
Abstract
Mitochondria are essential for tumor growth and progression. However, the heavy demand for mitochondrial activity in cancer leads to increased production of mitochondrial reactive oxygen species (mtROS), accumulation of mutations in mitochondrial DNA, and development of mitochondrial dysfunction. If left unchecked, excessive mtROS can damage and unfold proteins in the mitochondria to an extent that becomes lethal to the tumor. Cellular systems have evolved to combat mtROS and alleviate mitochondrial stress through a quality control mechanism called the mitochondrial unfolded protein response (UPRmt). The UPRmt system is composed of chaperones and proteases, which promote protein folding or eliminate mitochondrial proteins damaged by mtROS, respectively. UPRmt is conserved and activated in cancer in response to mitochondrial stress to maintain mitochondrial integrity and support tumor growth. In this review, we discuss how mitochondria become dysfunctional in cancer and highlight the tumor-promoting functions of key components of the UPRmt.
Similar content being viewed by others


Introduction
As cancer cells undergo uncontrolled proliferation, these cells develop a number of vulnerabilities that require protection by key support systems [1, 2]. Mitochondria, in particular, contribute to growth and survival throughout the various stages of cancer [3]. However, mitochondria also undergo genetic alterations resulting in a dysfunctional electron transport chain, which generate excessive levels of mitochondrial reactive oxygen species (mtROS) (Fig. 1) [4,5,6]. Under physiological conditions and during the early stages of disease, mitochondria produce moderate levels of mtROS that are beneficial to cellular growth and survival. However, as mitochondrial dysfunction worsens, levels of mtROS can exceed the tolerable threshold and become lethal to tumor cells [7,8,9]. The mtROS promote unfolding and aggregation of mitochondrial proteins, leaving mitochondria in an increasingly fragile and dysfunctional state [4,5,6].

Mitochondrial dysfunction develops in aging cells and cancer cells. Increased mitochondrial activity in cancer cells leads to an increased production of mtROS. Over time, mtROS damage mtDNA and cause the accumulation of mutations in mtDNA. This leads to further increases in mtROS, which eventually give way to mitochondrial dysfunction. Thereafter, a vicious cycle occurs in which mitochondrial dysfunction increases mtROS and leads to a deeper state of mitochondrial dysfunction. Cancer cells can utilize dysfunctional mitochondria to promote tumor growth and progression
The mitochondrial unfolded protein response (UPRmt), a mitochondrial stress response observed in C. elegans and mammalian system [10, 11], serves as an important support system in cancer by maintaining mitochondrial integrity and promoting tumor growth [12, 13]. UPRmt activates a series of chaperones and proteases that alleviate the damaging effects of mtROS. Increasing evidence suggests that the UPRmt is conserved between C. elegans and mammals [14].
The current review elaborates the development of mitochondrial dysfunction in cancer and discusses how mitochondrial dysfunction and elevated mtROS benefit tumor growth. We discuss the ability of UPRmt to prevent functional decline of mitochondria in supporting tumor growth and progression (Figs. 1 and 2). In examining how UPRmt preserves mitochondrial health, we discuss the individual functions of UPRmt components, their associations with clinical outcomes, and their tumor-promoting roles.

Mitochondrial unfolded protein response (UPRmt) is proposed to mediate mitochondrial maintenance in cancer. Cancer cells develop mitochondrial dysfunction due to increased accumulation of mtDNA and mtROS relative to non-malignant cells. Although mtROS can be beneficial to cell growth, excess mtROS can damage proteins and further propagate oxidative and proteotoxic stress in the mitochondria. The UPRmt system refolds proteins so that they return to their proper conformation or cleaves such proteins. This preserves mitochondrial integrity and prevents mitochondrial apoptosis
Non-oncogene addiction
Targeting of oncogenes has always been an enticing approach in cancer therapy [15]. Oncogenes such as rat sarcoma virus (RAS) [16], myelocytomatosis (MYC) [17], and epidermal growth factor receptor (EGFR) [18] serve as key drivers of tumor initiation and growth [19, 20]. However, we often overlook the importance of support systems that maintain the tumorigenic state. As cancer cells undergo rapid proliferation, cancer cells bear genetic alterations that produce multiple stress phenotypes including DNA damage stress, mitotic stress, metabolic stress, oxidative stress, and proteotoxic stress [1, 2]. These vulnerabilities, if left unchecked, can be lethal to tumor viability. In order to overcome these challenges, stress response pathways are recruited to alleviate stress, allow cell survival, and promote tumor progression. The genes associated with stress response may not necessarily have the classical features of oncogenes, such as activating mutations or overexpression that can directly induce carcinogenesis, and are therefore referred to as non-oncogenes [21]. Nonetheless, these non-oncogenes are fundamental to tumor maintenance and the increased reliance of cancer cells upon these non-oncogenes is referred to as “non-oncogene addiction” [21].
The current review focuses on the vulnerability of mitochondria in cancer cells. Extensive ROS production by mitochondria along with other events such as alterations in the antioxidant system leads to oxidative stress in cancer cells [22]. Reactive oxygen species (ROS) then induce the unfolding/misfolding and aggregation of proteins within the mitochondrion to propagate proteotoxic stress [23, 24]. Genes of a mitochondria-specific stress response, known as the UPRmt, relieve the constant oxidative and proteotoxic stress present in mitochondria. In doing so, activation of UPRmt functions as a form of non-oncogene addiction in cancer. The following section describes how mitochondria become dysfunctional in cancer, yet continue to contribute to cancer progression, and highlights the specific vulnerabilities that pressure mitochondria to increasingly rely upon UPRmt.
Mitochondrial dysfunction and cancer
Mitochondria are key organelles critical for fulfilling the bioenergetic and biosynthetic needs of the cellular system [25]. Mitochondria contain their own genome, which encodes for 13 proteins, 22 tRNAs, and 2 rRNAs [26]. All 13 mitochondrial gene-encoded proteins are part of the oxidative phosphorylation (OXPHOS) Complexes in the electron transport chain (ETC) that facilitate ATP production [27]. Mitochondria are highly dynamic networks that perform fusion and fission to: allow communication and distribution of resources between mitochondrial compartments, preserve genomic integrity, and maintain mitochondrial homeostasis [28, 29]. The mitochondrial metabolism also generates oncometabolites such as (R)-2-hydroxyglutarate (2-HG) via isocitrate dehydrogenase (IDH) mutation in acute myeloid leukemia (AML), succinate via mutation succinate dehydrogenase (SDH), and fumarate via mutation in fumarate hydratase (FH), which promote tumor growth and progression [3, 30, 31]. The following sections will elaborate on how the accumulation of mutations in mitochondrial DNA (mtDNA) and increases in mitochondrial reactive oxygen species (mtROS) contribute to the development of mitochondrial dysfunction in cancer [23], and how mitochondrial dysfunction propagates the growth and survival of cancer cells (Figs. 1 and 2).
Mitochondrial ROS production and mitochondrial mutagenesis
Mitochondria are a major source of ROS, due to the activity of protein complexes of the ETC [4, 32]. Within the ETC, the transfer of electrons between complexes is coupled with the movement of protons across the inner mitochondrial membrane to generate an electrochemical proton gradient that drives ATP synthesis [33]. During this process, electrons can leak from the ETC complexes and interact with oxygen to form mtROS such as superoxide, hydrogen peroxide, and hydroxyl radicals [4, 5]. In addition, enzymes of the tricarboxylic acid (TCA) cycle located in the mitochondrial matrix also contribute to mtROS production. For example, 2-oxoglutarate dehydrogenase, branched-chain 2-oxoacid dehydrogenase, pyruvate dehydrogenase complexes, glycerol-3-phosphate dehydrogenase (mGPDH), electron-transferring flavoprotein–ubiquinone oxidoreductase (ETF-QOR), dihydroorotate dehydrogenase (DHODH), and p66shc/cytochrome c system produce superoxide and hydrogen peroxide [34, 35]. These highly reactive species can promote oxidative damage to surrounding proteins, lipids, and DNA [36], but are rapidly quenched by antioxidant enzymes under most circumstances [37].
As an organism ages, mtDNA suffers extensive oxidative damage due to the proximity of mtDNA to mtROS production [38]. The lack of protection by histones and deficiency in DNA repair mechanisms relative to nuclear DNA further lead to the vulnerability of mtDNA [39]. As a result, mtDNA undergoes a greater rate of mutations compared to nuclear DNA. The accumulation of mutations, including point mutations, insertions, deletions, and alterations in mtDNA copy number [40], further exacerbates mtROS generation, leading to a self-enforcing cycle of mtDNA damage and mtROS production [41]. These ongoing activities ultimately result in a state of mitochondrial dysfunction in which various functions of the mitochondria, including electron transfer by the ETC and ATP production, are impaired during the aging process [6, 42]. Cancer cells develop mitochondrial dysfunction in a similar fashion (Fig. 1) [6]. The increased mitochondrial activity required by cancer cells leads to increased production of mtROS. This results in elevated levels of oxidative damage and mtDNA mutations, leading to mitochondrial dysfunction. A vicious cycle develops in which mitochondrial dysfunction aggravates mtROS generation, leading to further mitochondrial dysfunction (Fig. 1). However, as we will discuss in the following sections, cancer cells use the mitochondrial dysfunction to their advantage throughout the progression of cancer [43].
The role of mitochondria in tumorigenesis
Although the link between mitochondrial dysfunction and tumorigenesis is not fully clear, some evidence points to the role of mitochondrial dysfunction and oxidative stress in malignant transformation. Severe mutations in the tRNALys gene of mtDNA (m.8363G > A) that critically disrupt the ETC do not support tumorigenesis, but mutations in the MT-ND1 (m.3460G > A), MT-ND4 (m.11778G > A) and MT-ND6 (m.14484 T > C) of mtDNA that mildly impair the ETC promote mild mitochondrial dysfunction and support tumorigenesis [44]. Mutations in the ATP synthase subunit 6 gene (ATP6) in mtDNA can increase superoxide production from Complexes I, II, and III to provide an advantage in early tumor growth [45]. The T8993G mutation in ATP6 specifically favors tumorigenicity in prostate cancer (PCa) [46]. A heteroplasmic mutation in the NADH dehydrogenase subunit 5 gene (ND5), identified in colorectal tumors [47], disrupts the synthesis of the ND5 subunit of Complex I and subsequently hinders the proper assembly of Complex I. This causes an increase in mtROS levels and enhanced tumorigenicity [48]. Furthermore, increases in mtROS due to the Kirsten rat sarcoma virus (KRAS) drive the formation of pancreatic precancerous lesions [49]. Overall, key features of mitochondrial dysfunction, mutations in mtDNA and increases in mtROS, appear to support tumor development as early as the tumor initiation stage.
Mitochondrial ROS and cellular signaling
Mitochondrial ROS show beneficial effects in cancer cells via direct induction of multiple signaling events for cell growth. For example, hydrogen peroxide can inactivate the tumor suppressor phosphatase and tensin homolog (PTEN) by oxidizing its essential cysteine residue in various types of cells [50, 51]. This leads to the accumulation of phosphatidylinositol 3,4,5-trisphosphate (PIP3) followed by signaling through the Ak strain transforming (AKT) pathway that promotes the growth and survival of cancer cells [50, 52, 53]. Hydrogen peroxide also drives tumorigenesis via the AMP-activated protein kinase (AMPK) pathway [54] and can inactivate Cdk1-opposing phosphatases to allow cyclin-dependent kinase 1 (Cdk1)-mediated mitotic progression [55]. Production of mtROS from Complex III of the ETC is essential for KRAS-mediated mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signaling for cell proliferation and tumorigenicity [56]. Additionally, mtROS can trigger protein kinase D1 (PKD1), thereby activating transcription factors nuclear factor kappa B1 (NF-κB1) and NF-κB2 to upregulate EGFR signaling [49]. These direct roles of mtROS in activating proliferative pathways highlight the tumor-promoting role of increased mtROS production by dysfunctional mitochondria.
