
Abstract
Targeting nucleotide metabolism can not only inhibit tumor initiation and progression but also exert serious side effects. With in-depth studies of nucleotide metabolism, our understanding of nucleotide metabolism in tumors has revealed their non-proliferative effects on immune escape, indicating the potential effectiveness of nucleotide antimetabolites for enhancing immunotherapy. A growing body of evidence now supports the concept that targeting nucleotide metabolism can increase the antitumor immune response by (1) activating host immune systems via maintaining the concentrations of several important metabolites, such as adenosine and ATP, (2) promoting immunogenicity caused by increased mutability and genomic instability by disrupting the purine and pyrimidine pool, and (3) releasing nucleoside analogs via microbes to regulate immunity. Therapeutic approaches targeting nucleotide metabolism combined with immunotherapy have achieved exciting success in preclinical animal models. Here, we review how dysregulated nucleotide metabolism can promote tumor growth and interact with the host immune system, and we provide future insights into targeting nucleotide metabolism for immunotherapeutic treatment of various malignancies.
Similar content being viewed by others


Introduction
Nucleotides are the main building blocks of genetic materials and are composed of purines (adenine and guanine) and pyrimidines (thymine, uracil, and cytosine). They are essential substances for the biosynthesis of DNA and RNA, cell signaling, enzyme regulation, and metabolism. Cancer cells must synthesize and utilize large amounts of energy and nucleotides for DNA and RNA, and upregulated de novo nucleotide metabolism enables cells to proliferate rapidly; therefore, nucleotide metabolism is a potential target for cancer treatment. Although numerous efforts to target this attractive metabolic pathway have been reported, the key enzymes and regulatory mechanisms involved in nucleotide metabolism remain unclear. All classical antitumor drugs inhibiting nucleotide synthesis are based on analogs of tumor nucleotide metabolites and have previously served as chemotherapies in cancer treatment [1, 2]. However, due to their lack of specificity for tumor cell nucleotide metabolism, these drugs also inhibit the metabolic processes of normal cells, causing serious side effects [3, 4]. Therefore, more in-depth study of the regulatory processes of nucleotide metabolism has important theoretical and clinical significance. Moreover, recent studies have shown that abnormal nucleotide metabolism not only accelerates the development of tumors but also alters the normal immune response in the tumor microenvironment (TME), indicating the potential effectiveness of targeting nucleotide metabolism to enhance immunotherapy [5,6,7]. This review provides an overview of nucleotide metabolism and its role in cancer and emphasizes that nucleotide metabolism is a therapeutic target not only for chemotherapy but also for enhancing the efficacy of cancer immunotherapy.
Nucleotide biosynthesis and degradation
Nucleotide metabolism includes nucleotide biosynthesis and degradation to maintain nucleotide homeostasis [8]. Proliferating cells acquire nutrients (mainly glucose, glutamine, and CO2) to generate energy to drive anabolism of nucleic acids, while nucleotides also need to be replenished at certain rates consistent with nucleotide biosynthesis [9]. Although nucleotides can be taken up via salvage pathways, the de novo biosynthesis pathway remains the main pathway through which most dividing cells synthesize nucleotides and their related metabolites [10, 11].
De novo nucleotide metabolism is regulated by several critical metabolic genes and encoded enzymes (Table 1). These essential metabolic enzymes play important roles in maintaining nucleotide biosynthesis [12]. Purine biosynthesis, in which purine nucleotides are synthesized directly by the addition of a pyrophosphate at C-1 of the ribose sugar, differs from pyrimidine biosynthesis in many ways [13]. Purine biosynthesis begins with ribose-5-phosphate converted to phosphoribosyl pyrophosphate (PRPP), and several ATP equivalents are required to activate PRPP [9]. The enzyme involved in this step is PRPP synthetase, encoded by the gene PRPS (Table 1). The rate-limiting step in this pathway is the second step, wherein PRPP is catalyzed by PPAT to bind with glutamine, causing the formation of 5-phosphoribosylamine along with the release of pyrophosphate [14,15,16]. The next steps characterized as ATP-dependent include several reactions in which inosine monophosphate (IMP) is converted and biosynthesized from 5-phosphoribosylamine, and glycinamide ribonucleotide transformylase (GART) plays a key role in maintaining its biosynthesis [17]. IMP serves as a precursor to adenosine monophosphate (AMP) and guanosine monophosphate (GMP) synthesis [17]. During the synthesis of AMP and GMP from IMP, adenylosuccinate synthetase (ADSS) and Inosine monophosphate dehydrogenase (IMPDH) are essential catalysts of the conversion of IMP into succinyl adenosine 5'-monophosphate (sAMP) and xanthosine monophosphate (XMP) through several kinetic intermediates [18,19,20].
In the de novo pyrimidine biosynthesis pathway, the pyrimidine ring structure is assembled through a 6-step process with L-glutamine and L-aspartate as precursors, which are transformed into dihydroorotate in the initial steps [9]. The trifunctional proteins carbamoyl phosphate synthetase, aspartyl transcarbamoylase, and dihydroorotase (CAD) are associated with the enzymatic activities of the first three reactions [41, 42]. As another well-known rate-limiting enzyme of pyrimidine biosynthesis, dihydroorotate dehydrogenase (DHODH) catalyzes dihydroorotate into orotate and derives mitochondrial electron transport and oxygen consumption [43,44,45, 53]. The final two steps of the de novo pyrimidine biosynthetic pathway are catalyzed by uridine monophosphate synthetase (UMPS), a bifunctional enzyme that includes orotate phosphoribosyltransferase and orotidine monophosphate (OMP) decarboxylase [8, 42, 47]. The first reaction is initiated from orotate to form orotidine-5P, while orotidine-5P is converted into uridine-5-phosphate in the second step [54, 55]. Uridine-5-phosphate constitutes the building block of the subsequent reactions of pyrimidine biosynthesis [9].
The common pathways of both purine and pyrimidine nucleotide biosynthesis include several reactions. Generally, the transformation and homeostasis between nucleoside triphosphate and nucleoside monophosphate are controlled by ecto-nucleoside triphosphate diphosphohydrolase-1 (ENTPD1) (Table 1). ENTPD1 (also known as CD39) and ecto-5-nucleotidase (NT5E, also known as CD73) are critical mediators among these regulators [56]. CD73 converts AMP to adenosine with phosphate, whereas CD39 can hydrolyze nucleoside-5-triphosphates into nucleoside-5-monophosphate and its products (Table 1). CD39 and CD73 play essential roles in maintaining nucleotide metabolism, while they regulate immune responses via substrate levels of extracellular ATP and adenosine with tumor-promoting and tumor-suppressing effects [57]. Ribose-5-monophosphate and deoxyribose-5-monophosphate are further catalyzed to compose nucleosides and deoxynucleosides mediated by CD73 and CD39 [58]. Furthermore, purine nucleoside phosphorylase (PNP), a ubiquitously expressed homotrimer, catalyzes the reversible phosphorolysis of nucleosides to generate the corresponding purine and pyrimidine base and ribose 1-phosphate, which are converted into purines and pyrimidines [59].
In addition to the de novo biosynthesis pathway, the salvage pathway, which uses free bases that are derived endogenously from the turnover of nucleic acids or exogenously from dietary intake, can generate purines and pyrimidines [60]. The relative importance of salvage versus de novo synthesis likely depends on the growth conditions and on the specific tissue. As the exact steps involved in recycling are only known for purine bases, the final products of the salvage pathway of purines are AMP, IMP, and GMP [59, 60]. During the salvage pathway, ubiquitous PNPs play a key role in catalyzing hypoxanthine–guanine phosphoribosyltransferase (HGPRT) to synthesize the monophosphates (MPs) of inosine (Ino) and guanosine (Guo) [61]. Ribo- and deoxyribonucleosides are converted to the PNP pathway to form only ribonucleotides mediated by adenosine deaminase (ADA) [61]. Uridine–cytidine kinases (UCK1 and UCK2), rate-limiting enzymes involved in the salvage pathway of pyrimidine-nucleotide biosynthesis, convert uridine and cytidine to their corresponding MPs [62].
Nucleotide degradation is another important step in maintaining the homeostasis of nucleotides. Purine nucleotides undergo degradation processes in which nucleotides are converted into nucleosides with the catalysis of nucleotidase in the first step. Adenosine initiates deamination and is catalyzed to Ino and Guo, which are further converted to hypoxanthine and guanine [63]. In the last two steps, hypoxanthine is degraded into uric acid mediated by xanthine dehydrogenase (XDH), and uric acid is then excreted from the body [64, 65]. For pyrimidine catabolism, some pyrimidine molecules (e.g., TMP and dUMP) are sequentially dephosphorylated to their respective bases and converted into open chain amino acids [8]. Dihydropyrimidine dehydrogenase (DPD), a rate-limiting enzyme encoded by DPYD, not only initiates the pyrimidine catabolic pathway but also is involved in fluorouracil (5-FU) catabolism [8, 66, 67]. Uridine and thymidine are cleaved and metabolized via amino acids to NH3 and CO2 mediated by uridine phosphorylase (UPP1) and thymidine phosphorylases (TYMP), respectively [47].
