
Abstract
Chemotherapy combined with or without targeted therapy is the fundamental treatment for metastatic colorectal cancer (mCRC). Due to the adverse effects of chemotherapeutic drugs and the biological characteristics of the tumor cells, it is difficult to make breakthroughs in traditional strategies. The immune checkpoint blockades (ICB) therapy has made significant progress in the treatment of advanced malignant tumors, and patients who benefit from this therapy may obtain a long-lasting response. Unfortunately, immunotherapy is only effective in a limited number of patients with microsatellite instability—high (MSI-H), and segment initial responders can subsequently develop acquired resistance. From September 4, 2014, the first anti-PD-1/PD-L1 drug Pembrolizumab was approved by the FDA for the second-line treatment of advanced malignant melanoma. Subsequently, it was approved for mCRC second-line treatment in 2017. Immunotherapy has rapidly developed in the past 7 years. The in-depth research of the ICB treatment indicated that the mechanism of colorectal cancer immune-resistance has become gradually clear, and new predictive biomarkers are constantly emerging. Clinical trials examining the effect of immune checkpoints are actively carried out, in order to produce long-lasting effects for mCRC patients. This review summarizes the treatment strategies for mCRC patients, discusses the mechanism and application of ICB in mCRC treatment, outlines the potential markers of the ICB efficacy, lists the key results of the clinical trials, and collects the recent basic research results, in order to provide a theoretical basis and practical direction for immunotherapy strategies.
Similar content being viewed by others


Introduction
Colorectal cancer currently ranks third in cancer incidence and second in cancer-related death globally. In 2020, there were 1,931,590 new cases of colorectal cancer worldwide and 935,173 deaths, accounting for 10.0% and 9.4% of the total number of cancer incidences and deaths, respectively [1]. In recent years, the research on the comprehensive treatment of colorectal cancer has continued to increase, and combination therapy with a targeted agent is the main treatment method for metastatic colorectal cancer (mCRC) patients [2, 3]. Whereas medication regimen is developing continually, it is difficult to make breakthroughs in mCRC therapy, prognosis remaining poor with a median overall survival (mOS) of only 25–30 months [4, 5]. Immunotherapy has achieved significant curative effects in the treatment of solid tumors, notably in melanoma and non-small cell lung cancer (NSCLC) [6, 7]. Immune checkpoint blockade (ICB) has enabled certain patients to obtain long-lasting benefits and have significantly improved disease prognosis. According to the latest follow-up data, the mOS of advanced melanoma was 72.1 months with Nivolumab plus Ipilimumab [8].These agents are used as the first-line treatment for patients with advanced solid tumor [9, 10]. However, inhibition of programmed death-1 (PD-1) or programmed cell death-ligand 1 (PD-L1) therapy has limited effects in the treatment of colorectal cancer. Until 2017, the first anti-PD-1 drug Pembrolizumab was approved by the Food and Drug Administration (FDA) as a second-line treatment for mCRC patients with microsatellite instability—high (MSI-H). However, there are only a few patients with dMMR/MSH-H (about 15% of colorectal cancer patients, 4% of patients with mCRC), and part of them enter into the stage of immune resistance soon [11,12,13].
Mutations in the DNA mismatch repair (MMR) genes can cause defects in the repair function during DNA replication, which leads to the occurrence of MSI. Both DNA deficient mismatch repair (dMMR) and MSI-H can lead to the accumulation of DNA mutations in tumor cells and results in generation of sufficient tumor neoantigens to enhance tumor immunogenicity, which can induce strong T cell and tumor immune response [14,15,16]. By contrast, mismatch repair-proficient (pMMR) colorectal tumor cells express weak immunogenicity and infiltrate a limited number immune cells, which makes it difficult to induce an adequate immune response [17, 18]. Therefore, ICB therapy is ineffective in such patients. In order to increase immunotherapy sensitivity, combined treatments are required to enhance tumor immunogenicity. With the development of research, the mechanism of immunotherapy resistance has been studied more thoroughly in mCRC. In addition to MSI, numerous biomarkers have been discovered and have been used to guide tumor immunotherapy, such as tumor mutation burden (TMB) [19, 20] and PD-L1 expression [21, 22]. However, due to the heterogeneity of the tumors, these markers are commonly used in other solid tumors and exhibit limited value in mCRC. It has been shown that MSS mCRC patients with POLE mutations can achieve an ideal immunotherapeutic effect [23]. More effective biomarkers are required for clinical use. The theory of tumor immunotherapy is complex, which is the result of multiple factors, temporal and spatial heterogeneity, and network co-regulation. A single theory or a single indicator cannot adequately explain the mechanism of immunotherapy. According to the genetic background, tumor microenvironment (TME), and the cell metabolism of mCRC patients, various models including CMS and CIRC subtypes have been proposed to guide the immunotherapy and comprehensive treatment of mCRC patients [24, 25].
Differences in the TME and in individual gene expression lead to diverse effects of immunotherapy. To date, a significant number of immunotherapy-related clinical trials have produced satisfying results which have led to the approval of Pembrolizumab and Nivolumab for mCRC treatment by the FDA [10, 26,27,28,29,30]. Other clinical trials, either ongoing or scheduled to initiate, are exploring the potential of activating inactive tumors [(“cold”) into (“hot”) tumors]. The clarification of the immune resistant mechanism is the premise to design innovative immunotherapeutic strategies. The present study focused on the following aspects: the current treatment opinions of mCRC, the mechanism of resistance ICB, the identification of potential biomarkers of the immune response, the key achievements of the latest clinical trials, and the breakthrough results of the preclinical studies. The analysis of this information aimed to demonstrate the application and treatment potential of the ICB therapy in mCRC.
Treatment opinions of advanced colorectal cancer
Surgery is the primary treatment used for the majority of CRC patients. The majority of CRC patients with distant metastasis or recurrence do not receive radical resection. Surgery can only solve the tumor complications, such as intestinal obstruction, perforation, and bleeding, but it is not helpful for improving the survival of the patients. Comprehensive therapy, including chemotherapy, radiotherapy, immunotherapy, and targeted therapy, has become the main treatment opinion for these patients. Experts generally advise CRC patients to detect the mutations of KRAS and NRAS and guide targeted tumor therapy. They are also advised to assess the BRAF V600E status so as to stratify disease prognosis. In addition, MMR/MSI is recommended for all CRC patients to stratify prognosis and guide immunotherapy [2, 3]. Until the 2000s, 5-fluorouracil (5-FU) was the only alternative drug for advanced CRC, and the median survival time was no more than 1 year. In 1998 and 2002, irinotecan and oxaliplatin were approved by FDA to combine fluoropyrimidine for the treatment of advanced CRC, nearly doubling the survival. Then, combined with targeted drug (anti-VEGF, anti-EGFR or TKI), the median survival surpassed 2 years [5]. Regrettably, the large randomized phase III trial found that chemotherapy combined with two targeted drugs cannot further improve survival, but increase intolerable toxicity [31, 32]. Therefore, the traditional treatment of advanced CRC has entered the bottleneck again and is difficult to break through. Of note, Nivolumab and Pembrolizumab have been approved by the FDA for the treatment of mCRC [33, 34], due to their excellent performance [27]. ICB therapy is recommended for mCRC patients with dMMR/MSI-H, but immunotherapy resistance is observed in patients with pMMR/MSS [27]. According to previous studies, the proportion of dMMR/MSI-H decreases with the increase in the tumor stage. MSI is noted in approximately 9–21% of stage II and 4.7–11% of stage III tumors [35]. Its incidence is even lower in stage IV CRC (approximately 2.1–4%) [12, 36,37,38] (Fig. 1). Therefore, the application of immunotherapy in mCRC is very limited.

Proportion of dMMR/MSI-H in different tumor stages
The application of ICB therapy in CRC is limited
In Keynote-016, mCRC patients failed in prior standard treatment were divided into three cohorts based on their MMR status (MSI-H/dMMR CRC cohort, MSI-H/dMMR non-CRC cohort, and MSI-H/pMMR CRC cohort). Each cohort was provided with 10 mg/kg Pembrolizumab every 2 weeks, and the primary endpoint was objective response rate (ORR). The ORR was 40% (MSI-H/dMMR CRC cohort), 71% (MSI-H/dMMR non-CRC cohort), and 0% (MSI-H/pMMR CRC cohort), respectively [27]. It is deduced that mCRC patients with dMMR can benefit from PD-1/PD-L1 inhibitor, while the ICB therapy is ineffective in patients with pMMR.
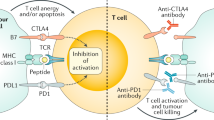
ICB therapy can activate the immune response of tumor lesions and repair the existing immune response by targeting the tumor-induced immune deficiency [39, 40]. It has been approved for the use in melanoma, NSCLC, head and neck squamous cell carcinoma and other malignant tumors and results in optimal clinical effects [22, 41,42,43]. Currently, the majority of mCRC patients are considered to be resistant to ICB therapy. The induction of tumor immune response involves omnifarious aspects, including tumor antigen presentation, T cell activation, T cell infiltration, and T cell recognition, which ultimately activate tumor cell killing. Any defect in these processes can lead to primary or acquired immune resistance [44]. The resistance mechanism of immunotherapy is extremely complex and is related to genetic factors and previous treatment of the patients. It is generally believed that immunotherapy resistance of CRC may be related to the following reasons: insufficient tumor antigen presentation, tumor antigen presentation damage, T cell exclusion, and immunosuppressive signaling in the TME (summarized in Fig. 2).

Mechanism of therapeutic resistance in ICB treatment. The reasons can be summarized as follows: insufficient tumor antigen presentation, tumor antigen presentation damage, T cell exclusion, and immunosuppressive signaling in the TME. The corresponding clinical trials are also marked in the figure
Absence or loss of tumor antigens
Neoantigen is a new antigen encoded by mutated genes of tumor cells, which is an abnormal peptide mainly generated by gene point mutations, deletion mutations, and gene fusions. Its structure is different from that expressed by normal cells [45, 46]. The tumor antigen is the target of the immune system and aims to recognize cancer cells. It is the starting step of the antitumor immune response, which is notably important for the antitumor effect of ICB. Tumor-associated antigens (TAAs) are specific proteins that are overexpressed in tumor cells. They are also expressed in normal cells and can be detected by immune cells in order to trigger an immune response [47]. TAAs limit the ability of immune cells to recognize and induce relative weak specific immune response. CEA is an important TAA of CRC and is frequently expressed on the surface of the majority of mCRC cells. It induces immune tolerance owing to its occurrence in the embryonic stage [48]. To address this disadvantage, the T cell bispecific antibody (TCB) has been employed to strengthen T cell engagement in TME. CEA-TCB induces an interaction of cancer cells with T cells via binding to CEA and CD3. In a phase I trial (NCT02650713), combination of CEA-TCB with ICB in CEA-positive solid tumors indicated a partial response (PR) of 20% (MSS status) and a stable disease (SD) of 50% [49]. In addition, under the pressure of antitumor immunity, the tumor antigens are reduced or lost. This process is termed antigenic modulation and enables tumor cells to escape immune recognition and killing. There is insufficient mutant load to express tumor antigens that produce focused CD8+ T cell responses. These tumors do not respond to ICB therapy due to a lack of T cells specific for distinct tumor antigens [46]. In fact, tumors with a high mutational load and overexpression of tumor neoantigens, such as melanoma, head and neck, NSCLC and MSI tumors, are generally more sensitive to ICB therapy. The majority of the CRC patients exhibited a relatively low mutational load. In order to solve the problem of tumor antigen deficiency or defect, tumor vaccines have become a major focus of investigation. The purpose is to introduce tumor antigens (including tumor cells, tumor-related proteins or peptides, and genes expressing tumor antigens) into patients, enhance immunogenicity, activate the patient’s own immune system, and induce the body’s immune response, so as to control or eliminate the tumors [50]. In April 2010, the FDA approved the cancer vaccine PROVENGE® for the treatment of metastatic castration-resistant prostate cancer. PROVENGE® became the first and, to date, the only therapeutic cancer vaccine [51]. From October 20, 2021, 2,119 tumor vaccine-related clinical trials have been registered on the clinicaltrials.gov Web site. However, the majority of the phase III clinical trials ended in failure due to nonsignificant improvement in the OS of the patients [52]. However, the development of tumor vaccines is still one of the major breakthroughs to improve immunotherapy, along the whole omics development path, gradually from cell to protein and then specific to gene, and now it has entered into the stage of nucleic acid vaccine. It is also a transition from a general tumor vaccine to a tumor personalized vaccine for precise treatment, as well as the transition from TAA to tumor-specific antigens (TSAs) [53].
Tumor antigen presentation damage
Tumor antigens are displayed on the cell surface through major histocompatibility complex class I (MHC-I) molecules. Lack of antigen presentation causes tumor cells to induce tolerance toward T cells, which includes the two following parts: a. Tumor antigen is absorbed by dendritic cells (DCs) and cross-presented to initiate CD8+ T cell activation; b. the antigen is directly presented by the tumor cells so that the activated CD8+ T cells can recognize and kill them [54]. Tumor cells can use diverse escape mechanisms to evade the immune recognition from these two steps. Lost or low expression of MHC-I molecules has been reported on the surface of tumor cells, which results in the obstacle of tumor antigen presentation and the inability to provide the first signal for T cell activation [55]. Previous studies have shown that an antigen-specific T cell level close to 1% is only likely to initiate an effective antitumor response. However, current studies have shown that the antigen presentation level of the majority of cancer cells is very low or even absent, resulting in a weak immune response.