Metabolite formation and tumor growth
Mitochondrial dysfunction increases levels of high-mobility group box 1 (HMGB1) [57]. HMGB1 is upregulated in cancer and is released into the extracellular space to allow interaction with the receptor for advanced glycation end products (RAGE) [58]. This induces a signaling pathway in which RAGE phosphorylates Complex I to enhance ATP production [59]. Moderate mitochondrial defects due to mtDNA mutations promote integration of glutamine into the TCA cycle by conversion of glutamine to glutamate, thereby fueling the TCA cycle for the production of ATP [60]. During inflammation-associated pancreatic tumor development, an increase in mitochondrial fatty acid β-oxidation occurs [61]. Elevated fatty acid β-oxidation activity has been observed in tumor spheres and is associated with increased NADH and FADH2 [62]. Therefore, increased fatty acid β-oxidation-mediated generation of ROS and altered energy metabolism in mitochondria promote tumor growth and progression. Overall, dysfunctional mitochondria can increase the production of ATP-generating metabolites to meet the increased metabolic demands of tumor growth.
Tumor microenvironment and inflammation
Tumor cells in the tumor microenvironment (TME) recruit non-malignant cells to promote tumor growth and further propagate mitochondrial dysfunction [63, 64]. Among the non-malignant cells of the TME, inflammatory immune cells contribute to mitochondrial dysfunction by directly releasing ROS into the TME [65, 66] or secreting pro-inflammatory cytokines [67]. For example, macrophages and neutrophils can release various forms of ROS to damage the DNA of neighboring cells [68]. Likewise, immune cells can secrete factors such as tumor necrosis factor alpha (TNF-α), interleukin 1 beta (IL-1β), and interferon-gamma (IFN-γ) to alter mitochondrial membrane potential, inhibit the ETC, increase proton leak, and ultimately stimulate the production of mtROS [69,70,71,72,73]. This underscores the idea that mitochondrial dysfunction does not always occur due to the activity of tumor cells alone, but may involve non-tumor cells within the TME.
Adaptation to hypoxia
As tumors grow, the demand for oxygen and nutrition exceeds the availability of sufficient vasculature for supporting tumor proliferation. This leads to a state of hypoxia in various malignancies [74]. In response, hypoxia-inducible factor-1α (HIF-1α) signaling is activated to continue the growth and survival of the tumor [75]. Normally, HIF-1α is destabilized by prolyl hydroxylases (PHDs) in the presence of oxygen [76]. However, as oxygen levels decline during hypoxia, PHD activity is inhibited and HIF-1α is stabilized [74]. During hypoxic conditions, mitochondria generate excess mtROS from Complex III of the ETC, promoting the stabilization of HIF-1α. [77, 78]. Even respiration incompetent cancer cells, due to defects in Complex III, can still produce increased mtROS to stabilize HIF-1α during hypoxia [79]. Altogether, this indicates that dysfunctional mitochondria, despite impairments in ETC, assist in HIF-1α stabilization through the production of mtROS.
Mitochondrial DNA mutations and resistance to cell death
Mitochondrial dysfunction also contributes to deficits in the cell death pathways. Various mutations in mtDNA modulate the apoptotic response. For example, partial deletions in mtDNA offer protection against mitochondria-initiated cell death [80]. The previously mentioned mutations in ATP6 and ND5 raise the apoptotic threshold of tumor-forming cells, allowing resistance against oxidative stress-induced cell death [45, 48]. In vivo, a combination of mtDNA mutations in ND4, ATP6, and 16S rRNA genes or in the COI and ND3 genes confers resistance to 5-fluorouracil- and cisplatin-induced apoptosis [81]. In ovarian cancer, mutations in ND4 lead to acquired chemoresistance against paclitaxel and carboplatin [82]. Gefitinib-mediated mitochondrial dysfunction in non-small cell lung cancer (NSCLC) cells is proposed to aid in the development of drug resistance [83, 84]. The G10398A substitution in ND3 produces apoptotic resistance to etoposide [85]. Although counterintuitive, numerous mutations in the mitochondrial genome actually enhance the survival of cancer cells [86, 87].
Mitochondrial DNA mutations, mtROS, and metastasis
Metastasis is a process observed in late-stage disease in which tumor cells disseminate to secondary sites. Metastasis is the leading cause of mortality in patients [88, 89]. Mutations in mtDNA, particularly in the ND genes, enhance the metastatic activity of cancer cells. In breast cancer, the G10398A substitution in ND3 increases the number of metastatic foci in the lungs of mice [85]. Similarly, missense mutations in breast cancer such as C12084T in the ND4 gene and A13966G in the ND5 gene are associated with defects in mitochondrial respiration and augmented metastatic potential independent of ROS overproduction [90]. Highly metastatic breast cancer cells exhibit downregulation of mitochondrial transmembrane protein 126A (TMEM126A), which results in mitochondrial dysfunction, as shown by altered mitochondrial membrane potential and increased mtROS [91]. This decrease in TMEM126A has been shown to promote epithelial–mesenchymal transition (EMT), extracellular matrix (ECM) remodeling, cell adhesion, and increased lung metastasis [91]. In lung cancer, the G13289A mutation in ND5 increases invasive activity [92]. G13997A and 13885insC mutations in ND6 produce defects in Complex I of the ETC, leading to an overproduction of mtROS and an increase in the metastatic potential of lung carcinoma cells [93]. Likewise, ND6 nonsense and missense mutations are associated with higher rates of lymph node metastases in human lung cancer by promoting migratory and invasive activities [94]. The 13885insC mutation in ND6 is associated with the overexpression of metastasis-related genes and promotes metastasis in various cancers [95]. In addition, a study of cancer stem cells demonstrates that a subset of these cells maintain elevated mtROS, which serve to activate MAPK and promote EMT in order to increase metastatic potential [62]. Even as tumors approach late-stage disease, dysfunctional mitochondria are able to assist in tumor development at secondary sites.
Overall, mitochondria contribute to various stages of cancer development and progression despite their dysfunctional state [3]. Moreover, cancer cells are able to take advantage of the mtDNA mutations and elevated mtROS levels associated with mitochondrial dysfunction to promote growth and survival. However, as cancer cells continue to propagate in a highly proliferative manner, mitochondrial dysfunction can worsen and become lethal (Figs. 1 and 2) [6]. The resulting increases in oxidative stress due to mtROS lead to further proteotoxic stress, which is marked by oxidative damage, protein unfolding, and protein aggregation [36, 96]. This leaves mitochondria in a fragile state and pushes cells to the brink of cell death. To adjust to elevated mtROS, cancer cells maintain oxidative status at a level that benefits, but does not become toxic to cancer cells [7,8,9].
Relieving mitochondrial stress through antioxidants
Mitochondria rely upon antioxidants to quench mtROS. Antioxidant enzymes such as superoxide dismutase (SOD), catalase, and glutathione peroxidase scavenge excess mtROS [37]. SODs allow the dismutation of superoxide radicals into hydrogen peroxide and oxygen. Catalases and peroxidases further process hydrogen peroxide into water [37]. An enhanced antioxidant response is commonly observed in cancer [97]. However, levels of individual antioxidants can vary and are, at times, lower in tumors compared to normal tissue [98,99,100,101,102,103]. As mtROS levels exceed the capacity of the antioxidant system due to mitochondrial dysfunction, another mechanism must be at play in order to preserve mitochondrial health.
Relieving mitochondrial stress through the mitochondrial unfolded protein response (UPRmt)
As mentioned, chronic oxidative stress due to inherent mitochondrial dysfunction leads to proteotoxic stress: unfolding, misfolding, and aggregation of mitochondrial proteins. If left unresolved, the accumulation of protein aggregates can further increase oxidative stress, leading to a feedback loop that propagates mitochondrial dysfunction [104]. For example, disruption of proteins in the ETC enhances the leakage of electrons and leads to increased mtROS [105]. The following section will discuss a mitochondrial stress response, consisting of chaperones and proteases, which halts this harmful feedback loop by directly targeting proteotoxicity in mitochondria (Fig. 2).
Early signs of a mitochondrial stress response
Signs of a mitochondrial stress response involving chaperones and proteases emerged as early as 1996, when Martinus et al. discovered that generating mitochondria-specific stress, by depletion of mtDNA, induces the transcriptional activation of mitochondrial chaperones’ heat shock protein 60 (HSP60) and heat shock protein 10 (HSP10) in rat hepatoma cells [11]. Further evidence of this mitochondrial stress response was shown by a study in which the accumulation of protein aggregates in the mitochondrial matrix induced the transcriptional upregulation of mitochondria-specific genes that code for chaperones HSP60 and HSP10, as well as the protease, caseinolytic protease (ClpP) [106]. Altogether, these chaperones and proteases were shown to reduce mitochondrial protein aggregation (Fig. 3) [106].

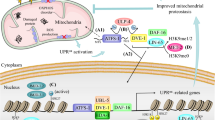
Hypothetical model of the mitochondrial unfolded protein response (UPRmt) signaling in cancer. Mitochondrial dysfunction increases mtROS (oxidative stress), which damages proteins in the mitochondria and cause the unfolding and aggregation of mitochondrial proteins (proteotoxic stress). In response, the transcription factor ATF5 induces the upregulation of mitochondrial components to ease proteotoxic stress. Chaperones HSP60, HSP10, and mtHSP70 mediate the refolding of proteins into their proper conformation. Proteases LONP1 and ClpP cleave and dispose of any additional damaged proteins that did not undergo processing by HSPs. Together, this system maintains mitochondrial integrity in the face of continuous oxidative and proteotoxic stress in cancer
Various proteins involved in the mitochondrial stress response are encoded by the nuclear DNA [11, 106] and must be imported into the mitochondria [107, 108]. Therefore, mitochondria-to-nucleus communication during mitochondrial stress plays a critical role in maintaining mitochondrial homeostasis or functions. This process is known as mitochondrial retrograde signaling, in which mitochondria send a signal to the nucleus in order to regulate nuclear gene expression and allow mitochondrial homeostasis (Fig. 3) [109, 110].
C. elegans as a model to study UPRmt
The C. elegans model has been integral in understanding this mitochondria-specific retrograde stress response, now known as the UPRmt. During UPRmt, defective protein folding in mitochondria transactivates hsp-60 and hsp-6, which encode chaperones HSP60 and mitochondrial HSP70 (mtHSP70), respectively [111], as well as clpp-1, which encodes ClpP. [112]. Using C. elegans, Nargund et al. demonstrated that the transcription factor ATFS-1 is key to activating the UPRmt response [113]. ATFS-1 contains both a nuclear localization sequence (NLS) and mitochondrial targeting sequence (MTS). Under non-stress conditions, ATFS-1 is imported into the mitochondria and is rapidly degraded by the protease, Lon peptidase 1 (LONP1). However, during mitochondrial stress, proteotoxicity impairs protein import into the mitochondria and, consequently, ATFS-1 accumulates in the nuclei to induce the expression of a broad variety of genes involved in mitochondrial stress, including hsp-60 [113].