Key modulators of nucleotide metabolism
Several metabolic enzymes involved in nucleotide metabolism regulate the pathway at mainly the enzyme level. However, the regulation of nucleotide biosynthesis is also controlled by negative feedback of substrate levels such as Pi, purine and pyrimidine analogs [68]. Purine biosynthesis is inhibited by AMP, GMP and Pi, which act on PRPP synthetase, and by adenosine and Guo mono, di or triphosphates (AXP and GXP) at two sites on the PRPP amidotransferase [14, 15]. A key metabolic enzyme involved in pyrimidine biosynthesis, CAD, is controlled by negative feedback of UTP binding to the CPSII domain of CAD and activated by PRPP [9, 69]. CTP synthase catalyzes the transfer of amide nitrogen from glutamine metabolism to UTP to form CTP [70]. Therefore, the activity of CTP synthase regulates the UTP and CTP pools and coordinates the production of pyrimidine and purine nucleotides.
Nucleotide metabolism is not only regulated by metabolic enzymes but also limited and controlled by nucleotide substrates or nucleotide metabolites. Previous studies have observed that acquiring nucleotide bases might be a metabolic bottleneck for cancer development and progression [8, 53, 71]. This provides the rationale for targeting nucleotide metabolism for cancer treatment therapies. Much effort has been made to investigate and explore cancer treatment by disrupting nucleotide metabolism. To date, many chemotherapeutics targeting nucleotide metabolism have been developed and approved for cancer treatments. Herein, it is necessary to investigate and explore the relationship between nucleotide metabolism and cancer, which might provide insights into cancer treatments.
Nucleotide metabolism and cancer
Multiple metabolic processes are altered in tumorigenesis and cancer progression [68, 72, 73]. The increased demand for nitrogen is regarded as one of the important metabolic hallmarks of cancer cells reported by Pavlova and Thompson [74]. Due to the biological capability of sustaining proliferative signaling in cancers, proliferating cells must synthesize essential nitrogen-containing molecules such as nucleotides [74, 75]. Nucleotide metabolism is considered the most critical link in tumorigenesis and cancer cell replication [76]. One reasonable explanation is that the TME cannot provide sufficient quantities or proportions of nucleotides unless proliferating cells upregulate integrated metabolism of nonessential amino acids, ribose, and one-carbon donors to synthesize these complex molecules [53]. Another potential mechanism is that cancer cells can utilize dysregulated nucleotide metabolism to enhance proliferation and progression [77]. For example, the catalytic activity of DPYD was essential for epithelial-mesenchymal transition (EMT) and cancer progression [78]. Moreover, for the process of nucleotide degradation, downregulated of XDH would contribute to the development and progression of hepatocellular carcinoma, breast cancer, and gastric cancer [79,80,81].
In diverse cancers, nucleotide metabolism is enriched to meet the demand of uncontrolled and rapid self-proliferation [74]. Meanwhile, upregulated nucleotide metabolism can lead to genomic instability and further carcinogenesis [82]. Several well-known oncogenes and tumor suppressor genes can regulate nucleotide metabolism by signaling pathways to influence tumor growth and progression [74, 83]. A well-known oncogene, C-myc, orchestrates nucleotide biosynthesis by upregulating the expression of numerous metabolic enzymes in nucleotide metabolism, such as CAD, TS, and IMPDH [84,85,86]. Wang et al. [87] indicated that CAD was upregulated in various cancers, including breast cancer, liver cancer, colon cancer with poor clinical outcomes. Furthermore, enriched expression of DHODH and other enzymes of the pyrimidine nucleotide production was found in the MYC-amplified neuroblastoma [88]. MYC is activated by proto-oncogene K-RAS and induces increased transcription of one raw material of nucleotide metabolism, ribose 5’-phosphate isomerase A (RPIA) [89]. It was found that IMPDH-dependent GTP synthesis was linked to MYC’s gene expression programs and suppression of ribosome biogenesis in small-cell lung cancer [90]. Furthermore, mutation of the well-known tumor suppressor gene p53 has been demonstrated to drive tumorigenesis and metastasis [91]. Reddy et al. [92] found that a nucleotide biosynthetic enzyme, guanosine 5'-monophosphate synthase (GMPS), is required for ubiquitin-specific protease 7 (USP7)-mediated stabilization of p53. Mutant p53 alleles can facilitate the expression of nucleotide enzymes such as IMPDH and GMPS [93]. Loss of p53 can activate mTOR complex 1 (mTORC1) to promote de novo pyrimidine and purine synthesis through activation of the CAD enzyme and induction of one-carbon metabolism [36, 94]. In addition, the transcription factor ATF3 can maintain the biosynthesis of purines and pyrimidines and inhibit differentiation in acute myeloid leukemia (AML) [95].
Hence, downregulating nucleotide metabolism could be an effective strategy to kill cancer cells or promote efficacy of cancer treatment. Zhou et al. indicated that inhibiting CDC-like kinase 3 (CLK3), a kinase regulated by C-myc, blocks the progression of cholangiocarcinoma through reprogramming nucleotide metabolism [96]. Blocking U2AF homology motif kinase 1 (UHMK1) could inhibit gastric cancer progression by downregulating the expression of purine metabolism-associated target genes [97]. Furthermore, deoxyuridine 5'-triphosphate nucleotidohydrolase inhibition could sensitize TNBC cell lines to fluoropyrimidines and anthracyclines through imbalanced nucleotide pools and increased DNA damage to improve efficacy of these chemotherapeutics [98]. Besides, DNA methyltransferase (DNMT) inhibitor gemcitabine could have synergic effects with PARPi to inhibit breast and ovarian cancers [99]. Binenbaum found that tumor-associated macrophages (TAMs) could release macrophage-derived exosomes, whereas miR-365 generated immunosuppressive effects [100]. miR-365 from exosomes can inactivate gemcitabine by upregulating the triphospho-nucleotide pool in cancer cells and activating cytidine deaminase [100]. One year later, Halbrook et al. provided another explanation for gemcitabine resistance [6]. They observed that TAM-released deoxycytidine, a pyrimidine metabolite, could hamper the antitumor effects of gemcitabine, which inhibits gemcitabine through molecular competition at the level of drug uptake and metabolism [6].
In addition to chemotherapy, the effectiveness of other cancer therapies was associated with altered nucleotide metabolism. As for target therapy, inhibition of DNPH1, a protein that eliminates cytotoxic nucleotide 5-hydroxymethyl-deoxyuridine (hmdU) monophosphate, can resensitize patients with resistance to PARP inhibitors [101]. Also, the lincNMR was found to be the first lncRNA to regulate nucleotide metabolism in cancer cells via maintaining activities of key enzymes essential for dNTP biosynthesis [102]. Knockdown of this lncRNA could induce decrease in cell proliferation, senescence, and colony formation [102]. Radiotherapy could decrease metabolites of nucleotide metabolism [103]. Glutamine synthetase and mucin1 were found to promote radiation resistance via facilitating nucleotide biosynthesis in cancer treatment [104, 105].
Chemotherapeutic agents disrupt nucleotide metabolism to suppress cancers
As discussed above, nucleotide metabolism plays a crucial role in carcinogenesis and cancer progression. Much effort has been devoted to cancer treatment by targeting nucleotide metabolism [1, 13, 106]. Drugs such as 5-FU and gemcitabine block nucleotide metabolism and are an important part of chemotherapy [107, 108]. To date, chemotherapy is the keystone treatment in the adjuvant setting in many types of cancer [109, 110]. There are over 20 approved nucleotide and nucleotide analogs used in cancer chemotherapies, which account for nearly 20% of all drugs in cancer treatment (Table 2). Therapeutic agents targeting nucleotide metabolism can be classified into three primary categories, including purine analogs, pyrimidine analogs, and metabolic enzymatic inhibitors, based on their structures and mechanisms [107, 111].
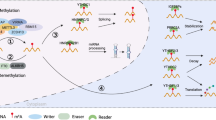
Purine analog antimetabolites include thiopurines, deoxypurines, arabinose purine analogs, and base-modified purine nucleosides [107]. Although thiopurines were introduced into the clinic in the early era of cancer chemotherapy, few representative drugs are known. In the early 1950s, Elion’s group discovered that 6-mercaptopurine and thioguanine, two of the earliest thiopurine analogs found, could hamper the growth of Lactobacillus casei [134]. In 1953, 6-mercaptopurine was proven to have effects in clinical trials by inhibiting the phosphorylation and hydrolysis of nucleosides and was approved by the FDA for the treatment of childhood leukemia (Fig. 1) [111,112,113]. Subsequently, thioguanine received approval for acute non-lymphocytic leukemias in 1966 [113, 115]. In addition, thiopurines have various clinical applications outside hematologic cancers, such as autoimmune diseases and organ transplantation rejection [139, 140]. Deoxyadenosine analogs have been found to be resistant to adenosine deaminase activity and may be phosphorylated into their triphosphate forms in the cell [141]. Cladribine, an approved deoxyadenosine analog, was used as a first-line monotherapy for hairy cell leukemia [123]. Clofarabine, a second-generation deoxyadenosine analog with more stability than first-generation drugs such as cladribine, was indicated for relapsed or refractory pediatric acute lymphoblastic leukemias [129, 130, 142]. Arabinose purine analogs consist of nelarabine and fludarabine. They were approved for the treatment of relapsed T cell acute lymphocytic leukemia, relapsed T cell lymphoblastic lymphoma, and chronic lymphocytic leukemia [121, 122, 133]. Base-modified purine nucleosides, including 8-chloro-adenosine, tocladesine, and forodesine, have not yet received approval from the FDA.