Various mechanisms have been proposed that disrupt antigen presentation in CRC, including interference with the process of proteasome processing of antigens, regulation of the function of transporter associated with antigen processing (TAPs), and obstruction of the expression of MHC structural components through gene mutations, which are notably found in MSI-H tumors [56, 57]. CRC patients with TAPs and MHC-I positive expression were accompanied by increased infiltration of CTLs, leading to subsequent tumor response [58]. β-2-microglobulin (β2M) plays a role in MHC transportation and stable expression on the cell surface. The loss of heterozygosity of β2M can affect the antigen presentation of MHC-I, which leads to melanoma resistance to T cell infiltration and induces primary and acquired ICB resistance [59, 60]. A MSI-H mCRC patient who possessed typical MSI-H molecular characteristics including high mutation load demonstrated disease progression during ICB therapy. Dung et al. surprisingly found that this patient had a loss of β2M biallelic genes, which may be an important reason for his primary resistance to ICB treatment [10]. In addition, the EZH2 inhibitor can overcome ICB treatment resistance by reducing the histone H3K27me3 modification on the β2M promoter [61, 62]. Methylation and histone acetylation can significantly affect the antigen processing and surface presentation of MHC. In lymphoma and melanoma models, both demethylating agents and histone deacetylating agents can increase MHC expression, resulting in increased infiltration of CD8+ T cells and subsequent induction of the antitumor response [63, 64].
T lymphocyte exclusion
T cells are the central link of the immune response, and the lack of T lymphocytes in the TME is a direct and fundamental cause of immunotherapy failure. The lack of tumor antigen or impairment of tumor antigen presentation described above can both affect the recognition of tumor cells by CD 8+ T lymphocytes and indirectly affect T lymphocytes infiltration into TME. Of note, a deficiency in T lymphocytes has been noted in the TME, which is termed T lymphocyte exclusion [65]. Differential expression of chemokine receptors is required for efficient T cell homing and recruitment in the TME [66]. In particular, CXCR3 has been identified as a chemokine receptor critical for T cell infiltration [67]. It has been shown that the Wnt/β-catenin signaling is frequently activated and associated with T cell exclusion in CRC, which is the major obstacle for immunotherapy [68]. Previous studies have shown that activation of the Wnt pathway and expression of nuclear β-catenin are inversely correlated with the infiltration of T cells in CRC tissues. ICRT14 is an inhibitor of β-catenin/T cell factor (TCF), which potently enhances T cell and natural killer cell (NK cell) infiltration. The expression of chemokine (C-X-C motif) ligand 9/10/11 (CXCL9/10/11) was inhibited by activation of the Wnt/β-catenin signaling, suggesting that suppression of β-catenin is expected to shift the colorectal cancer microenvironment into a T cell inflammatory phenotype and enhance the efficacy of immunotherapy [69,70,71]. Clinically, melanoma tumors with Wnt/β-catenin activation respond poorly to ICB, whereas they produce a strong response without Wnt/β-catenin mutations. Inhibitors of the Wnt/β-catenin pathway are intensively investigated in clinical trials and can be combined with ICB to overcome this pattern of primary resistance [72].
Furthermore, signal transducer and activator of transcription 3 (STAT3) can decrease the ability of CD8+ T cells to produce interferon in tumors-γ (IFN-γ). This in turn inhibits CXCL10 secretion by tumor-associated myeloid cells and prevents T cell recruitment. The mitogen-activated protein kinase (MAPK) signaling cascades upregulate the expression levels of the immunosuppressive cytokines vascular endothelial growth factor (VEGF) and interleukin 8 (IL-8), which inhibit T cell function and recruitment into the tumors [73, 74]. Inhibition of the MAPK cascade improves CD8+ T cell infiltration and may also sensitize tumors to ICB therapy [75]. This provides a strong rationale for combination therapy of multi-kinase inhibition and ICB. Similarly, loss of phosphatase and tensin homologue (PTEN) leads to activation of phosphatidylinositol 3-kinase (PI3K) signaling, associated with an increase in the expression of VEGF and reduction in CD8+ T cell infiltration [75]. Epigenetic alterations including DNA methylation and histone modifications have also been considered as an important mechanism of chemokine inhibition and tumor progression. Therefore, treatment with epigenetic modulators can increase chemokine expression and T cell infiltration in the TME.
T cell suppression in TME
CRC development is a complex multifactorial process, during which CRC cells and their surroundings constitute a specific TME. Cancer cells interact and co-evolve with TME, thereby promoting tumor initiation and progression. Several factors in the TME act to suppress immune function, including regulatory T cells (Tregs), IL-10, tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and related cytokines, which can affect ICB therapy and lead to drug resistance [76].
Previous studies have shown that resistance in ICB therapy can occur even with CD8+ T cell infiltration in the TME, which may be due to the lack of the IFN- γ response. It is known that, during antigen-specific immunity, IFN-γ is mainly secreted by CD8+ cytotoxic T cells and by CD4 Th1. The IFN- γ binding with the IFN-γ receptor (IFNGR) leads to Janus kinase 1 (JAK1) and Janus kinase 2 (JAK2) activation and subsequent recruitment and phosphorylation of STAT1 [77]. This complex translocates to the nucleus, where it activates interferon regulatory factor 1 (IRF1). The transcriptional activity of this factor ultimately leads to an IFN-γ-mediated antitumor effect, as well as increased PD-L1 expression [78,79,80,81]. Tumors that have a high mutational load are more likely to respond to ICB therapy. However, certain patients who do not respond despite a high mutational load can present with JAK1/JAK2 mutations. Similarly, functional loss of JAK1/JAK 2 mutations has been found in melanoma samples and melanoma cell lines, which fail to respond to IFN-γ signaling and result in lack of PD-L1 expression. Moreover, a CRISPR screen assay found that apelin receptor interacted with JAK1 and regulated IFN-γ response [82]. It was further suggested that activating mutations were present in protein tyrosine phosphatase non-receptor type 2 (PTPN2), which negatively regulated JAK1 and STAT1 signaling. These were both associated with resistance to ICB therapy caused by the reduced response to IFN-γ. Conversely, CRISPR-Cas9 genome editing can restore melanoma resistance to IFN-γ sensitivity owing to PTPN2 loss [83]. PD-L1 expression may reflect the IFN-γ response, and consequently PD-L1 expression can predict to some extent the clinical efficacy of ICB therapy. However, genetic mutations in IFN-γ signaling genes are uncommon in CRC patients and occur in less than 10% of patients with colorectal adenocarcinoma [84]. Loss of function alteration including JAK1 frameshift is noted in lower than 3% in MSS colon adenocarcinoma samples [85].
Therapy resistance of ICB occurs despite adequate CD8+ T cell infiltration and IFN-γ response when some certain non-tumor cells (NTCs) existed in TME. Tregs and MDSCs with the ability to modulate local immune functions are considered the representative of NTCs. When Tregs or MDSCs are present in the TME, they lead to a reduced immune response against tumor cells. Several studies have shown that depletion of MDSCs or Tregs in the TME can reverse ICB therapy resistance [86,87,88]. Tumor cells and their surrounding stroma can co-regulate the immunosuppressive microenvironment, which leads to resistance to ICB therapy. Therefore, journal of Cell has profiled the transcriptomic and genomic features of metastatic melanoma patients during their treatment with ICB [89]. During the enrichment of BRCA2 mutations, high expression levels of the DNA repair genes were observed in the responding patients. By contrast, tumors with the innate PD-1 resistance (IPRES) signature demonstrate upregulation of genes involved in the regulation of multiple biological processes, including local immunosuppressive genes (VEGF, IL-10), and genes involved in monocyte/macrophage/MDSC chemotaxis, angiogenesis, mesenchymal transition, and wound healing. It is important to note that MAPK-targeted therapies can induce a similar emergence of transcriptional signatures in melanoma, implying that mitogen-activated protein kinase kinase (MEK) inhibitor therapies may be cross-resistant with ICB therapies. IPRES signature is a transcriptomic representative of TME existing in various cancer types, including melanoma, renal clear cell carcinoma, and colon adenocarcinoma. The relevance between this pattern with CRC may provide a unique method to predict the ICB response. Interestingly, the intestinal flora microenvironment is associated with ICB treatment. There are more than 100 trillion bacteria in the human intestine, which form a complex microflora microenvironment that can regulate metabolism and immune function. Since 2011, one after another researches reported that the intestine microflora can function in immune modulation. In 2015, two articles were simultaneously published in the journal Science to discuss the use of ICB in melanoma [90, 91]. Two distinct groups reached the following similar conclusions: Antibiotics can disrupt the antitumor effects of ICB. Specifically, antibiotic-treated mice that were administered with deficient intestinal microflora could restore the ICB’ anticancer effects; patients with specific microbiota, such as Bacteroides and Bifidobacterium, exhibited improved outcomes in immunotherapy. This is the first time that the intestinal microflora was linked with the efficacy of ICB. Subsequently, significant research findings were published validating this theory. Routy et al. demonstrated that Akkermansia muciniphila existed in the majority of patients who were treated with ICB and could achieve remission [92]. Gopalakrishnan et al. demonstrated that the profile of intestinal microflora was associated with the efficacy of ICB therapy. Their study showed that the 30 patients who responded to ICB treatment exhibited a significantly different microflora from the 13 patients who did not respond, and the two specific bacteria, faecalibacterium and clostridiales, were prevalent in progression-free survival (PFS) patients [93, 94]. Recently, a phase I clinical trial (NCT03353402) examining the fecal microbiota transplantation (FMT) was performed in 10 ICB refractory metastatic melanoma patients. Two patients with a partial response (PR) and one patient with a complete response (CR, near complete disappearance of tumor cells) were observed. PFS crossed the 6-month milestone in all responders [95]. For mCRC, a clinical trial in MSS patients demonstrated that patients with high abundance of fusobacterium had significantly shorter PFS (median PFS = 2.0 versus 5.2 months; p = 0.002) [96]. A recent study indicated that fusobacterium was present in both primary tumors and liver metastases in mCRC. Its presence was significantly associated with poor prognosis [97]. Viaud et al. suggested that the metabolic functions of bacteria present in tumor cells may be responsible for the immune resistance of mice with colon cancer [98]. The aforementioned studies indicated that the disturbance of the intestinal flora microenvironment could promote the immune escape of tumor cells, leading to immunotherapy resistance. Therefore, the gut microbiota may be one potential factor influencing the response to immunotherapy in CRC.
Predictive biomarkers of response in ICB therapy
The reduced efficacy of ICB therapy in mCRC patients led to the subsequent investigation of this pathway and its contribution in the response to cancer therapy. Colorectal tumorigenesis is a multistage, multistep, and multigenetic process, with high degrees of genetic heterogeneity. During CRC progression, different key genes and different signaling pathways act at different stages. The current consensus molecular classification (CMS) of CRC approved by the academic community consists of the following five types (Fig. 3): CMS1 is an immune-activated type with MSI-H, which presents with mutations in mismatch repair genes and accounts for approximately 14%. CMS2 is a classical type with aberrant activation of the Wnt and Myc signaling pathways, which harbors significant somatic copy number variation and accounts for 37% of cases. CMS3 is a metabotropic type with a high rate of KRAS mutations, accounting for approximately 13%. CMS4 is the mesenchymal type with abnormal activation of the transforming growth factor β (TGF- β) signaling pathway, noted in approximately 23% of cases. Finally, 13% of the cases cannot be classified alone into any of the aforementioned categories and are classified as mixed type [24].

Consensus molecular classification (CMS) and the coordinate immune response cluster (CIRC) typing of CRC
The majority of the CMS1 tumors are located in the right colon, with deep local invasion and poor differentiation, while a limited number of cases develop distant metastasis. CMS1 has a low frequency of KRAS mutations, whereas BRAF is mutated at a high frequency. Its OS and PFS are poor. BRAF mutations represent poor responsiveness to standard chemotherapy and EGFR targeted therapy, and they are considered as a major reason for the poor prognosis of patients with this type. Different CMS subtypes of CRC exhibit different characteristics in their TME, which is important for developing treatment strategies. CMS1 TME exhibits abundant infiltration of CD8+ lymphocytes, CD68+ macrophages, and adequate expression of PD-1 and PD-L1. Optimal clinical outcomes can be achieved from ICB therapy. CMS4 is associated with an “immunosuppressive phenotype” and is presented in immune tolerance. CMS4 is infiltrated by Tregs, MDSCs, monocyte-derived cells, and T helper 17 (Th17) cells and is associated with high expression of CXCL12 and TGF-β. In this type of cells, TGF-β and angiogenesis-related factors play important roles in immune evasion. Preclinical and clinical studies have demonstrated that TGF-β inhibitors can reverse immune tolerance into immune sensitization [99]. The CMS2 and CMS3 CRC cells are described as “immune desert” and “cold” tumors, which are noted during primary resistance to immunotherapy [100]. Based on this evidence, different types of CMS patients require continued in-depth exploration of their mechanisms of immune tolerance in the search for more effective immunotherapeutic strategies [101,102,103].
