
Abstract
Mantle cell lymphoma is a B cell non-Hodgkin lymphoma (NHL), representing 2–6% of all NHLs and characterized by overexpression of cyclin D1. The last decade has seen the development of many novel treatment approaches in MCL, most notably the class of Bruton's tyrosine kinase inhibitors (BTKi). BTKi has shown excellent outcomes for patients with relapsed or refractory MCL and is now being studied in the first-line setting. However, patients eventually progress on BTKi due to the development of resistance. Additionally, there is an alteration in the tumor microenvironment in these patients with varying biological and therapeutic implications. Hence, it is necessary to explore novel therapeutic strategies that can be effective in those who progressed on BTKi or potentially circumvent resistance. In this review, we provide a brief overview of BTKi, then discuss the various mechanisms of BTK resistance including the role of genetic alteration, cancer stem cells, tumor microenvironment, and adaptive reprogramming bypassing the effect of BTK inhibition, and then provide a comprehensive review of current and emerging therapeutic options beyond BTKi including novel agents, CAR T cells, bispecific antibodies, and antibody–drug conjugates.
Similar content being viewed by others
Background
Mantle cell lymphoma (MCL) is a subtype of B cell non-Hodgkin lymphoma (NHL) characterized by overexpression of CCND1 and translocation t(11:14)(q13;q32) [1]. The most common type of MCL originates from mature B cells and is often found to become unstable and aggressive through accumulating mutations in genes related to cell cycle regulation, such as the DNA damage response pathway. They are often found to express SOX11 [2] and carry little to no immunoglobulin heavy variable (IGHV) somatic mutations [2] and include classical, blastoid, and pleomorphic variants of MCL. The second indolent subtype (10–15% cases, leukemic non-nodal variant) is less aggressive, carries IGHV somatic hypermutations [1], and is genetically stable with low to no SOX11 expression. Patients can potentially have asymptomatic disease with this subtype for extended periods [3]. Recently, the diagnosis-to-treatment interval (DTI) was shown to be prognostic patients with newly diagnosed MCL, wherein patients with short DTI (DTI ≤ 14 days) had worse outcomes and was strongly associated with adverse clinical factors [4]. While the outcomes of MCL have been conventionally poor, there has been improvement in survival in the past decade owing to the advent of novel therapies [5].
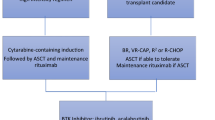
The treatment of MCL in the frontline setting largely relies on patient-specific factors such as age, overall performance status, and underlying co-morbidities. For the young, transplant-eligible patient, treatment generally consists of induction chemotherapy, consolidation with an autologous stem cell transplantation, and maintenance with rituximab for about three years. For induction chemotherapy, no specific chemotherapy regimen has been firmly established as the standard of care, and the treatment regimen used is variable based on the institution or physician's practice. However, it is generally accepted that the regimen should contain rituximab and cytarabine. Less toxic chemotherapy treatments are given for patients unfit for intensive chemotherapy, such as bendamustine/rituximab (BR) or R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone), with or without maintenance rituximab. However, patients eventually progress following frontline therapy, so establishing effective treatments for relapsed/refractory (R/R) MCL is important. In the past decade, BTK inhibitors (BTKi) have revolutionized the management of patients with R/R MCL. However, a significant proportion of patients eventually progress with poor post-BTKi relapse outcomes.
This review article focuses on three main aspects: (1) discuss the current BTKis approved for clinical use in the USA, (2) detail the various mechanisms of BTK resistance including the role of genetic alteration, cancer stem cells, tumor microenvironment, and adaptive reprogramming bypassing the effect of BTK inhibition, and (3) current and emerging therapeutic strategies beyond BTKi.
BTKi in MCL
Currently, there are four BTKis approved for MCL therapy. They are ibrutinib, acalabrutinib, zanubrutinib (all covalent BTKis), and pirtobrutinib (only approved non-covalent BTKi) [6,7,8,9,10]. Table 1 summarizes the BTKi currently approved for MCL, the study that led to their FDA approval, and the potential adverse effects reported on these studies.
Resistance to BTKis and approaches for targeting mutant BTK
Although the first-generation BTKi (ibrutinib) has shown encouraging therapeutic effects in MCL, nearly one-third of treated patients have ultimately been found to develop primary intrinsic resistance. Additionally, acquired resistance developed in nearly all patients [11]. Furthermore, those patients who developed ibrutinib resistance have typically had dismal clinical outcomes, with median overall survival (OS) of ~ 6 to 8 months even after salvage treatment [9, 12,13,14]. The primary resistance is often associated with sustained activation of the PI3K-AKT pathway or other genetic alterations providing an alternative activation of B cell receptor (BCR) signaling [15]. In contrast, secondary resistance in MCL patients is found in patients harboring a point mutation in the BTK gene (BTKC481S) which reduces the binding affinity of covalent BTKis to BTK [16]. Second-generation BTKis such as acalabrutinib, tirabrutinib (ONO/GS-4059), spebrutinib (CC-292), and zanubrutinib (BGB-3111), which are typically more sensitive than ibrutinib, work by binding covalently and irreversibly to ATP binding region within the kinase domain of BTK at cysteine 481 position, and therefore, a point mutation in BTK (BTKC481S) can prevent the activity of these agents [17, 18]. In addition to the BTKC481S mutation, the gain of function mutations in PLCG2 (R665W, L845F, S707Y) has also been attributed to the secondary mechanism of ibrutinib resistance. However, BTKC481S mutation is infrequent in practice, and mutations in PLCG2 are typically not observed in MCL [19, 20]. In order to address resistance against first and second-generation covalent BTKis, third-generation non-covalent (reversible) BTKis (those could bind to both wild-type and BTKC481S) and proteolysis-targeting chimeras (PROTACs) targeting BTK have been designed, and many of them are being investigated in preclinical/clinical studies or already approved for use [21].
Non-covalent BTKi
Pirtobrutinib (LOXO-305) blocks the ATP binding site of BTK and, unlike ibrutinib, shows no direct interaction with C481. A recent study from Gomez et al. described pharmacologic, biophysical, and structural attributes that detailed and differentiated pirtobrutinib from the current covalent BTKi (ibrutinib, zanubrutinib, and acalabrutinib). This preclinical study demonstrated differential binding of pirtobrutinib to both BTK and BTKC481 substitution mutants that prevented BTKY551 phosphorylation in the activation loop and inhibited BTK signaling in multiple B cell lymphoma cell lines and lymphoma xenograft tumor growth [22]. Pirtobrutinib is the first and only non-covalent reversible inhibitor that received FDA approval (27th January 2023) for R/R MCL patients. This is based on promising results in the BRUIN Phase 1/2 trial (NCT03740529) with an overall response rate (ORR) of 52% and complete response (CR) rate of 13% [23]. This study included 120 patients with MCL that were previously treated with a covalent BTKi—ibrutinib (67%), acalabrutinib (30%), and zanubrutinib (8%). Among these 120 MCL cases, 83% discontinued their last BTKi due to refractory or progressive disease. The trial data suggest that pirtobrutinib was well tolerated at all doses tested (the maximum tolerated dose was not reached). The updated data were presented in ASH 2022 meeting [24]. These data indicate that pirtobrutinib could be a potential treatment strategy to overcome covalent BTKi-associated resistance development in MCL. A phase 3 trial, BRUIN MCL-321 (NCT04662255) comparing pirtobrutinib monotherapy to the investigator's choice of covalent BTKi monotherapy (ibrutinib, acalabrutinib, or zanubrutinib) in MCL patients (n = 500) who received ≥ 1 prior line of systemic therapy that did not include a prior BTKi is currently ongoing [25].
Besides pirtobrutinib, other BTK-targeting non-covalent inhibitors are currently in the pipeline as well. Promising agents include fenebrutinib (GDC-0853) that inhibits BTK via forming hydrogen bonds with K430, M477, D539 of BTK and nemtabrutinib (MK-1026, formerly ARQ 531) that binds BTK via hydrogen bonds with E475, Y476 residues [26, 27]. These inhibitors have been tested in MCL, chronic lymphocytic leukemia (CLL), diffuse large B cell lymphoma (DLBCL), follicular lymphoma (FL), and other B cell malignancies [27, 28]. These agents have demonstrated acceptable safety profiles and efficacy against BTKC481S mutant B cell malignancies in phase 1 clinical trials (NCT01991184, NCT03162536). However, only a few MCL cases have been included in these studies. As such, due to the lack of convincing data on these agents in the MCL population, further comments on the efficacy of these inhibitors in MCL cannot be made at this time.
BTK-PROTAC
PROTAC strategy is a novel approach that degrades a target of interest by bringing it to the proximity of E3 ubiquitin–proteasome ligase (Cereblon or Von Hippel–Lindau) through an attached linker. Several BTK-targeting PROTAC have been synthesized to overcome ibrutinib resistance, which could potentially target both the mutant and wild-type BTK. However, most of these agents are in the preclinical stage of development, and no clinical data exist at this time [21]. DD-03–171, a BTK degrader, has shown anti-proliferative activity in MCL cells in vitro and the patient-derived xenograft (PDX) model [29]. Other BTK-PROTAC, such as MT-802 (derived from an ibrutinib warhead lacking the meta-acrylamide moiety), and advanced agents with improved pharmacokinetic properties, such as SJF620 and L18I, have also been tested for their efficacy to degrade BTK [30,31,32]. UBX-382 is another novel BTK degrader that inhibits both wild-type and BTK mutant tumor growth in DLBCL via targeting the BCR pathway [33]. Though BTK-PROTAC has shown anti-proliferative activity and selective degradation of BTK in DLBCL, only a limited number of studies are available in MCL. Furthermore, these BTK degraders are yet to go into clinical trials, and clinical data on them is still largely lacking.
BTK-independent BTKi resistance mechanism
Although non-covalent and covalent BTKis have been found to target BTK selectively, these inhibitors may not mitigate BTK-independent BTKi resistance mechanisms found in lymphoma. Below we discuss BTKi resistance mechanisms beyond their dependency on BTK status (expression or mutation).
Genetic cause of BTKi resistance in MCL
The mechanisms associated with genetic alterations leading to primary BTKi resistance include missense mutations or somatic DNA copy number alterations in high-risk genes, BCR signaling component alterations, mutations in DNA damage repair machinery, tumor suppressors, and NFkB pathway dysregulation. These genetic alterations are found to be present in MCLs at a higher rate than CLL at baseline, predisposing them to significantly higher treatment resistance. In search of BTKi resistance-associated gene expression signatures, Zhang et al. performed RNA sequencing of ibrutinib-sensitive and resistant MCL tumors and identified the differentially expressed genes involved in glycolysis, glutaminolysis, and mitochondrial biogenesis related to metabolic reprogramming of oxidative phosphorylation (OXPHOS)[34]. Importantly, activation of the OXPHOS pathway was identified as a driver of acquired ibrutinib resistance in MCL, and DNA methyl-transferase 3A (DNMT3A) was found to act as a mediator of mitochondrial biogenesis, which is required for OXPHOS activation in MCL. Considering genetic alterations beyond BTKC481S mutation as the primary cause, Rahal et al. demonstrated loss of function mutation in NF-κB inhibitors (TRAF2, TRAF3, and BIRC3) in ibrutinib-resistant MCL cell lines (n = 6) causing dependency of resistant cells on the MAP3K14 pathway which in turn activated the alternative NF-κB survival signaling. In contrast, ibrutinib-sensitive MCL cell lines (n = 4) displayed chronic activation of BCR signaling [35]. Furthermore, in the same study, mutations in TRAF3 and BIRC3 were further confirmed to be present in patients' tumors that did not respond to ibrutinib. In a phase-3 MCL3001 (RAY) trial (NCT01646021), Lenz et al. confirmed that primary ibrutinib resistance was associated with mutations in NF-κB inhibitor genes and the EGFR family of genes [36]. Loss of function mutation in BIRC3 has also been demonstrated to activate non-canonical NF-κB signaling in MCL, which is quite different from BCR-dependent classical NF-κB activation signaling [37]. Using 13 paired primary MCL tumors and a whole exome sequencing approach (WES), Chenglin et al. identified 18 recurrently mutated genes, including ATM, MEF2B, and MLL2 and novel mutation targets such as SIPR1 and CARD11. CARD11 is a scaffold protein that acts downstream of BTK signaling and functions by regulating NF-κB signaling. Investigation of 173 MCL samples identified that 5.5% of MCL cases harbor CARD11 mutation [38]. Genomic profiling of 24 MCL patients (phase 2 clinical trial; NCT02471391) who received ibrutinib treatment for four weeks, followed by venetoclax (a BCL2 inhibitor), identified genetic signatures between responders and non-responders. Five of 24 MCL patients did not respond to treatment and had 9p21.1–p24.3 loss and mutation in SMARCA2 genomic region (4/5) or deletions in ARID2 (3/5).
Furthermore, a mutation in the ATM gene was observed in patients who achieved CR [39]. Large-scale genomic data has provided the landscape of somatic mutations and clonal evolution of hotspot mutations in CCND1 (E36K, Y44D, and C47S) (cyclin D1 gene), leading to the accumulation of the cyclin D1 protein through its defective proteolysis [40]. Importantly, it was found that these CCND1 mutations are associated with ibrutinib resistance in MCL [34, 41].
Naeem et al. showed genetic causes beyond BTKC481S mutation as responsible for BTKi resistance in CLL. WES on primary CLL patients who experienced disease progression following pirtobrutinib treatment revealed the presence of a second-site BTK mutation (T474I) [42]. The primary cause of such BTKi resistance-associated genetic alterations could be intratumoral heterogeneity present at the time of diagnosis and modulation of the initial mutational profile at the progression of the disease [40, 43].
Cancer stem cells and BTKi resistance in MCL
Besides genetic causes, non-genetic molecular changes have been found to cause the development of intrinsic and acquired BTKi resistance. Other non-genetic mechanisms, such as PI3K-AKT-mTOR, non-canonical NFκB activation, or epigenetic gene dysfunctions that bypass the survival BCR signaling may also contribute to BTKi resistance. Modulation in tumor immune microenvironment (TIME) and the presence of cancer stem cells (CSCs) have also been found to be associated with BTKi resistance development [13, 21, 44]. Chen et al. identified the CD19-/CD45 + cellular population in primary MCL tumors with CSCs-like characteristics (high expression of the stem cell-specific genes Oct4 and Nanog) and were quiescent. Interestingly, even CD19-/CD45 + cells could form a complete heterogeneous tumor in immunocompromised mice compared to CD19+/CD45+ MCL cells [45]. Subsequently, Mathur et al. identified Wnt signaling as a critical oncogenic pathway activated in MCL CD19-/CD45+ CSCs cells (also called MCL-initiating cells or MCL-IC cells) and associated with ibrutinib resistance [46]. Interestingly, the mechanism of how CD19+ non-IC becomes IC is still largely unknown; interrogating the underlying mechanisms can identify rationale therapeutic targets. Though CD19-/CD45+ cells have been found to represent CSC characteristics in MCL tumors, no such molecular features of CSCs have been identified in DLBCL tumors [47], possibly due to the different histological makeup.