Historical development and breakthroughs in targeting nucleotide metabolism in cancer treatment. Targeting nucleotide metabolism in cancer treatment could be divided into two generations. In the Generation 1, targeting nucleotide metabolism was designed as chemotherapeutics to treat cancer. In the Generation 2, immunotherapy could enhance its efficacy with therapeutic agents blocking nucleotide metabolism
Pyrimidine analogs include fluorinated pyrimidines, azanucleosides, ribosugar-modified cytidine analogs, cytarabine, and its prodrugs [107]. Fluorinated pyrimidines contain 5-FU, capecitabine, floxuridine, and tipiracil hydrochloride (TAS-102) [107]. In 1954, Rutman et al. [68] observed that exogenous uracil is utilized for nucleic acid formation during the process of hepatic carcinogenesis in vivo. Based on this finding and previous understanding of thymidylate synthase, Heidelberger and colleagues synthesized fluorouracil [143]. Then, 5-FU received FDA approval in 1960 [144,145,146], and it currently has further extensive indications for various malignancies, such as gastrointestinal cancer, breast cancer, and renal cell cancer [108, 147]. In 1990, Hertel et al. [148] synthesized gemcitabine, a novel pyrimidine antimetabolite, and discovered its excellent antitumor activity in experimental tumor models. Gemcitabine was originally tested in hematological malignancies and found to have outstanding antitumor effects not only in hematological cancers but also in other solid tumors [107]. Currently, it has been approved by the FDA for ovarian, lung, breast and pancreatic cancer (Table 2). Floxuridine is converted into floxuridine-5’-monophosphate II mediated by thymidine kinase; thus, using floxuridine induces inhibition of thymidylate synthase, and it gained approval for metastatic colon and colorectal cancers from the FDA in 1970 [149]. TAS-102 refers to the combination of tipiracil hydrochloride and trifluorothymidine and was approved for colorectal cancer patients [150, 151]. In the category of azanucleosides, decitabine and azacytidine inhibit DNA methylation to achieve antitumor effects [131, 132, 134]. They received approval for myelodysplastic syndrome, known as preleukemia [152]. Gemcitabine, a representative drug of ribosugar-modified cytidine analogs, disrupts DNA biosynthesis through cell cycle arrest induced by “masked chain termination” [153]. Cytarabine was observed to have expected antitumor effects in vivo in the 1960s, tested rapidly in animal models and clinical trials, and subsequently received approval from the FDA [154].
Specific inhibitors target metabolic enzymes of nucleotide metabolism, providing a secondary mode of action that inhibits cell growth. However, most specific enzymatic blockers have not gained FDA approval and remain in phase I/II clinical trials. Enzymatic blockers of nucleotide metabolism can be further divided into purine, pyrimidine and general inhibitors. For purine enzymatic blockers, IMDPH has specific inhibitors, such as mizoribine, merimepodib and mycophenolic mofetil, which have not been approved for cancer therapy [36,37,38,39,40, 155]. A phase I clinical trial testing mycophenolic mofetil on pancreatic cancer has been completed (NCT00997958). A deoxyadenosine agent, pentostatin, targets ADA and gained FDA approval for hairy cell leukemia [29]. Pemetrexed, an antifolate, inhibits folate-dependent enzymes involved in the de novo biosynthesis of thymidine and purine nucleotides such as TS and GART. MLN4924, a structural analog of AMP, inhibits carcinogenesis by blocking the proteasomal degradation pathway [156]. It has been tested and proven to be safe and effective in many clinical trials on various cancers (e.g., melanoma, acute myeloid leukemia, and lymphoma) [157]. In contrast, there are more specific inhibitors of enzymes involved in pyrimidine nucleotide metabolism. As a key rate-limiting enzyme, DHODH is a target of several drugs, such as teriflunomide and leflunomide [158]. Although these two therapeutic agents have been reported to achieve antiproliferative effects on cancers such as multiple myeloma, NSCLC, and neuroblastomas, none to date has gained FDA approval for cancer [158, 159]. Another drug, gimeracil, has been shown to play an antineoplastic role by blocking DPYD to prevent the breakdown of 5-FU [48]. Furthermore, there are several inhibitors, such as CD73 and CD39, that target the common pathway of both purine and pyrimidine metabolism [160]. Anti-CD73 inhibitors include oleclumab, AB680, and adenosine 5'-(alpha, beta-methylene) diphosphate (APCP) [21, 58]. Oleclumab has shown safety and efficacy in combination with durvalumab in pancreatic cancers [161]. Oleclumab and other anti-CD73 monoclonal antibodies (mAbs) are currently being investigated in phase I/II clinical trials [21]. Similarly, pharmacological CD39 inhibitors, including sodium polyoxotungstate (POM-1), antisense oligonucleotides (ASOs), and TTX-030, have been evaluated as monotherapies and in combination with chemotherapy and/or immunotherapy in currently undergoing clinical trials (Table 3) [162]. Due to the biological and clinical significance of these two molecules, dual blockers have indicated a potential synergistic antitumor effect in several preclinical studies [22].
Based on the summary of therapeutic agents targeting nucleotide metabolism, this study demonstrates the significance and reliability of the general effectiveness and safety of targeting nucleotide metabolism as chemotherapy in cancers. These research results have not only greatly enriched the understanding of the regulatory mechanism of nucleotide metabolism in cancers but also provided insights into the clinical development of new specific therapeutic drugs. Although chemotherapy is still the keystone of all cancer treatment therapies to date in the adjuvant setting, the effectiveness of chemotherapy is hampered by drug resistance and adverse side effects [163, 164]. Resistance to chemotherapeutics is caused by several mechanisms, including gene mutations, chromosomal instability, and DNA repair [164]. These disadvantages of cytotoxic chemotherapy have forced scientists and clinicians to consider other systemic treatment therapies.
With continuous in-depth study of chemotherapy and cancers, our understanding of nucleotide metabolism in tumors has revealed their non-proliferative effects beyond their effects on cancer cell proliferation [8]. Dysregulated nucleotide metabolism has been found to alter the immune microenvironment and affect the host immune response [72, 165]. Altered immune components in the TME indicate the potential application of immunotherapy, which is considered one of most promising approaches to precisely killing tumors and maintaining the immune microenvironment in the era of precision medicine. Therefore, it is important to explore the interactions between nucleotide metabolism and cancer immunity to provide a theoretical basis for cancer treatments outside chemotherapy.
Interaction between nucleotide metabolism and cancer immunity
Nucleotide metabolism provides genetic materials and energy resources for immune system activation and proliferation. Dysregulated nucleotide metabolism induces inhibition or activation of the immune response [68]. Endogenous and exogenous nucleotides and their metabolites from host and microbial infection activate the immune system through several host receptors, such as Toll-like receptors (TLRs), RIG-like receptors (RLRs), NOD-like receptors (NLRs), purinergic receptors, and adenosine receptors [56, 166,167,168].
Purine analogs, such as released extracellular ATP or adenosine, activate purinergic receptors and adenosine receptors from immune cells to promote or inhibit the immune response [68, 169] (Fig. 2A). Adenosine acts on several adenosine receptors, including A1R, A2AR, A2BR, and A3R, and mediates its regulatory roles between nucleotide metabolism and the immune response. These four receptors have different affinities toward adenosine levels. A1R, A2AR and A3R more easily connect with accumulated adenosine, while A2BR can respond only when it meets high concentrations of adenosine in some pathological conditions [170]. Although A2BR can only be activated with relatively high concentrations of adenosine, blockade of A2BR can contribute to inhibiting the growth of tumors in vivo [171, 172]. Extracellular adenosines can act on A2AR in Treg cells and effector T cells, causing the activation of CD39, CD73, programmed cell death protein 1 (PD-1), and cytotoxic T lymphocyte antigen 4 (CTLA-4) on Treg cells and inhibiting the secretion of IL-2 and other cytokines [173, 174]. Specifically, the inhibitory mechanism of adenosine is mainly via inhibition of Ca2+ influx and nuclear factor of activated T cells (NFAT) stimulation [175]. Hence, adenosine inhibits effector T lymphocyte proliferation and the secretion of inflammatory cytokines and is therefore critical for both innate and adaptive immune responses [169]. In contrast, high levels of ATP can activate P2X on Treg cells to drive apoptosis and bind to P2X and P2Y receptors on effector T cells to facilitate their proliferation [173, 174]. Moreover, cancer-derived purine metabolites carried by exosomes work as potential contributors to tumor immune escape [176].