Another study by Nargund et al. used ChIP-Seq to identify 381 genes induced by ATFS-1 during mitochondrial stress [114]. Of these genes, ATFS-1 interacts with 70 of their corresponding promoters. Notably, ATFS-1 binds and suppresses mtDNA-encoded OXPHOS genes in both the nuclear and mitochondrial genomes. Concurrently, ATFS-1 binds promoters of NADH ubiquinone oxidoreductase assembly factors to aid in ETC complex. This indicates that ATFS-1 limits further transcription of OXPHOS genes while maintaining OXPHOS complexes already present, in order to optimize respiratory activity during mitochondrial stress [114]. Protein synthesis in the cytoplasm is also reduced during UPRmt [115], presumably to reduce cell stress. Speculations remained as to whether mammalian cells similarly utilize a regulated UPRmt system. The individual works of Nargund, Wu, Schulz, and Fiorese et al. began to fill this gap in knowledge [10, 14, 113, 116].
Transcription factor ATF5 and the mammalian model of UPR mt
Transcriptomic and proteomic analysis later revealed strong conservation in the UPRmt response between C. elegans, mice, and humans. Genes encoding chaperones (Hspd1, Hspe1, Hspa9) and proteases (Clpp, Lonp1) in C. elegans also associate with UPRmt homologs in mammals [14]. Over 400 genes were found to be activated in the mammalian UPRmt response [14, 113], which can be categorized by various functions including mitochondrial biogenesis, metabolism, protein folding quality control, and ROS detoxification [116]. Recently, Fiorese et al. identified activating transcription factor 5 (ATF5) as the mammalian homolog of ATFS-1 [10]. When ATF5 was expressed in worms lacking ATFS-1, it induced hsp-60 during mitochondrial stress but not during endoplasmic reticulum (ER) stress, indicating organelle specificity. In HEK293T cells, elevated mtROS activated ATF5-mediated transcription of the genes for HSP60, mtHSP70, and LONP1. Again, ER chaperones were not induced by ATF5 under these conditions. Similar to ATFS-1, ATF5 contains both an NLS and MTS, allowing ATF5 to shuttle between the mitochondria and nucleus during UPRmt. Knockdown of ATF5 in HEK293T reduced proliferation, mitochondrial respiration, and induction of HSP60, mtHSP70, and LONP1. Overall, this study highlights ATF5 as the transcription factor that specifically mediates mammalian UPRmt and is independent of the ER stress response [10].
Other transcription factors in UPR mt
In addition to ATF5, other transcription factors have been implicated in the UPRmt. Transcription factor ATF4 induces the expression of another transcription factor CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP) [117, 118]. CHOP, in turn, dimerizes with transcription factor C/EBPβ to function as a regulator of the mitochondrial stress response [106]. In breast cancer, misfolded proteins in the mitochondria activate estrogen receptor alpha (ERα), which increases the transcription of the intermembrane space protease OMI and increases the activities of the proteasome [119]. ERα-negative breast cancer cells respond to mitochondrial stress by increasing the expression of sirtuin deacetylase 3 (SIRT3), leading to the deacetylation of the transcription factor FOXO3a [120]. Forkhead box protein O3a (FOXO3a) then translocates to the nucleus to promote the transcription of antioxidants superoxide dismutase 2 (SOD2) and catalase [120]. Sirtuin deacetylase 7 (SIRT7) can alleviate protein folding stress in hematopoietic stem cells, partly by inducing the expression of canonical UPRmt components including HSP60, HSP10, and ClpP [121]. Another transcription factor, heat shock factor 1 (HSF1), has been shown to induce the expression of UPRmt chaperones in mammalian cells in response to mitochondria-specific stress [122].
Altogether, these other arms of UPRmt act in tandem to maintain the integrity of the mitochondria. However, the transcription factors ATF4, CHOP, C/EBPβ, ERα, and HSF1 are also activated during ER stress to promote endoplasmic reticulum unfolded protein response (UPRer) [117, 118, 123,124,125,126,127,128,129]. While overlap in the functions of ATF5 between UPRmt and UPRer is expected due to the known regulation of ATF5 by CHOP [130] and ATF5 having been shown to increase in expression due to ER stress [131, 132], studies continue to demonstrate the inability of ATF5 to induce chaperones during ER stress [10, 133]. Therefore, ATF5 remains to be the only mitochondria-specific transcription factor during mitochondrial stress and is viewed as the main regulator of UPRmt (Fig. 3).
Mitochondrial dysfunction is known to induce ATF5 transcripts [10, 134, 135] and transactivate UPRmt targets downstream of ATF5, due to the various stressors associated with mitochondrial dysfunction: mtDNA depletion [11, 111], accumulation of unfolded proteins in the mitochondria [106, 119], inhibition of ETC activity [10, 113, 136], and deficient expression of ETC components [111]. ATF5 has been evidenced to transactivate HSP60, mtHSP70, and LONP1 [10] and likely induces HSP10 and ClpP [137] during activation of mammalian UPRmt. Mitochondrial chaperones HSP60, mtHSP70, and HSP10 assist in protein folding and are particularly essential to reducing the aggregation of unfolded proteins during stress conditions [138]. Mitochondrial proteases LONP1 and ClpP degrade damaged proteins during this response [139, 140]. Digested peptides can then be transported by HAF-1, a mitochondrial inner-membrane-localized ABC transporter, from the mitochondrial matrix to the intermembrane space where they are believed to diffuse into the cytoplasm [141]. Between the mitochondrial chaperone and protease systems, it is believed that cells prefer to utilize chaperones before resorting to proteases in order to manage unfolded proteins in the mitochondria [142]. In light of this, we believe that mitochondrial dysfunction in cancer produces a state of chronic mitochondrial stress, which then constitutively activates ATF5 and upregulates the expression of the UPRmt components (Fig. 3).
Roles of the UPRmt components in cancer
UPRmt signaling plays a critical role in cancer [143] and requires further investigation to clearly indicate its impact on tumor growth and progression. While many studies, primarily utilizing C. elegans and sometimes mammalian kidney cells, have confirmed the presence and function of UPRmt during mitochondrial stress, the study of the UPRmt as a whole system has been lacking in cancer. However, individual components of UPRmt have displayed roles in tumor growth and survival. This is unsurprising due to the prevalence of mitochondrial dysfunction and oxidative stress throughout cancer development and progression. The following sections will discuss the known cancer-specific roles of ATF5 and key UPRmt proteins downstream of ATF5: HSP60, HSP10, mtHSP70, LONP1, and ClpP.
Activating transcription factor 5 (ATF5)
ATF5, part of the bZip family of transcription factors, contains a leucine zipper that allows dimerization with ATF5 and other transcription factors, and contains a basic N-terminal portion involved in DNA binding [144,145,146]. The structure also includes an MTS, nuclear export sequence (NES), and NLS. This allows ATF5 to translocate between different cell compartments [10]. ATF5 must homodimerize or form heterodimers with other transcription factors in order to function [146]. Binding partners of ATF5 include other ATF5 proteins, as well as C/EBPβ [147, 148].
ATF5 is upregulated in glioblastoma [149] as well as cancers of the breast [150], pancreas [151], rectum [152], and ovaries [153]. High ATF5 expression correlates with reduced survival in glioma [154] and lung cancer patients [155]. ATF5 expression can be induced by various forms of stress: heat shock [156], amino acid depletion [157], increases in ROS [10, 131, 158], inhibition of the proteasome [131], endoplasmic reticulum stress [131, 132], and radiation [155].
As a transcription factor, ATF5 regulates the expression of various anti-apoptotic genes in cancer. For example, ATF5 regulates Egr-1 expression in glioma and breast cancer cells to mediate proliferation and survival [159]. ATF5 transactivates BCL-2 in glioma and breast cancer cells to promote survival [160]. In glioblastoma, non-small cell lung cancer, and pancreatic cancer, ATF5 regulates the protein expression of deubiquitinase USP9X, which, in turn, stabilizes B cell lymphoma 2 (BCL-2) and myeloid leukemia 1 (MCL1) [161]. In addition, ATF5 regulates B cell lymphoma-extra large (Bcl-xL) expression in glioblastoma cells [161]. Although highly probable, studies have not yet shown the ability of ATF5 to regulate the expression of UPRmt components in cancer. In the following sections, the pro-tumor functions of downstream targets of ATF5 will be examined.
Heat shock protein 60 (HSP60)
Monomers of HSP60 arrange into stacked heptameric rings that form a complex with HSP10 and ATP to create a classical theorized “football-shaped” structure that can envelop misfolded or unfolded proteins and assemble them into their proper conformation [162,163,164]. This is particularly essential for mitochondrial proteins, as most of these proteins are produced outside of the mitochondria and must be imported into the mitochondria in an unfolded state [107, 108]. Interestingly, during the chaperonin reaction cycle active single- (half-football) and double-ring (football) complexes coexist for the proper folding of proteins [164]. The HSP60–HSP10 complex is crucial during high oxidative stress, as ROS can also directly oxidize mitochondrial proteins to promote protein aggregation [104, 165]. Indeed, protein aggregation has been shown to cause the accumulation of HSP60 in the mitochondria [166]. In addition, HSP60 can play a role in posttranslational modifications [167].
HSP60 is overexpressed in various cancers. Specifically, HSP60 is upregulated early in prostate carcinogenesis [168] and large bowel carcinoma [169], increases during the progression of hepatocarcinogenesis [170], and indicates a greater risk for progression of urothelial tumors of the bladder [171]. High HSP60 levels correlate with advanced tumor grade in PCa [172], pancreatic cancer [173], and large bowel carcinoma [174]. HSP60 is increased in the serum of patients with metastatic colorectal cancer [175], is associated with the presence of lymph node metastases in large bowel carcinoma [174], and correlates with deep invasion and lymph node metastasis in gastric cancer [176]. Furthermore, elevated HSP60 levels indicate reduced survival in patients with PCa [172], gastric cancer [176], and neuroblastoma [177].
Knockdown of HSP60 decreases cell proliferation in pancreatic cancer [173], ovarian cancer [178], breast cancer [179], and glioblastoma [180, 181]. And HSP60 knockdown inhibits tumor growth in xenograft models of pancreatic cancer [173] and glioblastoma [181]. HSP60 knockdown leads to severe deficiencies in mitochondrial functions, which hinder cell growth and survival. For example, in glioblastoma, reduction in HSP60 increases ROS, which in turn activates the AMPK pathway to inhibit protein translation and slow cell proliferation [180]. In ovarian cancer, knockdown of HSP60 suppresses pathways related to OXPHOS and alters metabolic pathways to allow accumulation of adenine, which activates the adenine-AMPK pathway, suppresses the mammalian target of rapamycin (mTOR) pathway, and inhibits cancer progression [178]. In pancreatic cancer, HSP60 knockdown reduces the expression of subunits in OXPHOS Complexes I, III, IV, and V, disrupts the formation of Complexes I and III, and blocks mitochondrial respiration and ATP production [173]. Consequently, diminished ATP deters the phosphorylation of Erk1/2 and promotes apoptotic activity [173]. As shown in various cancer types, HSP60 associates with cyclophilin D to inhibit cyclophilin D-mediated mitochondrial permeability transition [181]. As such, HSP60 knockdown triggers mitochondrial permeability transition, cytochrome c release, and loss of mitochondrial membrane potential [181]. In addition to direct mitochondrial functions, HSP60 regulates the expression and release of IL-8 in prostate and colon cancers, possibly via transforming growth factor-beta (TGF-β), to enhance cell survival [182].