Another molecular type used to describe the immunotherapy response in a population of CRC is the coordinate immune response cluster (CIRC) typing (Fig. 3), which primarily divides CRC patients into four groups, including groups A, B, C, and D, based on cluster expression of a gene set (Table 1) [25]. Group A patients exhibits high expression of CIRC genes and were characterized by MSI-H, and upregulation of several immune checkpoint genes, including CTLA4, PD-L1, LAG-3, and TIM3. In contrast to these observations, hypermutations in KRAS, BRAF, NRAS, TP53, PIK3CA, and PTEN, have been reported in the group of lower CIRC expression. This links the genetics and immunobiology of CRC carcinogenesis, providing a theoretical and practical basis for immune stratified therapy. The frequent mutations of MSI-H and POLE mutations were observed in Group A and were associated with high mutational load and high immune infiltration, which suggests that patients can benefit from ICB therapy. Although group D patients enriched in RAS mutations are resistant to ICB treatment, novel strategies have to be developed to address potential resistance in this patient population.
The aforementioned molecular typing is of great significance for the treatment of CRC patients. Recently, a clinical trial based on the CMS principle is ready to be performed for the assessment of the clinical application and prediction value [104, 105]. It is interesting to note that CMS1 and Group A are both immune-activated and respond well to ICB therapy. Moreover, BRAF mutation is frequently accompanied by MSI-H in these two types of CRCs [106]. BRAF mutation was found to be a negative prognostic marker in CRC, and it could promote the development of TME [107, 108]. Therefore, it was speculated that BRAF mutations may possess predictive value in immunotherapy. A meta-analysis was performed comparing the ORR of immunotherapy in MSI-H CRC patients with BRAF mutant and wild-type patients. The results indicated no significant differences in ORR in response to immunotherapy between BRAF mutant and wild-type patients. The results suggested that BRAF mutation did not exhibit predictive value in MSI-H mCRC immunotherapy [109]. However, immunotherapy, as an important complementary method, is still ineffective in the treatment of the majority of CRC patients. Significant investigations have been performed to address the ability of specific indicators to predict the efficacy of ICB therapy.
Microsatellite instability
The MMR genes encode the corresponding mismatch repair protein. Four principal genes are associated with genomic instability as follows: MLH1, MSH2, MSH6, and PMS2 [110]. Defects in DNA mismatch repair system can cause MSI [111]. Generally, the MSI-assay evaluates five selected microsatellite loci as follows: Bat-25, Bat-26, d5s346, d2s123, and d17s250. The MSI status was classified as high instability (MSI-H), low instability (MSI-L), and stability (MSS) [112]; MMR is divided into deficient mismatch repair and proficient mismatch repair. Clinically, dMMR is equivalent to MSI-H, pMMR to MSI-L or MSS [113]. Accumulation of mutated and unrepaired genes will cause MSI-H and induction of carcinogenesis. Approximately 15% of CRCs are caused due to activation of the MSI pathway [38, 111, 114, 115]. MSI-H is associated with abundant neoantigen production, which causes higher immunogenicity and potent immune responses [27]. In CRC, MSI is an excellent predictive biomarker, and as a result, the FDA has approved Pembrolizumab in the treatment of MSI-H/dMMR mCRC patients [116].
Tumor mutational burden
TMB is the number of total mutations per megabase of tumor cells in the coding region, which is used as another predictor of ICB treatment efficacy [117]. The positive correlation between TMB and the efficacy of ICB in immunotherapy was reported for the first time in 2014 [118]. The KEYNOTE-158 study demonstrated that high TMB was associated with better OS, and that TMB could be used as a potential pan-cancer biomarker [19]. In a prospective planned retrospective analysis of various solid tumor patients treated with Pembrolizumab, the ORR was 29% in the TMB high (TMB-H) group compared with only 6% noted in the non-TMB-H group. However, colorectal cancer was not included [19]. Based on this evidence, FDA-approved Pembrolizumab on June 16, 2020 for the treatment of refractory unresectable or metastatic solid tumors of TMB-H, defined by 10 mutations/megabase (muts/MB) based on the FoundationOne CDx assay. However, a latest multicenter, open-label, phase 2a multiple basket study in pan-cancers [119], which accounts for the majority of CRC, reported the value of TMB as a predictor of atezolizumab treatment. The results exhibited that the ORR of MSI-H and MSI-L in patients with TMB ≥ 16 mut/MB was 54.5% and 31.0%, respectively. The median PFS (mPFS) was 8.3 months and 5.6 months, respectively; and the median OS was NE and 19.8 months, respectively. In MSI-L population, the PFS of patients with TMB ≥ 16mut/MB was significantly better than that of patients with 10 ≤ TMB < 16 mut/MB (HR = 0.33, P < 0.0001)). As shown from the data of Aaron et al., MSS/TMB-H accounted for 5.36% (7, 972/148, 803) of cancer cases, while MSI-H only accounted for 1.46% (2, 179/148, 803); thus, it was initially speculated that the use of TMB could make more potential mCRC patients benefit from immunotherapy [120]. David et al. also found a similar phenomenon in patients with MSS [121]. Moreover, studies also found that TMB could be used as an independent biomarker for ORR, PFS, and OS in patients with MSI-H mCRC [122]. In general, the detection of TMB requires tumor specimens for sequencing, termed tTMB, while advanced-stage patients do not have conditional access to tissue samples. Therefore, various clinical centers have attempted to use blood samples to detect bTMB. Certain clinical trials demonstrated that bTMB was significantly associated with tTMB [123,124,125,126]. In a clinical trial of Durvalumab in combination with Tremelimumab in refractory mCRC, it was found that the MSS mCRC patients with bTMB ≥ 28 muts/MB benefitted from ICB treatment [127]. This evidence suggests that bTMB may be an efficacy predictor in the MSS mCRC population, although the specific cutoff value required further exploration and validation.
It is interesting to note that TMB metrics are also flawed, since certain patients with high TMB exist who do not respond to immunotherapy [128] or patients with low TMB who can also achieve optimal therapeutic responses [129,130,131]. This is due to the fact that a high TMB does not correspond to a high tumor neoantigen level. It is therefore clear that the mutation quality is much more important than the mutation quantity. However, tumor neoantigen burden (TNB) is an indicator that reflects the total neoantigen quantity in tumor cells, which can be used as an adjunct to the TMB indicator [132].
Expression levels of PD-L1
The predictive role of PD-L1 in the immunotherapy of solid tumors has been affirmed by various studies. It is generally believed that high expression of PD-L1 is associated with sufficient immune response and clinical benefits from ICB treatment [133]. However, the conclusions from multiple trials were not consistent. An additional study indicated no significant difference in the immune response rate between the PD-L1-positive subgroup (≥ 1%) and the negative subgroup (≤ 1%) [134]. Some researchers believe that PD-L1 expressing should be distinguished from different cells. In a 73 MSI CRC patients cohort, Overman et al. evaluated the relationship between the efficacy of Nivolumab and the expression of PD-L1 on tumor cells or immune cells. The results showed that there was no significant correlation between PD-L1 expression on tumor cells and immunotherapy response, but it was found that ORR with numerous expressions on immune cells was significantly improved [29]. Le et al. reported that PD-L1 expression was only observed in MSI patients, while it was previously speculated that MSS tumors with high PD-L1 expression may also respond to ICB treatment [27, 135]. Subsequently, 138 CRC patients were recruited in O'Neil's study to detect the expression levels of PD-L1. PD-L1-positive CRC patients received Pembrolizumab treatment, and only 4% (1/23) obtained PR. This was also noted in the MSI status [136]. Obviously, the real challenge of immunotherapy is to find biomarkers of MSS CRC, but the current research on PD-L1 has not broken through the dilemma [137]. The reason for this discrepancy among different clinical trials may be that immunohistochemistry was the most commonly used method to detect PD-L1. However, certain differences have been noted with regard to the immunohistochemical antibodies and the scoring systems adopted by different centers, which leads to the incompatibility of the results. The use of different antibodies resulted in significant differences in the evaluation of results [138]. In upper gastrointestinal cancer, the score of PD-L1 has been standardized, and the Combined Positive Score (CPS) > 1 is the critical value of pembrolizumab immune response [139]. It is worth mentioning that the standardized antibody (22C3 pharmDx IHC assay) is used in CPS [139]. Therefore, a unified standard should be formed for CRC so that the most suitable critical value can be determined. In addition, the uneven distribution of PD-L1 in tumors and stromal cells leads to the inconsistency between biopsy specimens and resected tissues [140]. Therefore, the lesions cannot be completely removed, and simple biopsy increases the probability of false negative results. Compared with single-core biopsy, multi-core biopsy is more sensitive to the detection of PD-L1 [140]. The expression levels of PD-L1 were significantly altered in the initial stage of the disease, during its progression and following treatment. Kelly et al. demonstrated that 50% of patients with advanced esophageal adenocarcinoma exhibited altered PD-L1 status (from negative to positive PD-L1 expression) following radiotherapy and chemotherapy [141]. In conclusion, additional prospective studies using unified standards to examine the tumor therapeutic response of CRC patients are required to explore and validate the efficacy of PD-L1.
IFN-γ
In the TME of CRC, tumor-infiltrating CD8+ T cells, NK cells, and NK T cells are the main producers of IFN-γ. IFN-γ can in turn prompt more CD8+ T cells and NK cells infiltration into TME [142]. IFN-γ promotes the MHC-I expression both in antigen-presenting cells (APCs) and in tumor cells, enhancing the antigen recognition function of CD 8+ T cells to kill tumor cells. Furthermore, IFN-γ boosts Th1 cells polarization and directly induces tumor cells apoptosis or non-apoptotic death [143, 144]. IFN-γ can also establish tumor cells dormancy and inhibit tumor cells proliferation by IFN-γ/STAT1 pathway [145] or non-STAT1 signaling [146]. Nevertheless, IFN-γ may also promote tumor immune evasion by promoting tumor cell dormancy under certain condition. IFN-γ can induce tumor antigen loss, recruit MDSCs and TAMs into the TME, and induce tumor immunoediting which results in tumor progression and relapse [147]. In addition, IFN-γ facilitates the expression of immunosuppressive molecules PD-L1, indoleamine 2,3-dioxygenase (IDO), and arginase in TME. Clinically, a relatively high level of IFN-γ was associated with response of immunotherapy [148, 149]. However, shortage or excess of IFN-γ signaling may also lead to immune resistance [81, 150].
Therefore, the role of IFN-γ in the TME is controversial (summarized in Fig. 4). Recently, Joseph et al. analyzed single-cell sequencing data of IFN-stimulated genes (ISGS) [151] in various cell populations in melanoma samples from the TCGA repository and the data demonstrated that the ISGS resistance signature-related genes (ISGS. RS) were mainly expressed in cancer cells. By contrast, IFNG.GS was predominantly expressed by immune cells within the TME, such as T cells, NK cells, and macrophages. Interestingly, a low IFNG.GS/ISG.RS ratio was associated with resistance to ICB treatment. However, a high IFNG.GS/ISG.RS ratio was associated with increased CD8+ T cell and NK cell activation, and high response to ICB immunotherapy. Particularly, the immunotherapeutic response predicted by this ratio was independent of TMB. Previous studies have shown that IFN-γ signaling released by tumor cells can limit the immune response. However, IFN-γ signaling from adaptive and innate immune cells can enhance immune responses. These two actions of the same process are completely opposite. This intriguing study may provide us with an explanation for the dual role of IFN-γ in tumor immunity. It also highlights the potential of administrating therapeutic strategies for patients with different IFN-γ status.

Controversial roles of IFN-γ in the TME of CRC. Antitumor: IFN-γ signaling from immune cells and a high IFNG.GS/ISG.RS ratio were associated with increased CD8+ T cells and NK cell activation, and high response to ICB immunotherapy. IFN-γ can prompt CD8+ T cells and NK cells infiltration into TME, promote the MHC-I expression, boost Th1 cells polarization, directly induce tumor cells apoptosis or non-apoptotic death, establish tumor cells dormancy and inhibit tumor cells proliferation by IFN-γ/STAT1 pathway or non-STAT1 signaling; immune evasion: IFN-γ signaling released by tumor cells and a low IFNG.GS/ISG.RS ratio were associated with resistance to ICB treatment. IFN-γ can induce tumor antigen loss, recruit MDSC and TAMs into the TME, induce tumor immunoediting, and facilitate the expression of PD-L1, IDO, and arginase in TME
Tumor-infiltrating lymphocytes (TILs)
TIL is an important component of the TME, and TILs have been associated with upregulation of PD-L1 expression and various clinical benefits. The antitumor effects of ICB require the participation of lymphocytes in the vicinity of the tumor. Therefore, the abundance of TILs can also be used as a marker to predict the efficacy of the ICB. Normally, immunohistochemical analysis is used to assess the infiltration of CD8+ T cells in the tumor tissues. As a co-stimulatory signal, a higher proportion of CD28+ in TIL cells generally predicts a higher response to therapy, which can also be detected by immunohistochemical analysis [152]. It was reported that only small fractions of the CD8+ TILs were sensitized to tumor antigens, whereas the majority of them did not. Further investigations revealed that the differential CD39 expression was a key factor in discriminating the sensitivity of CD8+ TILs [153]. CD39 is a molecule implicated in chronic immune cell stimulation and is often markedly upregulated in a variety of malignant solid tumors. CD8+ CD39+ TILs are consistent with the characteristics of chronic antigen persisting stimulation, indicating that the activity of this class of TILs is suppressed. They retain the antitumor capacity but require additional stimuli to uncouple suppression, while ICB can then serve this function. Therefore, the higher the proportion of CD39+ in CD8+ TILs, the more likely that the PD-1/PD-L1 signaling axis will be effective. Hence, CD39+ is also a potential marker [154]. In addition, tumor microenvironment immune types (TMITs) were constructed and classified into four groups to describe different TMEs, based on PD-L1 expression and TILs. TILs are characterized by CD8A mRNA expression and the cytolytic activity score (CYT, GZMA, according mRNA expression levels of GZMA and PRF1). This stratification underscores the importance of PD-L1 expression and TIL recruitment. PD-L1-positive TILs are classified as TMIT type I. ICB therapy can benefit patients with PD-L1-positive TILs [155]. Moreover, in CRC, the number of mutations or neoantigens was significantly higher in TIMT type I (high PD-L1 and CD8A/CYT) than in TIMT type II (low PD-L1 and CD8A/CYT) cancers [155]. TMIT stratification may serve as a supplementary method to distinguish “cold” from “hot” tumors and to develop optimal immunotherapeutic strategies. The examination of TMIT may predict the therapeutic response of more diverse tumors to immune strategies on the basis of quantification of immune infiltration using mRNA-seq analysis.