Interactions between nucleotide metabolism and host immunity. Cancer cells could release metabolites from nucleotide metabolism, such as ATP, adenosine to A Regulate immunoregulatory cells through adenosine and purinergic receptors; B In cancer cells, disrupted nucleotide pool could raise tumor immunogenicity; C Microbes release nucleoside analogs to regulate immunity. A2AR adenosine 2A receptor, P2X purinergic P2X receptor, P2Y purinergic P2Y receptor, TLRs Toll-like receptors, CD39 ecto-nucleoside triphosphate diphosphohydrolase-1, CD73 ecto-5-nucleotidase, M2 M2-type macrophage, MICA major histocompatibility complex class I-related chain A, NFAT nuclear factor of activated T cells, PTMB pyrimidine-rich transversion mutational bias, TAM tumor-associated macrophage
Furthermore, accumulated adenosine and ATP cause extensive immune inhibition and activation in similar manners on other components in the TME, such as natural killer (NK) cells, monocytes and neutrophils. ATP activates NK cells by P2 receptors and promotes proliferation and NK-mediated innate immunity, while CD73-derived adenosine stimulates A2AR to suppress antitumor immunity [177, 178]. Monocytes can polarize into macrophages with two different immunostimulatory characteristics. Extracellular ATP can act on TLRs, P2X, and P2Y of monocytes, subsequently inducing the polarization of M1 macrophages via NF-κB [169]. However, high levels of adenosine can block NF-κB, which promotes monocytes to polarize into M2 macrophages [169] (Fig. 2A). In neutrophils, adenosine stimulates A2AR via activation of the NF-kB pathway, limiting NK cell activation and IFNγ production but increasing TGF-β and IL-10 secretion [179]. Nils Ludwig et al. [176] found that head and neck squamous cell carcinoma (HNSCC) cells secrete purine metabolites in exosomes, whereas immunosuppressive adenosine and Ino are predominant. The amounts of shuttled purine metabolites in exosomes are significantly reduced with increased cancer stages and progression [176]. CD39 and CD73 are primarily involved in hydrolyzing proinflammatory ATP to generate immunosuppressive adenosine to regulate the host immune system [180]. In addition to mediating the rate-limiting step for conversion of extracellular ATP and adenosine, CD39 and CD73 promote angiogenesis of endothelial cells and lymphocyte adhesion to the endothelium, resulting in an increased risk of metastatic progression [181, 182]. In addition, oxidative stress controls Treg cell apoptosis, wherein it promotes the release and conversion of ATP to adenosine via CD39 and CD73 and mediates immunosuppression via the adenosine and A2A pathways [183]. In addition, cross talk between immune cells and cancer cells is mediated by products of nucleotide metabolism to some extent. Upregulated purine metabolism in cancer cells can induce increased expression of MICA, binding natural killer group 2D receptor (NKG2D) expressed in NK cells and inducing proliferation of NK cells and immune response [184].
Compared to purine and its analogs, there have been relatively fewer studies on the relationship between pyrimidine metabolism and immunity. A recent study had indicated that supplement of uridine diphosphate (UDP) could activate immune responses in vivo [185]. Lee et al. found that urea cycle dysregulation could enhance pyrimidine synthesis via changes in nitrogen metabolism and activation of CAD [186]. Excessive pyrimidines cause an increased pyrimidine/purine ratio and purine-to-pyrimidine transversion mutations (PTMB), which are associated with enhanced immunogenicity and a better response to immune checkpoint inhibitors [186]. Similarly, Keshet also indicated consistent findings that the IMDPH inhibitor mizoribine could block purine biosynthesis in ASS1-expressing tumors, disrupt the balance of nucleotide pools, release many immunoproteasomes, and subsequently generate PTMB [187]. Therefore, a dysregulated nucleotide pool raising tumor immunogenicity is another important mechanism between cancer nucleotide metabolism and cancer immunity (Fig. 2B).
Recently, the linkage between microbes and cancer immunity has raised interest among the scientific community, while nucleotide analogs play an important role in this linkage [5, 188,189,190] (Fig. 2C). Several microbes could act on the adenosine pathway to affect cancer immunity and enhance the efficacy of immune checkpoint blockage (ICB) therapies in animal models [5]. Specifically, Bifidobacterium pseudolongum, Lactobacillus johnsonii, and Olsenella species were found to promote ICB therapies by raising CD4+ and CD8+ T cell activation [5]. Among these three microbes, B. pseudolongum promoted the immune response by producing the metabolite Ino and directly acting on A2AR expressed in naïve T cells. Microbe-derived Ino could independently activate A2AR and upregulate the cAMP-PKA pathway to initiate Th1 differentiation and costimulate dendritic cells [5]. In addition, several favorable microbes can secrete and release c-di-AMP to induce the activation of monocytes [189], which activates the proliferation and secretion of adaptive immune cells such as NK cells and DC cells through the STING pathway [189]. Hence, based on this rationale, fecal microbiota transplantation from patients who are sensitive to ICB therapies could help to improve the efficacy of immunotherapy [189].
Indeed, targeting nucleotide metabolism can directly alleviate immune suppression; for example, secreted purines can directly bind inhibitory receptors on immune cells [191]. However, targeting nucleotide biosynthesis can inhibit the rapid proliferation of not only cancer cells but also immune cells [192]. Hence, this strategy exerts secondary effects to block the host immune response, as adaptive immunity depends on rapid proliferation of lymphocytes [192]. Therefore, integrated effects must be considered when developing therapeutic strategies that target nucleotide metabolism [165].
These findings highlight the solid association between nucleotide metabolism and antitumor immunity. Here, we elaborate the idea that antimetabolites or specific inhibitors targeting nucleotide metabolism promote infiltration of the immune microenvironment and facilitate the efficacy of immunotherapy, such as anti-PD-1 or CAR T cell treatment.
Targeting nucleotide metabolism could enhance the efficacy of cancer immunotherapy in experimental and clinical studies
As we mentioned above, there are close interactions between nucleotide metabolism and immunity, which provides rational potential for immunotherapy with antinucleotide metabolism agents in cancer patients. Immunotherapy, as a novel cancer treatment, has captured considerable attention across the oncology community in the past decade. It has made huge progress in several kinds of malignancies, such as non-small-cell lung cancer (NSCLC) [193], melanoma [194], lymphoma [195], and metastatic bladder cancer [196]. However, in some cold tumors, such as triple-negative breast cancer (TNBC), immune checkpoint blockade monotherapy does not seem to achieve our expectations [197]. Improving the efficacy of cancer immunotherapy in the clinic is an urgent problem for oncologists. To solve this conundrum, scientists have attempted to combine immunotherapy with other adjuvant treatment therapies, such as chemotherapy, radiotherapy, and targeted therapy, and have observed clinical benefits in various malignancies [198,199,200,201,202]. However, although olaparib accompanied with durvalumab could improve pathological completed rates of HER2-negative breast cancer patients in I-SPY 2 trial and radiotherapy was found to increase responses combined with immunotherapy in metastatic NSCLC, chemotherapy was still the major partner of immunotherapy [203,204,205].
Chemotherapy was considered immunosuppressive, causing neutropenia and lymphopenia and other adverse side effects [206]. However, Prehn first reviewed the relationship between the immune system and cancer progression and raised the concept of immunostimulation in 1971, which indicated the accelerated growth of tumors stimulated by immune factors [207]. Macpherson et al. further discussed the role of immunostimulation in immunochemotherapy and explained why combined immunotherapy and chemotherapy had synergistic effects on cancer treatment [208]. Immunotherapy promotes more active tumor cells into the cell cycle, while chemotherapy inhibiting the synthesis and integrity of nucleotides could achieve more profound antitumor effects [208]. Half a century ago, Stewart et al. [209] performed the first randomized trial of a combination of immunotherapy and chemotherapy in lung cancer patients. They observed that the third experimental group receiving methotrexate and immunization had longer disease-free survival than groups receiving chemotherapy monotherapy or single immunotherapy [209]. Another early randomized trial compared chemotherapy, immunotherapy, and immunochemotherapy in melanoma patients but failed to achieve the expected results [210].
In 2003, Nowak et al. [206, 211] made an important discovery that additional agents targeting nucleotide metabolism could have synergic effects in the combination of immunotherapy for cancer treatment (Fig. 1). They found that gemcitabine could raise CD4 and CD8 T-cell infiltration and subsequently promote antigen-specific cellular antitumor immunity [206, 211]. Since chemotherapy has generally been considered immunosuppressive in past decades, these findings provide insights into the synergy between cytotoxic chemotherapy and immunotherapy. Therefore, what induced synergism of immunochemotherapy could not be simply explained by blockade of more cancer cells into the S phase of the cell cycle. The chemotherapeutic agents mentioned above mainly targeted the different sections of nucleotide metabolism. Targeting nucleotide metabolism could drive the activation of adaptive immune responses, which could facilitate antitumor effects accompanied by immunotherapy. Taken together, nucleotide metabolism could be a promising target to improve the effectiveness of cancer immunotherapy.