Overexpression of HSP60 enhances the migratory and invasive abilities of FADU cells and promotes the development of metastatic nodules in the lung [183]. The increased β-catenin levels and transcriptional activity due to HSP60 overexpression are believed to underlie this metastatic phenotype. HSP60 directly interacts with β-catenin [183]. The transcription factor c‐MYC induces overexpression of HSP60, which causes the transformation of Rat1a cells [184].
HSP60 interacts with various cellular proteins to exert biological functions. HSP60 complexes inhibit clusterin to promote cell survival in neuroblastoma [185]. HSP60 can also form a complex with cell cycle and apoptosis regulator protein 2 (CCAR2) and bind the anti-apoptotic protein survivin to promote cell survival [177, 186]. HSP60 is involved in hepatocyte growth factor (HGF)-induced ERK activation to promote cell migration in hepatocellular carcinoma [187].
Although HSP60 is best known as a mitochondrial chaperone and is predominantly located in the mitochondria in non-malignant cells, non-chaperone activity of HSP60 has been reported to be present in the cytoplasm, plasma membrane, and extracellular space of cancer cells [188,189,190]. HSP60 accumulates in the cytoplasm during apoptosis [188]. Depending on the apoptotic stimuli, this can occur with or without mitochondrial release and HSP60 can display either pro-apoptotic or pro-survival properties, respectively [188]. As a pro-apoptotic protein, HSP60 localized in the cytoplasm assists in the cleavage and activation of procaspase 3 [188, 191]. As a pro-survival protein, cytoplasmic HSP60 is known to bind and restrain p53 to inhibit p53-dependent upregulation of pro-apoptotic Bax [192]. Additionally, cytoplasmic HSP60 can directly bind Bax/Bak to block their translocation to the mitochondria, and thus, interferes with apoptosis [193, 194]. Furthermore, cytoplasmic HSP60 plays a role in TNF-induced expression of SOD1 and Bfl-1/A1, and activation of IKK/NF-κB [195].
Various studies have observed HSP60 on the surface of cancer cells [190, 196,197,198]. HSP60 proteins localized in the cell membrane are suggested to play a role in the metastatic process [198]. For example, HSP60 can activate α3β1 integrin [199], a transmembrane receptor that promotes adhesion of breast cancer cells to metastatic sites [200, 201]. HSP60 at the cell surface can also be actively exported into the extracellular space by exosomes. Exosomes are nanometer-sized membrane vesicles containing protein, lipids, and DNA, which are released into the circulation and then taken up by cells in neighboring or distant areas as a means of cell–cell communication [202, 203]. A study by Campanella et al. demonstrated that HSP60 is present on the cell membrane and exosomal membrane and in the Golgi apparatus of cancer cells [189]. They propose a process by which HSP60 present at the cell membrane is internalized by lipid rafts and packaged into multivesicular bodies (MVB) for secretion via exosomes. The Golgi apparatus can assist in transferring cytoplasmic HSP60 into MVB or releasing them from the cell as free, soluble HSP60 [189]. HSP60 can be modified by glycosylation in the endoplasmic reticulum before release, presumably affecting the immunological properties of HSP60 [204]. Overall, exosomal release allows proximate and distant circulation of HSP60.
Interestingly, HSP60 is not overexpressed in all cancers and does not necessarily associate with a poor prognosis in patients. For example, HSP60 is downregulated in bronchial cancer [205], colorectal cancer [206], clear cell renal cell carcinoma [207], and hepatocellular carcinoma [208]. Interestingly, patients with esophageal squamous cell carcinoma [209], clear cell renal cell carcinoma [210], and hepatocellular carcinoma [208] experience better survival rates when their tumors display elevated expression of HSP60. Furthermore, overexpression of HSP60 suppresses cell proliferation in clear cell renal cell carcinoma [207] and inhibits invasive activity in hepatocellular carcinoma [208]. This highlights the multimodal functions of HSP60 across various cancers.
Heat shock protein 10 (HSP10)
Compared to HSP60, less is known about the HSP60 binding partner HSP10. HSP10 is overexpressed in astrocytoma [211], oral squamous cell carcinoma [212], nasopharyngeal carcinoma [213], large bowel carcinoma [169, 174]. HSP10 is upregulated early during prostate tumorigenesis [168], and levels of HSP10 have been shown to increase throughout the progression of large bowel carcinoma [169]. HSP10 correlates with pathological grade in oral squamous cell carcinoma [212] and nasopharyngeal carcinoma [213], lymph node metastasis in oral squamous cell carcinoma [212], nasopharyngeal carcinoma [213], and large bowel carcinoma [174], and recurrence in astrocytoma [211]. High HSP10 is associated with reduced overall survival in astrocytoma [211], oral squamous cell carcinoma [212], and nasopharyngeal carcinoma [213].
Mitochondrial heat shock protein 70 (mtHSP70)
Mitochondrial heat shock protein 70 (mtHSP70) is a member of the HSP70 chaperone family and is predominantly located in the mitochondria [214]. It contains a nucleotide-binding domain and a substrate-binding domain [215]. Similar to HSP60, mtHSP70 holds a role in housekeeping and mediates the refolding of unfolded proteins [216]. Multiple studies highlight the protective effect of mtHSP70 on cancer cells and thereby increasing malignancy in cancer.
The chaperone mtHSP70 is upregulated in melanoma [217] as well as cancers of the liver, kidney, thyroid, breast, brain, ovary, lung, and colon [218,219,220,221,222,223]. Expression levels of mtHSP70 correlate with various clinical features. For example, increased expression of mtHSP70 in breast cancer indicates higher histological grade and decreased survival [224]. Furthermore, mtHSP70 is increased in invasive ductal carcinoma relative to ductal carcinoma in situ and correlates with lymph node metastasis [224]. Similarly, in both hepatocellular carcinoma and non-small cell lung cancer, elevated levels of mtHSP70 are associated with advanced tumor stage, metastasis, and reduced overall survival [220, 223, 225].
Knockdown of mtHSP70 inhibits proliferation, migration, and invasion of various cancer lines [218, 222, 225]. Notably, studies in neuroblastoma indicate that reduction in mtHSP70 is associated with Drp-1-dependent mitochondrial fragmentation [226]. Furthermore, mtHSP70 knockdown leads to diminished levels of p‐ERK1/2, p‐c‐Raf [222], the receptor tyrosine kinase RET (rearranged during transfection), and anti-apoptotic proteins, Bcl-2, Bcl-xL, and Mcl-1 [218].
Overexpression of mtHSP70 induces malignant transformation of fibroblasts, enhancing proliferation and tumor formation in mice [227]. In breast cancer, augmenting mtHSP70 increases the levels of stemness markers such as ATP-binding cassette transporter G2 protein (ABCG2), octamer-binding transcription factor 4 (OCT-4), CD133 and enhances resistance to chemotherapy [228]. In addition, overexpression of mtHSP70 supports EMT transition and metastatic activity in breast cancer.
Elevated mtHSP70 downregulates epithelial markers, upregulates mesenchymal markers, and increases the migratory and invasive abilities of breast cancer cells [221, 228, 229].
Mitochondrial HSP70 can bind various proteins to produce an anti-apoptotic response. For example, mtHSP70 interacts with p53 in response to stressors such as cisplatin [230]. During stress, mtHSP70 binds and prevents nuclear accumulation of tumor suppressor p53, thereby suppressing the transactivation of pro-apoptotic gene targets [231,232,233]. In response to mtHSP70 knockdown, hepatocellular carcinoma cells undergo p53-mediated apoptosis [230]. On the other hand, in HepG2, a cell line that lacks mtHSP70-p53 interactions, interference with mtHSP70 expression has no effect on cell viability [230].
ERK phosphorylates HIF-1α to increase nuclear translocation and promote its transcriptional activity [234]. During hypoxia, if ERK is inactive and unable to phosphorylate HIF-1α, mtHSP70 binds and localizes HIF-1α to the outer membrane of the mitochondria [235]. HIF-1α then associates with voltage-dependent anion-selective channel 1 (VDAC1) and hexokinase-II (HK-II) to prevent apoptosis and promote survival. This mechanism has been shown to protect HeLa cells from etoposide- and doxorubicin-induced death [235].
Interactions between mtHSP70 and other protein targets have also been reported in cells with BRAF mutations. In this setting, deregulated MEK-ERK signaling increases interactions between adenine nucleotide translocase 3 (ANT3) and peptidyl-prolyl isomerase cyclophilin D, greatly increasing mitochondrial permeability and promoting mitochondria-mediated cell death [236]. However, mtHSP70 directly interacts with ANT3 to inhibit ANT3–cyclophilin D interactions and secure cell survival [236]. In addition, mtHSP70 can help overcome v-raf murine sarcoma viral oncogene homolog (BRAF) mutation-induced growth arrest signaling by inhibiting Raf-induced MEK/ERK activity through inhibition of MEK1/2 protein expression and ERK1/2 phosphorylation [217].
Interestingly, mtHSP70 has also been reported to form complexes with HSP60 [237], although the function of this complex remains to be fully elucidated. However, it is possible that this study has detected a transient interaction in which HSP60 is mediating the folding of mtHSP70 that has been recently imported into the mitochondria.
Lon peptidase (LONP1)
As mitochondrial proteins undergo folding, they are prone to aggregation. This is especially true in the presence of oxidative stress [238,239,240]. LONP1, which is composed of a hexagonal cylinder with a large, unfolding, and degradation chamber [241], acts as a protease that cleaves aggregates into short peptides that are cleared from the mitochondria. In doing so, LONP1 maintains mitochondrial proteostasis [142]. LONP1 does not appear to degrade folding intermediates of mitochondrial matrix proteins, indicating that mitochondria preferentially utilize chaperones to prevent aggregates and restore protein functions. Proteolytic actions are taken only as a final measure.
LONP1 is upregulated in melanoma [242], prostate cancer [243], pancreatic cancer [244], and colorectal cancer [242]. Notably, LONP1 increases during colorectal cancer progression and is particularly increased in colorectal samples with mutated p53 or β-catenin [245]. Additionally, hypoxia increases LONP1 expression in PCa [243]. High expression of LONP1 correlates with reduced overall survival in neuroblastoma, breast cancer, colorectal cancer, renal cell carcinoma [243], and metastatic cohorts of melanoma [242].
Knockdown of LONP1 leads to reduced proliferation in melanoma [242], colorectal cancer [242], pancreatic cancer [244], and PCa cells [243]. In vivo, LONP1 knockdown inhibits the growth of prostate [243] and colorectal tumors [242] and inhibits the formation of metastatic lesions from primary prostate [243] and melanoma tumors [242]. A deficiency in LONP1 expression in mice inhibits the formation and growth of azoxymethane- and dextran sulfate-induced colorectal tumors [242]. Similarly, these animals are also resistant to 12-dimethylbenzanthracene and tetradecanoylphorbol acetate (DMBA/TPA)-induced skin papilloma [242]. In addition, several have implicated LONP1 in gastric carcinogenesis. In response to H. pylori infection, LONP1 expression increases in gastric cancer cells [246], and LONP1 is necessary for H. pylori-induced gastric cell proliferation and promotes H. pylori-induced metabolic switch to glycolysis. [246].
The pro-tumor effects of LONP1 can be partially attributed to LONP1-mediated regulation of Bcl-2 in melanoma [242], cyclin D1 in pancreatic cancer [244], and β-catenin in colorectal cancer [245]. LONP1 also appears to have a role in EMT. As shown in pancreatic cancer cells, LONP1 knockdown increases the expression of the epithelial marker claudin-1 and decreases the mesenchymal marker vimentin as well as transcription factors snail and slug [244]. In addition, decreases in matrix metalloproteinase (MMP2), MMP9, and p-JNK are observed after LONP1 knockdown. Overall, these features are believed to contribute to a resulting decrease in migratory and invasive abilities of LONP1 knockdown cells [244].