Tumor microenvironment and BTKi resistance
TME has been found to facilitate tumor cell growth by providing bi-directional signaling between tumor cells and the stromal compartment, including cellular and soluble factors. The cellular milieu in TME includes mesenchymal stromal cells (MSCs), fibroblasts, endothelial cells, immune cells (Treg, NK cells, macrophages), and the soluble component includes cytokines and growth factors. A study by Medina et al. has shown that bone marrow-derived MSCs secrete B cell activating factor (BAFF) and protect MCL cells from BTK inhibition via activating canonical and non-canonical NFκB pathways [48]. A report from Zao et al. has identified that the interaction of TME and MCL cells can induce innate and acquired resistance to BTKis via activating the PI3K-mTOR pathway and integrin-β1 signaling [15]. Other reports have identified the importance of the PI3K pathway and integrin-VLA-4 signaling in facilitating BTKi resistance or enhanced focal adhesion kinase (FAK) or CXCR4 activity in MCL during MCL–stromal cell interaction [49,50,51]. Evasion of the immune system is also a potential resistance mechanism for BTKis. MCL cells can evade the immune system by downregulating the expression of surface antigens or by developing a hostile TME that inhibits the function of immune cells such as T cells. Balsas and colleagues directly linked SOX11 expression to immunosuppressive microenvironment characteristics in MCLs, including reduced expression of antigen presentation gene, T cell activation factors, or increased Treg cell infiltration in TME. As such, this immunosuppressive microenvironment could potentially be associated with BTKi resistance development in MCLs [52].
Targeting BTKi-resistant mechanism in MCL: beyond BTKi
Historically, MCL patients who progress after BTKi treatment (e.g., post-treatment with FDA-approved ibrutinib) have been found to have poor outcomes. Recently approved BTK-targeting non-covalent inhibitor pirtobrutinib has shown promising outcomes in R/R MCLs in BRUIN Phase 1/2 trial (NCT03740529) who progress on covalent BTKi. However, resistance development via activation of BTK-independent mechanism as mentioned earlier remains a significant hurdle toward the development of BTKi in MCL therapy. Therefore, therapies targeting beyond BTK are required for BTKi-resistant MCL tumors. In that regard, many small molecule inhibitors that target different oncogenes, some of whom have already received FDA approval for MCL treatment, are discussed and summarized in Table 2.
Proteasome inhibitors
Bortezomib, a first-in-class compound that reversibly interacts with 20S proteasome, received FDA approval as a second-line therapy for R/R MCLs based on the outcomes of a phase-2 clinical trial (PINNACLE). In this trial, 155 MCL patients were treated with bortezomib monotherapy with a median duration of response (DOR) of 9.2 months [53]. Following updated time-to-event data in this clinical trial, a median OS of 23.5 months was achieved [74]. However, although bortezomib produced promising initial outcomes, more than half of the patients were refractory. The discovery of next-generation proteasome inhibitors, including carfilzomib and ixazomib, has not improved the treatment outcomes as expected [75]. Several resistance mechanisms associated with bortezomib have been reported in MCL, including activation of NFkB signaling, accumulation of anti-apoptotic protein Mcl-1, and accumulation of casein kinase 2 (CK2, the serine/threonine kinase and activator of STAT3) [76,77,78]. A report from Patricia et al. has shown that bortezomib resistance development is associated with plasmacytic differentiation of MCL cells with upregulation of interferon regulatory factor 4 (IRF4) and CD38 and CD138 expression [79]. Notably, overexpression of SOX11 in MCL was identified as a master regulator for the expression of IRF4 and PAX5 and was also found to block terminal B cell differentiation [80]. In addition, bortezomib treatment in MCL induces the transcription of a zinc finger protein (PRDM1, Blimp1) required for NOXA-induced apoptosis in MCLs and PRDM1 expression, which are critical for bortezomib efficacy in MCL [81].
BCL2 inhibitors
BCL-2 is an anti-apoptotic protein frequently overexpressed in almost 90% of MCLs and identified to be amplified via 18q21 DNA copy gain locus leading to the overexpression of BCL2 [82]. Another study associated the high expression of BCL2 with the expression of LINK-A lncRNA in MCL [83]. Several BCL2-targeting agents have been discovered and are currently being investigated for MCL therapy. Venetoclax (ABT-199) is an oral BCL2-targeting agent that received FDA approval for CLL and SLL. Since MCL is also known to be a high BCL2 expression, the efficacy of venetoclax in MCL cases, either as a single agent or in combination therapies, has been investigated. In a phase-1 clinical trial without prior treatment, venetoclax as a single agent showed ORR in 75% of MCL (21/28) and CR in 21% of MCL (6/28) (NCT01328626) [54]. Another clinical trial using venetoclax as monotherapy on R/R MCLs (n = 28) and prior BTKi treatment showed ORR of 53% and CR of 18% [84]. Venetoclax has also been tested in high-risk MCLs that had progressed on BTKis or had multiple relapses on five prior therapies (n = 24). The ORR with venetoclax treatment was 50%, and a CR of 21% could be achieved [85]. Although venetoclax treatment as a single agent was effective in BTKi-resistant MCLs, other MCL cases found to be resistant to venetoclax have also been investigated. Patients who progressed on venetoclax treatment showed clonal evolution of genetic alterations of SMARCA4 and BCL2 and increased frequency of alterations in other genes (TP53, CDKN2A, KMT2D, CELSR3, CCND1, NOTCH2, and ATM) [85]. With a focus on synergistic relationships in MCLs, recent clinical trials seek to combine BCL2 inhibitors with BTKis or other targeting agents. In a multicenter retrospective cohort that evaluated the outcomes of patients with R/R MCL treated with venetoclax (n = 81), the authors reported an ORR of 40% with a median PFS of 3.7 months and OS of 12.5 months. In the study, 62% of the patients (n = 50) were treated with venetoclax monotherapy, 14% (n = 11) in combination with an anti-CD20 monoclonal antibody, and 20% (n = 16) with a BTKi or with other novel agents [86].
PI3K-Akt-mTOR inhibitors
The phosphoinositide 3-kinase (PI3K) signaling pathway is often found to be crucially deregulated in ibrutinib-resistant MCL cells. Wang et al. developed an ibrutinib-resistant patient-derived xenograft (PDX) model of MCL (MCL-PDX) through chronic exposure to ibrutinib. They identified constitutive activation of PI3K-AKT-mTOR signaling as a crucial survival pathway in ibrutinib-resistant MCL cells leading to tumor development. Tumor growth of ibrutinib-resistant MCL-PDX was inhibited by combined treatment of PI3K-δ-targeting agent idelalisib plus ibrutinib [87]. Similar to these MCL studies, our group and others have also developed ibrutinib-resistant DLBCL cells and identified upregulation of PI3K-AKT signaling in resistant cells, which can be overcome by selective PI3K isoform inhibitors treatment [88, 89]. Five PI3K-specific isoform-targeting inhibitors had received FDA approval for hematological malignancies such as CLL/SLL and FL. These are copanlisib (p110α/δ), idelalisib (p110δ), umbralisib (p110δ), duvelisib (p110δ/γ), and alpelisib (p110α). In a phase 1/1b clinical trial, umbralisib in combination with ibrutinib (n = 21) showed an ORR of 67% and a CR rate of 19% (NCT02268851) [90]. Copanlisib in combination with ibrutinib demonstrated an ORR of 87.5% and a CR rate of 50% (NCT03877055) [91]. Buparlisib (pan-PI3Kδ inhibitor) in combination with ibrutinib (n = 18 with 17 evaluated for response) showed an ORR of 94% and a CR rate of 76% (NCT02756247) [92]. In a phase 1 clinical trial (NCT00710528), idelalisib showed an ORR of 40% with a DOR of 2.7 months [55]. Parsaclisib, a selective p110δ inhibitor, showed therapeutic potential (phase-II CITADEL-205 trials) in R/R MCL patients. The ORR was 70% in those who received parsaclisib without prior BTKi (n = 108) and 25% in those who received prior BTKi (n = 53) [93, 94]. Notably, it was also found that dynamic feedback interaction between MCL cells and stromal cells also contributes to ibrutinib resistance development and reciprocal activation of PI3K-AKT-mTOR as well as Integrin-B1 signaling, which could be reversed by combined disruption of BCR signaling with ibrutinib and PI3K-AKT-mTOR axis with GS-1101 (p110δ inhibitor) [15]. However, loss of PTEN or feedback amplification of other PI3K isoforms, such as p110α, has impaired the efficacy of idelalisib in MCL [95,96,97]. Therefore, inhibitors targeting the dual isoforms of PI3K have been generated and tested in MCL, including KA2237 (p110β/δ) [98].
Immunomodulators
TME is one of the critical elements responsible for the R/R status of MCL. Immunomodulatory agents such as lenalidomide (Revlimid) directly influence tumor cells and various cellular compartments of the TME, including NK cells, stromal cells, and T cells, via activating antitumor immune responses [99, 100]. Lenalidomide exhibited antitumor activity in MCL cell lines by upregulating immune response genes, including CD40, CD58, and CD86, and inhibited IL6 production required for bone marrow-derived stromal cell activity [99, 101]. In 2013, lenalidomide was FDA-approved for R/R MCL based on results from MCL-001 (EMERGE; NCT00737529) and MCL-002 (SPRINT; NCT00875667) clinical trials for patients who were refractory to bortezomib treatment and were ineligible for intensive chemotherapy or stem cell transplantation. Lenalidomide-treated group had significantly improved progression-free survival (PFS) with a manageable safety profile [56, 57]. However, despite improved efficacy, lenalidomide treatment did not show promising results after BTKi treatment failure. In the MCL-004 trial (NCT02341781), MCL patients (n = 58) either relapsed, progressed, refractory, or intolerant to ibrutinib treatment had a cumulative ORR of 29% and CR of 14%, with a median DOR of 20 weeks. The ORR with lenalidomide monotherapy (n = 13) was 15% (n = 2), while the ORR with lenalidomide in combination with rituximab (n = 11) was 27% (n = 3) with CR rate of 9% (n = 1) [58]. The exact mechanism of lenalidomide resistance in BTKi-resistant MCL is unknown, but this could be attributed to the activation of PI3K-AKT signaling or other genetic alterations in MCL [102].
Immune checkpoint inhibitors
Immune checkpoint proteins, including Programmed Death 1 (PD-1) and its ligands PD-L1 and PD-L2, Lymphocyte Activation Gene 3 (LAG-3), CD200, Cytotoxic T Lymphocyte Activator 4 (CTLA-4), and CD47, are involved in tumor immunology and benefit tumor growth. Except for T cells, other immune cells rarely express these immune effectors [103].
CD47 and CD24
CD47 acts as a checkpoint that provides a "don't-eat-me" signal to macrophages via interaction with its surface protein SIRPα, resulting in immune evasion by the tumor cells. CD47 is overexpressed in cancer cells, which is the target of interest in MCL. Several monoclonal antibodies (Hu5F9-G4, AO-176, AK117, and CC-90002) and bispecific antibodies, including IBI322, PF-07257876 (target CD47 and PD-L1), IMM0306, CPO107 (targeting CD47 and CD20 at the same time), and TG-1801 (targeting both CD19 and CD47), are under investigation in a clinical trial for multiple lymphoid and solid tumor malignancies [104]. On the other hand, limited studies have focused on targeting CD47 in MCLs. So far, only three clinical trials are accruing patients, including a limited number of MCL cases (NCT04806035, NCT04599634, NCT05025800). A phase 1 clinical trial included 4 R/R MCL cases who received rituximab in combination with CD47-targeting ALX148 (decoy receptor fusion protein composed of SIRPα N-terminal D1 domain and been mutated for increasing its affinity for CD47 binding). However, ALX148 demonstrated excellent tolerability in MCL cases; only 2 out of 4 achieved partial response [105].
A recent study demonstrated that CD24 (a highly glycosylated cell adhesion protein–ligand; Siglec-10) expression but not the CD47 expression is associated with poor clinical response in MCL. Moreover, CD24 was also highly expressed in MCL cell lines where treatment of MCL cell lines with CD24-targeting antibody SN3 yielded 90% removal of MCL cells via phagocytosis by autologous macrophages. In addition, this study also identified that treatment with CD24-targeting antibody was superior to CD47-targeting antibody in MCL [106]. Many CD24-targeting agents, including monoclonal antibodies, chimeric antigen receptor (CAR) T cells, and bispecific antibodies, have been developed and tested in preclinical studies. Some are also in clinical trials for solid tumors [107, 108]. Studies about testing CD24-targeting agents' efficacy in MCL are minimal; therefore, more research is required in this area as MCL are also high CD24 expressers.
PD-L1/PD-1
Similar to CD47 or CD24, limited data are available in MCL for another important immune checkpoint PD-L1/PD-1 expression and targeted therapies. In a study by Yang et al., the highest PD-L1 expression was observed in DLBCL, followed by SLL, mucosa-associated lymphoid tissue lymphoma, and MCL, and the lowest expression was found in FL [109]. Compared to normal PBMCs, Wang et al. 2013 described that the percentage of PD-L1-expressing cells is high in primary MCL tumors and most MCL cell lines. The authors also reported that this high PD-L1 expression in MCL inhibited the T cell activity and proliferation, impaired antigen-specific T cell responses, and rendered MCL cells resistant to T cell-mediated cytolysis. Additionally, the inactive phenotype of T cells due to high PD-L1 expression was reversed by blocking PD-L1 expression on MCL cells [110]. Harrington et al. also demonstrated the constitutive expression of PD-L1 in primary MCL cells, whereas expression of other immune checkpoint genes, including PD-L2, LAG-3, and CTLA-4, was absent. Mechanistically, it was identified that both IFNγ and CD40:CD40L interaction between MCL cells and activated T cells in a co-culture condition regulates PD-L1 expression in MCL cells, which was attenuated by concurrent treatment with BTK or PI3K inhibition [111]. Expression and activity of PD-L1 in MCL are controversial. In a recent report by Ameli et al., neither PD-1 nor its ligand PD-L1 are relevant targets for MCL treatment. Using 79 formalin-fixed paraffin-embedded blocks of MCL and immunohistochemistry of PD-L1, Ameli et al. showed that only 3.8% of MCL are positive for PD-L1 expression [103]. This limited expression of PD-L1 also contributes toward the partial response of PD-L1/PD-1-targeting agents in MCL. Data from 81 R/R lymphoma patients (including 4 MCL) treated with nivolumab (anti-PD-1-targeting antibody) as a single agent showed no significant clinical response in MCL. Three of four MCL patients experienced stable disease; surprisingly, these patients were negative for PD-L1 expression [112]. PD-L1-targeting drugs, as a single therapeutic agent, have inferior outcomes in MCL; therefore, clinical trials evaluate potential combination strategies in MCL. For instance, pembrolizumab (KEYTRUDA, anti-PD-1) plus ibrutinib in a phase-2/3 trial (NCT03153202) and nivolumab plus lenalidomide (NCT03015896) are under investigation for MCL and other subtypes of lymphoma.
CD27
The other immune checkpoint proteins include CD27, a co-stimulatory molecule that negatively regulates T cell activation by engaging its ligand CD70. Notably, CD70 was identified as a direct target of the Sox11 gene and is overexpressed in SOX11post MCL, not in Sox11neg MCL. Sox11post MCL is associated with an immune imbalance with increased effector Treg cells in the TME [52]. Varlilumab (CDX-1127) is a CD27-targeting monoclonal antibody that could reverse the T cell exhaustion status and is being under investigation in a phase-2 clinical trial in combination with nivolumab for aggressive B cell lymphomas, including MCL cases (NCT01460134) [113].