Polymerase epsilon (POLE) mutations
POLE is the key enzyme involved in DNA synthesis and repair processes, and the proofreading role of POLE is essential for replication fidelity, ensuring the appropriate replication of the genome during the cell cycle [156]. After POLE mutations, DNA repair defects and genetic material errors cannot be repaired. Over time, a large number of mutations have accumulated, even up to 10 times that of MSI-H CRC [157]. A retrospective analysis indicated that 66 (1.0%) of 6,517 CRC patients exhibited somatic POLE mutations. The patients with POLE mutations were relatively young, mostly male, and exhibited mainly lesions at the right side. They were also in the early stage of the disease (stage II–III) during initial diagnosis. Approximately 1–2% of MSS CRC exhibits a POLE mutation, while the frequency can range between 5 and 7% in patients aged < 50 years. Previous study has pointed out that CRC patients with POLE mutations are often accompanied with high levels of TILs, upregulated PD-L1 expression, and increased expression of cytotoxic T cell markers and effector cytokines, suggesting enhanced tumor immunogenicity [158]. POLE mutation seems to be a novel predictor of the response to ICB treatment. Wang et al. demonstrated that POLE mutations were independent biomarkers for determining the efficacy of immunotherapy across multiple cancer types [159]. Notably, MSS CRC patients with POLE mutations exhibit durable clinical responses from ICB therapy [23, 160]. This suggested that POLE mutations were a promising indicator that can be used to improve the benefits of immunotherapy on MSS mCRC patients [161].
Neutrophil-to-lymphocyte ratio (NLR)
It is well established that tumor-related inflammation plays an important role in tumorigenesis, disease progression, and patient outcome. By contrast, systemic inflammation is associated with peripheral leukocyte alterations, which is manifested as alteration of the NLR. The NLR can predict the prognosis of patients with CRC and other solid tumors [162, 163]. Several retrospective studies have shown that high baseline NLR and an elevated NLR during treatment were significantly associated with poor outcomes, suggesting that NLR may be a potential predictive factor in patients who received ICB therapy [164,165,166,167,168]. In metastatic NSCLC, high NLR was associated with low response to immunotherapy and was an independent risk factor for poor prognosis [169]. Nevertheless, Jong et al. also indicated that the immune response was associated with a lower NLR at week 6 following immunotherapy, and a reduction in NLR during treatment was associated with longer PFS. As shown in previous studies from different centers, the specific baseline and the change range of NLR have not been previously demonstrated by large prospective studies. In addition, a limited number of studies have examined CRC patients. However, compared with other predictors, NLR is convenient for acquisition and monitoring, which is a research direction worth exploring.
Certain factors are closely associated with immune cell infiltration and the TME. However, their clinical value has not been confirmed and their importance is uncertain. We have compiled and listed these factors in Table 2. Importantly, the hyperprogression is a malignant phenomenon during ICB therapy. The related genes are sorted into Table 2.
The exploration of clinical strategies for ICB therapy and resistance in mCRC (summarized in Additional file 1: Table 1)
The important role of ICB therapy in MSI-H/dMMR CRC has received increasing attention, and certain drugs have been gradually approved and used in routine clinical practice (summarized in Fig. 5 and Table 3). However, 95% of mCRC patients are classified as MSS/pMMR. Therefore, the strategy to overcome immunotherapy resistance has been continuously explored. With regard to the immune microenvironment, MSS mCRC mostly belongs to the “immune-excluded tumor” and “immune desert tumor” classifications. The majority of the immune resistance mechanisms summarized above were noted in MSS mCRC, and the expression levels of cytotoxic cells, CD8+, Th1, Th2, follicular helper T cells, and T cell markers were significantly lower in MSS mCRC than those noted in MSI-H patients. Moreover, the TMB, percentage of missense or frameshift mutations, and the number of tumor neoepitopes were also significantly lower in MSI patients than those noted in MSI-H patients. The Keynote 016 study also demonstrated that MSS patients were largely refractory to immune monotherapy. However, the transformation of the “cold” MSS tumor into the “hot” tumor is an exploratory hotspot. Previous studies indicated that immunotherapy combined with MEK inhibitors or combined with anti-VEGF was not successful, whereas preliminary positive results have also been reported in MSS mCRC. (Selected results are summarized in Table 4.)

Timeline of ICB therapy in MSI-H/dMMR CRC
Immunotherapy combination with chemotherapy: issues that remain to be explored
The 2020 ESMO congress announced the progress of the Keynote-651 (NCT03374254) clinical trial, which is a multicenter, open-label, non-randomized phase Ib study aimed to assess the efficacy and toxicity of Pembrolizumab plus either mFOLFOX7 or FOLFIRI in mCRC [205, 206]. The results indicated an ORR of 58.1% in cohort B (Pembrolizumab plus mFOLFOX7) and an ORR of 15.6% in cohort D (Pembrolizumab plus FOLFIRI). Furthermore, a disease control rate (DCR) of 94% and 63% were noted, respectively. Pembrolizumab plus mFOLFOX7 or FOLFIRI demonstrated preliminary safety and efficacy in patients with MSS/pMMR mCRC. The METIMMOX study (NCT03388190) was reported at the ASCO congress, which compared the repeated sequential oxaliplatin-based chemotherapy (FLOX) combined Nivolumab versus FLOX alone as a first-line treatment of MSS mCRC. The results indicated that the mPFS of the FLOX plus Nivolumab group was 6.6 months (range, 0.5–20), whereas the ORR at 8 months reached 46.3%. This study suggested that FLOX therapy could convert MSS to an immunogenic state, allowing unresectable, previously untreated metastatic patients to achieve durable disease control following treatment with ICB [207]. The present study focused on the identification of predictive biomarkers of ICB responsiveness. Chemotherapy combined with immunotherapy has been the exploratory direction and the common method used by clinicians. Nevertheless, the aforementioned studies did not exhibit clear benefits, and additional investigations are required.
Immune combination MEK inhibitors: changes to be made
Previous studies have shown that inhibition of MEK activity can induce a transcriptional signature similar to immune resistance in melanoma, suggesting that MEK inhibitor therapies may be cross-resistant to ICB therapies. Recently, Obenauf et al. demonstrated that dendritic cells (DCs), which are the key cells of the immune system, have lower activity and reduced cell number in a melanoma mouse model resistant to anti-MEK therapy. Moreover, stimulation of DCs restored the response to immunotherapy [208]. IMblaze370 (NCT02788279) is a multicenter, open-label, randomized controlled clinical trial exploring the efficacy of PD-L1 inhibitors in combination with a MEK inhibitor regimen (Atezolizumab plus Cabozantinib). Phase Ib results indicated a modest response rate (8%) and disease control (31%). However, the final results indicated that the combination therapy exhibited a median OS (mOS) of 8.87 months (95% CI 7.00 to 10.61) and a mPFS of 1.91 months (95% CI 1.87 to 1.97), which was not significantly different compared with other monotherapy groups. Additional adverse effects were also noted [209]. To date, several clinical trials have produced invalid results from ICB combinations with MEK inhibitors in CRC. However, certain preclinical studies have shown that a simple combination therapy is a suboptimal approach, while rational dosing and sequencing administration contributes to improved survival in mouse models [210]. The regimen of short-term ICB treatment prior to administration of MEK inhibitors is suggested to reverse drug resistance, which is more effective in prolonging tumor shrinkage and preventing the development of drug resistance. Certainly, relevant therapeutic approaches await exploration in high-quality clinical trials.
Immunotherapy combined with anti-epidermal growth factor receptor (EGFR) therapy: Worth trying
The prognosis in all types of CMS4 tumor, with high APC and low BRAF mutation rates, is considerably poor [211]. The angiogenesis-associated pathway is aberrantly activated in this cancer type, promoting the hyperplasia and growth of metastatic tumor cells. The antiangiogenic drug Bevacizumab can significantly improve patient survival. A previous study indicated that Cetuximab exhibited a direct tumor killing effect, acting as an IgG1 monoclonal antibody that also exhibited antibody-dependent cell-mediated cytotoxicity (ADCC) effects. Cetuximab was also able to recruit anti-EGFR T cells as well as CD8+and CD3+ T cells. Moreover, this antibody can also increase PD-L1 expression and induce immunosuppression by possible synergism with ICB. The AVETUXIRI (NCT03608046) study investigated the combination of Bavencio with Cetuximab and Irinotecan in patients with refractory MSS mCRC who had failed prior standard therapy [212]. This study was stratified by the RAS mutation status into cohort A (RAS wild type) and cohort B (RAS mutated type). Initial findings indicated an ORR of 30% in cohort A, reaching its primary efficacy endpoint and proceeding to the phase II study. No PR was observed in cohort B, while both RAS wt and RAS mt groups exhibited DCRs of 60% (6/10) and 61.5% (8/13), respectively, mPFS of 4.2 and 3.8 months, and mOS of 12.7 and 14.0 months, respectively. The 6-month PFS rates were 40.0% and 38.5%, respectively. The 12-month OS rates were 53.3% and 57.7%, respectively. PR was not observed in the RAS mt cohort. However, optimal DCR, PFS, and OS data were also obtained in the RAS mt cohort, and the investigators established a RAS mt mCRC cohort with PFS as the primary endpoint in order to expand these research findings. In a single-arm, single-institution, phase I/II clinical trial (NCT04017650) [213], 26 patients with refractory MSS and BRAFV600E metastatic CRC were recruited and Encorafenib, Cetuximab, and Nivolumab were used in combination. The ORR of 45% (95% CI 23 to 68) and DCR of 95% (95% CI 75% to 100), the mPFS of 7.3 months (95% CI 5.5 to NA) and the mOS of 11.4 months (95%CI 7.6 to NA) were reported. This study reached the predetermined efficacy endpoint, indicating that this novel regimen is effective and well tolerated in the treatment of MSS, BRAFV600E mCRC.
Immunotherapy combinations with tyrosine kinase inhibitors (TKIs) are anticipated to be scheduled in the future
Preclinical studies have shown that antiangiogenic therapy may improve the TME, increase and activate effector immune cells, reduce immunosuppressive cells, and relieve immunosuppressive effects, which play a significant role in the synergism of immunotherapy. However, the clinical effects of immunotherapy combined with anti-VEGF agents are not optimal. The BACCI (NCT02873195) study aimed to evaluate the efficacy of Capecitabine and Bevacizumab combined with Atezolizumab or placebo as the third-line treatment for refractory mCRC patients. The mPFS and mOS in the experimental group were 4.37 (95%CI 4.07 to 6.41), 10.55 (95%CI 8.21 to NA), and 3.32 (95%CI 2.14 to 6.21) and 10.61 (95%CI 8.80 to NA) in the control group [214]. The difference was not statistically significant in MSS patients, and the control group did not significantly improve OS. Inhibition of angiogenesis has long been considered as a potential approach to reverse immunotherapy resistance. However, immunization in combination with Bevacizumab has been shown to be unsuccessful. Despite these findings, immunotherapy combined with TKI has achieved promising results. TKIs, which also possess antiangiogenic effects, can block the three targets of VEGFR, notablyVEGFR3. Tyrosine kinases enzymes phosphorylate specific amino acids on substrate enzymes, which affect signal transduction pathway. TKIs exhibit a wide range of target inhibition effects, and it has been recognized that they may also inhibit colony-stimulating factor-1 receptor (CSF-1R). Based on their antiangiogenic effects, they can reverse antitumor immune activity by blocking CSF-1R-mediated pathway to inhibit tumor immune-related macrophages. Multiple prospective studies have been conducted worldwide to explore the efficacy of ICB with TKI therapy in the treatment of MSS mCRC.