Alterations in LONP1 expression lead to various dysfunctions of the mitochondria. In PCa, knockdown of LONP1 associates with an accumulation of misfolded subunits of OXPHOS Complex II and V, reduced assembly of OXPHOS Complexes I, III, IV, and V, as well as inhibition of activities of OXPHOS Complexes I, II, and V [243]. Consequently, mitochondrial respiration and ATP production are decreased. [243]. Studies in gastric cancer demonstrate that LONP1 knockdown reduces mitochondrial respiration [246]. In melanoma, LONP1 knockdown inhibits the formation of OXPHOS Complexes I and III and reduces the activities of OXPHOS Complexes I, II, and III [242]. Consequently, cells experience an increase in mitochondrial fragmentation and ROS levels as well as a decrease in mitochondrial respiration and ATP.
Interestingly, overexpression of LONP1 also leads to deleterious effects. LONP1 overexpression in cancer cells lowers the activities of Complexes I, II, III, and IV in conjunction with reduced mitochondrial respiration [242]. A study in squamous cell carcinoma further demonstrates that while LONP1 overexpression induces mtROS production through Complex I, mtROS can activate Ras and MAPK signaling and promote the survival of these cells [247]. Additionally, LONP1 binds and stabilizes Hsp60–mtHsp70 complexes, particularly under oxidative stress conditions [248]. This interaction is proposed to inhibit apoptosis. However, knockdown of LONP1 has no effect on the individual expression levels of HSP60 and mtHSP70 [248].
Caseinolytic protease (ClpP)
CIpP forms a tetradecameric cylinder, similar to LONP1, which accepts protein substrates for degradation [249]. Notably, various adaptors can bind to ClpP to influence substrate selectivity [250]. In addition, accessory proteins deliver protein substrates to ClpP for degradation.
ClpP is upregulated in breast cancer, PCa, and acute myeloid leukemia [251,252,253]. High ClpP expression correlates with poor recurrence-free survival in breast cancer [251]. Elevated ClpP levels promote cisplatin resistance in cervical cancer cells by inhibiting the accumulation of cisplatin in these cells, and partly through increasing the expression of copper efflux pump ATP7A [254].
Knockdown of ClpP is associated with reduced proliferation, migration, and invasion of various cancer cells [251,252,253]. Xenograft studies demonstrate that ClpP knockdown suppresses the growth of PCa-derived liver metastases [252]. These characteristics are likely due to the observed reductions in the expression levels of cyclin A, cyclin B1, cyclin D1, and MMP7 [251, 252], as well as the inhibition of PI3K and AKT activation after ClpP knockdown [251]. Conversely, overexpression of ClpP increases migratory and invasive activity in breast cancer [251]. Interference with ClpP expression perturbs mitochondrial function, as evidenced by an increase in mtROS, accumulation of misfolded mitochondrial Complex II subunits, hyperoxidation of mitochondrial peroxiredoxin III, reduced Complex II activity but increased Complex V activity, and overall diminished mitochondrial respiration [252, 253].
Concluding remarks
Although mitochondria contribute to uncontrolled proliferation throughout the various stages of cancer [3], mitochondria are also highly vulnerable in cancer cells. Tumors generate increased levels of oxidative stress and proteotoxic stress that leave mitochondria in a fragile, dysfunctional state [4,5,6]. Overall, the literature supports the idea that UPRmt, a mitochondrial stress response observed in C. elegans [10], serves as an important support system in cancer to maintain mitochondrial health and promote tumor growth (Figs. 1, 2, 3).
Multiple questions regarding the regulation of UPRmt in the cancer setting remain. Specifically, the mechanism by which ATF5 regulates UPRmt in cancer must be further addressed. Under non-stress conditions in the C. elegans model, ATFS-1, the homolog of ATF5, accumulates in the mitochondria and is degraded by LONP1 [113]. When mitochondrial stress is present, mitochondrial import of ATFS-1 into the mitochondria is reduced. Instead, ATFS-1 accumulates in the nucleus where it can function as a transcription factor and upregulate components of UPRmt to alleviate mitochondrial stress. It is unknown if ATF5 similarly translocates from mitochondria to the nucleus in mammals. In addition, no study has investigated if ATF5 has a direct transcriptional role in the mitochondria, where it could bind mtDNA and directly regulate the expression of mitochondrial-encoded proteins.
Furthermore, it is important to recognize that ATF5 is likely not the sole transcription factor responsible for the activation of UPRmt in cancer. Although ATF5 appears to be the only mitochondria-specific mediator of UPRmt, it remains likely that ATF5 functions in parallel with other key transcription factors that contribute to both forms of UPR in the mitochondria and endoplasmic reticulum. This suggests cross talk between UPRmt and UPRer. The extent to which established transcription factors of UPRer can induce UPRmt relative to the mitochondria-specific ATF5 is unknown. As we continue to understand the specific roles of ATF5 and other members of UPRmt in cancer, we become better equipped to develop pharmacological agents that can target UPRmt as a novel form of cancer therapy.
Current findings clearly suggest that cancer cells are highly reliant on the UPRmt for growth and progression, and therefore, aggressive and resistant cancers such as PCa possess robust activation of UPRmt [255, 256], which could be targeted for developing novel therapies to cure cancer. Recently, we have identified an inhibitor of UPRmt, a promising new anticancer agent for cancer such as PCa. This unique UPRmt inhibitor does not rely on androgen receptor-mediated signaling and, thus, will establish a foundation for the development of novel therapies to cure resistant PCa irrespective of AR status. Therefore, targeting this longevity promoting function of mitochondria, the UPRmt, will be an attractive and feasible target for aggressive and metastatic cancers.
Availability of data and materials
Not applicable.
Abbreviations
- UPRmt :
-
Mitochondrial unfolded protein response
- mtROS:
-
Mitochondrial reactive oxygen species
- RAS:
-
Rat sarcoma virus
- MYC:
-
Myelocytomatosis
- EGFR:
-
Epidermal growth factor receptor
- PTEN:
-
Phosphatase and tensin homolog (PTEN)
- MAPK:
-
Mitogen-activated protein kinase
- AKT:
-
Ak strain transforming
- AMPK:
-
AMP-activated protein kinase
- ERK:
-
Extracellular signal-regulated kinase
- NF-κB:
-
Nuclear factor kappa B
- TMEM126A:
-
Transmembrane protein 126A
- BCL-2:
-
B cell lymphoma 2
- MCL-1:
-
Myeloid leukemia 1
- Bcl-xL:
-
B cell lymphoma-extra
- mTOR:
-
Mammalian target of rapamycin
- MMP:
-
Matrix metalloproteinase
- ROS:
-
Reactive oxygen species
- OXPHOS:
-
Oxidative phosphorylation
- ETC:
-
Electron transport chain
- mtDNA:
-
Mitochondrial DNA
- TCA:
-
Tricarboxylic acid
- PCa:
-
Prostate cancer
- ND5:
-
NADH dehydrogenase subunit 5 gene
- PIP3:
-
Phosphatidylinositol 3,4,5-trisphosphate
- CDK1:
-
Cyclin-dependent kinase 1
- PKD1:
-
Protein kinase D1
- HMGB1:
-
High-mobility group box 1
- RAGE:
-
Receptor for advanced glycation end products
- TME:
-
Tumor microenvironment
- HIF-1α :
-
Hypoxia-inducible factor-1α
- PHDs:
-
Prolyl hydroxylases
- NSCLC:
-
Non-small cell lung cancer
- EMT:
-
Epithelial–mesenchymal transition
- SOD:
-
Superoxide dismutase
- HSP60:
-
Heat shock protein 60
- HSP10:
-
Heat shock protein 10
- mtHSP70:
-
Mitochondrial heat shock protein 70
- ClpP:
-
Caseinolytic protease
- MTS :
-
Mitochondrial targeting sequence
- NLS:
-
Nuclear localization sequence
- LONP1:
-
Lon peptidase 1
- ATF5:
-
Activating transcription factor 5
- ER:
-
Endoplasmic reticulum
- CHOP:
-
C/EBP homologous protein
- ERα :
-
Estrogen receptor alpha
- SIRT3:
-
Sirtuin deacetylase 3
- HSF1:
-
Heat shock factor-1
- UPRer :
-
Endoplasmic reticulum unfolded protein response
- NES:
-
Nuclear export sequence
- CCAR2:
-
Cell cycle and apoptosis regulator protein 2
- HGF:
-
Hepatocyte growth factor
- MVB:
-
Multivesicular bodies
References
Hartwell LH, et al. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997;278(5340):1064–8.
Luo J, et al. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136(5):823–37.
Vyas S, et al. Mitochondria and cancer. Cell. 2016;166(3):555–66.
Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13.
Bigarella CL, et al. Stem cells and the impact of ROS signaling. Development. 2014;141(22):4206–18.
Lagouge M, Larsson NG. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J Intern Med. 2013;273(6):529–43.
Morrell CN. Reactive oxygen species: finding the right balance. Circ Res. 2008;103(6):571–2.
Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010;44(5):479–96.
Galadari S, et al. Reactive oxygen species and cancer paradox: to promote or to suppress? Free Radic Biol Med. 2017;104:144–64.
Fiorese CJ, et al. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr Biol. 2016;26(15):2037–43.
Martinus RD, et al. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur J Biochem. 1996;240(1):98–103.
Kenny TC, et al. Mitohormesis, UPR(mt), and the complexity of mitochondrial DNA landscapes in cancer. Cancer Res. 2019;79(24):6057–66.
Naresh NU, Haynes CM. Signaling and regulation of the mitochondrial unfolded protein response. Cold Spring Harb Perspect Biol. 2019;11(6):a033944.
Wu Y, et al. Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell. 2014;158(6):1415–30.
Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007;21(24):3214–31.
Fisher GH, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15(24):3249–62.
Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4(2):199–207.
Velu TJ, et al. Epidermal-growth-factor-dependent transformation by a human EGF receptor proto-oncogene. Science. 1987;238(4832):1408–10.
Weinstein IB. Cancer. Addiction to oncogenes—the Achilles heal of cancer. Science. 2002;297(5578):63–4.
Torti D, Trusolino L. Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: promises and perils. EMBO Mol Med. 2011;3(11):623–36.
Solimini NL, et al. Non-oncogene addiction and the stress phenotype of cancer cells. Cell. 2007;130(6):986–8.
Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014;2:17.
Haas RH. Mitochondrial dysfunction in aging and diseases of aging. Biology (Basel). 2019;8(2):48.
Kenny TC, et al. The mitochondrial unfolded protein response as a non-oncogene addiction to support adaptation to stress during transformation in cancer and beyond. Front Oncol. 2017;7:159.
van der Bliek AM, et al. Cell biology of the mitochondrion. Genetics. 2017;207(3):843–71.
Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta. 1999;1410(2):103–23.
Kuhlbrandt W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015;13:89.
Meyer JN, et al. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology. 2017;391:42–53.
Lackner LL. Shaping the dynamic mitochondrial network. BMC Biol. 2014;12:35.
Chandel NS. Metabolism of proliferating cells. Cold Spring Harb Perspect Biol. 2021;13(10):a040618.
Martinez-Reyes I, Chandel NS. Cancer metabolism: looking forward. Nat Rev Cancer. 2021;21(10):669–80.