Receptor tyrosine kinase-like orphan receptor 1
ROR1 was first identified to be overexpressed in CLL and then found to be overexpressed in several other lymphoma subtypes, including MCL [114, 115]. Importantly, ROR1 is absent in most normal adult tissues but overexpressed in other malignancies, required for tumor cell survival and metastasis, making it a suitable candidate for a therapeutic target [116, 117]. Many therapeutic agents, including small molecule inhibitors, CAR T cell products, and monoclonal antibodies targeting ROR1, have been developed and tested for their efficacy in MCL. The study from Ghaderi et al. identified that ROR1 is overexpressed in MCL cell lines and primary MCL tumors. Notably, treatment of MCL cells with ROR1-targeting small molecule inhibitor KAN0441571C inhibited ROR1 phosphorylation, non-canonical WNT signaling, and induced MCL cell death in a dose-dependent manner. In addition combining KAN0441571C with ibrutinib or other agents (venetoclax, idelalisib, everolimus, or bendamustine) showed a synergetic impact on MCL cell apoptosis [118]. Cirmtuzumab (UC-961), a humanized monoclonal antibody designed to inhibit ROR1 activity showed antitumor activity (inhibited MCL cell proliferation) in a preclinical model [119]. To enhance its antitumor activity, cirmtuzumab was conjugated to monomethyl auristatin E (MMAE) via a cleavable linker leading to the creation of zilovertamab vedotin (VLS-101 or MK-2140). Additional data pertaining to zilovertamab vedotin has been presented in the antibody–drug conjugate section below.
Chromatin modifiers
Chromatin modifiers, including histone acetyl-transferase (HATs), histone deacetylase (HDACs), and DNA methyl-transferase (DNMTs), are deregulated in many B cell malignancies and displayed genome-wide DNA/histone modifications such as acetylation/de-acetylation, hypo/hyper-methylation, at the regulatory elements [120].
HDAC inhibitors
Elevated expression of HDAC6 has been reported in B cell lymphoma compared to normal B cells, which is directly correlated to disease progression. Fimepinostat (CUDC-907) is a first-in-class oral small molecule inhibitor of HDAC and PI3K enzymes tested in MCL. A preclinical study using CUDC-907 as a targeting agent in primary MCL tumors and cell lines, including ibrutinib-resistant MCL-PDX model, demonstrated tumor regression and apoptosis of MCL cell lines via increasing histone acetylation in MCL [121]. Though this dual inhibitor has shown impressive anti-proliferative activity in ibrutinib-resistant MCL, for unknown reasons, this compound has not been tested in clinical trials for MCL (NCT01742988). Vorinostat (SAHA) is a 2nd generation HDAC inhibitor tested as a single agent or combined with many other MCL treatment regimens. In a preclinical study of MCL, vorinostat as a single agent inhibited R/R/ MCL cell growth and induced apoptosis. Importantly this inhibitor showed synergistic anti-proliferative activity when combined with CDK4/6 dual inhibitor palbociclib [122]. A phase 1 trial of vorinostat and bortezomib had a modest activity for previously untreated (n = 22) and prior bortezomib treatment (n = 4) MCL with a median PFS of 7.6 months and 1.8 months, respectively [123], suggesting that vorinostat should be studied with a different combinatorial agent for treatment of R/R MCL. Multiple clinical trials tested vorinostat in combination with other agents for MCL including, rituximab, ifosfamide, carboplatin, and etoposide (NCT00601718) and cladribine and rituximab (NCT00764517) [61]. Abexinostat (formerly PCI-24781) is a new broad-spectrum hydroxamate-based HDAC inhibitor that affects chromatin organization and gene transcription in MCL and induces apoptosis in lymphoma cell lines in a caspase and reactive oxygen species-dependent mechanisms [124, 125]. Based on preclinical findings, abexinostat was evaluated in a phase 1/2 clinical trial (NCT03939182, n = 11) wherein it demonstrated an ORR of 27.3% with a median PFS of 3.9 months [62]. Given this, abexinostat is currently being studied with ibrutinib in MCL. Romidepsin and belinostat (PXD101) are also pan-HDAC inhibitors showing preclinical activity in MCL cell lines. Further combination of bortezomib with romidepsin and belinostat induced potent mitochondrial membrane depolarization and apoptosis in xenograft mice model; thus, this combination could offer a new sensible approach for treating MCL [126].
PRMT5 inhibitors
Protein arginine methyl-transferase (PRMT5) is a type II arginine methyl-transferase that catalyzes the dimethylation of arginine residues on H3R8 and H4R3 of histone tails or other proteins. PRMT5 regulates multiple biological functions, including RNA processing, signal transduction, DNA damage response, and gene expression [127]. PRMT5 is overexpressed and dysregulated in MCL, including BTKi-resistant cells. Using ibrutinib-resistant MCL-PDX model, treatment with PRMT5 inhibitor PRT382 significantly reduced tumor burden and improved median survival in mouse models [128]. DNA damage repair genes such as ATM and TP53 are recurrently observed to be mutated in MCL, including CAR T cell therapies or those with intrinsic or acquired resistance to ibrutinib. Notably, ATM mutation in MCL led to the complete inactivation of ATM, which abrogated TP53 activation in response to DNA damage, allowing cells with unrepaired DNA to escape from TP53 surveillance [129, 130]. ATM mutated MCL cells are sensitive to PRMT5 inhibition, which was demonstrated using the ibrutinib-resistant MCL-PDX model. A recent report from Che et al. identified upregulated expression of PRMT5 in ibrutinib-resistant MCL tumors, which was associated with poor clinical outcomes. As PRMT5 is involved in epigenetic, post-transcriptional, and post-translational regulation of DNA damage response genes, PRMT5 inhibition by GSK3326595 induced downregulation of DNA damage genes (DNAPK, RAD51, NHEJ1) and induced oxidative stress markers leading to accumulation of DNA damage. Notably, in ATM-deficient MCL lines, PRMT5 inhibition by GSK3326595 resulted in more accumulated unrepaired DNA damage and attenuated MCL-PDX tumor growth. In addition, co-targeting MCL with PRMT5 inhibitor and ATR or CDK4 inhibitor had a synergistic response in both in vivo and in vitro MCL models [43]. A phase 1 clinical trial (NCT03886831) with PRT343 (a potent, selective, oral PRMT5 inhibitor) that included MCL and other malignancies with no available treatment options has completed accrual and awaiting read out. Many PRMT5 inhibitors, including GSK3326595, JNJ-64619178, and PRT811, have been developed, but most are being tested in other lymphoma subtypes or solid tumor malignancies [131]. Though PRMT5 inhibition emerged as an attractive therapeutic target in MCL, a recent study has also identified the development of primary or acquired resistance to PRMT5 inhibitors in MCL. PRMT5 inhibitor-resistant MCLs exhibited compensatory activation of multiple signaling pathways such as insulin receptors, PI3K, MAPK, and mTOR signaling in tumors, further using PRMT5 inhibitor (PRT-382) in combination with PI3K/mTORC1 and 2 (Omipalisib), or mTORC1 (Temsirolimus) or EIF1A (Silvestrol) could reverse this PRMT5 resistance in MCL [132].
DNMT inhibitors
DNA methylation is an essential epigenetic mechanism in normal and cancerous cells that directly controls DNA regulatory elements and gene expression. Notably, variation in the magnitude of DNA methylation could be used as an independent prognostic factor for MCL prognosis, which probably could read the expression of essential tumor suppressors or oncogenes [133]. A study from Xin et al. identified upregulated expression of DNMT1 in primary MCL tumors, which was co-associated with the activation of the Wnt/β-catenin pathway. Treatment of MCL cells with arsenic trioxide, a DNMT inhibitor, downregulated Wnt/β-catenin target genes and DNMT1 expression [134]. Other DNMT inhibitors, including azacitidine and decitabine, have been FDA-approved for treating acute myeloid leukemia and myelodysplastic syndrome, but these agents have shown limited efficacy and toxicity in MCL and other B cell lymphomas [135]. Furthermore, DNMT3A was identified as a mediator of OXPHOS pathway activation via mitochondrial biogenesis and thus associated with ibrutinib resistance in MCL. Thus, targeting DNMT3A with a low dose of decitabine, which degrades DNMT3A protein, synergized with IM156, an inhibitor of the mitochondrial complex, could overcome ibrutinib resistance in MCL.
Inhibitors of SUMOylation
SUMOylation is a post-translational modification of target proteins which is an essential step for the regulation of genomic integrity, gene expression, and intracellular signaling, which is deregulated in tumor cells. Selective inhibitor of SUMO-activating enzyme "Subasumstat" (TAK-981) identified to inhibit the growth of MCL cells when grown in stromal conditions and induced tumor regression in MCL-PDX model via inhibiting OXPHOS pathway and thus overcoming BTKi resistance mechanism. Furthermore, overexpression of the EGR1 gene was also identified to be upregulated in ibrutinib-resistant MCL cells associated with metabolic reprogramming and OXPHOS pathway deregulation in MCL, thus providing another strategy to target BTKi-resistant cells.
EZH2 inhibitors
Like DNMTs, histone methyl-transferases such as EZH2 (enhancer of zeste homolog-2 inhibitors) have emerged as an attractive therapeutic target in B cell and other solid tumor malignancies [136]. Baquero et al. assessed the EZH2 expression in 166 primary MCL, where 57 cases (38%) were positive for EZH2 expression and were associated with aggressive histologic variants (65% vs. 29%), high Ki-67 proliferation rate (72% vs. 19%), and p53 overexpression (43% vs. 2%) compared to EZH2 negative tumors. Surprisingly, EZH2 expression was not correlated to the expression of other PRC2 components (EED and SUZ12) and H3K27me3, but this was associated with inferior survival outcomes in MCL [137]. Mutations in the SET domain of the EZH2 gene that increased its tri-methylation activity are prevalent in other B cell lymphoma but have not been reported in the case of MCL, suggesting that high EZH2 expression in MCL is sufficient for augmenting oncogenic signaling [40, 138]. Multiple EZH2-targeting inhibitors (GSK343 or GSK126, or OR-S1) are being tested in MCL those have shown significant anti-proliferative activity in in vitro and in vivo MCL-PDX models [139, 140]. An open-labeled multicentric arm phase-1 study identifying EZH2 inhibitor (XNW5004) efficacy in R/R B cell lymphoma, including MCL. Tazemetostat (EZM6438) is a potent orally bioavailable EZH2 inhibitor that initially received FDA approval for treating epithelioid sarcoma and R/R FL. Tazemetostat is now in phase-1 clinical trial for MCL (NCT03010982, NCT03028103). Another study identified Fibroblast Growth Factor Receptor-1 (FGFR1) as a significant candidate upregulated in relapsed MCL patients and cell lines when cultured under the influence of bone marrow stromal cells [141]. Moreover, the loss of FGFR1 abrogated EZH2 expression, improved survival in vivo [141, 142], and provided an alternative therapeutic strategy for targeting R/R MCL. EZH2 inhibitor tazemetostat in combination with zanubrutinib or ibrutinib in an ibrutinib/zanubrutinib-resistant MCL model showed synergistic activity in the MCL xenograft model [143].
CDK4/6 inhibitors
Aberrant expression of cyclin D1 caused by a t(11;14)(q13;q32) chromosomal translocation is the hallmark of MCL. Cyclin D1 assembles CDK4/6 to phosphorylate retinoblastoma protein, releasing the E2F transcription factor to initiate oncogenic gene expression. As expected, the expression of cyclin D1 is significantly high in MCL cases compared to normal peripheral B cells. Expression of CDK4 but not CDK6 was elevated in MCL cells compared to peripheral B cells [144]. Three CDK4/6 inhibitors, palbociclib, abemaciclib, and ribociclib, received FDA approval for treatment of solid tumors [144]. Given the promising activity of CDK4/6 inhibitors in solid tumors and the high expression of cyclin D1/CDK4 in MCL, the efficacies of these agents were screened in clinical trials. In a phase-1 clinical trial (NCT00420056) by Leonard et al., CDK4/6 inhibitor palbociclib (PD0332991) was given to MCL patients (n = 17; 71% were at high/intermediate risk according to MCL International Prognostic Index score) that induced early G1 cells arrest and tumor regression in some patients. Five MCL patients achieved PFS time of > 1 years (18% ORR) with limited toxicities [63]. Given the modest clinical outcome achieved with palbociclib as a monotherapy, subsequent clinical trials were carried out of this agent in combination with other MCL-targeting agents. A phase-1 trial of palbociclib plus bortezomib was conducted (NCT01111188), including 19 MCL patients where an ORR of 24% (6% CR) with associated toxicities, including grade 3 neutropenia (63%) and thrombocytopenia (53%) [64]. A phase-1 trial was conducted where palbociclib was combined with ibrutinib (PALIBR) (NCT02159755). 27 MCL patients were treated with this combination; those appeared with improved outcomes (ORR of 67%, CDR of 37%, and 2-year PFS of 59%). The combination had an acceptable safety profile, including neutropenia (41%) and thrombocytopenia (30%), compared to previous palbociclib trials [65]. Other CDK inhibitors are also in clinical trials for MCL cases, including abemaciclib (22 R/R MCL, ORR of 23%), ribociclib (7 MCL, ORR 0%), AT7519M (12 MCL, ORR of 27%), and Flavopiridol (30 MCL, ORR of 11%) [144].
Other therapeutic agents in MCL
Chemokine receptors and adhesion molecules like integrin are required for both regular and malignant B cells for trafficking and homing to supportive tissue microenvironments, including secondary lymph nodes. Stromal cells constitutively express chemokines, such as CXCL12, CXCL13, and many more, guiding B cell homing and positioning within the lymph node compartment [145]. However, very few studies have disseminated the expression and function of adhesion molecules in MCL. Kurtova et al. examined chemokine receptors, adhesion molecule expression, and their functions in MCL cells [146]. MCL cells expressing high levels of CXCR4 and CXCR5 chemokine and VLA-4 adhesion molecules are required for adhesion and spontaneous migration of MCL cells beneath the MSCs layer and are associated with drug resistance [146]. Further study by Chen et al. identified that silencing of CXCR4 expression in MCL significantly reduced proliferation and adhesion to bone marrow stromal cells. Moreover, co-culturing of MCL cells with either stromal cells or condition media from stromal cells prevented apoptosis of MCL after ibrutinib treatment, suggesting that interaction with bone marrow stromal cells some have protective effects on MCL from therapeutic agents [147]. As chemokine and adhesion molecules contribute to BTKi or other drug resistance in MCL, therapies targeting these molecules have been developed, and their antitumor efficacies in MCL have been tested. A recent report identified that CXCR4 expression is an independent poor prognostic factor for MCL and can be a promising target for imaging and radioligand therapy [148]. Plerixafor (CXCR4 antagonist) and natalizumab (anti-VLA-4 antibody) are the agents that could inhibit the interaction of MCL to stromal cells keeping these MCL cells in a mobilized state; these mobilized MCL cells are more susceptible to standard therapies [149].
Identifying alternative oncogenic pathways in BTKi-resistant MCL has been limited to protein-coding genes. However, one study identified subsets of miRNAs that regulate the MAPK-ERK cascade, including miRs-221, 146a, 182, 342, and the let-7 family members were downregulated in ibrutinib-resistant MCL cells, thereby causing upregulation of MAPK-ERK signaling which can be targetable by MEK inhibitor (cobimetinib) [150].
Combination approaches with small molecule inhibitors to overcome BTKi resistance in MCL
While several agents targeting different MCL signaling as single agents have shown some activity, they are less effective in patients with BTKi-resistant MCL. Hence, combination approaches have now been studied to overcome resistance to BTKis in patients with R/R MCL. This section discusses various combination approaches for treating R/R MCL containing BTKis, or other agents tested in clinical trials.
Bortezomib has also shown anticancer activity in MCL preclinical model when combined with zanubrutinib [151]. A phase 1 clinical trial of bortezomib and ibrutinib has completed accrual in R/R MCL (NCT02356458). A phase 2 trial of bortezomib in combination with lenalidomide (CALGB 50501) only showed a modest ORR of 40% [152]. Combining bortezomib with rituximab, lenalidomide, and dexamethasone (DR2IVE) was well tolerated in ibrutinib-resistant MCL patients with an ORR of 100% and 3 out of 5 patients were still alive at the last follow-up [153]. However, this study was limited to a small number of MCL patients.