The REGONIVO (NCT03406871) study in Japan was a phase Ib study using nivolumab plus regorafenib in refractory MSS CRC and gastric cancer. This study demonstrated an ORR of 28% and a mPFS of 7.8 months (95% CI, 2.8 to NR) in mCRC, with a 1-year PFS rate of 41.7% and a 1-year OS rate of 68.0%, which were considerably higher than the data from previous studies in MSS CRC [215]. However, this study could not be repeated in the follow-up North American REGONIVO phase II study (NCT04126733), and an ORR of 7.1%, mPFS of 8 weeks, and a mOS of 52 weeks were reported [204]. The REGOTORI (NCT03946917) study indicated that 5 out of 33 evaluable patients treated with 80 mg regorafenib achieved a tumor response with an ORR of 15.2% (95% CI, 5.7%–32.7%) and a DCR of 36.4% (95% CI, 21.0% to 54.9). The ORR was higher in patients without liver metastasis than that noted in those with liver metastasis (30.0% vs. 8.7%). The ORR of patients with only lung metastasis (3/3, 100%) was considerably higher than that of those with liver metastasis alone (0/4, 0%), suggesting that ICB combined with TKI is an option for refractory MSS mCRC, notably for patients without liver metastasis or lung metastasis alone [96]. A phase Ib study (NCT03903705) was performed to evaluate the safety and preliminary efficacy of Fruquintinib in combination with GB 226 for the treatment of mCRC. The data indicated favorable results. The ORR in 12 patients with MSS mCRC was 25.0%, whereas the DCR was 75%, and the mPFS was 5.45 months (95% CI 1.84 to 9.66) [216]. Recently, the largest trial of ICB combined with regorafenib in the treatment of MSS CRC (NCT03657641) was reported [217]. The subjects were patients with chemotherapy failure, of which 78% of patients had liver metastasis. The mPFS was 2.0 (95% CI 1.8 to 3.5) months and the mOS was 10.9 (95% CI 5.3 to NR) months. In 16 patients (23%) with non-hepatic metastatic disease, PFS was 4.3 (95% CI 1.9 to 8.4) months. Unfortunately, the trial did not reach its primary endpoint, and biomarker analysis is currently being conducted to further explore the benefit population.
The efficacy of the dual immune checkpoint inhibitor combination therapy requires further validation
The CCTG CO.26 (NCT02870920) study evaluated the use of the dual ICB combination regimen as a post-line treatment for refractory mCRC. The experimental group received Durvalumab + Tremelimumab compared with the best supportive care (BSC) group, which was used as a control [218]. Notably, OS was significantly longer in the dual immunization group (6.6 months vs 4.1 months), whereas PFS was not prolonged (1.8 months vs. 1.9 months), and DCR was estimated to 22.6% and 6.6%, respectively. Subsequent analysis (excluding 2 patients with MSI-H) revealed that the median TMB was increased to 20.4, and the patients with a TMB > 28 MTs/MB could benefit more from dual immunotherapy, whereas high TMB in the BSC group was associated with a poor prognosis. The MEDITREME trial (NCT03202758) was a single-arm exploratory trial that enrolled 57 patients with RAS mutant, MSS, untreated mCRC, in which the investigators presumed that FOLFOX chemotherapy could induce immunogenic cell death and remove MDSCs, in order to potentiate the antitumor effects of immunotherapy. Enrolled patients received first-line therapy with FOLFOX in combination with Durvalumab and Tremelimumab, and the results indicated that the mPFS was not reached, with a 6-month PFS rate of 62.5% [219]. Another multicenter phase II study LCCC1632 (NCT03442569) met the primary study endpoint of a remission rate of 35% at 12 weeks [220]. The study was designed to evaluate the efficacy and safety of Ipilimumab and Nivolumab plus Panitumumab in patients with KRAS/NRAS/BRAF wild-type MSS mCRC. A total of 49 patients were evaluated with regard to the efficacy of treatment at 12 weeks, 34.7% of the patients (17/49) exhibited PR, 0 had CR, and 42.9% (21/49) exhibited stable disease (SD). The mPFS was 5.7 months (95% CI, 5.5–7.9 months). This chemotherapy-free immune combination targeted therapy regimen offers first-line hope for the chemotherapy-resistant advanced RAS/BRAF wild-type MSS mCRC patients. Of course, phase III studies are still required for confirmation. As an important new immune checkpoint, LAG-3 is structurally similar to CD4. It has four extracellular regions that bind to ligands, thus inducing immune cell failure and reducing cytokine secretion. An increase in reliable clinical data on the double blocking of LAG-3 and PD-1 has prompted people to focus on this combination immunotherapy. A clinical trial (NCT02720068) [221] demonstrated that the combination of LAG-3 inhibitor (Favezelimab) and Pembrolizumab exhibits good antitumor activity, especially in patients with PD-L1 CPS ≥ 1.
Future perspectives
Immune checkpoint inhibitor therapy is relatively less toxic than chemotherapy and targeted therapy. However, some of its unique adverse effects reduce its clinical efficacy, and certain rare adverse effects could be life-threatening, which result in skin, endocrine, hepatic, gastrointestinal, pulmonary, and skeletal muscle toxicity. The related aspects of this topic will not be covered in this review.
ICBs have been successfully used in the MSI-H CRC population. Pembrolizumab, Nivolumab, and Ipilimumab have been approved to be used in MSI-H refractory mCRC, and Pembrolizumab has been recommended as first-line therapy of treatment. More recently, clinical trials indicated that neoadjuvant immunotherapy may have the potential to become the standard therapy for CRC patients. In a stepwise exploration, ICBs hold promise as adjuvant therapies for patients with stage III CRC after resection. Moreover, the combination with specific drugs is expected to improve efficacy and attenuate associated toxicity. The urgent task is to find multiple biomarkers and formulate standardized scoring standards in order to screen population and benefit more patients. For the MSS population, which constitutes the majority of CRC patients, ICB monotherapy was ineffective. In addition to the microsatellite status, other potential biomarkers can be developed that can aid the identification of potential populations. More importantly, it is significant to develop measures that can turn “cold tumor” into “hot tumor” so as to expand the application scope of immunotherapy. This scope of research may provide a milestone in CRC treatment.
Availability of data and materials
The datasets used and analyzed during the current study are available within the manuscript and its additional files.
Abbreviations
- mCRC:
-
Metastatic colorectal cancer
- ICB:
-
Immune checkpoint blockades
- MSI-H:
-
Microsatellite instability—high
- mOS:
-
Median overall survival
- NSCLC:
-
Non-small cell lung cancer
- PD-1:
-
Programmed death-1
- PD-L1:
-
Programmed cell death-ligand 1
- FDA:
-
Food and Drug Administration
- dMMR:
-
Deficient mismatch repair
- pMMR:
-
Mismatch repair-proficient
- TMB:
-
Tumor mutation burden
- TME:
-
Tumor microenvironment
- 5-FU:
-
5-Fluorouracil
- TAAs:
-
Tumor-associated antigens
- TCB:
-
T cell bispecific antibody
- TSAs:
-
Tumor-specific antigens
- MHC-I:
-
Major histocompatibility complex class I
- TAPs:
-
Transporter associated with antigen processing
- β2M:
-
β-2-Microglobulin
- TCF:
-
T cell factor
- NK cell:
-
Natural killer cell
- STAT3:
-
Signal transducer and activator of transcription 3
- MAPK:
-
Mitogen-activated protein kinase
- VEGF:
-
Vascular endothelial growth factor
- IL-8:
-
Interleukin 8
- PTEN:
-
Phosphatase and tensin homologue
- PI3K:
-
Phosphatidylinositol 3-kinase
- Tregs:
-
Regulatory T cells
- TAMs:
-
Tumor-associated macrophages
- MDSCs:
-
Myeloid-derived suppressor cells
- IFNGR:
-
IFN-γ receptor
- JAK1/JAK2:
-
Janus kinase 1/Janus kinase 2
- IRF1:
-
Interferon regulatory factor 1
- PTPN2:
-
Protein tyrosine phosphatase non-receptor type 2
- NTCs:
-
Non-tumor cells
- IPRES:
-
Innate PD-1 resistance
- MEK:
-
Mitogen-activated protein kinase kinase
- PFS:
-
Progression-free survival
- FMT:
-
Fecal microbiota transplantation
- PR:
-
Partial response
- CR:
-
Complete response
- CMS:
-
Consensus molecular classification
- TGF- β:
-
Transforming growth factor β
- Th17:
-
T helper 17
- CIRC:
-
Coordinate immune response cluster
- MSI-H:
-
Microsatellite high instability
- MSI-L:
-
Microsatellite low instability
- MSS:
-
Microsatellite stability
- TMB-H:
-
TMB high
- muts/MB:
-
Mutations/megabase
- mPFS:
-
Median progression-free survival
- TNB:
-
Tumor neoantigen burden
- CPS:
-
Combined positive score
- APCs:
-
Antigen-presenting cells
- ISGS:
-
IFN-stimulated genes
- ISGS. RS:
-
ISGS resistance signature-related genes
- TILs:
-
Tumor-infiltrating lymphocytes
- TMITs:
-
Tumor microenvironment immune types
- POLE:
-
Polymerase epsilon
- DCR:
-
Disease control rate
- DCs:
-
Dendritic cells
- EGFR:
-
Epidermal growth factor receptor
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- TKIs:
-
Tyrosine kinase inhibitors
- CSF-1R:
-
Colony-stimulating factor 1 receptor
- BSC:
-
Best supportive care
- SD:
-
Stable disease
References
Siegel RL, Miller KD. Cancer statistics. CA Cancer J Clin. 2019;69:7–34. https://doi.org/10.3322/caac.21551.
The National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology, Rectal Cancer. Version 3.2021, <https://www.nccn.org/professionals/physician_gls/pdf/rectal.pdf> (2021).
The National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology, Colon Cancer. Version 2.2021, <https://www.nccn.org/professionals/physician_gls/pdf/colon.pdfc> ( 2021).
ESMO consensus guidelines for the management of patients with metastatic colorectal cancer, <https://www.annalsofoncology.org/article/S0923-7534(19)34754-4/pdf> (2016).
Chibaudel B, et al. Therapeutic strategy in unresectable metastatic colorectal cancer: an updated review. CA Cancer J Clin. 2015;7:153–69. https://doi.org/10.1177/1758834015572343.
Brahmer J, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373:123–35. https://doi.org/10.1056/NEJMoa1504627.
Robert C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–26. https://doi.org/10.1056/NEJMoa1104621.
Wolchok JD, et al. CheckMate 067: 6.5-year outcomes in patients (pts) with advanced melanoma. J Clin Oncol. 2021;39:9506–9506. https://doi.org/10.1200/JCO.2021.39.15_suppl.9506.
Antonia SJ, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med. 2017;377:1919–29. https://doi.org/10.1056/NEJMoa1709937.
Le DT, Durham JN. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13. https://doi.org/10.1126/science.aan6733.
Benatti P, et al. Microsatellite instability and colorectal cancer prognosis. Clin Cancer Res. 2005;11:8332–40. https://doi.org/10.1158/1078-0432.ccr-05-1030.
Venderbosch S, et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: a pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res. 2014;20:5322–30. https://doi.org/10.1158/1078-0432.ccr-14-0332.
André T, et al. Adjuvant Fluorouracil, leucovorin, and oxaliplatin in stage II to III colon cancer: updated 10-year survival and outcomes according to BRAF mutation and mismatch repair status of the MOSAIC study. J Clin Oncol. 2015;33:4176–87. https://doi.org/10.1200/jco.2015.63.4238.
Stadler ZK, et al. Reliable detection of mismatch repair deficiency in colorectal cancers using mutational load in next-generation sequencing panels. J Clin Oncol. 2016;34:2141–7. https://doi.org/10.1200/jco.2015.65.1067.
The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. https://doi.org/10.1038/nature11252.
Maby P, et al. Correlation between density of CD8+ T-cell infiltrate in microsatellite unstable colorectal cancers and frameshift mutations: a rationale for personalized immunotherapy. Can Res. 2015;75:3446–55. https://doi.org/10.1158/0008-5472.can-14-3051.
Marisa L, et al. The balance between cytotoxic T-cell lymphocytes and immune checkpoint expression in the prognosis of colon tumors. J Natl Cancer Inst. 2018. https://doi.org/10.1093/jnci/djx136.
Llosa NJ, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5:43–51. https://doi.org/10.1158/2159-8290.cd-14-0863.
Marabelle A, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21:1353–65. https://doi.org/10.1016/s1470-2045(20)30445-9.
Samstein RM, Lee CH, Shoushtari AN. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202–6. https://doi.org/10.1038/s41588-018-0312-8.
Cristescu R. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Nat Genet. 2018. https://doi.org/10.1126/science.aar3593.
Loboda A, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. Science (New York, NY). 2015;372:2018–28. https://doi.org/10.1056/NEJMoa1501824.
Gong J, Wang C, Lee PP, Chu P, Fakih M. Response to PD-1 blockade in microsatellite stable metastatic colorectal cancer harboring a POLE mutation. J Natl Compr Cancer Netw JNCCN. 2017;15:142–7. https://doi.org/10.6004/jnccn.2017.0016.
Guinney J, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–6. https://doi.org/10.1038/nm.3967.
Tejpar S, Lal N, Beggs AD, Willcox BE, Middleton GW. An immunogenomic stratification of colorectal cancer: Implications for development of targeted immunotherapy. Nat Med. 2015;4: e976052. https://doi.org/10.4161/2162402x.2014.976052.