Balaban RS, et al. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–95.
Huttemann M, et al. Regulation of mitochondrial oxidative phosphorylation through cell signaling. Biochim Biophys Acta. 2007;1773(12):1701–20.
Quinlan CL, et al. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J Biol Chem. 2014;289(12):8312–25.
Brand MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med. 2016;100:14–31.
Evans MD, et al. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res. 2004;567(1):1–61.
Weydert CJ, Cullen JJ. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat Protoc. 2010;5(1):51–66.
Wei YH, et al. Mitochondrial theory of aging matures–roles of mtDNA mutation and oxidative stress in human aging. Zhonghua Yi Xue Za Zhi (Taipei). 2001;64(5):259–70.
Alexeyev M, et al. The maintenance of mitochondrial DNA integrity–critical analysis and update. Cold Spring Harb Perspect Biol. 2013;5(5): a012641.
Larsen NB, et al. Nuclear and mitochondrial DNA repair: similar pathways? Mitochondrion. 2005;5(2):89–108.
Escames G, et al. Mitochondrial DNA and inflammatory diseases. Hum Genet. 2012;131(2):161–73.
Nicolson GL. Mitochondrial dysfunction and chronic disease: treatment with natural supplements. Integr Med (Encinitas). 2014;13(4):35–43.
Moro L. Mitochondrial dysfunction in aging and cancer. J Clin Med. 2019;8(11):1983.
Cruz-Bermudez A, et al. Enhanced tumorigenicity by mitochondrial DNA mild mutations. Oncotarget. 2015;6(15):13628–43.
Shidara Y, et al. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005;65(5):1655–63.
Petros JA, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A. 2005;102(3):719–24.
Polyak K, et al. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat Genet. 1998;20(3):291–3.
Park JS, et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum Mol Genet. 2009;18(9):1578–89.
Liou GY, et al. Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 2016;14(10):2325–36.
Kwon J, et al. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A. 2004;101(47):16419–24.
Garcia-Santamarina S, et al. Reversible cysteine oxidation in hydrogen peroxide sensing and signal transduction. Biochemistry. 2014;53(16):2560–80.
Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–50.
Chetram MA, Hinton CV. PTEN regulation of ERK1/2 signaling in cancer. J Recept Signal Transduct Res. 2012;32(4):190–5.
Zhou C, et al. Redox regulation by SOD2 modulates colorectal cancer tumorigenesis through AMPK-mediated energy metabolism. Mol Carcinog. 2020;59(5):545–56.
Lim JM, et al. Control of the pericentrosomal H2O2 level by peroxiredoxin I is critical for mitotic progression. J Cell Biol. 2015;210(1):23–33.
Weinberg F, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107(19):8788–93.
Qi L, et al. HMGB1 promotes mitochondrial dysfunction-triggered striatal neurodegeneration via autophagy and apoptosis activation. PLoS ONE. 2015;10(11): e0142901.
Tang D, et al. High-mobility group box 1 and cancer. Biochim Biophys Acta. 2010;1799(1–2):131–40.
Kang R, et al. The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene. 2014;33(5):567–77.
Chen Q, et al. Rewiring of glutamine metabolism is a bioenergetic adaptation of human cells with mitochondrial DNA mutations. Cell Metab. 2018;27(5):1007-1025 e5.
Khasawneh J, et al. Inflammation and mitochondrial fatty acid beta-oxidation link obesity to early tumor promotion. Proc Natl Acad Sci U S A. 2009;106(9):3354–9.
Wang C, et al. Elevated level of mitochondrial reactive oxygen species via fatty acid beta-oxidation in cancer stem cells promotes cancer metastasis by inducing epithelial-mesenchymal transition. Stem Cell Res Ther. 2019;10(1):175.
Balkwill FR, et al. The tumor microenvironment at a glance. J Cell Sci. 2012;125(Pt 23):5591–6.
Neagu M, et al. Inflammation and metabolism in cancer cell-mitochondria key player. Front Oncol. 2019;9:348.
Frenkel K. Carcinogen-mediated oxidant formation and oxidative DNA damage. Pharmacol Ther. 1992;53(1):127–66.
Shacter E, et al. Activated neutrophils induce prolonged DNA damage in neighboring cells. Carcinogenesis. 1988;9(12):2297–304.
Wang J, et al. Crosstalk between cancer and immune cells: Role of tumor-associated macrophages in the tumor microenvironment. Cancer Med. 2019;8(10):4709–21.
Trush MA, Kensler TW. An overview of the relationship between oxidative stress and chemical carcinogenesis. Free Radic Biol Med. 1991;10(3–4):201–9.
Hahn WS, et al. Proinflammatory cytokines differentially regulate adipocyte mitochondrial metabolism, oxidative stress, and dynamics. Am J Physiol Endocrinol Metab. 2014;306(9):E1033–45.
Lopez-Armada MJ, et al. Mitochondrial activity is modulated by TNFalpha and IL-1beta in normal human chondrocyte cells. Osteoarthritis Cartilage. 2006;14(10):1011–22.
El Jamal SM, et al. Interferon gamma-induced apoptosis of head and neck squamous cell carcinoma is connected to indoleamine-2,3-dioxygenase via mitochondrial and ER stress-associated pathways. Cell Div. 2016;11:11.
Cao Y, et al. Proinflammatory cytokines stimulate mitochondrial superoxide flashes in articular chondrocytes in vitro and in situ. PLoS ONE. 2013;8(6): e66444.
Yang D, et al. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp Eye Res. 2007;85(4):462–72.
Muz B, et al. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl). 2015;3:83–92.
Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29(5):625–34.
Huang LE, et al. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1998;95(14):7987–92.
Chandel NS, et al. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998;95(20):11715–20.
Chandel NS, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275(33):25130–8.
Bell EL, et al. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol. 2007;177(6):1029–36.
Kwong JQ, et al. The mitochondrial respiratory chain is a modulator of apoptosis. J Cell Biol. 2007;179(6):1163–77.
Mizutani S, et al. Mutations in the mitochondrial genome confer resistance of cancer cells to anticancer drugs. Cancer Sci. 2009;100(9):1680–7.
Guerra F, et al. Mitochondrial DNA mutation in serous ovarian cancer: implications for mitochondria-coded genes in chemoresistance. J Clin Oncol. 2012;30(36):e373–8.
Okon IS, et al. Gefitinib-mediated reactive oxygen specie (ROS) instigates mitochondrial dysfunction and drug resistance in lung cancer cells. J Biol Chem. 2015;290(14):9101–10.
Okon IS, Zou MH. Mitochondrial ROS and cancer drug resistance: Implications for therapy. Pharmacol Res. 2015;100:170–4.
Kulawiec M, et al. mtDNA G10398A variant in African-American women with breast cancer provides resistance to apoptosis and promotes metastasis in mice. J Hum Genet. 2009;54(11):647–54.
Gammage PA, Frezza C. Mitochondrial DNA: the overlooked oncogenome? BMC Biol. 2019;17(1):53.
Hopkins JF, et al. Mitochondrial mutations drive prostate cancer aggression. Nat Commun. 2017;8(1):656.
Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127(4):679–95.
Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12(8):895–904.
Imanishi H, et al. Mitochondrial DNA mutations regulate metastasis of human breast cancer cells. PLoS ONE. 2011;6(8): e23401.
Sun HF, et al. Loss of TMEM126A promotes extracellular matrix remodeling, epithelial-to-mesenchymal transition, and breast cancer metastasis by regulating mitochondrial retrograde signaling. Cancer Lett. 2019;440–441:189–201.
Dasgupta S, et al. Mitochondrial DNA mutations in respiratory complex-I in never-smoker lung cancer patients contribute to lung cancer progression and associated with EGFR gene mutation. J Cell Physiol. 2012;227(6):2451–60.
Ishikawa K, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320(5876):661–4.
Yuan Y, et al. Nonsense and missense mutation of mitochondrial ND6 gene promotes cell migration and invasion in human lung adenocarcinoma. BMC Cancer. 2015;15:346.
Koshikawa N, et al. Association of predicted pathogenic mutations in mitochondrial ND genes with distant metastasis in NSCLC and colon cancer. Sci Rep. 2017;7(1):15535.
Reichmann D, et al. Maintaining a healthy proteome during oxidative stress. Mol Cell. 2018;69(2):203–13.
Idelchik M, et al. Mitochondrial ROS control of cancer. Semin Cancer Biol. 2017;47:57–66.
Oberley TD, Oberley LW. Antioxidant enzyme levels in cancer. Histol Histopathol. 1997;12(2):525–35.
Cobanoglu U, et al. Erythrocyte catalase and carbonic anhydrase activities in lung cancer. Asian Pac J Cancer Prev. 2010;11(5):1377–82.
Oltra AM, et al. Antioxidant enzyme activities and the production of MDA and 8-oxo-dG in chronic lymphocytic leukemia. Free Radic Biol Med. 2001;30(11):1286–92.
Balasubramaniyan N, et al. Status of antioxidant systems in human carcinoma of uterine cervix. Cancer Lett. 1994;87(2):187–92.
Nishida S, et al. Manganese superoxide dismutase content and localization in human thyroid tumours. J Pathol. 1993;169(3):341–5.
Peddireddy V, et al. Assessment of 8-oxo-7, 8-dihydro-2’-deoxyguanosine and malondialdehyde levels as oxidative stress markers and antioxidant status in non-small cell lung cancer. Biomarkers. 2012;17(3):261–8.
Levy E, et al. Causative links between protein aggregation and oxidative stress: a review. Int J Mol Sci. 2019;20(16):3896.
Genova ML, Lenaz G. The interplay between respiratory supercomplexes and ROS in aging. Antioxid Redox Signal. 2015;23(3):208–38.
Zhao Q, et al. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21(17):4411–9.
Harbauer AB, et al. The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab. 2014;19(3):357–72.
Schmidt O, et al. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol. 2010;11(9):655–67.
Liu Z, Butow RA. Mitochondrial retrograde signaling. Annu Rev Genet. 2006;40:159–85.
Ryan MT, Hoogenraad NJ. Mitochondrial-nuclear communications. Annu Rev Biochem. 2007;76:701–22.
Yoneda T, et al. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci. 2004;117(Pt 18):4055–66.
Haynes CM, et al. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell. 2007;13(4):467–80.
Nargund AM, et al. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337(6094):587–90.
Nargund AM, et al. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol Cell. 2015;58(1):123–33.
Baker BM, et al. Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS Genet. 2012;8(6): e1002760.
Schulz AM, Haynes CM. (2015) UPR(mt)-mediated cytoprotection and organismal aging. Biochim Biophys Acta. 1847;11:1448–56.
Nishitoh H. CHOP is a multifunctional transcription factor in the ER stress response. J Biochem. 2012;151(3):217–9.
Harding HP, et al. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5(5):897–904.
Papa L, Germain D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J Cell Sci. 2011;124(Pt 9):1396–402.
Papa L, Germain D. SirT3 regulates the mitochondrial unfolded protein response. Mol Cell Biol. 2014;34(4):699–710.
Mohrin M, et al. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science. 2015;347(6228):1374–7.
Katiyar A, et al. HSF1 is required for induction of mitochondrial chaperones during the mitochondrial unfolded protein response. FEBS Open Bio. 2020;10(6):1135–48.
Horibe T, Hoogenraad NJ. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS ONE. 2007;2(9): e835.
Pirkkala L, et al. Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J. 2001;15(7):1118–31.