To illustrate the recent progress on therapeutic agents targeting nucleotide metabolism in cancer treatment, we classify them into four categories based on the nucleotide metabolism pathway as follows: (A) targeting purine and pyrimidine pathways, (B) blocking DNA synthesis, (C) inhibiting the adenosine pathway, and (D) fecal microbiota transplantation (Fig. 3).

Therapeutic strategies to exploit the nucleotide metabolism–immunity interplay in the clinic. A Targeting purine or pyrimidine pathways; B blocking DNA synthesis; C inhibiting adenosine pathway; D fecal microbiota transplantation. CAD carbamoyl phosphate synthetase, aspartyl transcarbamoylase, and dihydroorotase, IMPDH inosine monophosphate dehydrogenase, MDSCs myeloid-derived suppressive cells, A2AR adenosine 2A receptor, CD39 ecto-nucleoside triphosphate diphosphohydrolase-1, ASOs antisense oligonucleotides, CD73 ecto-5-nucleotidase
Directly targeting purine and pyrimidine metabolism could be an effective strategy for enhancing cancer immunotherapy (Fig. 3A). As mentioned above, mizoribine could inhibit purine synthesis and promote the release of the immunoproteasome [187]. This could induce an increased response of autologous CD8+ T cells to anti-PD1 therapies in specific ASS1-expressing cancers [187]. Furthermore, numerous studies have demonstrated that blocking DHODH could efficiently inhibit the growth of various malignancies, including glioblastoma stem cells [41], acute myeloid leukemia [46], and small-cell lung cancer [212]. However, few studies have investigated whether DHODH inhibitors have synergistic effects with immunotherapy. A DHODH inhibitor, P1788, was identified and found that DHODH inhibition could enhance cellular antitumor immunity via increased interferon signaling [213]. This study provided insights into enhancing innate immunity through blockade of de novo pyrimidine biosynthesis. Another recent study indicated that afatinib, a kind of EGFR-tyrosine kinase inhibitor (TKI), could be a potential agent to enhance the efficacy of ICB therapies by targeting another important rate-limiting enzyme of nucleotide metabolism, CAD [214]. Blocking CAD and nucleotide metabolism in cytotoxic T cells causes immune suppression in the short term, but the proliferation of CD8+ T cells unexpectedly rebounds following long-term treatment [214].
Targeting the common pathway of both purine and pyrimidine metabolism is generally considered chemotherapy to arrest tumor cells in the cell cycle. Transformation between ribose-5-monophosphate and ribose-5-triphosphate is an essential step in complete nucleotide biosynthesis. Common chemotherapeutic agents, such as gemcitabine, capecitabine, and 5-FU, have been indicated in numerous malignancies, but recent studies have observed the potential to have synergistic effects with immunotherapy (Fig. 3B). Gemcitabine was found to enhance antitumor immunity via increased antigen cross-presentation, T lymphocyte expansion, and infiltration in solid tumors [206, 211]. Furthermore, scientists observed that gemcitabine and 5-FU could selectively kill myeloid-derived suppressor cells (MDSCs), and other immune components in the TME, such as T cells, B cells, NK cells, or DC cells, remained unchanged [215]. 5-FU could induce IFN-γ production by tumor-specific CD8+ T cells infiltrating the tumor and promote T cell-dependent antitumor responses by eliminating MDSCs in vivo [215]. Similarly, capecitabine, an oral prodrug of 5-FU, enhances immunotherapy efficacy in glioblastoma [216]. In a phase 0/I dose-escalation clinical trial, Peereboom et al. [216] proved the effectiveness and tolerance of additional metronomic capecitabine with bevacizumab in glioblastoma patients. The addition of capecitabine increased cytotoxic immune infiltrations by inhibiting MDSCs and facilitated immunotherapy. In addition to these chemotherapeutic agents, DNMT inhibitors contribute to an increase in the activation and cytolytic activity of CD8+ T cells [217]. Decitabine upregulates T cell activation and promotes T cell-based immunotherapy in lung cancer in vivo models [218]. Similarly, 5-azacytidine increases CD45+ immune cells, CD8+ T cells, and NK cells [219]. A combination of DNMT inhibitors plus the immune checkpoint inhibitor anti-PD-1 enhances antitumor effects in ovarian cancer [219]. In addition, a phase I trial confirmed the safety and efficacy of additional fludarabine, and targeting the ribonucleoside-diphosphate reductase large subunit, the DNA polymerase alpha catalytic subunit, increased the efficacy of CAR T cell immunotherapy in patients with neuroblastoma [132].
Adenosine and its analogs have been extensively recognized as key metabolites in modulating the immune microenvironment [57, 220]. Adenosine receptor expression is negatively correlated with immune infiltration and prognosis in various cancers [221,222,223,224]. Adenosine receptors are considered perfect targets by several inhibitors, such as DPCPX and AZD4635, to activate immunosuppressive adenosine signaling [220, 225] (Fig. 3C). An A2AR antagonist, AZD4635 could prompt T cell proliferation and interferon gamma production and reduce the tumor load in multiple myeloma (MM) models [220]. In addition, another A2AR inhibitor, DPCPX could upregulate PD-L1 via the transcription factor ATF3 [225]. Authors also proved the potential benefits of the synergism of DPCPX and a PD-1 mAb in NSCLC or melanoma models [225]. Moreover, another A2AR antagonist, CPI-144, could enhance the efficacy of anti-PD-L1 or anti-CTLA-4 treatment in preclinical models [226]. Recently, Giuffrida et al. [227, 228] found that deletion of A2AR by CRISPR/Cas9 or shRNA could promote antitumor immunity with CAR T immunotherapy in vivo. Furthermore, extracellular nucleotides were found to stimulate purinergic receptors to induce chemotaxis and adhesion of lung cancer cells [229]. This evidence highlights the potential applications of agents targeting adenosine–adenosine receptors combined with immunotherapy for cancer treatment.
Given the critical position of CD39 and CD73 in downmodulating effector antitumor immunity through the generation of adenosine, strategies targeting these central mediators could enhance cancer immunotherapy. In human peripheral blood, CD39 and CD73 are extensively expressed on immune subsets, especially in B cells and CD4+ T cells [21]. CD39 antagonists hamper the proliferation of CD4+ and CD8+ T cells via an adenosine-dependent pattern and ATPase activity in vivo [230]. Furthermore, POM-1, a specific inhibitor of CD39, increases IFN-gamma or IL-2 secretion upon anti-CD3/CD28 stimulation and enhances antitumor immunity in vitro [231]. POM-1 also effectively suppresses metastases by activating NK cells when used in combination with immunotherapy, such as anti-PD-1, anti-CTLA-4 and IL-2 [232]. Kashyap and coworkers demonstrated that ASOs lead to improved CD8+ T cell proliferation and reduced Treg and tumor-associated macrophages [233] (Fig. 3C).
Regarding CD73, previous studies have indicated that CD73 promotes tumor metastasis by blocking the function of NK cells [234]. Previous studies have observed its prognostic value and found that CD73 expression is conversely associated with prognosis and antitumor immunity in TNBC and renal cell carcinoma [235, 236]. An early phase of a clinical trial proved that oleclumab significantly altered several immune subpopulations in the TIME, including increased CD8+ T cells and activated macrophages [237]. Data from single-cell RNA sequencing have also indicated that CD73 is a specific immunotherapeutic target for facilitating ICB therapies in glioblastoma (GBM) [238]. Anti-CD73 enhances the activity of anti-CTLA4 mAbs through activation of the T-cell response in in vivo cancer models such as colon cancer, prostate cancer, melanoma, and glioblastoma [238,239,240]. A combinational approach of CD39 and CD73 inhibition synergistically enhances antitumor immunity. As immune cells infiltrating the tumor coexpress CD39 and CD73 in association with other coinhibitory molecules (e.g., CTLA4 and PD-L1), dual blockade of both CD39 and CD73 has been proposed with the aim of controlling the immunosuppressive role of adenosine signaling while minimizing the side effects of ICB [22, 220, 241,242,243].
As we mentioned above, microbes could modulate host immunity through release of different metabolites, such as Ino and c-di-AMP (Fig. 2C). Hence, many preclinical studies had revealed that the potential effectiveness of fecal microbiota transplantation (FMT) from responders to overcome resistance to immunotherapy [244,245,246]. Based on this rationale, scientist designed and evaluated FMT in clinical trials; they had demonstrated that FMT together with anti-PD-1 could treat refractory melanoma patients [247, 248]. Therefore, FMT could be considered to improve the efficacy of immunotherapy in other solid tumors, which were previously regarded insensitivity to immunotherapy. Extraction of microbe-release nucleotide analogs with immunoregulatory functions such as Ino and c-di-AMP and exogenous supplement of these materials might be helpful in improving immunotherapy, warranting further exploration and validation.
Targeting a specific purine or pyrimidine metabolic pathway can induce a nucleotide pool imbalance by decreasing the biochemistry levels of one pool relative to the other [165]. Imbalance of nucleotide pools between purines and pyrimidines could generate PTMB and subsequently increase neoantigens and immunogenicity as mentioned above [186, 187, 249]. An integrated study of clinical and genomic data found that higher somatic tumor mutational burden (TMB) was correlated with better survival and response to ICB therapies across multiple cancer types [250]. Therefore, it is possible to induce more mutations by disrupting the nucleotide-pool balance to raise the efficacy of immunotherapy during cancer treatment, warranting further exploration and validation.
