandhi L, Galvao VR, Leow CC, Doi T. Safety and immunogenicity of LY3415244, a bispecific antibody against TIM-3 and PD-L1, in patients with advanced solid tumors. Clin Cancer Res. 2021;27(10):2773–81.
Klein C, Schaefer W, Regula JT, Dumontet C, Brinkmann U, Bacac M, Umana P. Engineering therapeutic bispecific antibodies using crossmab technology. Methods. 2019;154:21–31.
Herrera-Camacho I, Anaya-Ruiz M, Perez-Santos M, Millan-Perez Pena L, Bandala C, Landeta G. Cancer immunotherapy using anti-TIM3/PD-1 bispecific antibody: a patent evaluation of EP3356411A1. Expert Opin Ther Pat. 2019;29(8):587–93.
Zhu B, Dai T, Liu R, Suarez A, Liban T, Zhang B, Karow M, Sheng J, Sheng Z, Lv B. Abstract 6368: preclinical pharmacology and safety studies of ZG005: an anti-PD-1/TIGIT bispecific mAb in a phase I clinical trial for advanced tumors. Cancer Res. 2023;83(7_Supplement):6368–6368.
Yang F, Zhao L, Wei Z, Yang Y, Liu J, Li Y, Tian X, Liu X, Lu X, Sui J. A cross-species reactive TIGIT-blocking antibody Fc dependently confers potent antitumor effects. J Immunol. 2020;205(8):2156–68.
Agenus and bristol myers squibb announce exclusive global license for agenus’ anti-TIGIT bispecific antibody program. [https://www.globenewswire.com/news-release/2021/05/18/2231478/8060/en/Agenus-and-Bristol-Myers-Squibb-Announce-Exclusive-Global-License-for-Agenus-Anti-TIGIT-Bispecific-Antibody-Program.html]
Min J, Huang Z, Pang X, Zhong T, Jin C, Chen N, Xia D, Zhang P, Wang ZM, Xia Y, Li B. 486P AK130, a first-in-class Fc-mutant anti-TIGIT antibody fused with TGF-βRII protein, elicits potent anti-tumor efficacy in pre-clinical studies. Ann Oncol. 2022;33:S762.
Dirix L, Triebel F. AIPAC: a phase IIb study of eftilagimod alpha (IMP321 or LAG-3Ig) added to weekly paclitaxel in patients with metastatic breast cancer. Future Oncol. 2019;15(17):1963–73.
Maruhashi T, Sugiura D, Okazaki IM, Okazaki T. LAG-3: from molecular functions to clinical applications. J Immunother Cancer. 2020;8(2): e001014.
Atkinson V, Khattak A, Haydon A, Eastgate M, Roy A, Prithviraj P, Mueller C, Brignone C, Triebel F. Eftilagimod alpha, a soluble lymphocyte activation gene-3 (LAG-3) protein plus pembrolizumab in patients with metastatic melanoma. J Immunother Cancer. 2020;8(2): e001681.
Lu J, Jiang G. The journey of CAR-T therapy in hematological malignancies. Mol Cancer. 2022;21(1):194.
Qiao Y, Chen J, Wang X, Yan S, Tan J, Xia B, Chen Y, Lin K, Zou F, Liu B, He X, Zhang Y, Zhang X, Zhang H, Wu X, Lu L. Enhancement of CAR-T cell activity against cholangiocarcinoma by simultaneous knockdown of six inhibitory membrane proteins. Cancer Commun (Lond). 2023;43(7):788–807.
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, Qayed M, De Moerloose B, Hiramatsu H, Schlis K, Davis KL, Martin PL, Nemecek ER, Yanik GA, Peters C, Baruchel A, Boissel N, Mechinaud F, Balduzzi A, Krueger J, June CH, Levine BL, Wood P, Taran T, Leung M, Mueller KT, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–48.
Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, Boesteanu AC, Wang Y, O’Connor RS, Hwang WT, Pequignot E, Ambrose DE, Zhang C, Wilcox N, Bedoya F, Dorfmeier C, Chen F, Tian L, Parakandi H, Gupta M, Young RM, Johnson FB, Kulikovskaya I, Liu L, Xu J, Kassim SH, Davis MM, Levine BL, Frey NV, Siegel DL, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24(5):563–71.