Ibrutinib in combination with venetoclax has been studied in three clinical trials in MCL. The AIM trial (NCT02471391) included 23 R/R MCL patients, with 50% exhibiting altered TP53 genes. Despite this, combining ibrutinib with venetoclax in this cohort showed an ORR of 71% [154], suggesting that the combination of ibrutinib and venetoclax-based treatment approach was highly active. In SYMPATICO (phase 3 trial enrolled 352 MCL patients, NCT03112174), the combination of venetoclax and ibrutinib demonstrated an ORR of 81% and CR of 31% at a median follow-up of 31 months [68]. In another study by Portell et al. ibrutinib 420 mg daily in combination with venetoclax at 200 mg daily (which is lower than the doses used in the AIM or SYMPATICO trials) provided comparable benefits with ORR of 83% and a CR rate of 42% (NCT02419560) [69]. The synergistic results of combining these two agents in clinical trials could be due to the mutual targeting of the common pathway by the respective agents. Regarding this, Li et al. identified that BTK expression was positively correlated with BCL2 expression, as targeting BTK by short hairpin RNA led to downregulating BCL2 and other anti-apoptotic gene expressions [155]. In addition, combining venetoclax and ibrutinib showed enhanced dephosphorylation of AKT or BTK and more PARP cleavage in MCL [156]. A BTKi, ibrutinib, or its 2nd generation agents do not target mutant BTK. Therefore, BTK degraders have been synthesized, showing profound preclinical activity [21]. BTK degrader Nx-2127 also had synergistic anti-neoplastic activity when combined with BCL2 inhibitors at low doses.
CG-806 (luxeptinib) is a non-covalent kinase inhibitor targeting BCR-associated kinases LYN, SYK, and BTK, which are now under investigation in clinical trials [157]. CG-806 inhibited both wild-type and mutant BTKC481S. The study from Thieme et al. demonstrated the promising activity of CG-806 in the MCL-PDX model via disrupting BCR signaling networks [158]; thus, this agent can be a potential alternative molecule to be tested in MCL cases.
Bromodomain and extra-terminal (BET) family of proteins recognize the acetylated lysine on histone and regulate transcription of many oncogenes, including genes involved in the BCR pathway, BLNK, PAX5, Myc, and IKAROS family in MCL [159]. Notably, treatment with BRD4 inhibitor I-BET151 as a single agent inhibited MCL cell line proliferation in a dose-dependent manner [159]. Furthermore, bromodomain antagonist JQ1 has been tested in MCL cell lines, inhibiting the MYC gene and expression of NFkB target genes. In addition, JQ1 treatment showed a synergistic association in inducing apoptosis of the ibrutinib-resistant MCL cells when combined with another agent such as ibrutinib or panobinostat (pan-histone deacetylase inhibitor) or palbociclib (CDK4/6 inhibitor) or ABT-199 [160].
PI3Kδ Inhibitor zandelisib, combined with the BTKi zanubrutinib in patients with R/R MCL (n = 17), showed improved efficacy [161]. An ORR was 76% with a CR of 35%, the preliminary median PFS was 10.4 months, and very few patients discontinued the treatment due to adverse events reported [161].
Combination approaches with monoclonal antibodies for the treatment of MCL
Rituximab was studied in combination with ibrutinib and was shown to be safe and tolerable in patients with R/R MCL. In an open-label, phase 2 trial in R/R MCL, rituximab in combination with ibrutinib resulted in higher responses with ORR and CR of 88% and 44%, respectively [70]. Rituximab was also studied in combination with venetoclax, and lenalidomide in the Nordic Lymphoma Group NLG-MCL7 (VALERIA) trial. This study included BTKi-resistant MCL and demonstrated an ORR of 40% (n = 6) with 4 patients achieving CR [162]. Rituximab combination with anti-CD74-targeting antibody milatuzumab has been tested successfully in the preclinical MCL models [163].
Obinutuzumab (GA101) is a CD20-targeting humanized antibody that has demonstrated efficacy in the MCL preclinical models [164]. This antibody has non-fucosylated sugars on the Fc portion and was designed to overcome mechanisms of resistance to rituximab. Obinutuzumab as monotherapy in R/R MCL (n = 15) showed ORR of 27% in the GAUGUIN phase-2 trial (NCT00517530) [71]. Subsequently obinutuzumab was studied in combination with ibrutinib and venetoclax in relapsed and untreated MCL patients (n = 48) (OAsIs; a phase-1/2 trial) (NCT02558816). This combination was well tolerated and showed CR of 67% in relapsed and 86.6% in untreated MCL patients [72]. This combination can be considered a possible salvage therapy for ibrutinib-resistant MCL patients [165]. However, despite high initial response rates in the OAsIs trial nearly 1/3 of patients relapsed [166]. In order to identify factors leading to resistance in the OAsIs trial, single-cell RNA sequencing and targeted DNA sequencing of patients' tumor samples were performed (n = 12 at baseline and n = 5 at relapse) that revealed a gain of function mutation in the CARD11 gene [166]. Of note, while CARD11mut tumor cells were minute (0.0005%) at the onset of treatment, all the cells carried a heterozygous mutation at the time of relapse. By integrating DNA sequencing data with single-cell RNA sequencing data, the authors identified the CARD11 associated gain of function mutation named "OAsIs-R" signature that was also predictive for OS/PFS in MCL patients treated with conventional chemotherapy. Furthermore, BCL2A1 overexpression was identified as the top gene of the OAsIs-R signature, which can be targetable by MALT1 protease inhibition along with BCL2 inhibition in a synergistic fashion [166].
A third-generation CD20-targeting monoclonal antibody ublituximab (TG-1101) is a glycoengineered monoclonal antibody that has improved antibody-dependent cell-mediated cytotoxicity than rituximab. Ublituximab has been studied in combination with ibrutinib in R/R MCL (n = 15) and demonstrated an ORR of 87% with 33% CR [167].
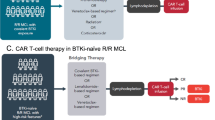
CAR T cell therapy in R/R MCL
CAR T cell therapy is an exciting new avenue for treating solid and liquid tumors, wherein host T cells are genetically engineered to express artificial receptors specific to target tumor-specific cell surface antigens.
CD19-directed CAR T
CD19-directed CAR T cells have shown impressive outcomes in B cell lymphoma treatment when given either as monotherapy or in combination with other treatment regimens. Brexucabtagene autoleucel (Tecartus, KTE-X19) received FDA approval based on ZUMA-2 phase-2 trial for R/R MCL after chemotherapy and BTKi [168]. This study enrolled 74 R/R MCL patients where 62% of patients had primary BTKi resistance, 26% had a relapse after an initial response to BTKi therapy, 7% experienced relapse after stopping BTKi therapy, and 4% were intolerant of BTKi (had adverse events). The study had an ORR of 85%, with a CR of 59%. Adverse events include cytokine release syndrome (CRS) in 15% of MCL patients, cytopenias in 94%, and infections in 32% of cases [169]. A long-term follow-up (3 years) of the pivotal ZUMA-2 study of KTE-X19 has recently been reported. With a median follow-up of 35.6 months, the ORR for 68 treated patients was 91%, and CR was 68% (NCT02601313) [170]. These data, representing the most extended follow-up of CAR T cell therapy in patients with MCL, suggest that KTE-X19 induced durable long-term responses with safety profiles in patients with R/R MCL. Data from the Descar-T French registry (LYSA Group) has put forward the first results of KTE-X19 in R/R MCL who failed after at least one line of chemo-immunotherapy or BTKi treatment. 47 MCL patients were infused with the KTE-X19 CAR T product. The ORR was 88% with a CR of 61.9%, CRS was noted in 78.7% of patients, and neurotoxicity was observed in 48.9% [171]. Another real-world experience from the United States lymphoma CAR T consortium by Wang et al. presented data from 93 R/R MCL patients infused with the KTE-X19 CAR T product, where an ORR of 86% with 64% CR was achieved. CRS in 88% and neurotoxicity in 58% of patients were reported. [172] Real-world outcomes data of Brexucabtagene autoleucel [173], where 82 MCL patients R/R to BTKi were enrolled, showed a median follow-up of 9.1 months, ORR was 89.6% (83.3% CR, 6.3% PR).
Lisocabtagene maraleucel (JCAR017), modified to have 4-1BB as a co-stimulatory domain, has also been studied in 32 R/R MCL in the TRANSCEND NHL 001 phase-2 trial, and the ORR was 84%, including 59% CR [174]. CRS was noted in 50%, and neurologic events were present in 28% [174]. Ying et al. showed the efficacy and safety of Relmacabtagene autoleucel, a CD19-directed CAR T product in 11 R/R MCL in China [175]. Based on three months follow-up analysis, an ORR of 81% and CR of 54.5% with a low grade ≥ 3 CRS incidence were noted. A recent study on AUTO1 CD19-targeting CAR T cells (designed to reduce toxicity and improves engraftment) showed a 100% ORR rate in MCL (n = 3). A large cohort and multicentric study is required to evaluate its efficacy and toxicity further in R/R MCL patients [176]. A phase-1 single centric ENABLE clinical trial (NCT04049513) has been initiated using third-generation CD19 CAR (WZTL-002) incorporating the intracellular signaling domains of CD28 and Toll-like receptor 2 (TLR2) to identify a safety dose of R/R B cell NHL patients including MCL [177]. Other phase-1 study evaluating BAFFR-targeting CAR T cells in various B-NHL including MCL (NCT05370430).
ROR1-targeting CAR T
Multiple therapeutic modalities have been developed to target ROR1 in hematological and solid tumor malignancies. PRGN-3007 UltraCAR-T is a first-in-class investigational multigenic, autologous CAR T cell therapy developed on Precigen's UltraCAR-T platform, which has been engineered to express a ROR1-targeting CAR receptor, a membrane-bound interleukin-15 (mbIL15) for enhanced in vivo expansion and persistence, a kill switch to conditionally eliminate CAR T cells for improved safety profile and intrinsic blockade of PD-1 gene expression. PRGN-3007 UltraCAR-T started its Phase-1 clinical trial for ROR1-positive hematological malignancies and solid tumors, including triple-negative breast cancer (NCT05694364). Preclinical studies using PRGN-3007 UltraCAR-T have so far shown a significant reduction in PD-1 expression, increased ROR-specific tumor cell cytotoxicity, and inflammatory cytokine production upon co-culture with ROR1 + PD-L1 + tumors, effectively reducing tumor burden in xenograft models with long-term persistence of PRGN-3007 UltraCAR-T in tumor-bearing mice [178].
CD37-targeting CAR T
CD37 is a tetraspanin protein expressed in various B cell lymphomas, including MCL, and has been found to mediate tumor survival signaling [179]. GEN3009 is a CD37-targeting biparatopic antibody in a phase-1 clinical trial for multiple B cell lymphomas, including R/R MCL (NCT04358458). Besides therapeutic antibodies, CD37-targeting CAR T cells have also been designed into a third-generation lentiviral plasmid backbone with a CD8 hinge and 4-1BB co-stimulatory domain. CD37 CAR T cells have been preclinically tested for efficacy in B cell lymphomas which exhibited robust effector functions, Th1-type cytokines expression, and tumor clearance in the MCL-PDX model. Additionally, a bispecific CAR targeting CD19 and CD37 has also been developed to respond to either a single or both targets, and there was no discernible difference in cytotoxicity to CD19 or CD37 found [179]. Due to promising outcomes of CD37-targeting CAR T cells in preclinical in vitro and in vivo studies, a clinical trial exploring its efficacy in MCL cases will be the next logical step. Various other modified universal CAR products, such as WZTL-002 and SYNCAR-001, are under phase-1 clinical evaluations and are listed in Table 3.
Combination strategies with or after CAR T cell therapy
Despite the impressive outcomes of CAR T cell therapy, limitations such as toxicities (CRS and neurological), unavailability of robust CAR T cell expansion, failed engraftments, and resistance to CAR T cells have come up as potential hindrances to its growth [182]. These limitations can be overcome to a certain extent by prolonging BTKi treatment (≥ 5 cycles) before collection of autologous T cells or combining BTKi along with CAR T cell therapy which could reduce the immunosuppression markers (PD-1) on CAR T cells, prolonging the duration of remission [183, 184]. A preclinical study using MCL cell lines and mice xenograft model showed that combining ibrutinib with CD19 CAR T cell provided long-term remission of 80–100%, compared to 0–20% for the CAR T cell therapy-only treatment group [185]. The efficacy of ibrutinib and tisagenlecleucel (anti-CD19 CAR T product) was explored in confirmed BTKi-resistant MCL (n = 21) [186]. Twenty patients (44% of patients had a mutation in the TP53 gene) were infused with the CAR T product, and 75% of patients experienced CRS; 11/15 (73%) grade 1–2 with ORR at 13 months was 90% with CR of 80% [186]. To understand the oncogenic axis of dual-resistance to BTKi and CAR T therapy axis, Jiang et al. performed single-cell sequencing using 39 longitudinal samples from 15 MCL patients sequentially treated with BTKi and CAR T cell therapy [187]. Further bioinformatics and functional analysis identified that the HSP90-MYC-CDK9 axis was associated with dual-resistance development. Treatment with an HSP90 inhibitor (PU-H71 or 17-AAG) and a combination of CDK9 inhibitors (AZD4573) induced impressive anti-MCL activity both in vitro and in vivo the MCL-PDX model [187]. Using a xenograft model, the antitumor activity of LP-284 (a novel DNA-damaging agent) was evaluated in MCL, including those resistant to BTKi, bortezomib, or venetoclax [188]. LP-284 demonstrated antitumor activity with increased DNA damage in MCL cells [188]. A phase-2 clinical trial (NCT04484012) is underway to combine CD19 CAR with acalabrutinib in R/R MCLs.
Besides BTKis, BCL2-targeting agents combined with CAR T cell therapy have improved outcomes in preliminary and preclinical studies. However, these combinations have not yet been tested in clinical trials and are an avenue for further investigation for MCL treatment [189].
Identifying other B cell surface-specific markers or markers expressed by malignant B cell provided further development of new CAR T cell products that could reverse the previous CD19-directed CAR T cell therapy resistance (due to CD19 target loss) and other limitations [190]. Clinical trials are investigating the efficacy of CD20-targeting CAR T cells (NCT03277729).
Other CAR constructs include bispecific anti-CD19/CD20 CAR T cells (LV20.19) showed an ORR of 100% at 92 days with CR of 92% in phase-1 clinical trial of B cell lymphoma patients, including seven heavily pretreated MCL cases (NCT03019055) [181]. This data from NCT04186520, enrolling 10 MCL patients received LV20.19 CAR T cells, day 28 ORR was 100% (CR = 60% and PR = 40%) without any relapse at a median follow-up of 18 months and only 10% of patients had grade 1–2 CRS (no grade 3 + events was observed) [191]. Besides CAR T cell, modified CAR NK (NKX019), targeting CD19 is under clinical investigation for MCL (NCT05020678). Other NK cells-based CAR products including PCAR-119 or CAR.CD19-CD28-zeta-2A-iCasp9-IL15-transduced cord blood NK cells were enrolled in clinical trials, but recent data showed these therapeutic agents have been discontinued for unknown reasons.