The above evidence demonstrates that targeting nucleotide metabolism (cytotoxic chemotherapy or specific enzyme inhibitors) can facilitate immunotherapy in numerous cancer types, even in several well-known cold tumors. After exploration of the safety and efficacy of therapeutic agents targeting nucleotide metabolism, clinical trials could be performed, especially in advanced or progressed diseases after conventional first-line therapies.
Future clinical trials of cancer immunotherapy with additional nucleotide antimetabolites
After a conceptual breakthrough in immunotherapy with nucleotide antimetabolites, some clinical trials testing the safety and efficacy in cancer patients have been initiated or completed. Table 3 summarizes currently undergoing and completed clinical trials on cancer immunotherapy with additional nucleotide-metabolic targets versus immunotherapy monotherapy from ClinicalTrials.gov. In total, 22 clinical trials were identified, and approximately 30% (7/22) of the studies were terminated or completed (Table 3). Therefore, most of the clinical trials are still in phase I/II and recruiting cancer patients from different cancer types and stages. Anti-PD-1 or anti-PD-L1 agents, including durvalumab, pembrolizumab, and nivolumab, are preferable choices for immunotherapy combined with nucleotide antimetabolites (Table 3). The included cancer patients have several “hot tumors,” such as NSCLC, melanoma, and hematological malignancies, with high sensitivity to immunotherapy [251, 252].
Among seven terminated or completed trials, only one trial has reported results [253, 254]. This phase I trial explored the efficacy and safety of NY-ESO-1T cells in synovial sarcoma patients; specifically, cohort 3 received immunotherapy plus only cyclophosphamide and cohort 4 received the same regimen with additional fludarabine (NCT01343043). The responses between these two cohorts did not seem to be significantly different. One important reason we considered was the difference in the dosage of cyclophosphamide (cohort 3: 1800 mg/m2/day × 3 days vs. cohort 4: 0.600 mg/m2/day × 3 days) [254].
In these currently undergoing and completed clinical trials, physicians attempted to compare the efficacy of additional therapeutic agents targeting nucleotide metabolism with immunotherapy versus immunotherapy monotherapy. Highly immune-sensitive tumors, such as NSCLC and melanoma, were preferable for inclusion. To our surprise, some cold tumors, such as TNBC and squamous lung cancer, were selected in further clinical trials.
Although much evidence from preclinical studies has demonstrated the potential application of additional nucleotide antimetabolites, the results of phase I/II clinical trials have indicated that physicians need to consider cautiously the selection of immunotherapy agents, nucleotide antimetabolites and included cancer patients to achieve the expectations. Furthermore, the accompanying incidence rates of adverse events may increase with the addition of chemotherapeutic agents. Therefore, clinicians must manipulate more health care for patients receiving immunotherapy with antimetabolites targeting nucleotide metabolism.
Conclusion
The tight linkage between nucleotide metabolism and cancer immunity is discussed and summarized. Specifically, purine and its related metabolites, such as adenosine and ATP, are critical for the activation or suppression of innate and adaptive immune responses. Pyrimidine or purine metabolism dysregulation independently affects the pyrimidine versus purine ratio, whereas the ratio correlates with gene mutation and tumor immunogenicity. Furthermore, microbes can release nucleoside analogs to regulate the immune microenvironment, indicating potential effectiveness of fecal microbiota transplantation. Nucleotide metabolism is dynamically positioned within the cancer-immune cycle, and targeting this pathway proves the potential to enhance the effectiveness of immunotherapy. Targeting nucleotide metabolism in combination with immunotherapy agents could achieve promising synergistic effects on various cancers, warranting further validation in future clinical trials.
Availability of data and materials
Not applicable.
Abbreviations
- 5-FU:
-
Fluorouracil
- ADA:
-
Adenosine deaminase
- ADSS:
-
Adenylosuccinate synthetase
- AML:
-
Acute myeloid leukemia
- AMP:
-
Adenosine monophosphate
- APCP:
-
Adenosine 5'-(alpha, beta-methylene) diphosphate
- ASOs:
-
Antisense oligonucleotides
- CAD:
-
Carbamoyl phosphate synthetase, aspartyl transcarbamoylase, and dihydroorotase
- CLK3:
-
CDC-like kinase 3
- CTLA-4:
-
Cytotoxic T lymphocyte antigen 4
- DHODH:
-
Dihydroorotate dehydrogenase
- DNMT:
-
DNA methyltransferase
- DPD:
-
Dihydropyrimidine dehydrogenase
- EMT:
-
Epithelial-mesenchymal transition
- ENTPD1/CD39:
-
Ecto-nucleoside triphosphate diphosphohydrolase-1
- FMT:
-
Fecal microbiota transplantation
- GART:
-
Glycinamide ribonucleotide transformylase
- GBM:
-
Glioblastoma
- GMP:
-
Guanosine monophosphate
- GMPS:
-
Guanosine 5'-monophosphate synthase
- HGPRT:
-
Hypoxanthine–guanine phosphoribosyltransferase
- hmdU:
-
Nucleotide 5-hydroxymethyl-deoxyuridine
- HNSCC:
-
Head and neck squamous cell carcinoma
- ICB:
-
Immune checkpoint blockage
- IMP:
-
Inosine monophosphate
- IMPDH:
-
Inosine monophosphate dehydrogenase
- MDSCs:
-
Myeloid-derived suppressor cells
- MM:
-
Multiple myeloma
- MPs:
-
Monophosphates
- mTORC1:
-
MTOR complex 1
- NFAT:
-
Nuclear factor of activated T cells
- NKG2D:
-
Natural killer group 2D receptor
- NLRs:
-
NOD-like receptors
- NSCLC:
-
Non-small-cell lung cancer
- NT4E/CD73:
-
Ecto-5-nucleotidase
- OMP:
-
Orotidine monophosphate
- PD-1:
-
Programmed cell death protein 1
- PNP:
-
Purine nucleoside phosphorylase
- POM-1:
-
Sodium polyoxotungstate
- PRPP:
-
Phosphoribosyl pyrophosphate
- PTMB:
-
Purine-to-pyrimidine transversion mutations
- RLRs:
-
RIG-like receptors
- RPIA:
-
Ribose 5'-phosphate isomerase A
- sAMP:
-
Succinyl adenosine 5'-monophosphate
- TAMs:
-
Tumor-associated macrophages
- TKI:
-
Tyrosine kinase inhibitor
- TLRs:
-
Toll-like receptors
- TMB:
-
Tumor mutational burden
- TME:
-
Tumor microenvironment
- TNBC:
-
Triple-negative breast cancer
- TYMP:
-
Thymidine phosphorylases
- UCK:
-
Uridine–cytidine kinases
- UDP:
-
Uridine diphosphate
- UHMK1:
-
U2AF homology motif kinase 1
- UMPS:
-
Uridine monophosphate synthetase
- UPP1:
-
Uridine phosphorylase
- XDH:
-
Xanthine dehydrogenase
- XMP:
-
Xanthosine monophosphate
References
Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: the mitotic checkpoint, adaptation, and cell death. Cancer Cell. 2005;8(1):7–12.
Butters DJ, Ghersi D, Wilcken N, Kirk SJ, Mallon PT. Addition of drug/s to a chemotherapy regimen for metastatic breast cancer. Cochrane Database Syst Rev. 2010;11:CD003368.
Shapiro CL, Recht A. Side effects of adjuvant treatment of breast cancer. N Engl J Med. 2001;344(26):1997–2008.
Schein PS, Winokur SH. Immunosuppressive and cytotoxic chemotherapy: long-term complications. Ann Intern Med. 1975;82(1):84–95.
Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science. 2020;369(6510):1481–9.
Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee HJ, et al. Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab. 2019;29(6):1390-9.e6.
Kepp O, Bezu L, Yamazaki T, Di Virgilio F, Smyth MJ, Kroemer G, et al. ATP and cancer immunosurveillance. EMBO J. 2021;40:e108130.
Siddiqui A, Ceppi P. A non-proliferative role of pyrimidine metabolism in cancer. Mol Metab. 2020;35:100962.
Lane AN, Fan TW. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015;43(4):2466–85.
Ipata PL, Balestri F. The functional logic of cytosolic 5’-nucleotidases. Curr Med Chem. 2013;20(34):4205–16.
Austin WR, Armijo AL, Campbell DO, Singh AS, Hsieh T, Nathanson D, et al. Nucleoside salvage pathway kinases regulate hematopoiesis by linking nucleotide metabolism with replication stress. J Exp Med. 2012;209(12):2215–28.
Huang Z, Xie N, Illes P, Di Virgilio F, Ulrich H, Semyanov A, et al. From purines to purinergic signalling: molecular functions and human diseases. Signal Transduct Target Ther. 2021;6(1):162.