Zolov SN, Rietberg SP, Bonifant CL. Programmed cell death protein 1 activation preferentially inhibits CD28.CAR-T cells. Cytotherapy. 2018;20(10):1259–66.
Galon J, Rossi J, Turcan S, Danan C, Locke FL, Neelapu SS, Miklos DB, Bartlett NL, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, Timmerman J, Reagan PM, Lekakis LJ, Unabia S, Go WY, Wiezorek JS, Bot A. Characterization of anti-CD19 chimeric antigen receptor (CAR) T cell-mediated tumor microenvironment immune gene profile in a multicenter trial (ZUMA-1) with axicabtagene ciloleucel (axi-cel, KTE-C19). J Clin Oncol. 2017;35(15_suppl):3025–3025.
Liu X, Zhang Y, Cheng C, Cheng AW, Zhang X, Li N, Xia C, Wei X, Liu X, Wang H. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017;27(1):154–7.
Schoutrop E, Poiret T, El-Serafi I, Zhao Y, He R, Moter A, Henriksson J, Hassan M, Magalhaes I, Mattsson J. Tuned activation of MSLN-CAR T cells induces superior antitumor responses in ovarian cancer models. J Immunother Cancer. 2023;11(2): e005691.
Blaeschke F, Ortner E, Stenger D, Mahdawi J, Apfelbeck A, Habjan N, Weisser T, Kaeuferle T, Willier S, Kobold S, Feuchtinger T. Design and evaluation of Tim-3-CD28 checkpoint fusion proteins to improve anti-CD19 CAR T-cell function. Front Immunol. 2022;13: 845499.
Lee YH, Lee HJ, Kim HC, Lee Y, Nam SK, Hupperetz C, Ma JSY, Wang X, Singer O, Kim WS, Kim SJ, Koh Y, Jung I, Kim CH. PD-1 and TIGIT downregulation distinctly affect the effector and early memory phenotypes of CD19-targeting CAR T cells. Mol Ther. 2022;30(2):579–92.
Jackson Z, Hong C, Schauner R, Dropulic B, Caimi PF, de Lima M, Giraudo MF, Gupta K, Reese JS, Hwang TH, Wald DN. Sequential single-cell transcriptional and protein marker profiling reveals TIGIT as a marker of CD19 CAR-T cell dysfunction in patients with Non-Hodgkin Lymphoma. Cancer Discov. 2022;12(8):1886–903.
Jiang VC, Hao D, Jain P, Li Y, Cai Q, Yao Y, Nie L, Liu Y, Jin J, Wang W, Lee HH, Che Y, Dai E, Han G, Wang R, Rai K, Futreal A, Flowers C, Wang L, Wang M. TIGIT is the central player in T-cell suppression associated with CAR T-cell relapse in mantle cell lymphoma. Mol Cancer. 2022;21(1):185.
Yang F, Zhang F, Ji F, Chen J, Li J, Chen Z, Hu Z, Guo Z. Self-delivery of TIGIT-blocking scFv enhances CAR-T immunotherapy in solid tumors. Front Immunol. 2023;14:1175920.
Zhao S, Wang C, Lu P, Lou Y, Liu H, Wang T, Yang S, Bao Z, Han L, Liang X, Ma C, Gao L. Switch receptor T3/28 improves long-term persistence and antitumor efficacy of CAR-T cells. J Immunother Cancer. 2021;9(12): e003176.
Lee WS, Ye Z, Cheung AMS, Goh YPS, Oh HLJ, Rajarethinam R, Yeo SP, Soh MK, Chan EHL, Tan LK, Tan SY, Chuah C, Chng WJ, Connolly JE, Wang CI. Effective killing of acute myeloid leukemia by TIM-3 targeted chimeric antigen receptor T cells. Mol Cancer Ther. 2021;20(9):1702–12.
Zhou S, Liu M, Ren F, Meng X, Yu J. The landscape of bispecific T cell engager in cancer treatment. Biomark Res. 2021;9(1):38.