Zhang HH, et al. Sirtuin-3 (SIRT3) protects pancreatic beta-cells from endoplasmic reticulum (ER) stress-induced apoptosis and dysfunction. Mol Cell Biochem. 2016;420(1–2):95–106.
Shin J, et al. SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell Rep. 2013;5(3):654–65.
Livezey M, et al. A new role for estrogen receptor alpha in cell proliferation and cancer: activating the anticipatory unfolded protein response. Front Endocrinol (Lausanne). 2018;9:325.
Zhou Z, et al. Estrogen receptor alpha protects pancreatic beta-cells from apoptosis by preserving mitochondrial function and suppressing endoplasmic reticulum stress. J Biol Chem. 2018;293(13):4735–51.
Chiribau CB, et al. Molecular symbiosis of CHOP and C/EBP beta isoform LIP contributes to endoplasmic reticulum stress-induced apoptosis. Mol Cell Biol. 2010;30(14):3722–31.
Teske BF, et al. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol Biol Cell. 2013;24(15):2477–90.
Zhou D, et al. Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J Biol Chem. 2008;283(11):7064–73.
Torres-Peraza JF, et al. Protective neuronal induction of ATF5 in endoplasmic reticulum stress induced by status epilepticus. Brain. 2013;136(Pt 4):1161–76.
Wang YT, et al. Cardioprotection by the mitochondrial unfolded protein response requires ATF5. Am J Physiol Heart Circ Physiol. 2019;317(2):H472–8.
Tyynismaa H, et al. Mitochondrial myopathy induces a starvation-like response. Hum Mol Genet. 2010;19(20):3948–58.
Dogan SA, et al. Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. 2014;19(3):458–69.
Runkel ED, et al. Surveillance-activated defenses block the ROS-induced mitochondrial unfolded protein response. PLoS Genet. 2013;9(3): e1003346.
Smyrnias I, et al. Cardioprotective effect of the mitochondrial unfolded protein response during chronic pressure overload. J Am Coll Cardiol. 2019;73(14):1795–806.
Beissinger M, Buchner J. How chaperones fold proteins. Biol Chem. 1998;379(3):245–59.
Quiros PM, et al. Lon protease: a key enzyme controlling mitochondrial bioenergetics in cancer. Mol Cell Oncol. 2014;1(4): e968505.
Gottesman S, et al. The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev. 1998;12(9):1338–47.
Young L, et al. Role of the ABC transporter Mdl1 in peptide export from mitochondria. Science. 2001;291(5511):2135–8.
Bezawork-Geleta A, et al. LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Sci Rep. 2015;5:17397.
Deng P, Haynes CM. Mitochondrial dysfunction in cancer: potential roles of ATF5 and the mitochondrial UPR. Semin Cancer Biol. 2017;47:43–9.
Baxevanis AD, Vinson CR. Interactions of coiled coils in transcription factors: where is the specificity? Curr Opin Genet Dev. 1993;3(2):278–85.
Ellenberger TE, et al. The GCN4 basic region leucine zipper binds DNA as a dimer of uninterrupted alpha helices: crystal structure of the protein-DNA complex. Cell. 1992;71(7):1223–37.
Vinson C, et al. Classification of human B-ZIP proteins based on dimerization properties. Mol Cell Biol. 2002;22(18):6321–35.
Angelastro JM, et al. Regulated expression of ATF5 is required for the progression of neural progenitor cells to neurons. J Neurosci. 2003;23(11):4590–600.
Sun X, et al. Dominant-negative ATF5 compromises cancer cell survival by targeting CEBPB and CEBPD. Mol Cancer Res. 2020;18(2):216–28.
Feldheim J, et al. Expression of activating transcription factor 5 (ATF5) is increased in astrocytomas of different WHO grades and correlates with survival of glioblastoma patients. Onco Targets Ther. 2018;11:8673–84.
Monaco SE, et al. The transcription factor ATF5 is widely expressed in carcinomas, and interference with its function selectively kills neoplastic, but not nontransformed, breast cell lines. Int J Cancer. 2007;120(9):1883–90.
Hu M, et al. Interference with ATF5 function enhances the sensitivity of human pancreatic cancer cells to paclitaxel-induced apoptosis. Anticancer Res. 2012;32(10):4385–94.
Kong X, et al. Overexpression of activating transcription factor 5 in human rectal cancer. Exp Ther Med. 2011;2(5):827–31.
Chen A, et al. ATF5 is overexpressed in epithelial ovarian carcinomas and interference with its function increases apoptosis through the downregulation of Bcl-2 in SKOV-3 cells. Int J Gynecol Pathol. 2012;31(6):532–7.
Sheng Z, et al. A genome-wide RNA interference screen reveals an essential CREB3L2-ATF5-MCL1 survival pathway in malignant glioma with therapeutic implications. Nat Med. 2010;16(6):671–7.
Ishihara S, et al. Activating transcription factor 5 enhances radioresistance and malignancy in cancer cells. Oncotarget. 2015;6(7):4602–14.
Wang H, et al. ATF5 promotes cell survival through transcriptional activation of Hsp27 in H9c2 cells. Cell Biol Int. 2007;31(11):1309–15.
Watatani Y, et al. Amino acid limitation induces expression of ATF5 mRNA at the post-transcriptional level. Life Sci. 2007;80(9):879–85.
Pei Z, et al. Transcriptomic and functional pathways analysis of ascorbate-induced cytotoxicity and resistance of Burkitt lymphoma. Oncotarget. 2016;7(39):63950–9.
Liu DX, et al. p300-Dependent ATF5 acetylation is essential for Egr-1 gene activation and cell proliferation and survival. Mol Cell Biol. 2011;31(18):3906–16.
Dluzen D, et al. BCL-2 is a downstream target of ATF5 that mediates the prosurvival function of ATF5 in a cell type-dependent manner. J Biol Chem. 2011;286(9):7705–13.
Karpel-Massler G, et al. A synthetic cell-penetrating dominant-negative ATF5 peptide exerts anticancer activity against a broad spectrum of treatment-resistant cancers. Clin Cancer Res. 2016;22(18):4698–711.
Levy-Rimler G, et al. The effect of nucleotides and mitochondrial chaperonin 10 on the structure and chaperone activity of mitochondrial chaperonin 60. Eur J Biochem. 2001;268(12):3465–72.
Okamoto T, et al. Functional structure and physiological functions of mammalian wild-type HSP60. Arch Biochem Biophys. 2015;586:10–9.
Gomez-Llorente, Y. et al. (2020) Structural basis for active single and double ring complexes in human mitochondrial Hsp60-Hsp10 chaperonin. Nat Commun 11 (1), 1916.
Avery SV. Molecular targets of oxidative stress. Biochem J. 2011;434(2):201–10.
Sarangi U, et al. Hsp60 chaperonin acts as barrier to pharmacologically induced oxidative stress mediated apoptosis in tumor cells with differential stress response. Drug Target Insights. 2013;7:35–51.
Marino Gammazza A, et al. Doxorubicin anti-tumor mechanisms include Hsp60 post-translational modifications leading to the Hsp60/p53 complex dissociation and instauration of replicative senescence. Cancer Lett. 2017;385:75–86.
Cappello F, et al. Immunohistochemical evaluation of PCNA, p53, HSP60, HSP10 and MUC-2 presence and expression in prostate carcinogenesis. Anticancer Res. 2003;23(2B):1325–31.
Rappa F, et al. Quantitative patterns of Hsps in tubular adenoma compared with normal and tumor tissues reveal the value of Hsp10 and Hsp60 in early diagnosis of large bowel cancer. Cell Stress Chaperones. 2016;21(5):927–33.
Lim SO, et al. Expression of heat shock proteins (HSP27, HSP60, HSP70, HSP90, GRP78, GRP94) in hepatitis B virus-related hepatocellular carcinomas and dysplastic nodules. World J Gastroenterol. 2005;11(14):2072–9.
Mano R, et al. Heat shock proteins 60 and 70 are associated with long-term outcome of T1-stage high-grade urothelial tumors of the bladder treated with intravesical Bacillus Calmette-Guerin immunotherapy. Urol Oncol. 2018;36(12):531 e9-531 e17.
Castilla C, et al. Immunohistochemical expression of Hsp60 correlates with tumor progression and hormone resistance in prostate cancer. Urology. 2010;76(4):1017 e1-1026.
Zhou C, et al. Oncogenic HSP60 regulates mitochondrial oxidative phosphorylation to support Erk1/2 activation during pancreatic cancer cell growth. Cell Death Dis. 2018;9(2):161.
Cappello F, et al. The expression of HSP60 and HSP10 in large bowel carcinomas with lymph node metastase. BMC Cancer. 2005;5:139.
Vocka M, et al. Novel serum markers HSP60, CHI3L1, and IGFBP-2 in metastatic colorectal cancer. Oncol Lett. 2019;18(6):6284–92.
Li XS, et al. Heat shock protein 60 overexpression is associated with the progression and prognosis in gastric cancer. PLoS ONE. 2014;9(9): e107507.
Kim W, et al. CCAR2/DBC1 and Hsp60 Positively Regulate Expression of Survivin in Neuroblastoma Cells. Int J Mol Sci. 2019;20(1):131.
Guo J, et al. HSP60-regulated mitochondrial proteostasis and protein translation promote tumor growth of ovarian cancer. Sci Rep. 2019;9(1):12628.
Chalmers SA, et al. A role for HMGB1, HSP60 and Myd88 in growth of murine mammary carcinoma in vitro. Cell Immunol. 2013;282(2):136–45.
Tang H, et al. Down-regulation of HSP60 suppresses the proliferation of glioblastoma cells via the ROS/AMPK/mTOR pathway. Sci Rep. 2016;6:28388.
Ghosh JC, et al. Heat shock protein 60 regulation of the mitochondrial permeability transition pore in tumor cells. Cancer Res. 2010;70(22):8988–93.
Kumar S, et al. Hsp60 and IL-8 axis promotes apoptosis resistance in cancer. Br J Cancer. 2019;121(11):934–43.
Tsai YP, et al. Interaction between HSP60 and beta-catenin promotes metastasis. Carcinogenesis. 2009;30(6):1049–57.
Tsai YP, et al. Direct regulation of HSP60 expression by c-MYC induces transformation. FEBS Lett. 2008;582(29):4083–8.
Chaiwatanasirikul KA, Sala A. The tumour-suppressive function of CLU is explained by its localisation and interaction with HSP60. Cell Death Dis. 2011;2: e219.
Kim W, et al. Mitochondrial CCAR2/DBC1 is required for cell survival against rotenone-induced mitochondrial stress. Biochem Biophys Res Commun. 2017;485(4):782–9.
Lin CY, et al. Oxidation of heat shock protein 60 and protein disulfide isomerase activates ERK and migration of human hepatocellular carcinoma HepG2. Oncotarget. 2016;7(10):11067–82.
Chandra D, et al. Cytosolic accumulation of HSP60 during apoptosis with or without apparent mitochondrial release: evidence that its pro-apoptotic or pro-survival functions involve differential interactions with caspase-3. J Biol Chem. 2007;282(43):31289–301.
Campanella C, et al. The odyssey of Hsp60 from tumor cells to other destinations includes plasma membrane-associated stages and Golgi and exosomal protein-trafficking modalities. PLoS ONE. 2012;7(7): e42008.
Campanella C, et al. Heat shock protein 60 levels in tissue and circulating exosomes in human large bowel cancer before and after ablative surgery. Cancer. 2015;121(18):3230–9.
Samali A, et al. Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J. 1999;18(8):2040–8.