Andre T, et al. Pembrolizumab versus chemotherapy for microsatellite instability-high/mismatch repair deficient metastatic colorectal cancer: the phase 3 KEYNOTE-177 Study. J Clin Oncol. 2020;38:LBA4–LBA4. https://doi.org/10.1200/JCO.2020.38.18_suppl.LBA4.
Le DT, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20. https://doi.org/10.1056/NEJMoa1500596.
Overman MJ, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36:773–9. https://doi.org/10.1200/jco.2017.76.9901.
Overman MJ, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–91. https://doi.org/10.1016/s1470-2045(17)30422-9.
Lenz HJ, Van Cutsem E. First-line nivolumab plus low-dose ipilimumab for microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: the phase II CheckMate 142 study. J Clin. 2021. https://doi.org/10.1200/jco.21.01015.
Hecht JR, et al. A randomized phase IIIB trial of chemotherapy, bevacizumab, and panitumumab compared with chemotherapy and bevacizumab alone for metastatic colorectal cancer. J Clin Oncol. 2009;27:672–80. https://doi.org/10.1200/jco.2008.19.8135.
Tol J, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360:563–72. https://doi.org/10.1056/NEJMoa0808268.
André T, et al. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. New Engl J Med. 2020;383:2207–18. https://doi.org/10.1056/NEJMoa2017699.
Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. N Engl J Med. 2019;25:3753–8. https://doi.org/10.1158/1078-0432.ccr-18-4070.
Roth AD, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60–00 trial. J Clin Oncol. 2010;28:466–74. https://doi.org/10.1200/jco.2009.23.3452.
Asaka S, et al. Microsatellite instability-low colorectal cancer acquires a KRAS mutation during the progression from Dukes’ A to Dukes’ B. Carcinogenesis. 2009;30:494–9. https://doi.org/10.1093/carcin/bgp017.
Koopman M, et al. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br J Cancer. 2009;100:266–73. https://doi.org/10.1038/sj.bjc.6604867.
Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138:2073-2087.e2073. https://doi.org/10.1053/j.gastro.2009.12.064.
Dong H, et al. Tumor-associated B7–H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. https://doi.org/10.1038/nm730.
Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375:1767–78. https://doi.org/10.1056/NEJMra1514296.
Topalian SL, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020–30. https://doi.org/10.1200/jco.2013.53.0105.
Massard C, et al. Safety and efficacy of durvalumab (MEDI4736), an anti-programmed cell death ligand-1 immune checkpoint inhibitor, in patients with advanced urothelial bladder cancer. J Clin Oncol. 2016;34:3119–25. https://doi.org/10.1200/jco.2016.67.9761.
Schadendorf D, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33:1889–94. https://doi.org/10.1200/jco.2014.56.2736.
Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–71. https://doi.org/10.1146/annurev-immunol-031210-101324.
Parkhurst MR, Robbins PF, Tran E, Prickett TD. Unique neoantigens arise from somatic mutations in patients with gastrointestinal cancers. Cancer Discov. 2019;9:1022–35. https://doi.org/10.1158/2159-8290.cd-18-1494.
Lo W, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Cancer Discov. 2014;515:577–81. https://doi.org/10.1038/nature13988.
Slansky JE, Spellman PT. Alternative splicing in tumors—a path to immunogenicity? Nature. 2019;380:877–80. https://doi.org/10.1056/NEJMcibr1814237.
Foon KA, et al. Immune response to the carcinoembryonic antigen in patients treated with an anti-idiotype antibody vaccine. J Clin Investig. 1995;96:334–42. https://doi.org/10.1172/jci118039.
Tabernero J, et al. Phase Ia and Ib studies of the novel carcinoembryonic antigen (CEA) T-cell bispecific (CEA CD3 TCB) antibody as a single agent and in combination with atezolizumab: preliminary efficacy and safety in patients with metastatic colorectal cancer (mCRC). J Clin Oncol. 2017;35:3002–3002. https://doi.org/10.1200/JCO.2017.35.15_suppl.3002.
Saxena M, van der Burg SH, Melief CJM, Bhardwaj N. Therapeutic cancer vaccines. Nat Rev Cancer. 2021;21:360–78. https://doi.org/10.1038/s41568-021-00346-0.
Kantoff PW, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. Nat Rev Cancer. 2010;363:411–22. https://doi.org/10.1056/NEJMoa1001294.
Schreiber TH, Raez L, Rosenblatt JD, Podack ER. Tumor immunogenicity and responsiveness to cancer vaccine therapy: the state of the art. N Engl J Med. 2010;22:105–12. https://doi.org/10.1016/j.smim.2010.02.001.
Blass E, Ott PA. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol. 2021;18:215–29. https://doi.org/10.1038/s41571-020-00460-2.
Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7–24. https://doi.org/10.1038/s41577-019-0210-z.
Rock KL, Reits E, Neefjes J. Present yourself! By MHC class I and MHC class II molecules. Nat Rev Immunol. 2016;37:724–37. https://doi.org/10.1016/j.it.2016.08.010.
Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol. 2000;74:181–273. https://doi.org/10.1016/s0065-2776(08)60911-6.
Kloor M, et al. Immunoselective pressure and human leukocyte antigen class I antigen machinery defects in microsatellite unstable colorectal cancers. Can Res. 2005;65:6418–24. https://doi.org/10.1158/0008-5472.can-05-0044.
Watson NF, et al. Immunosurveillance is active in colorectal cancer as downregulation but not complete loss of MHC class I expression correlates with a poor prognosis. Int J Cancer. 2006;118:6–10. https://doi.org/10.1002/ijc.21303.
D’Urso CM, et al. Lack of HLA class I antigen expression by cultured melanoma cells FO-1 due to a defect in B2m gene expression. J Clin Investig. 1991;87:284–92. https://doi.org/10.1172/jci114984.
Zaretsky JM, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375:819–29. https://doi.org/10.1056/NEJMoa1604958.
Zhou L, Mudianto T, Ma X, Riley R, Uppaluri R. Targeting EZH2 enhances antigen presentation, antitumor immunity, and circumvents anti-PD-1 resistance in head and neck cancer. Clin Cancer Res. 2020;26:290–300. https://doi.org/10.1158/1078-0432.ccr-19-1351.
Ennishi D, et al. Molecular and genetic characterization of MHC deficiency identifies EZH2 as Therapeutic target for enhancing immune recognition. Cancer Discov. 2019;9:546–63. https://doi.org/10.1158/2159-8290.cd-18-1090.
Gascoyne RD, et al. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Discov. 2009;69:8693–9. https://doi.org/10.1158/0008-5472.can-09-1456.
Serrano A, et al. Rexpression of HLA class I antigens and restoration of antigen-specific CTL response in melanoma cells following 5-aza-2’-deoxycytidine treatment. Int J Cancer. 2001;94:243–51. https://doi.org/10.1002/ijc.1452.
Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science (New York, NY). 2015;348:74–80. https://doi.org/10.1126/science.aaa6204.
Zhang Y, Guan XY, Jiang P. Cytokine and chemokine signals of T-cell exclusion in tumors. Front Immunol. 2020;11: 594609. https://doi.org/10.3389/fimmu.2020.594609.
Chow MT, et al. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity. 2019;50:1498-1512.e1495. https://doi.org/10.1016/j.immuni.2019.04.010.
Grasso CS, et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov. 2018;8:730–49. https://doi.org/10.1158/2159-8290.cd-17-1327.
Hsu L, et al. β-Catenin inhibition shapes tumor immunity and synergizes with immunotherapy in colorectal cancer. Cancer Discov. 2020;9:1809947. https://doi.org/10.1080/2162402x.2020.1809947.
Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer. 2018;18:139–47. https://doi.org/10.1038/nrc.2017.117.
Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17:559–72. https://doi.org/10.1038/nri.2017.49.
Duchartre Y, Kim YM, Kahn M. The Wnt signaling pathway in cancer. Crit Rev Oncol Hematol. 2016;99:141–9. https://doi.org/10.1016/j.critrevonc.2015.12.005.
Liu C, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res. 2013;19:393–403. https://doi.org/10.1158/1078-0432.ccr-12-1626.
Loi S, et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin Cancer Res. 2016;22:1499–509. https://doi.org/10.1158/1078-0432.ccr-15-1125.
Hu-Lieskovan S, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci Transl Med. 2015;7:279ra241. https://doi.org/10.1126/scitranslmed.aaa4691.
Ge P, et al. Profiles of immune cell infiltration and immune-related genes in the tumor microenvironment of colorectal cancer. Biomed Pharmacother Biomed Pharmacother. 2019;118:109228. https://doi.org/10.1016/j.biopha.2019.109228.
Darnell JE Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science (New York, NY). 1994;264:1415–21. https://doi.org/10.1126/science.8197455.
Garcia-Diaz A, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–201. https://doi.org/10.1016/j.celrep.2017.04.031.
Benci JL, et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. 2016;167:1540-1554.e1512. https://doi.org/10.1016/j.cell.2016.11.022.
Bifulco CB, Urba WJ. Unmasking PD-1 resistance by next-generation sequencing. N Engl J Med. 2016;375:888–9. https://doi.org/10.1056/NEJMe1606042.
Gao J, et al. Loss of IFN-γ Pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167:397-404.e399. https://doi.org/10.1016/j.cell.2016.08.069.
Patel SJ, et al. Identification of essential genes for cancer immunotherapy. Nature. 2017;548:537–42. https://doi.org/10.1038/nature23477.
Manguso RT, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547:413–8. https://doi.org/10.1038/nature23270.
Shin DS, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7:188–201. https://doi.org/10.1158/2159-8290.cd-16-1223.
Albacker LA, et al. Loss of function JAK1 mutations occur at high frequency in cancers with microsatellite instability and are suggestive of immune evasion. PLoS ONE. 2017;12: e0176181. https://doi.org/10.1371/journal.pone.0176181.
Steinberg SM, et al. Myeloid cells that impair immunotherapy are restored in melanomas with acquired resistance to BRAF inhibitors. Can Res. 2017;77:1599–610. https://doi.org/10.1158/0008-5472.can-16-1755.
ArceVargas F, et al. Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity. 2017;46:577–86. https://doi.org/10.1016/j.immuni.2017.03.013.
Taylor NA, et al. Treg depletion potentiates checkpoint inhibition in claudin-low breast cancer. J Clin Investig. 2017;127:3472–83. https://doi.org/10.1172/jci90499.
Hugo W, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. https://doi.org/10.1016/j.cell.2016.02.065.
Vétizou M, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science (New York, NY). 2015;350:1079–84. https://doi.org/10.1126/science.aad1329.
Sivan A, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science (New York, NY). 2015;350:1084–9. https://doi.org/10.1126/science.aac4255.
Routy B, Le Chatelier E. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–7. https://doi.org/10.1126/science.aan3706.
Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The Influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Science (New York, NY). 2018;33:570–80. https://doi.org/10.1016/j.ccell.2018.03.015.
Gopalakrishnan V, Spencer CN. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103. https://doi.org/10.1126/science.aan4236.
Baruch EN. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science (New York, NY). 2021;371:602–9. https://doi.org/10.1126/science.abb5920.
Wang F, et al. Regorafenib plus toripalimab in patients with metastatic colorectal cancer: a phase Ib/II clinical trial and gut microbiome analysis. Cell Rep Med. 2021;2: 100383. https://doi.org/10.1016/j.xcrm.2021.100383.
Yu T, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Science (New York, NY). 2017;170:548-563.e516. https://doi.org/10.1016/j.cell.2017.07.008.
Viaud S, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science (New York, NY). 2013;342:971–6. https://doi.org/10.1126/science.1240537.
Kim TW, et al. Efficacy and safety of vactosertib and Pembrolizumab combination in patients with previously treated microsatellite stable metastatic colorectal cancer. J Clin Oncol. 2021;39:3573–3573. https://doi.org/10.1200/JCO.2021.39.15_suppl.3573.
Lal N, et al. KRAS Mutation and consensus molecular subtypes 2 and 3 are independently associated with reduced immune infiltration and reactivity in colorectal cancer. Clin Cancer Res. 2018;24:224–33. https://doi.org/10.1158/1078-0432.ccr-17-1090.
Menter DG, et al. Back to the colorectal cancer consensus molecular subtype future. Curr Gastroenterol Rep. 2019;21:5. https://doi.org/10.1007/s11894-019-0674-9.
Loree JM, et al. Classifying colorectal cancer by tumor location rather than sidedness highlights a continuum in mutation profiles and consensus molecular subtypes. Clin Cancer Res. 2018;24:1062–72. https://doi.org/10.1158/1078-0432.ccr-17-2484.
Lee MS, Menter DG, Kopetz S. Right versus left colon cancer biology: integrating the consensus molecular subtypes. J Natl Compr Cancer Netw JNCCN. 2017;15:411–9. https://doi.org/10.6004/jnccn.2017.0038.
Smeby J, et al. CMS-dependent prognostic impact of KRAS and BRAFV600E mutations in primary colorectal cancer. Ann Oncol. 2018;29:1227–34. https://doi.org/10.1093/annonc/mdy085.
Eide PW, Bruun J, Lothe RA, Sveen A. CMScaller: an R package for consensus molecular subtyping of colorectal cancer pre-clinical models. Sci Rep. 2017;7:16618. https://doi.org/10.1038/s41598-017-16747-x.