T cell engaging bispecific/trispecific antibodies
T cell-engaging bispecific antibodies are an emerging cancer immunotherapy class that has shown promise in treating several types of cancer, including MCL. These antibodies comprise two binding sites for different antigens: one recognizes the tumor-specific antigen, and the other binds to an epitope on T cells, typically CD3 receptor, which is required to bring T cells close to tumor cells and activate the T cell cytotoxic activity. The advantage of bispecific antibodies over monoclonal antibodies as therapeutic entities includes the direct cell-mediated killing of tumor cells via T cells, high affinity, and reduced treatment cost. There are many bispecific antibodies targeting B cell malignancies that are currently FDA-approved. Bispecific antibodies under clinical investigations in MCL are listed in Table 4. The familiar markers in all B cell malignancies are CD19 and CD20, and many bispecific antibodies targeting these antigens have been generated, and some have shown impressive activity against MCL in clinical trials.
CD19/CD3-targeting bispecific antibody
Blinatumomab (Blincyto, AMG103, MT103) is a CD19-targeting bispecific antibody widely investigated in B cell lymphoma, including MCL. In a phase-1 clinical trial with 24 R/R MCL patients, blinatumomab, when given as a single agent, resulted in an ORR of 71% [192] in R/R MCL, which was higher than the ORR response achieved in DLBCL (55%). In the long-term follow-up studies including 13 MCL patients, the median OS was 4.6 years, PFS was 6.7 months, and the treatment-free survival (TFS) was 7.6 months [195]. Another bispecific antibody NVG-111 is a T cell engager with CD3 binding arm for T cells while simultaneously targeting ROR1-expressing malignant cells. NVG-111 is currently under evaluation in a phase-1 trial for B cell malignancies, including R/R MCL (NCT04763083).
CD20/CD3-targeting bispecific antibody
CD20-targeting bispecific antibody mosunetuzumab (Lunsumio) has been evaluated for efficacy in a first-in-human phase-1/2 trial (NCT02500407) that includes 13 MCL patients. ORR of 30.8% (4/13), including 23% CR (3/13), was achieved [196]. Glofitamab (RG6026) is a 2:1 configuration bispecific antibody with a monovalent binding site for CD3 of T cells and bivalent binding to CD20 on B cells. A recent phase-1/2 clinical trial that included 37 R/R MCLs, 64.9% of whom had received prior BTKi treatment and 1000 mg or 2000 mg of glofitamab with Obinutuzumab showed ORR of 83.8% and 73% CR was achieved [197]. Glofitamab was tolerated well, with neurologic adverse events of grades 1–2 occurring in 19 patients (51.4%), and the most frequently reported adverse events were CRS in 75.7% of MCL patients [197]. There is another ongoing phase-2 clinical trial (NCT04703686) where glofitamab is evaluated for R/R B cell lymphoma, including MCL, for patients who have progressed on CAR T cell therapy. Epcoritamab (GEN3013) is a CD20-targeting and T cell engaging bispecific antibody which has been tested in multiple B cell lymphoma subtypes, including 4 MCL patients with responses observed in 50% (2/4) MCL patients and 25% was CR (1/4) (NCT03625037) [193]. Odronextamab (REGN1979) is a CD20/CD3-targeting bispecific antibody modified from an IgG4-base to reduce Fc binding. This is still under investigation in a clinical trial for MCL. Preliminary results for other heavily pretreated B cell lymphoma patients showed a durable response with an ORR of 57.9%. Other bispecific-based intervention recruiting in clinical trials for multiple B-NHL includes IGM-2323, engineered to contain ten high-affinity binding domains for CD20 and one binding domain for CD3 (NCT04082936). Plamotamab (XmAb13676) is another IgG1 bispecific anti-CD20/CD3 antibody in clinical trials under investigation for MCL (NCT02924402).
CD20/CD37-targeting bispecific antibody
PSB202 is a novel anti-CD20/CD37-targeting antibody engineered by combining an Fc-enhanced humanized type II anti-CD20 IgG1 (PSB102) and a humanized anti-CD37 IgG1 (PSB107). PSB202 is in a phase-1 clinical trial for multiple B cell malignancies, including MCL cases (NCT05003141).
Trispecific T cell activating antibodies
Besides bispecific antibodies, there have several trispecific T cell activating (TriTAC) antibodies been developed, such as HPN328 (DLL3 targeting), NM21-1480 (anti-PDL-1/anti-4-1BB/anti-HSA), HPN217 (BCMA targeting), but till date as per study suggest these TriTAC have been investigated in multiple solid tumors and multiple myeloma.
Antibody–drug conjugates
Antibody–drug conjugates (ADCs) comprise a revolutionary cancer treatment strategy designed to target tumor cells more successfully and precisely when used alongside systemic cytotoxic chemotherapy. Monoclonal antibodies tailored for a tumor-associated antigen are combined with highly effective anticancer medicines (payloads or warheads) in their structure by a chemical linker. These antibodies can efficiently release cytotoxic payloads to tumor cells while sparing normal cells, lowering their unrequired toxicity.
Zilovertamab vedotin
Zilovertamab vedotin (VLS-101 or MK-2140), a ROR1 ADC, was designed to improve the efficacy of ROR1-targeting antibody cirmtuzumab by linking it to MMAE via a cleavable linker. Zilovertamab vedotin had shown significant antitumor activity in MCL cell lines and in ex vivo primary patient samples. The ROR1-drug conjugate was also tested in previously treated MCL in a phase 1 trial (NCT03833180) that enrolled 15 MCL case with an ORR of 47% (4 PR and 3 CR) [198]. Zilovertamab vedotin was studied in combination with ibrutinib in a phase 1/2 clinical trial which enrolled 27 R/R and treatment naïve MCL patients. Zilovertamab was well tolerated and demonstrated an ORR of 85.2% (40.7% CR, 44.4% PR) with a median DOR of 34.1 months [199]. Zilovertamab vedotin in combination with venetoclax, induced almost total tumor regression in xenograft studies, showing the high combined cytotoxicity of these two drugs [200]. ROR1 is also significantly elevated in CD19-targeting CAR T relapsed MCL tumors. Importantly, in the PDX model, VLS-101 treatment significantly induced regression of MCL tumors resistant to ibrutinib, venetoclax, or CAR T cell therapy, suggesting that targeting ROR1 could be a feasible approach in the treatment of ROR1 positive MCL tumors, particularly those with failure to prior MCL therapies [201].
Polatuzumab vedotin
Polatuzumab vedotin is an ADC-targeting cell surface receptor CD79B expressed by all B cell lymphomas. Polatuzumab vedotin received FDA approval based on study GO29365 (NCT02257567), in patients with R/R DLBCL. Polatuzumab vedotin-based combinatorial approaches are currently in clinical trials. These include polatuzumab vedotin + venetoclax, rituximab, and hyaluronidase (NCT04659044), polatuzumab vedotin + bendamustine and rituximab (NCT04913103), autologous stem cell transplant followed by polatuzumab vedotin ( NCT04491370), and polatuzumab vedotin and mosunetuzumab, a phase 1/2 study in patients with atleast 2 prior lines of systemic therapy in R/R MCL and other B-NHL (NCT03671018).
Loncastuximab tesirine
Loncastuximab tesirine (ADCT-402) is an anti-CD19 monoclonal antibody that has been humanized and is attached to a toxin called pyrrolobenzodiazepine dimer. In the phase 1 with dose expansion study in R/R B-NHL, 183 patients were evaluable for assessment of the safety, clinical efficacy, drug kinetics, and immunogenicity of loncastuximab tesirine. Overall, 15 MCL patients were included with an ORR of 46.7% [202].
Other ADCs
AGS67E, another ADC-targeting CD37, is currently undergoing clinical testing. In a phase I study in patients with R/R B- and T cell NHL (n = 2 MCL), the ORR was 22% [203]. Inotuzumab ozogamicin is a CD22-directed humanized monoclonal antibody conjugated to the cytotoxin, and calicheamicin. It was tested in combination with R-GDP (rituximab, gemcitabine dexamethasone cisplatin) in 13 MCL patients with three patients achieving CR and five PR [204]. SGN-CD70A targeting CD70 has also been studied in patients with R/R CD70-positive NHL (n = 20). Modest antitumor activity was noted in the study (1 CR and 3 PRs) and the applicability of SGN-CD70A was limited by the frequency and severity of thrombocytopenia [205]. There are no ADCs that are currently FDA-approved for treatment in R/R MCL. While the early data appears promising with some of the ADCs, additional research is required to further understand their effectiveness in patients with R/R MCL. Table 5 lists the important ADCs that are currently in clinical trials for treatment of R/R MCL.
Conclusions and future direction
The advent of BTKi revolutionized the treatment landscape of MCL; however, the emergence of resistance and the poor outcomes for those progressing on BTKi remains a major concern. With a better understanding of the MCL biology, several small molecule inhibitors targeting the various BCR signaling pathways and ADCs have been developed (see Fig. 1) that have shown promising activity even in patients who are resistant or progressed on BTKi. In the last few years, CAR T cell therapies and bispecific antibodies have significantly changed the clinical trajectory with good outcomes even in high-risk MCL patients.

A summary of various MCL-targeting agents, including BTKi and other small molecular inhibitors, antibody–drug conjugates, chimeric antigen receptor T cells, bispecific antibodies, and other immune modulators
Although there are a plethora of therapies available for treating patients with BTKi-resistant MCL, sequencing of these agents remains a challenge. We envision a future where not all MCL patients receive the same therapies, in the same sequence, but rather where understanding tumor mutational profile, cellular pathways that drive proliferation, and the tumor immune microenvironment may inform treatment decisions paving the way for a personalized treatment approach.
Availability of data and materials
Not applicable.
Abbreviations
- MCL:
-
Mantle cell lymphoma
- CLL:
-
Chronic lymphocytic leukemia
- DLBCL:
-
Diffuse large B cell lymphoma
- FL:
-
Follicular lymphoma
- BTKi:
-
Bruton's tyrosine kinase inhibitors
- CAR:
-
Chimeric antigen receptor
- ADC:
-
Antibody–drug conjugates
- ORR:
-
Overall response rate
- PFS:
-
Progression-free survival
- CR:
-
Complete response
- PR:
-
Partial response
- DOR:
-
Duration of response
- CT:
-
Clinical trial
- NHL:
-
Non-Hodgkin lymphoma
- PDX:
-
Patient-derived xenograft
- PROTAC:
-
Proteolysis-targeting chimera
- TIME:
-
Tumor immune microenvironment
- TME:
-
Tumor microenvironment
References
Rimokh R, Berger F, Delsol G, Digonnet I, Rouault JP, Tigaud JD, et al. Detection of the chromosomal translocation t (11; 14) by polymerase chain reaction in mantle cell lymphomas. 1994.
Beà S, Amador V. Role of SOX11 and genetic events cooperating with cyclin D1 in mantle cell lymphoma. Curr Oncol Rep. 2017;19:1–10.
Maddocks K. Update on mantle cell lymphoma. Blood J Am Soc Hematol. 2018;132(16):1647–56.
Epperla N, Switchenko J, Bachanova V, Gerson JN, Barta SK, Gordon MJ, et al. Impact of diagnosis to treatment interval in patients with newly diagnosed mantle cell lymphoma. Blood Adv. 2023;7(11):2287–96.
Epperla N, Hamadani M, Fenske TS, Costa LJ. Incidence and survival trends in mantle cell lymphoma. Br J Haematol. 2017;181(5):703–6.
Hanel W, Epperla N. Emerging therapies in mantle cell lymphoma. J Hematol Oncol. 2020;13:1–18.
Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507–16.
Wang M, Rule S, Zinzani PL, Goy A, Casasnovas O, Smith SD, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet. 2018;391(10121):659–67.
Song Y, Zhou K, Zou D, Zhou J, Hu J, Yang H, et al. Treatment of patients with relapsed or refractory mantle-cell lymphoma with zanubrutinib, a selective inhibitor of Bruton’s tyrosine KinaseZanubrutinib for relapsed/refractory MCL. Clin Cancer Res. 2020;26(16):4216–24.
Mato AR, Shah NN, Jurczak W, Cheah CY, Pagel JM, Woyach JA, et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): a phase 1/2 study. Lancet. 2021;397(10277):892–901.
Martin P. Ibrutinib–a new standard treatment for relapsed mantle cell lymphoma? Lancet. 2016;387(10020):728–9.
Jain P, Kanagal-Shamanna R, Zhang S, Ahmed M, Ghorab A, Zhang L, et al. Long-term outcomes and mutation profiling of patients with mantle cell lymphoma (MCL) who discontinued ibrutinib. Br J Haematol. 2018;183(4):578–87.
Nakhoda S, Vistarop A, Wang YL. Resistance to Bruton tyrosine kinase inhibition in chronic lymphocytic leukaemia and non-Hodgkin lymphoma. Br J Haematol. 2023;200(2):137–49.
Epperla N, Hamadani M, Cashen AF, Ahn KW, Oak E, Kanate AS, et al. Predictive factors and outcomes for ibrutinib therapy in relapsed/refractory mantle cell lymphoma—a “real world” study. Hematol Oncol. 2017;35(4):528–35.
Zhao X, Lwin T, Silva A, Shah B, Tao J, Fang B, et al. Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nat Commun. 2017;8:14920.
Zain R, Vihinen M. Structure-function relationships of covalent and non-covalent BTK inhibitors. Front Immunol. 2021;12:694853.
Correction: Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 2019;9(11):1629.
Cheng S, Guo A, Lu P, Ma J, Coleman M, Wang YL. Functional characterization of BTK(C481S) mutation that confers ibrutinib resistance: exploration of alternative kinase inhibitors. Leukemia. 2015;29(4):895–900.
Eyre TA, Cheah CY, Wang ML. Therapeutic options for relapsed/refractory mantle cell lymphoma. Blood. 2022;139(5):666–77.
Smith CIE, Burger JA. Resistance mutations to BTK inhibitors originate from the NF-kappaB but not from the PI3K-RAS-MAPK arm of the B cell receptor signaling pathway. Front Immunol. 2021;12:689472.
George B, Chowdhury SM, Hart A, Sircar A, Singh SK, Nath UK, et al. Ibrutinib resistance mechanisms and treatment strategies for B-cell lymphomas. Cancers (Basel). 2020;12(5):1328.
Gomez EB, Ebata K, Randeria HS, Rosendahl MS, Cedervall EP, Morales TH, et al. Preclinical characterization of pirtobrutinib, a highly selective, noncovalent (reversible) BTK inhibitor. Blood. 2023;142(1):62–72.
Cohen JB, Shah NN, Alencar AJ, Gerson JN, Patel MR, Fakhri B, et al. MCL-133 pirtobrutinib, a highly selective, non-covalent (reversible) BTK inhibitor in previously treated mantle cell lymphoma: updated results from the phase 1/2 BRUIN study. Clin Lymphoma Myeloma Leuk. 2022;22(Suppl 2):S394–5.
Wang ML, Shah NN, Jurczak W, Zinzani PL, Eyre TA, Cheah CY, et al. Efficacy of pirtobrutinib in covalent BTK-inhibitor pre-treated relapsed/refractory mantle cell lymphoma: additional patients and extended follow-up from the phase 1/2 BRUIN study. Blood. 2022;140(Supplement 1):9368–72.
Ito R, Eyre TA, Shah NN, Gouill SL, Dreyling M, Vandenberghe E, et al. MCL-135 BRUIN MCL-321, a phase 3 open-label, randomized study of pirtobrutinib versus investigator choice of BTK Inhibitor in patients with previously treated, btk inhibitor naive mantle cell lymphoma (trial in progress). Clin Lymphoma Myeloma Leuk. 2022;22(Suppl 2):S395–6.