Ghosh JC, et al. Hsp60 regulation of tumor cell apoptosis. J Biol Chem. 2008;283(8):5188–94.
Kirchhoff SR, et al. Cytosolic heat shock protein 60, apoptosis, and myocardial injury. Circulation. 2002;105(24):2899–904.
Chang AY, et al. Heat shock protein 60 in rostral ventrolateral medulla reduces cardiovascular fatality during endotoxaemia in the rat. J Physiol. 2006;574(Pt 2):547–64.
Chun JN, et al. Cytosolic Hsp60 is involved in the NF-kappaB-dependent survival of cancer cells via IKK regulation. PLoS ONE. 2010;5(3): e9422.
Shin BK, et al. Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J Biol Chem. 2003;278(9):7607–16.
Feng H, et al. Stressed apoptotic tumor cells stimulate dendritic cells and induce specific cytotoxic T cells. Blood. 2002;100(12):4108–15.
Piselli P, et al. Different expression of CD44, ICAM-1, and HSP60 on primary tumor and metastases of a human pancreatic carcinoma growing in scid mice. Anticancer Res. 2000;20(2A):825–31.
Barazi HO, et al. Identification of heat shock protein 60 as a molecular mediator of alpha 3 beta 1 integrin activation. Cancer Res. 2002;62(5):1541–8.
Tawil NJ, et al. Integrin alpha3beta1 can promote adhesion and spreading of metastatic breast carcinoma cells on the lymph node stroma. Int J Cancer. 1996;66(5):703–10.
Lundstrom A, et al. The role of alpha2 beta1 and alpha3 beta1 integrin receptors in the initial anchoring of MDA-MB-231 human breast cancer cells to cortical bone matrix. Biochem Biophys Res Commun. 1998;250(3):735–40.
Lemmon SK, Traub LM. Sorting in the endosomal system in yeast and animal cells. Curr Opin Cell Biol. 2000;12(4):457–66.
Zhang Y, et al. Exosomes: biogenesis, biologic function and clinical potential. Cell Biosci. 2019;9:19.
Hayoun D, et al. HSP60 is transported through the secretory pathway of 3-MCA-induced fibrosarcoma tumour cells and undergoes N-glycosylation. FEBS J. 2012;279(12):2083–95.
Cappello F, et al. Immunopositivity of heat shock protein 60 as a biomarker of bronchial carcinogenesis. Lancet Oncol. 2005;6(10):816.
Ruan W, et al. HSP60, a protein downregulated by IGFBP7 in colorectal carcinoma. J Exp Clin Cancer Res. 2010;29:41.
Tang H, et al. Downregulation of HSP60 disrupts mitochondrial proteostasis to promote tumorigenesis and progression in clear cell renal cell carcinoma. Oncotarget. 2016;7(25):38822–34.
Zhang J, et al. Hsp60 exerts a tumor suppressor function by inducing cell differentiation and inhibiting invasion in hepatocellular carcinoma. Oncotarget. 2016;7(42):68976–89.
Faried A, et al. Expression of heat-shock protein Hsp60 correlated with the apoptotic index and patient prognosis in human oesophageal squamous cell carcinoma. Eur J Cancer. 2004;40(18):2804–11.
Teng R, et al. HSP60 silencing promotes Warburg-like phenotypes and switches the mitochondrial function from ATP production to biosynthesis in ccRCC cells. Redox Biol. 2019;24: 101218.
Fan W, et al. Elevated expression of HSP10 protein inhibits apoptosis and associates with poor prognosis of astrocytoma. PLoS ONE. 2017;12(10): e0185563.
Feng J, et al. High expression of heat shock protein 10 (Hsp10) is associated with poor prognosis in oral squamous cell carcinoma. Int J Clin Exp Pathol. 2017;10(7):7784–91.
Feng J, et al. Increased expression of heat shock protein (HSP) 10 and HSP70 correlates with poor prognosis of nasopharyngeal carcinoma. Cancer Manag Res. 2019;11:8219–27.
Wadhwa R, et al. An Hsp70 family chaperone, mortalin/mthsp70/PBP74/Grp75: what, when, and where? Cell Stress Chaperones. 2002;7(3):309–16.
Amick J, et al. Crystal structure of the nucleotide-binding domain of mortalin, the mitochondrial Hsp70 chaperone. Protein Sci. 2014;23(6):833–42.
Kaul SC, et al. Three faces of mortalin: a housekeeper, guardian and killer. Exp Gerontol. 2007;42(4):263–74.
Wu PK, et al. A mortalin/HSPA9-mediated switch in tumor-suppressive signaling of Raf/MEK/extracellular signal-regulated kinase. Mol Cell Biol. 2013;33(20):4051–67.
Starenki D, et al. Mortalin (GRP75/HSPA9) upregulation promotes survival and proliferation of medullary thyroid carcinoma cells. Oncogene. 2015;34(35):4624–34.
Starenki D, et al. Mortalin (GRP75/HSPA9) Promotes Survival and Proliferation of Thyroid Carcinoma Cells. Int J Mol Sci. 2019;20(9):2069.
Yi X, et al. Association of mortalin (HSPA9) with liver cancer metastasis and prediction for early tumor recurrence. Mol Cell Proteomics. 2008;7(2):315–25.
Wadhwa R, et al. Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int J Cancer. 2006;118(12):2973–80.
Hu Y, et al. Oncogenic role of mortalin contributes to ovarian tumorigenesis by activating the MAPK-ERK pathway. J Cell Mol Med. 2016;20(11):2111–21.
Sun J, et al. Mortalin overexpression predicts poor prognosis in early stage of non-small cell lung cancer. Tumour Biol. 2017;39(3):1010428317695918.
Jin H, et al. The clinicopathological significance of Mortalin overexpression in invasive ductal carcinoma of breast. J Exp Clin Cancer Res. 2016;35:42.
Liu LX, et al. Mortalin stabilizes CD151-depedent tetraspanin-enriched microdomains and implicates in the progression of hepatocellular carcinoma. J Cancer. 2019;10(25):6199–206.
Park SJ, et al. Down-regulation of mortalin exacerbates Abeta-mediated mitochondrial fragmentation and dysfunction. J Biol Chem. 2014;289(4):2195–204.
Kaul SC, et al. Malignant transformation of NIH3T3 cells by overexpression of mot-2 protein. Oncogene. 1998;17(7):907–11.
Yun CO, et al. Relevance of mortalin to cancer cell stemness and cancer therapy. Sci Rep. 2017;7:42016.
Na Y, et al. Stress chaperone mortalin contributes to epithelial-mesenchymal transition and cancer metastasis. Cancer Res. 2016;76(9):2754–65.
Lu WJ, et al. Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ. 2011;18(6):1046–56.
Wadhwa R, et al. Inactivation of tumor suppressor p53 by mot-2, a hsp70 family member. J Biol Chem. 1998;273(45):29586–91.
Wadhwa R, et al. Hsp70 family member, mot-2/mthsp70/GRP75, binds to the cytoplasmic sequestration domain of the p53 protein. Exp Cell Res. 2002;274(2):246–53.
Kaul SC, et al. An N-terminal region of mot-2 binds to p53 in vitro. Neoplasia. 2001;3(2):110–4.
Mylonis I, et al. Atypical CRM1-dependent nuclear export signal mediates regulation of hypoxia-inducible factor-1alpha by MAPK. J Biol Chem. 2008;283(41):27620–7.
Mylonis I, et al. Mortalin-mediated and ERK-controlled targeting of HIF-1alpha to mitochondria confers resistance to apoptosis under hypoxia. J Cell Sci. 2017;130(2):466–79.
Wu PK, et al. Mortalin (HSPA9) facilitates BRAF-mutant tumor cell survival by suppressing ANT3-mediated mitochondrial membrane permeability. Sci Signal. 2020;13(622):eaay1478.
Wadhwa R, et al. Identification and characterization of molecular interactions between mortalin/mtHsp70 and HSP60. Biochem J. 2005;391(Pt 2):185–90.
Ben-Zvi A, et al. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci U S A. 2009;106(35):14914–9.
Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22(11):1427–38.
Tyedmers J, et al. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol. 2010;11(11):777–88.
Cha SS, et al. Crystal structure of Lon protease: molecular architecture of gated entry to a sequestered degradation chamber. EMBO J. 2010;29(20):3520–30.
Quiros PM, et al. ATP-dependent Lon protease controls tumor bioenergetics by reprogramming mitochondrial activity. Cell Rep. 2014;8(2):542–56.
Ghosh JC, et al. Akt phosphorylation of mitochondrial Lonp1 protease enables oxidative metabolism and advanced tumor traits. Oncogene. 2019;38(43):6926–39.
Liu C, et al. Inhibition of LONP1 suppresses pancreatic cancer progression Via c-Jun N-terminal kinase pathway-meditated epithelial-mesenchymal transition. Pancreas. 2019;48(5):629–35.
Gibellini L, et al. LonP1 differently modulates mitochondrial function and bioenergetics of primary versus metastatic colon cancer cells. Front Oncol. 2018;8:254.
Luo B, et al. ATP-dependent Lon protease contributes to helicobacter pylori-induced gastric carcinogenesis. Neoplasia. 2016;18(4):242–52.
Cheng CW, et al. Overexpression of Lon contributes to survival and aggressive phenotype of cancer cells through mitochondrial complex I-mediated generation of reactive oxygen species. Cell Death Dis. 2013;4: e681.
Kao TY, et al. Mitochondrial Lon regulates apoptosis through the association with Hsp60-mtHsp70 complex. Cell Death Dis. 2015;6: e1642.
Wang J, et al. The structure of ClpP at 2.3 A resolution suggests a model for ATP-dependent proteolysis. Cell. 1997;91(4):447–56.
Neher SB, et al. Distinct peptide signals in the UmuD and UmuD’ subunits of UmuD/D’ mediate tethering and substrate processing by the ClpXP protease. Proc Natl Acad Sci U S A. 2003;100(23):13219–24.
Luo J, et al. ClpP regulates breast cancer cell proliferation, invasion and apoptosis by modulating the Src/PI3K/Akt signaling pathway. PeerJ. 2020;8: e8754.
Seo JH, et al. The mitochondrial unfoldase-peptidase complex ClpXP controls bioenergetics stress and metastasis. PLoS Biol. 2016;14(7): e1002507.
Cole A, et al. Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2015;27(6):864–76.
Zhang Y, Maurizi MR. (2016) Mitochondrial ClpP activity is required for cisplatin resistance in human cells. Biochim Biophys Acta. 1862;2:252–64.
Kumar R, et al. A mitochondrial unfolded protein response inhibitor suppresses prostate cancer growth in mice via HSP60. J Clin Invest. 2022;132(13):e149906.
Shkedi A, et al. Selective vulnerabilities in the proteostasis network of castration-resistant prostate cancer. Cell Chem Biol. 2022;29(3):490-501 e4.
Acknowledgements
Not applicable
Funding
This work was supported by the National Cancer Institute (NCI) of the National Institutes of Health under award number R01CA246437 and the American Cancer Society under award number MBG-21-048-01-MBG to D.C. and in part by NCI Center support grant P30CA016056 to the Roswell Park Comprehensive Cancer Center.
Author information
Authors and Affiliations
Contributions
JRI and DC wrote the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests\
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Inigo, J.R., Chandra, D. The mitochondrial unfolded protein response (UPRmt): shielding against toxicity to mitochondria in cancer. J Hematol Oncol 15, 98 (2022). https://doi.org/10.1186/s13045-022-01317-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-022-01317-0