Colle R, et al. Immunotherapy and patients treated for cancer with microsatellite instability. Sci Rep. 2017;104:42–51. https://doi.org/10.1016/j.bulcan.2016.11.006.
Kopetz S, et al. Randomized Trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG S1406). J Clin Oncol. 2021;39:285–94. https://doi.org/10.1200/jco.20.01994.
Jo J. New molecular targeted therapy of metastatic colorectal cancer. Ewha Med J. 2021;44:11–8. https://doi.org/10.12771/emj.2021.44.1.11.
Park R, Silva LLD, Lee S, Saeed A. Impact of BRAF mutations on prognosis and immunotherapy response in microsatellite instability/mismatch repair deficient metastatic colorectal cancer: a systematic review and meta-analysis. J Clin Oncol. 2021;39:3557–3557. https://doi.org/10.1200/JCO.2021.39.15_suppl.3557.
Takamochi K, et al. DNA mismatch repair deficiency in surgically resected lung adenocarcinoma: microsatellite instability analysis using the Promega panel. Lung Cancer (Amsterdam, Netherlands). 2017;110:26–31. https://doi.org/10.1016/j.lungcan.2017.05.016.
Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558–61. https://doi.org/10.1038/363558a0.
Boland CR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Can Res. 1998;58:5248–57.
Cicek MS, et al. Quality assessment and correlation of microsatellite instability and immunohistochemical markers among population- and clinic-based colorectal tumors results from the Colon Cancer Family Registry. J Mol Diagn JMD. 2011;13:271–81. https://doi.org/10.1016/j.jmoldx.2010.12.004.
Aaltonen LA, et al. Clues to the pathogenesis of familial colorectal cancer. Science (New York, NY). 1993;260:812–6. https://doi.org/10.1126/science.8484121.
Hampel H, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med. 2005;352:1851–60. https://doi.org/10.1056/NEJMoa043146.
Lemery S, Keegan P, Pazdur R. First FDA approval agnostic of cancer site: when a biomarker defines the indication. N Engl J Med. 2017;377:1409–12. https://doi.org/10.1056/NEJMp1709968.
Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377:2500–1. https://doi.org/10.1056/NEJMc1713444.
Snyder A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–99. https://doi.org/10.1056/NEJMoa1406498.
Friedman CF, Hainsworth JD, Kurzrock R. Atezolizumab treatment of tumors with high tumor mutational burden from MyPathway, a multicenter, open-label, phase 2a multiple basket study. Cancer Discov. 2021. https://doi.org/10.1158/2159-8290.cd-21-0450.
Goodman AM, Sokol ES, Frampton GM. Microsatellite-stable tumors with high mutational burden benefit from immunotherapy. Cancer Immunol Res. 2019;7:1570–3. https://doi.org/10.1158/2326-6066.cir-19-0149.
Fabrizio DA, et al. Beyond microsatellite testing: assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoint inhibition. Cancer Immunol Res. 2018;9:610–7. https://doi.org/10.21037/jgo.2018.05.06.
Schrock AB, et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann Oncol. 2019;30:1096–103. https://doi.org/10.1093/annonc/mdz134.
Araujo DV, et al. Applications of circulating tumor DNA in a cohort of phase I solid tumor patients treated with immunotherapy. JNCI Cancer Spectrum. 2021;5:pkaa122. https://doi.org/10.1093/jncics/pkaa122.
Aggarwal C, et al. Baseline plasma tumor mutation burden predicts response to pembrolizumab-based therapy in patients with metastatic non-small cell lung cancer. Clin Cancer Res. 2020;26:2354–61. https://doi.org/10.1158/1078-0432.ccr-19-3663.
Rittmeyer A, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet (London, England). 2017;389:255–65. https://doi.org/10.1016/s0140-6736(16)32517-x.
Fehrenbacher L, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet (London, England). 2016;387:1837–46. https://doi.org/10.1016/s0140-6736(16)00587-0.
Chen EX, et al. Effect of combined immune checkpoint inhibition vs best supportive care alone in patients with advanced colorectal cancer: the canadian cancer trials group CO.26 study. JAMA Oncol. 2020;6:831–8. https://doi.org/10.1001/jamaoncol.2020.0910.
Lombardi G, Barresi V. Pembrolizumab activity in recurrent high-grade gliomas with partial or complete loss of mismatch repair protein expression: a monocentric, observational and prospective pilot study. Cancers. 2020. https://doi.org/10.3390/cancers12082283.
Gromeier M, et al. ATIM-27 Tumor mutational burden predicts response to oncolytic polio/rhinovirus recombinant (PVSRIPO) in malignant glioma patients: assessment of transcriptional and immunological correlates. Neuro Oncol. 2019;21:vi7–vi7. https://doi.org/10.1093/neuonc/noz175.026.
Zagonel V, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. Cancers. 2016;34:2206–11. https://doi.org/10.1200/jco.2016.66.6552.
Blaeschke F, et al. Low mutational load in pediatric medulloblastoma still translates into neoantigens as targets for specific T-cell immunotherapy. Cytotherapy. 2019;21:973–86. https://doi.org/10.1016/j.jcyt.2019.06.009.
Wang P, Chen Y, Wang C. Beyond tumor mutation burden: tumor neoantigen burden as a biomarker for immunotherapy and other types of therapy. Front Oncol. 2021;11: 672677. https://doi.org/10.3389/fonc.2021.672677.
Reck M, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823–33. https://doi.org/10.1056/NEJMoa1606774.
Sharma P, et al. Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): a multicentre, open-label, two-stage, multi-arm, phase 1/2 trial. Lancet Oncol. 2016;17:1590–8. https://doi.org/10.1016/s1470-2045(16)30496-x.
Le DT, et al. A blueprint to advance colorectal cancer immunotherapies. Cancer Immunol Res. 2017;5:942–9. https://doi.org/10.1158/2326-6066.cir-17-0375.
O’Neil BH, et al. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with advanced colorectal carcinoma. PLoS ONE. 2017;12: e0189848. https://doi.org/10.1371/journal.pone.0189848.
Alexander PG, McMillan DC, Park JH. A meta-analysis of CD274 (PD-L1) assessment and prognosis in colorectal cancer and its role in predicting response to anti-PD-1 therapy. Crit Rev Oncol Hematol. 2021;157: 103147. https://doi.org/10.1016/j.critrevonc.2020.103147.
Lee KS, et al. Programmed cell death ligand-1 protein expression and CD274/PD-L1 gene amplification in colorectal cancer: implications for prognosis. Cancer Sci. 2018;109:2957–69. https://doi.org/10.1111/cas.13716.
Kulangara K, et al. Clinical utility of the combined positive score for programmed death ligand-1 expression and the approval of pembrolizumab for treatment of gastric cancer. Cancer Sci. 2019;143:330–7. https://doi.org/10.5858/arpa.2018-0043-OA.
Munari E, et al. PD-L1 expression heterogeneity in non-small cell lung cancer: defining criteria for harmonization between biopsy specimens and whole sections. J Thoracic Oncol. 2018;13:1113–20. https://doi.org/10.1016/j.jtho.2018.04.017.
Kelly RJ, et al. The dynamic and transient immune microenvironment in locally advanced esophageal adenocarcinoma post chemoradiation. Ann Surg. 2018;268:992–9. https://doi.org/10.1097/sla.0000000000002410.
Du W, Frankel TL, Green M, Zou W. IFNγ signaling integrity in colorectal cancer immunity and immunotherapy. doi:https://doi.org/10.1038/s41423-021-00735-3 (2021).
Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, Borden EC. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. 2003;8(3):237–49. https://doi.org/10.1023/a:1023668705040.
Li P, et al. Interferon-γ induces autophagy with growth inhibition and cell death in human hepatocellular carcinoma (HCC) cells through interferon-regulatory factor-1 (IRF-1). Cancer Lett. 2012;314:213–22. https://doi.org/10.1016/j.canlet.2011.09.031.
Kortylewski M, et al. Interferon-gamma-mediated growth regulation of melanoma cells: involvement of STAT1-dependent and STAT1-independent signals. J Invest Dermatol. 2004;122:414–22. https://doi.org/10.1046/j.0022-202X.2004.22237.x.
Liu Y, et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-γ-induced immunologic dormancy of tumor-repopulating cells. Nat Commun. 2017;8:15207. https://doi.org/10.1038/ncomms15207.
Aqbi HF, Wallace M, Sappal S, Payne KK, Manjili MH. IFN-γ orchestrates tumor elimination, tumor dormancy, tumor escape, and progression. J Leukoc Biol. 2018. https://doi.org/10.1002/jlb.5mir0917-351r.
Jiang X, et al. Lycopene improves the efficiency of anti-PD-1 therapy via activating IFN signaling of lung cancer cells. Cancer Cell Int. 2019;19:68. https://doi.org/10.1186/s12935-019-0789-y.
Boutsikou E, et al. Tumour necrosis factor, interferon-gamma and interleukins as predictive markers of antiprogrammed cell-death protein-1 treatment in advanced non-small cell lung cancer: a pragmatic approach in clinical practice. Ther Adv Med Oncol. 2018;10:1758835918768238. https://doi.org/10.1177/1758835918768238.
Pai CS, et al. Clonal deletion of tumor-specific T cells by interferon-γ confers therapeutic resistance to combination immune checkpoint blockade. Immunity. 2019;50:477-492.e478. https://doi.org/10.1016/j.immuni.2019.01.006.
Benci JL, et al. Opposing functions of interferon coordinate adaptive and innate immune responses to cancer immune checkpoint blockade. Cell. 2019;178:933-948.e914. https://doi.org/10.1016/j.cell.2019.07.019.
Duraiswamy J, et al. Myeloid antigen-presenting cell niches sustain antitumor T cells and license PD-1 blockade via CD28 costimulation. Cancer Cell. 2021;39:1623-1642.e1620. https://doi.org/10.1016/j.ccell.2021.10.008.
Yeong J, et al. Intratumoral CD39(+)CD8(+) T cells predict response to programmed cell death protein-1 or programmed death ligand-1 blockade in patients with NSCLC. J Thoracic Oncol. 2021;16:1349–58. https://doi.org/10.1016/j.jtho.2021.04.016.
Krishna S, Lowery FJ. Stem-like CD8 T cells mediate response of adoptive cell immunotherapy against human cancer. Science. 2020;370:1328–34. https://doi.org/10.1126/science.abb9847.
Chen YP, et al. Genomic analysis of tumor microenvironment immune types across 14 solid cancer types: immunotherapeutic implications. Science (New York, NY). 2017;7:3585–94. https://doi.org/10.7150/thno.21471.
Prindle MJ, Loeb LA. DNA polymerase delta in DNA replication and genome maintenance. Environ Mol Mutagen. 2012;53:666–82. https://doi.org/10.1002/em.21745.
Heitzer E, Tomlinson I. Replicative DNA polymerase mutations in cancer. Curr Opin Genet Dev. 2014;24:107–13. https://doi.org/10.1016/j.gde.2013.12.005.
Domingo E, et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: a retrospective, pooled biomarker study. Lancet Gastroenterol Hepatol. 2016;1:207–16. https://doi.org/10.1016/s2468-1253(16)30014-0.
Wang F, et al. Evaluation of POLE and POLD1 mutations as biomarkers for immunotherapy outcomes across multiple cancer types. JAMA Oncol. 2019;5:1504–6. https://doi.org/10.1001/jamaoncol.2019.2963.
Mehnert JM, et al. Immune activation and response to pembrolizumab in POLE-mutant endometrial cancer. J Clin Investig. 2016;126:2334–40. https://doi.org/10.1172/jci84940.
Ciardiello D, et al. Immunotherapy of colorectal cancer: Challenges for therapeutic efficacy. Cancer Treat Rev. 2019;76:22–32. https://doi.org/10.1016/j.ctrv.2019.04.003.
Haram A, et al. The prognostic value of neutrophil-to-lymphocyte ratio in colorectal cancer: a systematic review. J Surg Oncol. 2017;115:470–9. https://doi.org/10.1002/jso.24523.
Qian S, Golubnitschaja O, Zhan X. Chronic inflammation: key player and biomarker-set to predict and prevent cancer development and progression based on individualized patient profiles. Epma J. 2019;10:365–81. https://doi.org/10.1007/s13167-019-00194-x.
Fan X, et al. Inflammatory markers predict survival in patients with advanced gastric and colorectal cancers receiving anti-PD-1 therapy. EPMA J. 2021;9: 638312. https://doi.org/10.3389/fcell.2021.638312.
Chen S, et al. Prognostic value of baseline and change in neutrophil-to-lymphocyte ratio for survival in advanced non-small cell lung cancer patients with poor performance status receiving PD-1 inhibitors. Transl Lung Cancer Res. 2021;10:1397–407. https://doi.org/10.21037/tlcr-21-43.
Ueda T, et al. Baseline neutrophil-to-lymphocyte ratio (NLR) is associated with clinical outcome in recurrent or metastatic head and neck cancer patients treated with nivolumab. Acta Otolaryngol. 2020;140:181–7. https://doi.org/10.1080/00016489.2019.1699250.