Byrd JC, Smith S, Wagner-Johnston N, Sharman J, Chen AI, Advani R, et al. First-in-human phase 1 study of the BTK inhibitor GDC-0853 in relapsed or refractory B-cell NHL and CLL. Oncotarget. 2018;9(16):13023–35.
Burkart M, Karmali R. Relapsed/refractory mantle cell lymphoma: beyond BTK inhibitors. J Pers Med. 2022;12(3):376.
Lewis KL, Cheah CY. Non-covalent BTK inhibitors-the new BTKids on the block for B-cell malignancies. J Pers Med. 2021;11(8):764.
Dobrovolsky D, Wang ES, Morrow S, Leahy C, Faust T, Nowak RP, et al. Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood. 2019;133(9):952–61.
Buhimschi AD, Armstrong HA, Toure M, Jaime-Figueroa S, Chen TL, Lehman AM, et al. Targeting the C481S ibrutinib-resistance mutation in Bruton’s tyrosine kinase using PROTAC-mediated degradation. Biochemistry. 2018;57(26):3564–75.
Jaime-Figueroa S, Buhimschi AD, Toure M, Hines J, Crews CM. Design, synthesis and biological evaluation of Proteolysis Targeting Chimeras (PROTACs) as a BTK degraders with improved pharmacokinetic properties. Bioorg Med Chem Lett. 2020;30(3):126877.
Sun Y, Ding N, Song Y, Yang Z, Liu W, Zhu J, et al. Degradation of Bruton’s tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia. 2019;33(8):2105–10.
Lim YS, Yoo SM, Patil V, Kim HW, Kim HH, Suh B, et al. Orally bioavailable BTK PROTAC active against wild-type and C481 mutant BTKs in human lymphoma CDX mouse models. Blood Adv. 2023;7(1):92–105.
Zhang L, Yao Y, Zhang S, Liu Y, Guo H, Ahmed M, et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci Transl Med. 2019;11:491.
Rahal R, Frick M, Romero R, Korn JM, Kridel R, Chan FC, et al. Pharmacological and genomic profiling identifies NF-kappaB-targeted treatment strategies for mantle cell lymphoma. Nat Med. 2014;20(1):87–92.
Lenz G, Balasubramanian S, Goldberg J, Rizo A, Schaffer M, Phelps C, et al. Sequence variants in patients with primary and acquired resistance to ibrutinib in the phase 3 MCL3001 (RAY) trial. J Clin Oncol. 2016;34(15_suppl):7570.
Vidal-Crespo A, Rodriguez V, Matas-Cespedes A, Lee E, Rivas-Delgado A, Gine E, et al. The Bruton tyrosine kinase inhibitor CC-292 shows activity in mantle cell lymphoma and synergizes with lenalidomide and NIK inhibitors depending on nuclear factor-kappaB mutational status. Haematologica. 2017;102(11):e447–51.
Wu C, de Miranda NF, Chen L, Wasik AM, Mansouri L, Jurczak W, et al. Genetic heterogeneity in primary and relapsed mantle cell lymphomas: Impact of recurrent CARD11 mutations. Oncotarget. 2016;7(25):38180–90.
Agarwal R, Chan YC, Tam CS, Hunter T, Vassiliadis D, Teh CE, et al. Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat Med. 2019;25(1):119–29.
Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci USA. 2013;110(45):18250–5.
Mohanty A, Sandoval N, Das M, Pillai R, Chen L, Chen RW, et al. CCND1 mutations increase protein stability and promote ibrutinib resistance in mantle cell lymphoma. Oncotarget. 2016;7(45):73558–72.
Naeem A, Utro F, Wang Q, Cha J, Vihinen M, Martindale S, et al. Pirtobrutinib targets BTK C481S in ibrutinib-resistant CLL but second-site BTK mutations lead to resistance. Blood Adv. 2023;7(9):1929–43.
Che Y, Liu Y, Yao Y, Hill HA, Li Y, Cai Q, et al. Exploiting PRMT5 as a target for combination therapy in mantle cell lymphoma characterized by frequent ATM and TP53 mutations. Blood Cancer J. 2023;13(1):27.
Wang H, Zhang W, Yang J, Zhou K. The resistance mechanisms and treatment strategies of BTK inhibitors in B-cell lymphoma. Hematol Oncol. 2021;39(5):605–15.
Chen Z, Ayala P, Wang M, Fayad L, Katz RL, Romaguera J, et al. Prospective isolation of clonogenic mantle cell lymphoma-initiating cells. Stem Cell Res. 2010;5(3):212–25.
Mathur R, Sehgal L, Braun FK, Berkova Z, Romaguerra J, Wang M, et al. Targeting Wnt pathway in mantle cell lymphoma-initiating cells. J Hematol Oncol. 2015;8:63.
Song S, Li Y, Zhang K, Zhang X, Huang Y, Xu M, et al. Cancer Stem cells of diffuse large B cell lymphoma are not enriched in the CD45(+)CD19(-) cells but in the ALDH(high) cells. J Cancer. 2020;11(1):142–52.
Medina DJ, Goodell L, Glod J, Gelinas C, Rabson AB, Strair RK. Mesenchymal stromal cells protect mantle cell lymphoma cells from spontaneous and drug-induced apoptosis through secretion of B-cell activating factor and activation of the canonical and non-canonical nuclear factor kappaB pathways. Haematologica. 2012;97(8):1255–63.
Guan J, Huang D, Yakimchuk K, Okret S. p110alpha Inhibition overcomes stromal cell-mediated ibrutinib resistance in mantle cell lymphoma. Mol Cancer Ther. 2018;17(5):1090–100.
Rudelius M, Rosenfeldt MT, Leich E, Rauert-Wunderlich H, Solimando AG, Beilhack A, et al. Inhibition of focal adhesion kinase overcomes resistance of mantle cell lymphoma to ibrutinib in the bone marrow microenvironment. Haematologica. 2018;103(1):116–25.
Balsas P, Palomero J, Eguileor A, Rodriguez ML, Vegliante MC, Planas-Rigol E, et al. SOX11 promotes tumor protective microenvironment interactions through CXCR4 and FAK regulation in mantle cell lymphoma. Blood. 2017;130(4):501–13.
Balsas P, Veloza L, Clot G, Sureda-Gomez M, Rodriguez ML, Masaoutis C, et al. SOX11, CD70, and Treg cells configure the tumor-immune microenvironment of aggressive mantle cell lymphoma. Blood. 2021;138(22):2202–15.
Fisher RI, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006;24(30):4867–74.
Davids MS, Roberts AW, Kenkre VP, Wierda WG, Kumar A, Kipps TJ, et al. Long-term follow-up of patients with relapsed or refractory non-hodgkin lymphoma treated with venetoclax in a phase I. First-in-Human Study Clin Cancer Res. 2021;27(17):4690–5.
Kahl BS, Spurgeon SE, Furman RR, Flinn IW, Coutre SE, Brown JR, et al. A phase 1 study of the PI3Kdelta inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood. 2014;123(22):3398–405.
Goy A, Sinha R, Williams ME, Kalayoglu Besisik S, Drach J, Ramchandren R, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol. 2013;31(29):3688–95.
Trneny M, Lamy T, Walewski J, Belada D, Mayer J, Radford J, et al. Lenalidomide versus investigator’s choice in relapsed or refractory mantle cell lymphoma (MCL-002; SPRINT): a phase 2, randomised, multicentre trial. Lancet Oncol. 2016;17(3):319–31.
Wang M, Schuster SJ, Phillips T, Lossos IS, Goy A, Rule S, et al. Observational study of lenalidomide in patients with mantle cell lymphoma who relapsed/progressed after or were refractory/intolerant to ibrutinib (MCL-004). J Hematol Oncol. 2017;10(1):171.
Chong EA, Alanio C, Svoboda J, Nasta SD, Landsburg DJ, Lacey SF, et al. Pembrolizumab for B-cell lymphomas relapsing after or refractory to CD19-directed CAR T-cell therapy. Blood. 2022;139(7):1026–38.
Salles G, Gopal AK, Minnema MC, Wakamiya K, Feng H, Schecter JM, et al. Phase 2 study of daratumumab in relapsed/refractory mantle-cell lymphoma, diffuse large B-cell lymphoma, and follicular lymphoma. Clin Lymphoma Myeloma Leuk. 2019;19(5):275–84.
Budde LE, Zhang MM, Shustov AR, Pagel JM, Gooley TA, Oliveira GR, et al. A phase I study of pulse high-dose vorinostat (V) plus rituximab (R), ifosphamide, carboplatin, and etoposide (ICE) in patients with relapsed lymphoma. Br J Haematol. 2013;161(2):183–91.
Evens AM, Balasubramanian S, Vose JM, Harb W, Gordon LI, Langdon R, et al. A phase I/II multicenter, open-label study of the oral histone deacetylase inhibitor abexinostat in relapsed/refractory lymphoma. Clin Cancer Res. 2016;22(5):1059–66.
Leonard JP, LaCasce AS, Smith MR, Noy A, Chirieac LR, Rodig SJ, et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood. 2012;119(20):4597–607.
Martin P, Ruan J, Furman R, Rutherford S, Allan J, Chen Z, et al. A phase I trial of palbociclib plus bortezomib in previously treated mantle cell lymphoma. Leuk Lymphoma. 2019;60(12):2917–21.
Martin P, Bartlett NL, Blum KA, Park S, Maddocks K, Ruan J, et al. A phase 1 trial of ibrutinib plus palbociclib in previously treated mantle cell lymphoma. Blood. 2019;133(11):1201–4.
Wang M, Popplewell LL, Collins RH Jr, Winter JN, Goy A, Kaminski MS, et al. Everolimus for patients with mantle cell lymphoma refractory to or intolerant of bortezomib: multicentre, single-arm, phase 2 study. Br J Haematol. 2014;165(4):510–8.
Robak T, Huang H, Jin J, Zhu J, Liu T, Samoilova O, et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med. 2015;372(10):944–53.
Wang M, Ramchandren R, Chen R, Karlin L, Chong G, Jurczak W, et al. Concurrent ibrutinib plus venetoclax in relapsed/refractory mantle cell lymphoma: the safety run-in of the phase 3 SYMPATICO study. J Hematol Oncol. 2021;14(1):179.
Portell CA, Wages NA, Kahl BS, Budde LE, Chen RW, Cohen JB, et al. Dose-finding study of ibrutinib and venetoclax in relapsed or refractory mantle cell lymphoma. Blood Adv. 2022;6(5):1490–8.
Wang ML, Lee H, Chuang H, Wagner-Bartak N, Hagemeister F, Westin J, et al. Ibrutinib in combination with rituximab in relapsed or refractory mantle cell lymphoma: a single-centre, open-label, phase 2 trial. Lancet Oncol. 2016;17(1):48–56.
Morschhauser FA, Cartron G, Thieblemont C, Solal-Celigny P, Haioun C, Bouabdallah R, et al. Obinutuzumab (GA101) monotherapy in relapsed/refractory diffuse large b-cell lymphoma or mantle-cell lymphoma: results from the phase II GAUGUIN study. J Clin Oncol. 2013;31(23):2912–9.
Le Gouill S, Morschhauser F, Chiron D, Bouabdallah K, Cartron G, Casasnovas O, et al. Ibrutinib, obinutuzumab, and venetoclax in relapsed and untreated patients with mantle cell lymphoma: a phase 1/2 trial. Blood. 2021;137(7):877–87.
Stephens DM, Huang Y, Ruppert AS, Walker JS, Canfield D, Cempre CB, et al. Selinexor combined with ibrutinib demonstrates tolerability and safety in advanced B-cell malignancies: a phase I study. Clin Cancer Res. 2022;28(15):3242–7.
Goy A, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, et al. Bortezomib in patients with relapsed or refractory mantle cell lymphoma: updated time-to-event analyses of the multicenter phase 2 PINNACLE study. Ann Oncol. 2009;20(3):520–5.
Lee HJ, Badillo M, Romaguera J, Wang M. A phase II study of carfilzomib in the treatment of relapsed/refractory mantle cell lymphoma. Br J Haematol. 2019;184(3):460–2.
Perez-Galan P, Roue G, Villamor N, Campo E, Colomer D. The BH3-mimetic GX15-070 synergizes with bortezomib in mantle cell lymphoma by enhancing Noxa-mediated activation of Bak. Blood. 2007;109(10):4441–9.
Yang DT, Young KH, Kahl BS, Markovina S, Miyamoto S. Prevalence of bortezomib-resistant constitutive NF-kappaB activity in mantle cell lymphoma. Mol Cancer. 2008;7:40.
Manni S, Brancalion A, Mandato E, Tubi LQ, Colpo A, Pizzi M, et al. Protein kinase CK2 inhibition down modulates the NF-kappaB and STAT3 survival pathways, enhances the cellular proteotoxic stress and synergistically boosts the cytotoxic effect of bortezomib on multiple myeloma and mantle cell lymphoma cells. PLoS ONE. 2013;8(9):e75280.
Perez-Galan P, Mora-Jensen H, Weniger MA, Shaffer AL 3rd, Rizzatti EG, Chapman CM, et al. Bortezomib resistance in mantle cell lymphoma is associated with plasmacytic differentiation. Blood. 2011;117(2):542–52.
Vegliante MC, Palomero J, Perez-Galan P, Roue G, Castellano G, Navarro A, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood. 2013;121(12):2175–85.
Desai S, Maurin M, Smith MA, Bolick SC, Dessureault S, Tao J, et al. PRDM1 is required for mantle cell lymphoma response to bortezomib. Mol Cancer Res. 2010;8(6):907–18.
Bea S, Salaverria I, Armengol L, Pinyol M, Fernandez V, Hartmann EM, et al. Uniparental disomies, homozygous deletions, amplifications, and target genes in mantle cell lymphoma revealed by integrative high-resolution whole-genome profiling. Blood. 2009;113(13):3059–69.
Zhang Y, Lu P, Zhou Y, Zhang L. Inhibition of LINK-A lncRNA overcomes ibrutinib resistance in mantle cell lymphoma by regulating Akt/Bcl2 pathway. PeerJ. 2021;9:e12571.
Eyre TA, Walter HS, Iyengar S, Follows G, Cross M, Fox CP, et al. Efficacy of venetoclax monotherapy in patients with relapsed, refractory mantle cell lymphoma post BTK inhibitor therapy. Haematologica. 2018.
Zhao S, Kanagal-Shamanna R, Navsaria L, Ok CY, Zhang S, Nomie K, et al. Efficacy of venetoclax in high risk relapsed mantle cell lymphoma (MCL) - outcomes and mutation profile from venetoclax resistant MCL patients. Am J Hematol. 2020;95(6):623–9.
Sawalha Y, Goyal S, Switchenko JM, Romancik JT, Kamdar M, Greenwell IB, et al. A multicenter analysis of the outcomes with venetoclax in patients with relapsed mantle cell lymphoma. Blood Adv. 2023;7(13):2983–93.
Zhang L, Nomie K, Zhang H, Bell T, Pham L, Kadri S, et al. B-cell lymphoma patient-derived xenograft models enable drug discovery and are a platform for personalized therapy. Clin Cancer Res. 2017;23(15):4212–23.
Jain N, Singh S, Laliotis G, Hart A, Muhowski E, Kupcova K, et al. Targeting phosphatidylinositol 3 kinase-beta and -delta for Bruton tyrosine kinase resistance in diffuse large B-cell lymphoma. Blood Adv. 2020;4(18):4382–92.
Kapoor I, Li Y, Sharma A, Zhu H, Bodo J, Xu W, et al. Resistance to BTK inhibition by ibrutinib can be overcome by preventing FOXO3a nuclear export and PI3K/AKT activation in B-cell lymphoid malignancies. Cell Death Dis. 2019;10(12):924.