Christopherson RI, Lyons SD, Wilson PK. Inhibitors of de novo nucleotide biosynthesis as drugs. Acc Chem Res. 2002;35(11):961–71.
Fridman A, Saha A, Chan A, Casteel DE, Pilz RB, Boss GR. Cell cycle regulation of purine synthesis by phosphoribosyl pyrophosphate and inorganic phosphate. Biochem J. 2013;454(1):91–9.
Smith JL. Glutamine PRPP amidotransferase: snapshots of an enzyme in action. Curr Opin Struct Biol. 1998;8(6):686–94.
Nagase O. Investigations on pantothenic acid and its related compounds. IV. Chemical studies. 3. Syntheses of D-pantetheine 4’-phosphate and N-D-pantothenoyl-L-cysteine 4’-phosphate. Chem Pharm Bull (Tokyo). 1967;15(5):648–54.
Robinson AD, Eich ML, Varambally S. Dysregulation of de novo nucleotide biosynthetic pathway enzymes in cancer and targeting opportunities. Cancer Lett. 2020;470:134–40.
Zhao H, Chiaro CR, Zhang L, Smith PB, Chan CY, Pedley AM, et al. Quantitative analysis of purine nucleotides indicates that purinosomes increase de novo purine biosynthesis. J Biol Chem. 2015;290(11):6705–13.
Gooding JR, Jensen MV, Dai X, Wenner BR, Lu D, Arumugam R, et al. Adenylosuccinate is an insulin secretagogue derived from glucose-induced purine metabolism. Cell Rep. 2015;13(1):157–67.
Knight RD, Mangum J, Lucas DL, Cooney DA, Khan EC, Wright DG. Inosine monophosphate dehydrogenase and myeloid cell maturation. Blood. 1987;69(2):634–9.
Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. 2017;276(1):121–44.
Perrot I, Michaud HA, Giraudon-Paoli M, Augier S, Docquier A, Gros L, et al. Blocking antibodies targeting the CD39/CD73 immunosuppressive pathway unleash immune responses in combination cancer therapies. Cell Rep. 2019;27(8):2411-25.e9.
Moesta AK, Li XY, Smyth MJ. Targeting CD39 in cancer. Nat Rev Immunol. 2020;20(12):739–55.
Hammami A, Allard D, Allard B, Stagg J. Targeting the adenosine pathway for cancer immunotherapy. Semin Immunol. 2019;42:101304.
Dummer R, Duvic M, Scarisbrick J, Olsen EA, Rozati S, Eggmann N, et al. Final results of a multicenter phase II study of the purine nucleoside phosphorylase (PNP) inhibitor forodesine in patients with advanced cutaneous T-cell lymphomas (CTCL) (Mycosis fungoides and Sezary syndrome). Ann Oncol Off J Eur Soc Med Oncol. 2014;25(9):1807–12.
Gandhi V, Kilpatrick JM, Plunkett W, Ayres M, Harman L, Du M, et al. A proof-of-principle pharmacokinetic, pharmacodynamic, and clinical study with purine nucleoside phosphorylase inhibitor immucillin-H (BCX-1777, forodesine). Blood. 2005;106(13):4253–60.
Balakrishnan K, Nimmanapalli R, Ravandi F, Keating MJ, Gandhi V. Forodesine, an inhibitor of purine nucleoside phosphorylase, induces apoptosis in chronic lymphocytic leukemia cells. Blood. 2006;108(7):2392–8.
Lewis AL, Guicherit OM, Datta SK, Hanten GR, Kellems RE. Structure and expression of the murine muscle adenylosuccinate synthetase gene. J Biol Chem. 1996;271(37):22647–56.
Sarvaria A, Topp Z, Saven A. Current therapy and new directions in the treatment of hairy cell leukemia: a review. JAMA Oncol. 2016;2(1):123–9.
Terao M, Romao MJ, Leimkuhler S, Bolis M, Fratelli M, Coelho C, et al. Structure and function of mammalian aldehyde oxidases. Arch Toxicol. 2016;90(4):753–80.
Kelemen LE, Terry KL, Goodman MT, Webb PM, Bandera EV, McGuire V, et al. Consortium analysis of gene and gene-folate interactions in purine and pyrimidine metabolism pathways with ovarian carcinoma risk. Mol Nutr Food Res. 2014;58(10):2023–35.
Qian X, Li X, Tan L, Lee JH, Xia Y, Cai Q, et al. Conversion of PRPS hexamer to monomer by AMPK-mediated phosphorylation inhibits nucleotide synthesis in response to energy stress. Cancer Discov. 2018;8(1):94–107.
Li B, Li H, Bai Y, Kirschner-Schwabe R, Yang JJ, Chen Y, et al. Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat Med. 2015;21(6):563–71.
Welin M, Grossmann JG, Flodin S, Nyman T, Stenmark P, Tresaugues L, et al. Structural studies of tri-functional human GART. Nucleic Acids Res. 2010;38(20):7308–19.
Zaza G, Yang W, Kager L, Cheok M, Downing J, Pui CH, et al. Acute lymphoblastic leukemia with TEL-AML1 fusion has lower expression of genes involved in purine metabolism and lower de novo purine synthesis. Blood. 2004;104(5):1435–41.
Valvezan AJ, Turner M, Belaid A, Lam HC, Miller SK, McNamara MC, et al. mTORC1 couples nucleotide synthesis to nucleotide demand resulting in a targetable metabolic vulnerability. Cancer Cell. 2017;32(5):624-38.e5.
Buey RM, Ledesma-Amaro R, Velazquez-Campoy A, Balsera M, Chagoyen M, de Pereda JM, et al. Guanine nucleotide binding to the Bateman domain mediates the allosteric inhibition of eukaryotic IMP dehydrogenases. Nat Commun. 2015;6:8923.
Tong X, Smith J, Bukreyeva N, Koma T, Manning JT, Kalkeri R, et al. Merimepodib, an IMPDH inhibitor, suppresses replication of Zika virus and other emerging viral pathogens. Antiviral Res. 2018;149:34–40.
Sintchak MD, Fleming MA, Futer O, Raybuck SA, Chambers SP, Caron PR, et al. Structure and mechanism of inosine monophosphate dehydrogenase in complex with the immunosuppressant mycophenolic acid. Cell. 1996;85(6):921–30.
Takebe N, Cheng X, Wu S, Bauer K, Goloubeva OG, Fenton RG, et al. Phase I clinical trial of the inosine monophosphate dehydrogenase inhibitor mycophenolate mofetil (cellcept) in advanced multiple myeloma patients. Clin Cancer Res Off J Am Assoc Cancer Res. 2004;10(24):8301–8.
Wang X, Yang K, Wu Q, Kim LJY, Morton AR, Gimple RC, et al. Targeting pyrimidine synthesis accentuates molecular therapy response in glioblastoma stem cells. Sci Transl Med. 2019;11(504):eaau4972.
Graves LM, Guy HI, Kozlowski P, Huang M, Lazarowski E, Pope RM, et al. Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature. 2000;403(6767):328–32.
Peters GJ, Schwartsmann G, Nadal JC, Laurensse EJ, van Groeningen CJ, van der Vijgh WJ, et al. In vivo inhibition of the pyrimidine de novo enzyme dihydroorotic acid dehydrogenase by brequinar sodium (DUP-785; NSC 368390) in mice and patients. Can Res. 1990;50(15):4644–9.
Klotz L, Eschborn M, Lindner M, Liebmann M, Herold M, Janoschka C, et al. Teriflunomide treatment for multiple sclerosis modulates T cell mitochondrial respiration with affinity-dependent effects. Sci Transl Med. 2019;11(490):eaao5563.
Grisar J, Aringer M, Koller MD, Stummvoll GH, Eselbock D, Zwolfer B, et al. Leflunomide inhibits transendothelial migration of peripheral blood mononuclear cells. Ann Rheum Dis. 2004;63(12):1632–7.
Sykes DB, Kfoury YS, Mercier FE, Wawer MJ, Law JM, Haynes MK, et al. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell. 2016;167(1):171-86.e15.
Loffler M, Fairbanks LD, Zameitat E, Marinaki AM, Simmonds HA. Pyrimidine pathways in health and disease. Trends Mol Med. 2005;11(9):430–7.
Sakata K, Someya M, Matsumoto Y, Tauchi H, Kai M, Toyota M, et al. Gimeracil, an inhibitor of dihydropyrimidine dehydrogenase, inhibits the early step in homologous recombination. Cancer Sci. 2011;102(9):1712–6.
Johnson MR, Wang K, Diasio RB. Profound dihydropyrimidine dehydrogenase deficiency resulting from a novel compound heterozygote genotype. Clin Cancer Res Off J Am Assoc Cancer Res. 2002;8(3):768–74.
Gardiner SJ, Begg EJ, Robinson BA. The effect of dihydropyrimidine dehydrogenase deficiency on outcomes with fluorouracil. Adverse Drug React Toxicol Rev. 2002;21(1–2):1–16.
Laliberte J, Momparler RL. Human cytidine deaminase: purification of enzyme, cloning, and expression of its complementary DNA. Can Res. 1994;54(20):5401–7.