Cortes-Selva D, Casneuf T, Vishwamitra D, Stein S, Perova T, Skerget S, Ramos E, van Steenbergen L, De Maeyer D, Boominathan R, Lau O, Davis C, Banerjee A, Stephenson T, Uhlar CM, Kobos R, Goldberg JD, Pei L, Trancucci D, Girgis S, Wang Lin SX, Wu LS, Moreau P, Usmani S, Bahlis NJ, Van De Donk NW, Verona R. Teclistamab, a B-cell maturation antigen (BCMA) × CD3 bispecific antibody, in patients with relapsed/refractory multiple myeloma (RRMM): correlative analyses from majesTEC-1. Blood. 2022;140(Supplement 1):241–3.
Ma J, Mo Y, Tang M, Shen J, Qi Y, Zhao W, Huang Y, Xu Y, Qian C. Bispecific antibodies: from research to clinical application. Front Immunol. 2021;12: 626616.
Kang J, Sun T, Zhang Y. Immunotherapeutic progress and application of bispecific antibody in cancer. Front Immunol. 2022;13:1020003.
Wood H. Parkinson disease: LAG3 facilitates cell-to-cell spread of alpha-synuclein pathology. Nat Rev Neurol. 2016;12(12):678.
Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, Zheng XX, Strom TB, Kuchroo VK. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6(12):1245–52.
Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, Levine Z, Beiman M, Dassa L, Achdout H, Stern-Ginossar N, Tsukerman P, Jonjic S, Mandelboim O. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci. 2009;106(42):17858–63.
Chew V, Lai L, Pan L, Lim CJ, Li J, Ong R, Chua C, Leong JY, Lim KH, Toh HC, Lee SY, Chan CY, Goh BKP, Chung A, Chow PKH, Albani S. Delineation of an immunosuppressive gradient in hepatocellular carcinoma using high-dimensional proteomic and transcriptomic analyses. Proc Natl Acad Sci. 2017;114(29):E5900–9.
Yan W, Liu X, Ma H, Zhang H, Song X, Gao L, Liang X, Ma C. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut. 2015;64(10):1593–604.
Li C, Chen X, Yu X, Zhu Y, Ma C, Xia R, Ma J, Gu C, Ye L, Wu D. Tim-3 is highly expressed in T cells in acute myeloid leukemia and associated with clinicopathological prognostic stratification. Int J Clin Exp Pathol. 2014;7(10):6880–8.
Liu L, Chang YJ, Xu LP, Zhang XH, Wang Y, Liu KY, Huang XJ. Reversal of T cell exhaustion by the first donor lymphocyte infusion is associated with the persistently effective antileukemic responses in patients with relapsed AML after allo-HSCT. Biol Blood Marrow Transplant. 2018;24(7):1350–9.
Guillerey C, Harjunpaa H, Carrie N, Kassem S, Teo T, Miles K, Krumeich S, Weulersse M, Cuisinier M, Stannard K, Yu Y, Minnie SA, Hill GR, Dougall WC, Avet-Loiseau H, Teng MWL, Nakamura K, Martinet L, Smyth MJ. TIGIT immune checkpoint blockade restores CD8(+) T-cell immunity against multiple myeloma. Blood. 2018;132(16):1689–94.
Wang P, Chen Y, Long Q, Li Q, Tian J, Liu T, Wu Y, Ding Z. Increased coexpression of PD-L1 and TIM3/TIGIT is associated with poor overall survival of patients with esophageal squamous cell carcinoma. J Immunother Cancer. 2021;9(10): e002836.
Acknowledgements
Not applicable.
Funding
This work is supported by grants from the National Natural Science Foundation of China (Nos. 82293630, 82293632, 82070152, 82000108) and Guangdong Basic and Applied Basic Research Foundation (2020A1515110310).
Author information
Authors and Affiliations
Contributions
LTC, YCL, and JXT performed the selection of literature, drafted the manuscript, and prepared the figures. LX helped to arrange the studies, participated in the abstract and conclusion writing, and revised the manuscript. YQL carried out the design of this review and revised the manuscript. All authors contributed to this manuscript. All authors read and approved the final version manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cai, L., Li, Y., Tan, J. et al. Targeting LAG-3, TIM-3, and TIGIT for cancer immunotherapy. J Hematol Oncol 16, 101 (2023). https://doi.org/10.1186/s13045-023-01499-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-023-01499-1