Moschetta M, Uccello M. Dynamics of neutrophils-to-lymphocyte ratio predict outcomes of PD-1/PD-L1 blockade. BioMed Res Int. 2017. https://doi.org/10.1155/2017/1506824.
Diem S, et al. Neutrophil-to-Lymphocyte ratio (NLR) and Platelet-to-Lymphocyte ratio (PLR) as prognostic markers in patients with non-small cell lung cancer (NSCLC) treated with nivolumab. Biomed Res Int. 2017;111:176–81. https://doi.org/10.1016/j.lungcan.2017.07.024.
Cao D, Xu H, Xu X, Guo T, Ge W. A reliable and feasible way to predict the benefits of Nivolumab in patients with non-small cell lung cancer: a pooled analysis of 14 retrospective studies. Oncoimmunology. 2018;7: e1507262. https://doi.org/10.1080/2162402x.2018.1507262.
Burgers PM. Polymerase dynamics at the eukaryotic DNA replication fork. J Biol Chem. 2009;284:4041–5. https://doi.org/10.1074/jbc.R800062200.
Lotan TL, Antonarakis ES. CDK12 deficiency and the immune microenvironment in prostate cancer. Clin Cancer Res. 2021;27:380–2. https://doi.org/10.1158/1078-0432.ccr-20-3877.
Wu YM, et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell. 2018;173:1770-1782.e1714. https://doi.org/10.1016/j.cell.2018.04.034.
Khemlina G, Ikeda S, Kurzrock R. The biology of Hepatocellular carcinoma: implications for genomic and immune therapies. Mol Cancer. 2017;16:149. https://doi.org/10.1186/s12943-017-0712-x.
Horn S, et al. Tumor CDKN2A-associated JAK2 loss and susceptibility to immunotherapy resistance. J Natl Cancer Inst. 2018;110:677–81. https://doi.org/10.1093/jnci/djx271.
Riaz N, Havel JJ. Recurrent SERPINB3 and SERPINB4 mutations in patients who respond to anti-CTLA4 immunotherapy. Nat Genet. 2016;48:1327–9. https://doi.org/10.1038/ng.3677.
Weinhold N, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Nat Genet. 2017;171:934-949.e916. https://doi.org/10.1016/j.cell.2017.09.028.
Dong ZY, et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res. 2017;23:3012–24. https://doi.org/10.1158/1078-0432.ccr-16-2554.
Scheel AH, et al. PD-L1 expression in non-small cell lung cancer: correlations with genetic alterations. Oncoimmunology. 2016;5: e1131379. https://doi.org/10.1080/2162402x.2015.1131379.
Wang Z, et al. Comutations in DNA damage response pathways serve as potential biomarkers for immune checkpoint blockade. Can Res. 2018;78:6486–96. https://doi.org/10.1158/0008-5472.can-18-1814.
Teo MY, et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD-1/PD-L1 blockade in advanced urothelial cancers. J Clin Oncol. 2018;36:1685–94. https://doi.org/10.1200/jco.2017.75.7740.
Gao Y, et al. Fusobacterium nucleatum enhances the efficacy of PD-L1 blockade in colorectal cancer. Signal Transduct Target Ther. 2021;6:398. https://doi.org/10.1038/s41392-021-00795-x.
Wu X, et al. CMTM6 expression in M2 macrophages is a potential predictor of PD-1/PD-L1 inhibitor response in colorectal cancer. Cancer Immunol Immunother. 2021;70:3235–48. https://doi.org/10.1007/s00262-021-02931-6.
Akbani R. Genomic classification of cutaneous melanoma. Cancer Immunol Immunother CII. 2015;161:1681–96. https://doi.org/10.1016/j.cell.2015.05.044.
Johnson DB, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res. 2016;4:959–67. https://doi.org/10.1158/2326-6066.cir-16-0143.
Kato S, et al. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res. 2017;23:4242–50. https://doi.org/10.1158/1078-0432.ccr-16-3133.
Borghaei H, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627–39. https://doi.org/10.1056/NEJMoa1507643.
Koyama S, et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Can Res. 2016;76:999–1008. https://doi.org/10.1158/0008-5472.can-15-1439.
Skoulidis F, Goldberg ME. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018;8:822–35. https://doi.org/10.1158/2159-8290.cd-18-0099.
Skoulidis F, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5:860–77. https://doi.org/10.1158/2159-8290.cd-14-1236.
Kim Y, et al. Comprehensive clinical and genetic characterization of hyperprogression based on volumetry in advanced non-small cell lung cancer treated with immune checkpoint inhibitor. J Thoracic Oncol. 2019;14:1608–18. https://doi.org/10.1016/j.jtho.2019.05.033.
Peng W, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6:202–16. https://doi.org/10.1158/2159-8290.cd-15-0283.
Sui Q, et al. Dickkopf 1 impairs the tumor response to PD-1 blockade by inactivating CD8+ T cells in deficient mismatch repair colorectal cancer. J Immunother Cancer. 2021. https://doi.org/10.1136/jitc-2020-001498.
Liu G, et al. Upregulation of LAGE3 correlates with prognosis and immune infiltrates in colorectal cancer: a bioinformatic analysis. J Immunother Cancer. 2020;85: 106599. https://doi.org/10.1016/j.intimp.2020.106599.
Jiang J, et al. Integrated genomic analysis identifies a genetic mutation model predicting response to immune checkpoint inhibitors in melanoma. Cancer Med. 2020;9:8498–518. https://doi.org/10.1002/cam4.3481.
Wagner NB, Forschner A, Leiter U, Garbe C, Eigentler TK. S100B and LDH as early prognostic markers for response and overall survival in melanoma patients treated with anti-PD-1 or combined anti-PD-1 plus anti-CTLA-4 antibodies. Cancer Med. 2020;119:339–46. https://doi.org/10.1002/cam4.3481.
Cona MS, et al. Combination of baseline LDH, performance status and age as integrated algorithm to identify solid tumor patients with higher probability of response to anti PD-1 and PD-L1 monoclonal antibodies. Cancers. 2019. https://doi.org/10.3390/cancers11020223.
Zuazo-Ibarra M, et al. Senescent CD4 T cells unequivocally identify primary resistance and risk of hyperprogression to PD-L1/PD-1 immune checkpoint blockade in lung cancer. Biorxiv. 2018. https://doi.org/10.1101/320176.
Xiong D, et al. Immunogenomic landscape contributes to hyperprogressive disease after anti-PD-1 immunotherapy for cancer. iScience. 2018;9:258–77. https://doi.org/10.1016/j.isci.2018.10.021.
Kirchberger S, et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med. 2013;210:917–31. https://doi.org/10.1084/jem.20122308.
Koyama S, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. https://doi.org/10.1038/ncomms10501.
Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med. 2015;212:1125–37. https://doi.org/10.1084/jem.20142237.
Yu J, et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med. 2021;27:152–64. https://doi.org/10.1038/s41591-020-1131-x.
Lee J, et al. Immunological Insights into liver metastasis associated resistance to checkpoint blockade cancer immunotherapy. J Immunol. 2018;200:122–6.
Fakih M, et al. Single-arm, phase 2 study of regorafenib plus nivolumab in patients with mismatch repair-proficient (pMMR)/microsatellite stable (MSS) colorectal cancer (CRC). J Clin Oncol. 2021;39:3560–3560. https://doi.org/10.1200/JCO.2021.39.15_suppl.3560.
Kim R, et al. 608P—Pembrolizumab (pembro) plus mFOLFOX or FOLFIRI in patients with metastatic colorectal cancer (mCRC): KEYNOTE-651 cohorts B and D. Ann Oncol. 2019;30:v229–30. https://doi.org/10.1093/annonc/mdz246.085.
Kim R, et al. 493P - Pembrolizumab (pembro) plus mFOLFOX7 or FOLFIRI in patients (pts) with metastatic colorectal cancer (mCRC): updated results from KEYNOTE-651 cohorts B and D. Ann Oncol. 2020;suppl_4:S409–61.
Ree AH, et al. Repeat sequential oxaliplatin-based chemotherapy (FLOX) and nivolumab versus FLOX alone as first-line treatment of microsatellite-stable (MSS) metastatic colorectal cancer (mCRC): initial results from the randomized METIMMOX study. J Clin Oncol. 2021;39:3556–3556. https://doi.org/10.1200/JCO.2021.39.15_suppl.3556.
Haas L, et al. Acquired resistance to anti-MAPK targeted therapy confers an immune-evasive tumor microenvironment and cross-resistance to immunotherapy in melanoma. Nat Cancer. 2021;2:693–708. https://doi.org/10.1038/s43018-021-00221-9.
Eng C, et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2019;20:849–61. https://doi.org/10.1016/s1470-2045(19)30027-0.
Wang Y, et al. Anti-PD-1/L1 lead-in before MAPK inhibitor combination maximizes antitumor immunity and efficacy. Cancer Cell. 2021;39:1375-1387.e1376. https://doi.org/10.1016/j.ccell.2021.07.023.
Ubink I, et al. Histopathological and molecular classification of colorectal cancer and corresponding peritoneal metastases. Br J Surg. 2018;105:e204–11. https://doi.org/10.1002/bjs.10788.
Van Den Eynde M, et al. Interim analysis of the AVETUXIRI Trial: Avelumab combined with cetuximab and irinotecan for treatment of refractory microsatellite stable (MSS) metastatic colorectal cancer (mCRC)—a proof of concept, open-label, nonrandomized phase IIa study. J Clin Oncol. 2021;39:80–80. https://doi.org/10.1200/JCO.2021.39.3_suppl.80.
Morris VK, et al. Phase I/II trial of encorafenib, cetuximab, and nivolumab in patients with microsatellite stable, BRAFV600E metastatic colorectal cancer. J Clin Oncol. 2022;40:12–12. https://doi.org/10.1200/JCO.2022.40.4_suppl.012.
Mettu NB, et al. 533PD - BACCI: A phase II randomized, double-blind, multicenter, placebo-controlled study of capecitabine (C) bevacizumab (B) plus atezolizumab (A) or placebo (P) in refractory metastatic colorectal cancer (mCRC): an ACCRU network study. Ann Oncol. 2019;30:v203. https://doi.org/10.1093/annonc/mdz246.011.
Fukuoka S, et al. Regorafenib plus nivolumab in patients with advanced gastric or colorectal cancer: an open-label, dose-escalation, and dose-expansion phase Ib trial (REGONIVO, EPOC1603). J Clin Oncol. 2020;38:2053–61. https://doi.org/10.1200/jco.19.03296.
Bai Y, et al. A phase ib trial of assessing the safety and preliminary efficacy of a combination therapy of geptanolimab (GB 226) plus fruquintinib in patients with metastatic colorectal cancer (mCRC). J Clin Oncol. 2021;39:e15551–e15551. https://doi.org/10.1200/JCO.2021.39.15_suppl.e15551.
Barzi A, et al. Phase I/II study of regorafenib (rego) and pembrolizumab (pembro) in refractory microsatellite stable colorectal cancer (MSSCRC). J Clin Oncol. 2022;40:15–15. https://doi.org/10.1200/JCO.2022.40.4_suppl.015.
Chen EX, et al. CCTG CO.26: Updated analysis and impact of plasma-detected microsatellite stability (MSS) and tumor mutation burden (TMB) in a phase II trial of durvalumab (D) plus tremelimumab (T) and best supportive care (BSC) versus BSC alone in patients (pts) with refractory metastatic colorectal carcinoma (rmCRC). J Clin Oncol. 2019;37:3512–3512. https://doi.org/10.1200/JCO.2019.37.15_suppl.3512.
Ghiringhelli F, et al. Durvalumab and tremelimumab in combination with FOLFOX in patients with RAS-mutated, microsatellite-stable, previously untreated metastatic colorectal cancer (MCRC): results of the first intermediate analysis of the phase Ib/II MEDETREME trial. J Clin Oncol. 2020;38:3006–3006. https://doi.org/10.1200/JCO.2020.38.15_suppl.3006.
Lee MS, et al. Phase II study of ipilimumab, nivolumab, and panitumumab in patients with KRAS/NRAS/BRAF wild-type (WT) microsatellite stable (MSS) metastatic colorectal cancer (mCRC). J Clin Oncol. 2021;39:7–7. https://doi.org/10.1200/JCO.2021.39.3_suppl.7.
Garralda E, et al. A phase 1 first-in-human study of the anti-LAG-3 antibody MK4280 (favezelimab) plus pembrolizumab in previously treated, advanced microsatellite stable colorectal cancer. J Clin Oncol. 2021;39:3584–3584. https://doi.org/10.1200/JCO.2021.39.15_suppl.3584.
Acknowledgements
Not applicable
Funding
This study was supported by the National Natural Science Foundation of China (82003060, 81772599, and 81972260).
Author information
Authors and Affiliations
Contributions
XL and JW conceived and designed the research study; JW, SL, and ZZ drew figures and tables; JW, QL, YY, and RZ collected and updated clinical trial data. JW wrote the manuscript and XL revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This research was approved by the Ethics Committee of Fudan University Shanghai Cancer Center.
Consent for publication
All patients involved in our study obtained written consent for publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Summary of Clinical Trials in This Paper.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Weng, J., Li, S., Zhu, Z. et al. Exploring immunotherapy in colorectal cancer. J Hematol Oncol 15, 95 (2022). https://doi.org/10.1186/s13045-022-01294-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-022-01294-4