Davids MS, Kim HT, Nicotra A, Savell A, Francoeur K, Hellman JM, et al. Umbralisib in combination with ibrutinib in patients with relapsed or refractory chronic lymphocytic leukaemia or mantle cell lymphoma: a multicentre phase 1–1b study. Lancet Haematol. 2019;6(1):e38–47.
Qualls D, Lam HY, Whiting K, Kumar A, Matasar M, Owens C, et al. A phase 1 trial of copanlisib plus ibrutinib in relapsed/refractory mantle cell lymphoma. Blood Adv. 2022;6(18):5262–6.
Stewart CM, Michaud L, Whiting K, Nakajima R, Nichols C, De Frank S, et al. Phase I/Ib study of the efficacy and safety of buparlisib and ibrutinib therapy in MCL, FL, and DLBCL with serial cell-free DNA monitoring. Clin Cancer Res. 2022;28(1):45–56.
Mehta A, Trněný M, Walewski J, Ribrag V, Dartigeas C, Christensen JH, et al. Efficacy and safety of parsaclisib in patients with relapsed or refractory mantle cell lymphoma not previously treated with a BTK inhibitor: primary analysis from a phase 2 study (CITADEL-205). Blood. 2021;138(Supplement 1):382.
Zinzani PL, Delwail V, Paneesha S, Rule S, Martin Garcia-Sancho A, Marin-Niebla A, et al. Phase 2 study evaluating the efficacy and safety of parsaclisib in patients with relapsed or refractory mantle cell lymphoma previously treated with ibrutinib (CITADEL-205). Blood. 2020;136(Supplement 1):43–4.
Rudelius M, Pittaluga S, Nishizuka S, Pham TH, Fend F, Jaffe ES, et al. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood. 2006;108(5):1668–76.
Psyrri A, Papageorgiou S, Liakata E, Scorilas A, Rontogianni D, Kontos CK, et al. Phosphatidylinositol 3’-kinase catalytic subunit alpha gene amplification contributes to the pathogenesis of mantle cell lymphoma. Clin Cancer Res. 2009;15(18):5724–32.
Iyengar S, Clear A, Bodor C, Maharaj L, Lee A, Calaminici M, et al. P110alpha-mediated constitutive PI3K signaling limits the efficacy of p110delta-selective inhibition in mantle cell lymphoma, particularly with multiple relapse. Blood. 2013;121(12):2274–84.
Al-Mansour M. Treatment landscape of relapsed/refractory mantle cell lymphoma: an updated review. Clin Lymphoma Myeloma Leuk. 2022;22(11):e1019–31.
Ioannou N, Jain K, Ramsay AG. Immunomodulatory drugs for the treatment of B cell malignancies. Int J Mol Sci. 2021;22(16):8572.
Desai M, Newberry K, Ou Z, Wang M, Zhang L. Lenalidomide in relapsed or refractory mantle cell lymphoma: overview and perspective. Ther Adv Hematol. 2014;5(3):91–101.
Zhang L, Yang J, Qian J, Li H, Romaguera JE, Kwak LW, et al. Role of the microenvironment in mantle cell lymphoma: IL-6 is an important survival factor for the tumor cells. Blood. 2012;120(18):3783–92.
Roue G, Sola B. Management of drug resistance in mantle cell lymphoma. Cancers (Basel). 2020;12(6):1565.
Ameli F, Shajareh E, Mokhtari M, Kosari F. Expression of PD1 and PDL1 as immune-checkpoint inhibitors in mantle cell lymphoma. BMC Cancer. 2022;22(1):848.
Jiang Z, Sun H, Yu J, Tian W, Song Y. Targeting CD47 for cancer immunotherapy. J Hematol Oncol. 2021;14(1):180.
Kim TM, Lakhani N, Gainor J, Kamdar M, Fanning P, Squifflet P, et al. A phase 1 study of ALX148, a CD47 blocker, in combination with rituximab in patients with non-hodgkin lymphoma. Blood. 2019;134(Supplement_1):1953.
Freile JA, Ustyanovska Avtenyuk N, Corrales MG, Lourens HJ, Huls G, van Meerten T, et al. CD24 is a potential immunotherapeutic target for mantle cell lymphoma. Biomedicines. 2022;10(5):1175.
Klapdor R, Wang S, Morgan M, Dork T, Hacker U, Hillemanns P, et al. Characterization of a novel third-generation anti-CD24-CAR against ovarian cancer. Int J Mol Sci. 2019;20(3):660.
Panagiotou E, Syrigos NK, Charpidou A, Kotteas E, Vathiotis IA. CD24: a novel target for cancer immunotherapy. J Pers Med. 2022;12(8):1235.
Yang J, Hu G. Significance of PD-L1 in the diagnosis and treatment of B-cell malignant lymphoma. Oncol Lett. 2019;17(3):3382–6.
Wang L, Qian J, Lu Y, Li H, Bao H, He D, et al. Immune evasion of mantle cell lymphoma: expression of B7–H1 leads to inhibited T-cell response to and killing of tumor cells. Haematologica. 2013;98(9):1458–66.
Harrington BK, Wheeler E, Hornbuckle K, Shana’ah AY, Youssef Y, Smith L, et al. Modulation of immune checkpoint molecule expression in mantle cell lymphoma. Leuk Lymphoma. 2019;60(10):2498–507.
Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase Ib study. J Clin Oncol. 2016;34(23):2698–704.
Wasiuk A, Testa J, Weidlick J, Sisson C, Vitale L, Widger J, et al. CD27-mediated regulatory T cell depletion and effector T cell costimulation both contribute to antitumor efficacy. J Immunol. 2017;199(12):4110–23.
Baskar S, Kwong KY, Hofer T, Levy JM, Kennedy MG, Lee E, et al. Unique cell surface expression of receptor tyrosine kinase ROR1 in human B-cell chronic lymphocytic leukemia. Clin Cancer Res. 2008;14(2):396–404.
Karvonen H, Niininen W, Murumagi A, Ungureanu D. Targeting ROR1 identifies new treatment strategies in hematological cancers. Biochem Soc Trans. 2017;45(2):457–64.
Balakrishnan A, Goodpaster T, Randolph-Habecker J, Hoffstrom BG, Jalikis FG, Koch LK, et al. Analysis of ROR1 protein expression in human cancer and normal tissues. Clin Cancer Res. 2017;23(12):3061–71.
Hojjat-Farsangi M, Moshfegh A, Daneshmanesh AH, Khan AS, Mikaelsson E, Osterborg A, et al. The receptor tyrosine kinase ROR1–an oncofetal antigen for targeted cancer therapy. Semin Cancer Biol. 2014;29:21–31.
Ghaderi A, Zhong W, Okhovat MA, Aschan J, Svensson A, Sander B, et al. A ROR1 small molecule inhibitor (KAN0441571C) induced significant apoptosis of mantle cell lymphoma (MCL) cells. Pharmaceutics. 2022;14(10):2238.
Yu J, Chen Y, Chen L, Zhang L, Rassenti LZ, Widhopf GF 2nd, et al. Cirmtuzumab inhibits ibrutinib-resistant, Wnt5a-induced Rac1 activation and proliferation in mantle cell lymphoma. Oncotarget. 2018;9(37):24731–6.
Sermer D, Pasqualucci L, Wendel HG, Melnick A, Younes A. Emerging epigenetic-modulating therapies in lymphoma. Nat Rev Clin Oncol. 2019;16(8):494–507.
Guo H, Zeng D, Zhang H, Bell T, Yao J, Liu Y, et al. Dual inhibition of PI3K signaling and histone deacetylation halts proliferation and induces lethality in mantle cell lymphoma. Oncogene. 2019;38(11):1802–14.
Chaturvedi NK, Hatch ND, Sutton GL, Kling M, Vose JM, Joshi SS. A novel approach to eliminate therapy-resistant mantle cell lymphoma: synergistic effects of Vorinostat with Palbociclib. Leuk Lymphoma. 2019;60(5):1214–23.
Yazbeck V, Shafer D, Perkins EB, Coppola D, Sokol L, Richards KL, et al. A phase II trial of bortezomib and vorinostat in mantle cell lymphoma and diffuse large B-cell lymphoma. Clin Lymphoma Myeloma Leuk 2018;18(9):569–75 e1.
Markozashvili D, Pichugin A, Barat A, Camara-Clayette V, Vasilyeva NV, Lelievre H, et al. Histone deacetylase inhibitor abexinostat affects chromatin organization and gene transcription in normal B cells and in mantle cell lymphoma. Gene. 2016;580(2):134–43.
Bhalla S, Balasubramanian S, David K, Sirisawad M, Buggy J, Mauro L, et al. PCI-24781 induces caspase and reactive oxygen species-dependent apoptosis through NF-kappaB mechanisms and is synergistic with bortezomib in lymphoma cells. Clin Cancer Res. 2009;15(10):3354–65.
Paoluzzi L, Scotto L, Marchi E, Zain J, Seshan VE, O’Connor OA. Romidepsin and belinostat synergize the antineoplastic effect of bortezomib in mantle cell lymphoma. Clin Cancer Res. 2010;16(2):554–65.
Chen Y, Shao X, Zhao X, Ji Y, Liu X, Li P, et al. Targeting protein arginine methyltransferase 5 in cancers: roles, inhibitors and mechanisms. Biomed Pharmacother. 2021;144:112252.
Sloan S, Brown F, Chung JH, Prouty A, Wheeler E, Harrington BK, et al. Targeting PRMT5 to circumvent acquired ibrutinib resistance in mantle cell lymphoma. Blood. 2019;134(Supplement_1):4065.
Mareckova A, Malcikova J, Tom N, Pal K, Radova L, Salek D, et al. ATM and TP53 mutations show mutual exclusivity but distinct clinical impact in mantle cell lymphoma patients. Leuk Lymphoma. 2019;60(6):1420–8.
Cheng Q, Chen J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle. 2010;9(3):472–8.
Rosenthal AC, Munoz JL, Villasboas JC. Clinical advances in epigenetic therapies for lymphoma. Clin Epigenet. 2023;15(1):39.
Long M, Koirala S, Sloan S, Brown F, Tallada S, Hinterschied C, et al. Acquired resistance to PRMT5 inhibition in mantle cell lymphoma is associated with compensatory activation of multiple signaling pathways. Blood. 2022;140(Supplement 1):3103–4.
Queiros AC, Beekman R, Vilarrasa-Blasi R, Duran-Ferrer M, Clot G, Merkel A, et al. Decoding the DNA methylome of mantle cell lymphoma in the light of the entire B cell lineage. Cancer Cell. 2016;30(5):806–21.
Li XY, Li Y, Zhang L, Liu X, Feng L, Wang X. The antitumor effects of arsenic trioxide in mantle cell lymphoma via targeting Wnt/beta-catenin pathway and DNA methyltransferase-1. Oncol Rep. 2017;38(5):3114–20.
Ribeiro ML, Reyes-Garau D, Armengol M, Fernandez-Serrano M, Roue G. Recent advances in the targeting of epigenetic regulators in B-cell non-hodgkin lymphoma. Front Genet. 2019;10:986.
Eich ML, Athar M, Ferguson JE 3rd, Varambally S. EZH2-Targeted therapies in cancer: hype or a reality. Cancer Res. 2020;80(24):5449–58.
Martinez-Baquero D, Sakhdari A, Mo H, Kim DH, Kanagal-Shamanna R, Li S, et al. EZH2 expression is associated with inferior overall survival in mantle cell lymphoma. Mod Pathol. 2021;34(12):2183–91.
Yang P, Zhang W, Wang J, Liu Y, An R, Jing H. Genomic landscape and prognostic analysis of mantle cell lymphoma. Cancer Gene Ther. 2018;25(5–6):129–40.
Demosthenous C, Gupta SK, Sun J, Wang Y, Troska TP, Gupta M. Deregulation of polycomb repressive complex-2 in mantle cell lymphoma confers growth advantage by epigenetic suppression of cdkn2b. Front Oncol. 2020;10:1226.
Kagiyama Y, Fujita S, Shima Y, Yamagata K, Katsumoto T, Nakagawa M, et al. CDKN1C-mediated growth inhibition by an EZH1/2 dual inhibitor overcomes resistance of mantle cell lymphoma to ibrutinib. Cancer Sci. 2021;112(6):2314–24.
Sircar A, Singh S, Xu-Monette ZY, Coyle KM, Hilton LK, Chavdoula E, et al. Exploiting the fibroblast growth factor receptor-1 vulnerability to therapeutically restrict the MYC-EZH2-CDKN1C axis-driven proliferation in Mantle cell lymphoma. Leukemia. 2023. https://doi.org/10.1038/s41375-023-02006-8
Sircar A, Singh S, Young KH, Epperla N, Sehgal L. Abstract 1422: Fibroblast growth factor receptor-1 is a novel therapeutic target in mantle cell Lymphoma. Cancer Res. 2023;83(7_Supplement):1422.
Keats JA, Lee A, Cunniff JC, Chen W, Mehovic R, Estanek V, et al. Abstract 1161: EZH2 inhibitor tazemetostat demonstrates activity in preclinical models of Bruton’s tyrosine kinase inhibitor-resistant relapsed/refractory mantle cell lymphoma. Can Res. 2021;81(131):1161.
Lee C, Huang X, Di Liberto M, Martin P, Chen-Kiang S. Targeting CDK4/6 in mantle cell lymphoma. Ann Lymphoma. 2020;4:1.
Okada T, Ngo VN, Ekland EH, Forster R, Lipp M, Littman DR, et al. Chemokine requirements for B cell entry to lymph nodes and Peyer’s patches. J Exp Med. 2002;196(1):65–75.
Kurtova AV, Tamayo AT, Ford RJ, Burger JA. Mantle cell lymphoma cells express high levels of CXCR4, CXCR5, and VLA-4 (CD49d): importance for interactions with the stromal microenvironment and specific targeting. Blood. 2009;113(19):4604–13.
Chen Z, Teo AE, McCarty N. ROS-induced CXCR4 signaling regulates mantle cell lymphoma (MCL) cell survival and drug resistance in the bone marrow microenvironment via autophagy. Clin Cancer Res. 2016;22(1):187–99.
Kwon D, Takata K, Zhang Z, Chong L, Fraser B, Zeisler J, et al. Targeting refractory mantle cell lymphoma for imaging and therapy using C-X-C chemokine receptor type 4 radioligands. Clin Cancer Res. 2022;28(8):1628–39.
Micallef IN, Stiff PJ, Nademanee AP, Maziarz RT, Horwitz ME, Stadtmauer EA, et al. Plerixafor plus granulocyte colony-stimulating factor for patients with non-Hodgkin lymphoma and multiple myeloma: long-term follow-up report. Biol Blood Marrow Transplant. 2018;24(6):1187–95.
Kersy O, Salmon-Divon M, Gomes da Silva M, Moita AF, Cabecadas J, Klener P Jr, et al. Inhibition of MAPK-ERK signaling pathway overcomes microrna-mediated ibrutinib resistance in mantle cell lymphoma. Blood. 2022;140(Supplement 1):3109–10.
Wang X, Fei Y, Liu X, Zhang T, Li W, Jia X, et al. Bortezomib enhances the anti-cancer effect of the novel Bruton’s tyrosine kinase inhibitor (BGB-3111) in mantle cell lymphoma expressing BTK. Aging (Albany NY). 2021;13(17):21102–21.
Morrison VA, Jung SH, Johnson J, LaCasce A, Blum KA, Bartlett NL, et al. Therapy with bortezomib plus lenalidomide for relapsed/refractory mantle cell lymphoma: final results of a phase II trial (CALGB 50501). Leuk Lymphoma. 2015;56(4):958–64.
Srour SA, Lee HJ, Nomie K, Ye H, Chen W, Oriabure O, et al. Novel chemotherapy-free combination regimen for ibrutinib-resistant mantle cell lymphoma. Br J Haematol. 2018;181(4):561–4.
Tam CS, Anderson MA, Pott C, Agarwal R, Handunnetti S, Hicks RJ, et al. Ibrutinib plus venetoclax for the treatment of mantle-cell lymphoma. N Engl J Med. 2018;378(13):1211–23.
Li Y, Bouchlaka MN, Wolff J, Grindle KM, Lu L, Qian S, et al. FBXO10 deficiency and BTK activation upregulate BCL2 expression in mantle cell lymphoma. Oncogene. 2016;35(48):6223–34.
Zhao X, Bodo J, Sun D, Durkin L, Lin J, Smith MR, et al. Combination of ibrutinib with ABT-199: synergistic effects on proliferation inhibition and apoptosis in mantle cell lymphoma cells through perturbation of BTK, AKT and BCL2 pathways. Br J Haematol. 2015;168(5):765–8.
Zhang H, Local A, Benbatoul K, Folger P, Sheng S, Tsai C-Y, et al. CG ’806, a first-in-class non-covalent pan-FLT3/BTK inhibitor, exerts superior potency against B-cell malignant cells. Blood. 2017;130(11):5200.
Thieme E, Liu T, Bruss N, Roleder C, Lam V, Wang X, et al. Dual BTK/SYK inhibition with CG-806 (luxeptinib) disrupts B-cell receptor and Bcl-2 signaling networks in mantle cell lymphoma. Cell Death Dis. 2022;13(3):246.
Tsukamoto T, Nakahata S, Sato R, Kanai A, Nakano M, Chinen Y, et al. BRD4-regulated molecular targets in mantle cell lymphoma: insights into targeted therapeutic approach. Cancer Genomics Proteomics. 2020;17(1):77–89.
Sun B, Shah B, Fiskus W, Qi J, Rajapakshe K, Coarfa C, et al. Synergistic activity of BET protein antagonist-based combinations in mantle cell lymphoma cells sensitive or resistant to ibrutinib. Blood. 2015;126(13):1565–74.
Soumerai JD, Diefenbach CS, Samaniego F, Kumar A, Tsai ML, Asch AS, et al. Safety and efficacy of the PI3Kδ inhibitor zandelisib in combination with the BTK inhibitor zanubrutinib in patients with relapsed/refractory (R/R) follicular lymphoma (FL) or mantle cell lymphoma (MCL). Blood. 2022;140(Supplement 1):189–91.
Jerkeman M, Kolstad A, Hutchings M, Pasanen A, Meriranta L, Niemann CU, et al. Venetoclax, lenalidomide and rituximab for patients with relapsed or refractory mantle cell lymphoma—the Nordic Lymphoma Group NLG-MCL7 (VALERIA) Phase Ib-II trial. Blood. 2022;140(Supplement 1):184–5.
Alinari L, Yu B, Christian BA, Yan F, Shin J, Lapalombella R, et al. Combination anti-CD74 (milatuzumab) and anti-CD20 (rituximab) monoclonal antibody therapy has in vitro and in vivo activity in mantle cell lymphoma. Blood. 2011;117(17):4530–41.
Chiron D, Bellanger C, Papin A, Tessoulin B, Dousset C, Maiga S, et al. Rational targeted therapies to overcome microenvironment-dependent expansion of mantle cell lymphoma. Blood. 2016;128(24):2808–18.
Zhou X, Steinhardt MJ, Dull J, Krummenast F, Danhof S, Meckel K, et al. Obinutuzumab and venetoclax induced complete remission in a patient with ibrutinib-resistant non-nodal leukemic mantle cell lymphoma. Eur J Haematol. 2020;104(4):352–5.
Decombis S, Bellanger C, Le Bris Y, Boulet D, Jardine J, Dousset C, et al. CARD11 and BCL2A1 join forces to promote resistance to ibrutinib/venetoclax combination in lymphoma patients. Blood. 2022;140(Supplement 1):2044–5.
Sawas A, Farber CM, Schreeder MT, Khalil MY, Mahadevan D, Deng C, et al. A phase 1/2 trial of ublituximab, a novel anti-CD20 monoclonal antibody, in patients with B-cell non-Hodgkin lymphoma or chronic lymphocytic leukaemia previously exposed to rituximab. Br J Haematol. 2017;177(2):243–53.
Deshpande A, Wang Y, Munoz J, Jain P. Brexucabtagene autoleucel: a breakthrough in the treatment of mantle cell lymphoma. Drugs Today (Barc). 2022;58(6):283–98.
Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382(14):1331–42.
Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. Three-year follow-up of KTE-X19 in patients with relapsed/refractory mantle cell lymphoma, including high-risk subgroups, in the ZUMA-2 study. J Clin Oncol. 2023;41(3):555–67.
Herbaux C, Bret C, Di Blasi R, Bachy E, Beauvais D, Gat E, et al. Kte-X19 in relapsed or refractory mantle-cell lymphoma, a “real-life” study from the descar-T registry and lysa group. Blood. 2021;138(Supplement 1):743.
Wang Y, Jain P, Locke FL, Munoz J, Maurer MJ, Beitinjaneh A, et al. Brexucabtagene autoleucel for relapsed/refractory mantle cell lymphoma: real world experience from the US lymphoma CAR T consortium. Blood. 2021;138(Supplement 1):744.
O’Reilly MA, Sanderson R, Wilson W, Burns D, Besley C, Creasey T, et al. Brexucabtagene autoleucel for relapsed/refractory mantle cell lymphoma: real-world outcomes in the United Kingdom. Blood. 2022;140(Supplement 1):7519–21.
Palomba ML, Gordon LI, Siddiqi T, Abramson JS, Kamdar M, Lunning MA, et al. Safety and preliminary efficacy in patients with relapsed/refractory mantle cell lymphoma receiving lisocabtagene maraleucel in transcend NHL 001. Blood. 2020;136(Supplement 1):10–1.
Ying Z, Zhou K, Li L, Li W, Yang S, Liu Y, et al. Preliminary safety and efficacy of relmacabtagene autoleucel (relma-cel) in adults with relapsed/refractory mantle cell lymphoma (r/r MCL) in China. Blood. 2022;140(Supplement 1):7470–1.
Qin H, Dong Z, Wang X, Cheng WA, Wen F, Xue W, et al. CAR T cells targeting BAFF-R can overcome CD19 antigen loss in B cell malignancies. Sci Transl Med. 2019;11(511):eaaw414.
George P, Dasyam N, Giunti G, Mester B, Bauer E, Andrews B, et al. Third-generation anti-CD19 chimeric antigen receptor T-cells incorporating a TLR2 domain for relapsed or refractory B-cell lymphoma: a phase I clinical trial protocol (ENABLE). BMJ Open. 2020;10(2):e034629.
Chan T, Scott SP, Du M, Bolinger C, Poortman C, Shepard L, et al. Preclinical evaluation of Prgn-3007, a non-viral, multigenic, autologous ROR1 Ultracar-T ® therapy with novel mechanism of intrinsic pd-1 blockade for treatment of hematological and solid cancers. Blood. 2021;138(Supplement 1):1694.
Scarfo I, Ormhoj M, Frigault MJ, Castano AP, Lorrey S, Bouffard AA, et al. Anti-CD37 chimeric antigen receptor T cells are active against B- and T-cell lymphomas. Blood. 2018;132(14):1495–506.
Minson A, Hamad N, Butler JP, Westerman DA, Ritchie D, Blombery P, et al. A Phase II, open-label, single arm trial to assess the efficacy and safety of the combination of tisagenlecleucel and ibrutinib in mantle cell lymphoma (TARMAC). Blood. 2020;136(Supplement 1):34–5.
Shah NN, Johnson BD, Schneider D, Zhu F, Szabo A, Keever-Taylor CA, et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat Med. 2020;26(10):1569–75.
Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019;16(6):372–85.
Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. 2016;127(9):1117–27.
Qin JS, Johnstone TG, Baturevych A, Hause RJ, Ragan SP, Clouser CR, et al. Antitumor potency of an anti-CD19 chimeric antigen receptor T-cell therapy, lisocabtagene maraleucel in combination with ibrutinib or acalabrutinib. J Immunother. 2020;43(4):107–20.
Ruella M, Kenderian SS, Shestova O, Fraietta JA, Qayyum S, Zhang Q, et al. The addition of the BTK inhibitor ibrutinib to anti-CD19 chimeric antigen receptor T cells (CART19) improves responses against mantle cell lymphoma. Clin Cancer Res. 2016;22(11):2684–96.
Minson A, Hamad N, Cheah CY, Tam CS, Blombery P, Westerman DA, et al. Time-limited ibrutinib and tisagenlecleucel is highly effective in the treatment of patients with relapsed or refractory mantle cell lymphoma, including those with TP53 mutated and btki-refractory disease: first report of the tarmac study. Blood. 2022;140(Supplement 1):181–3.
Jiang VC, Yan F, Jordan AA, Che Y, Liu Y, Cai Q, et al. Targeting the HSP90-MYC-CDK9 axis to overcome dual resistance to BTK inhibition and CAR-T therapy in mantle cell lymphoma. Blood. 2022;140(Supplement 1):3512–3.
Zhou J, Kulkarni A, Love C, Happ L, Sturtevant D, Biyani N, et al. Development of a potent DNA damaging agent LP-284 for treatment of mantle cell lymphoma. Blood. 2022;140(Supplement 1):4940–1.
Yang M, Wang L, Ni M, Neuber B, Wang S, Gong W, et al. Pre-sensitization of malignant B cells through venetoclax significantly improves the cytotoxic efficacy of CD19.CAR-T cells. Front Immunol. 2020;11:608167.
Bukhari A, El Chaer F, Koka R, Singh Z, Hutnick E, Ruehle K, et al. Rapid relapse of large B-cell lymphoma after CD19 directed CAR-T-cell therapy due to CD-19 antigen loss. Am J Hematol. 2019;94(10):E273–5.
Shah NN, Furqan F, Szabo A, Neumann J, Hari P, Schneider D, et al. Results from a phase 1/2 study of tandem, bispecific anti-CD20/Anti-CD19 (LV20.19) CAR T-cells for mantle cell lymphoma. Blood. 2022;140(11):9318–9.
Goebeler ME, Knop S, Viardot A, Kufer P, Topp MS, Einsele H, et al. Bispecific T-cell engager (BiTE) antibody construct blinatumomab for the treatment of patients with relapsed/refractory non-Hodgkin lymphoma: final results from a phase I study. J Clin Oncol. 2016;34(10):1104–11.
Hutchings M, Mous R, Clausen MR, Johnson P, Linton KM, Chamuleau MED, et al. Dose escalation of subcutaneous epcoritamab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an open-label, phase 1/2 study. Lancet. 2021;398(10306):1157–69.
Bannerji R, Arnason JE, Advani RH, Brown JR, Allan JN, Ansell SM, et al. Odronextamab, a human CD20xCD3 bispecific antibody in patients with CD20-positive B-cell malignancies (ELM-1): results from the relapsed or refractory non-Hodgkin lymphoma cohort in a single-arm, multicentre, phase 1 trial. Lancet Haematol. 2022;9(5):e327–39.
Topp MS, Gokbuget N, Zugmaier G, Stein AS, Dombret H, Chen Y, et al. Long-term survival of patients with relapsed/refractory acute lymphoblastic leukemia treated with blinatumomab. Cancer. 2021;127(4):554–9.
Budde LE, Assouline S, Sehn LH, Schuster SJ, Yoon SS, Yoon DH, et al. Single-agent mosunetuzumab shows durable complete responses in patients with relapsed or refractory B-cell lymphomas: phase I dose-escalation study. J Clin Oncol. 2022;40(5):481–91.
Phillips TJ, Dickinson M, Morschhauser F, Bachy E, Crump M, Trněný M, et al. Glofitamab monotherapy induces high complete response rates in patients with heavily pretreated relapsed or refractory mantle cell lymphoma. Blood. 2022;140(Supplement 1):178–80.
Wang ML, Barrientos JC, Furman RR, Mei M, Barr PM, Choi MY, et al. Zilovertamab Vedotin targeting of ROR1 as therapy for lymphoid cancers. NEJM Evidence. 2022;1(1):EVIDoa2100001.
Lee HJ, Choi MY, Siddiqi T, Rhodes JM, Wierda WG, Isufi I, et al. Phase 1/2 study of zilovertamab and ibrutinib in mantle cell lymphoma (MCL), chronic lymphocytic leukemia (CLL), or marginal zone lymphoma (MZL). Blood. 2022;140(Supplement 1):566–8.
Chu Y, Zhou X, Wang X. Antibody-drug conjugates for the treatment of lymphoma: clinical advances and latest progress. J Hematol Oncol. 2021;14(1):88.
Jiang VC, Liu Y, Jordan A, McIntosh J, Li Y, Che Y, et al. The antibody drug conjugate VLS-101 targeting ROR1 is effective in CAR T-resistant mantle cell lymphoma. J Hematol Oncol. 2021;14(1):132.
Hamadani M, Radford J, Carlo-Stella C, Caimi PF, Reid E, O’Connor OA, et al. Final results of a phase 1 study of loncastuximab tesirine in relapsed/refractory B-cell non-Hodgkin lymphoma. Blood. 2021;137(19):2634–45.
Sawas A, Savage KJ, Perez R, Advani RH, Butturini A, Lackey J, et al. A phase 1 study of the Anti-CD37 antibody-drug conjugate AGS67E in advanced lymphoid malignancies. Interim results. Blood. 2015;126(23):3976.
Sangha R, Davies A, Dang NH, Ogura M, MacDonald DA, Ananthakrishnan R, et al. Phase 1 study of inotuzumab ozogamicin combined with R-GDP for the treatment of patients with relapsed/refractory CD22+ B-cell non-Hodgkin lymphoma. J Drug Assess. 2017;6(1):10–7.
Phillips T, Barr PM, Park SI, Kolibaba K, Caimi PF, Chhabra S, et al. A phase 1 trial of SGN-CD70A in patients with CD70-positive diffuse large B cell lymphoma and mantle cell lymphoma. Invest New Drugs. 2019;37(2):297–306.
Acknowledgements
We thank several colleagues whose contributions made an important discovery in the BTKi resistance mechanism and new therapeutic interventions for mantle cell lymphoma. The institutional (CSIR-CDRI) communication number for this article is 10651.
Funding
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Numbers R01CA282483 (LS), 5KL2TR2734 (LS), IRP (Young investigator award to LS) and ASH Global Research Award; N.J., American Society of Hematology.
Author information
Authors and Affiliations
Contributions
NE was involved in the conception of the study. NE, NJ, and LS were involved in the study design. NJ, MM, UJ, IS, and SMC were involved in the data collection and preparing the first draft of the manuscript. NE and LS reviewed the manuscript and provided critical comments. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The authors permit the Journal of Hematology and Oncology to publish this work.
Competing interests
NE: Research funding from Beigene, Speakers Bureau for Beigene, Incyte, and Novartis, and Honoraria/consulting/ad boards for Merck, ADC Therapeutics, Lilly, and Novartis. The other co-authors do not have no relevant competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jain, N., Mamgain, M., Chowdhury, S.M. et al. Beyond Bruton’s tyrosine kinase inhibitors in mantle cell lymphoma: bispecific antibodies, antibody–drug conjugates, CAR T-cells, and novel agents. J Hematol Oncol 16, 99 (2023). https://doi.org/10.1186/s13045-023-01496-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-023-01496-4