Olou AA, King RJ, Yu F, Singh PK. MUC1 oncoprotein mitigates ER stress via CDA-mediated reprogramming of pyrimidine metabolism. Oncogene. 2020;39(16):3381–95.
Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell. 2017;168(4):657–69.
Jones ME. Pyrimidine nucleotide biosynthesis in animals: genes, enzymes, and regulation of UMP biosynthesis. Annu Rev Biochem. 1980;49:253–79.
Losson R, Lacroute F. Plasmids carrying the yeast OMP decarboxylase structural and regulatory genes: transcription regulation in a foreign environment. Cell. 1983;32(2):371–7.
Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. 2017;17(12):765.
Boison D, Yegutkin GG. Adenosine metabolism: emerging concepts for cancer therapy. Cancer Cell. 2019;36(6):582–96.
Jeffrey JL, Lawson KV, Powers JP. Targeting metabolism of extracellular nucleotides via inhibition of ectonucleotidases CD73 and CD39. J Med Chem. 2020;63(22):13444–65.
Camici M, Garcia-Gil M, Pesi R, Allegrini S, Tozzi MG. Purine-metabolising enzymes and apoptosis in cancer. Cancers. 2019;11(9):1354.
Wang L. Mitochondrial purine and pyrimidine metabolism and beyond. Nucleosides Nucleotides Nucleic Acids. 2016;35(10–12):578–94.
Bzowska A, Kulikowska E, Shugar D. Purine nucleoside phosphorylases: properties, functions, and clinical aspects. Pharmacol Ther. 2000;88(3):349–425.
Yu S, Li X, Guo X, Zhang H, Qin R, Wang M. UCK2 upregulation might serve as an indicator of unfavorable prognosis of hepatocellular carcinoma. IUBMB Life. 2019;71(1):105–12.
Holzer H, Duntze W. Metabolic regulation by chemical modification of enzymes. Annu Rev Biochem. 1971;40:345–74.
Bontemps F, Van den Berghe G, Hers HG. Pathways of adenine nucleotide catabolism in erythrocytes. J Clin Investig. 1986;77(3):824–30.
Bortolotti M, Polito L, Battelli MG, Bolognesi A. Xanthine oxidoreductase: one enzyme for multiple physiological tasks. Redox Biol. 2021;41:101882.
Fragoulakis V, Roncato R, Fratte CD, Ecca F, Bartsakoulia M, Innocenti F, et al. Estimating the effectiveness of DPYD genotyping in italian individuals suffering from cancer based on the cost of chemotherapy-induced toxicity. Am J Hum Genet. 2019;104(6):1158–68.
Offer SM, Fossum CC, Wegner NJ, Stuflesser AJ, Butterfield GL, Diasio RB. Comparative functional analysis of DPYD variants of potential clinical relevance to dihydropyrimidine dehydrogenase activity. Can Res. 2014;74(9):2545–54.
Ishii KJ, Akira S. Potential link between the immune system and metabolism of nucleic acids. Curr Opin Immunol. 2008;20(5):524–9.
Evans DR, Guy HI. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J Biol Chem. 2004;279(32):33035–8.
Zalkin H. CTP synthetase. Methods Enzymol. 1985;113:282–7.
Aird KM, Zhang G, Li H, Tu Z, Bitler BG, Garipov A, et al. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep. 2013;3(4):1252–65.
Ma J, Zhong M, Xiong Y, Gao Z, Wu Z, Liu Y, et al. Emerging roles of nucleotide metabolism in cancer development: progress and prospect. Aging (Albany NY). 2021;13(9):13349–58.
Kohnken R, Kodigepalli KM, Wu L. Regulation of deoxynucleotide metabolism in cancer: novel mechanisms and therapeutic implications. Mol Cancer. 2015;14:176.
Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23(1):27–47.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Kumar V. Adenosine as an endogenous immunoregulator in cancer pathogenesis: where to go? Purinergic Signal. 2013;9(2):145–65.
Aird KM, Zhang R. Nucleotide metabolism, oncogene-induced senescence and cancer. Cancer Lett. 2015;356(2 Pt A):204–10.
Shaul Yoav D, Freinkman E, Comb William C, Cantor Jason R, Tam Wai L, Thiru P, et al. Dihydropyrimidine accumulation is required for the epithelial-mesenchymal transition. Cell. 2014;158(5):1094–109.
Linder N, Haglund C, Lundin M, Nordling S, Ristimäki A, Kokkola A, et al. Decreased xanthine oxidoreductase is a predictor of poor prognosis in early-stage gastric cancer. J Clin Pathol. 2006;59(9):965–71.
Linder N, Lundin J, Isola J, Lundin M, Raivio KO, Joensuu H. Down-regulated xanthine oxidoreductase is a feature of aggressive breast cancer. Clin Cancer Res. 2005;11(12):4372–81.
Sun Q, Zhang Z, Lu Y, Liu Q, Xu X, Xu J, et al. Loss of xanthine oxidoreductase potentiates propagation of hepatocellular carcinoma stem cells. Hepatology. 2020;71(6):2033–49.
Burhans WC, Weinberger M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007;35(22):7545–56.
Pelletier J, Riano-Canalias F, Almacellas E, Mauvezin C, Samino S, Feu S, et al. Nucleotide depletion reveals the impaired ribosome biogenesis checkpoint as a barrier against DNA damage. EMBO J. 2020;39(13):e103838.
Eberhardy SR, Farnham PJ. c-Myc mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J Biol Chem. 2001;276(51):48562–71.
Liu YC, Li F, Handler J, Huang CR, Xiang Y, Neretti N, et al. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS ONE. 2008;3(7):e2722.
Mannava S, Grachtchouk V, Wheeler LJ, Im M, Zhuang D, Slavina EG, et al. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle. 2008;7(15):2392–400.
Wang H, Wang X, Xu L, Zhang J, Cao H. High expression levels of pyrimidine metabolic rate-limiting enzymes are adverse prognostic factors in lung adenocarcinoma: a study based on The Cancer Genome Atlas and Gene Expression Omnibus datasets. Purinergic Signal. 2020;16(3):347–66.
Yu Y, Ding J, Zhu S, Alptekin A, Dong Z, Yan C, et al. Therapeutic targeting of both dihydroorotate dehydrogenase and nucleoside transport in MYCN-amplified neuroblastoma. Cell Death Dis. 2021;12(9):821.
Ciou SC, Chou YT, Liu YL, Nieh YC, Lu JW, Huang SF, et al. Ribose-5-phosphate isomerase A regulates hepatocarcinogenesis via PP2A and ERK signaling. Int J Cancer. 2015;137(1):104–15.
Huang F, Huffman KE, Wang Z, Wang X, Li K, Cai F, et al. Guanosine triphosphate links MYC-dependent metabolic and ribosome programs in small-cell lung cancer. J Clin Investig. 2021;131(1):e139929.
Kim MP, Li X, Deng J, Zhang Y, Dai B, Allton KL, et al. Oncogenic KRAS recruits an expansive transcriptional network through mutant p53 to drive pancreatic cancer metastasis. Cancer Discov. 2021;11(8):2094–111.
Reddy BA, van der Knaap JA, Bot AG, Mohd-Sarip A, Dekkers DH, Timmermans MA, et al. Nucleotide biosynthetic enzyme GMP synthase is a TRIM21-controlled relay of p53 stabilization. Mol Cell. 2014;53(3):458–70.
Kollareddy M, Dimitrova E, Vallabhaneni KC, Chan A, Le T, Chauhan KM, et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat Commun. 2015;6:7389.
Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351(6274):728–33.
Di Marcantonio D, Martinez E, Kanefsky JS, Huhn JM, Gabbasov R, Gupta A, et al. ATF3 coordinates serine and nucleotide metabolism to drive cell cycle progression in acute myeloid leukemia. Mol Cell. 2021;81(13):2752–64.
Zhou Q, Lin M, Feng X, Ma F, Zhu Y, Liu X, et al. Targeting CLK3 inhibits the progression of cholangiocarcinoma by reprogramming nucleotide metabolism. J Exp Med. 2020;217(8):e20191779.
Feng X, Ma D, Zhao J, Song Y, Zhu Y, Zhou Q, et al. UHMK1 promotes gastric cancer progression through reprogramming nucleotide metabolism. EMBO J. 2020;39(5):e102541.
Davison C, Morelli R, Knowlson C, McKechnie M, Carson R, Stachtea X, et al. Targeting nucleotide metabolism enhances the efficacy of anthracyclines and anti-metabolites in triple-negative breast cancer. NPJ Breast Cancer. 2021;7(1):38.
Pulliam N, Fang F, Ozes AR, Tang J, Adewuyi A, Keer H, et al. An effective epigenetic-PARP inhibitor combination therapy for breast and ovarian cancers independent of BRCA mutations. Clin Cancer Res Off J Am Assoc Cancer Res. 2018;24(13):3163–75.
Binenbaum Y, Fridman E, Yaari Z, Milman N, Schroeder A, Ben David G, et al. Transfer of miRNA in macrophage-derived exosomes induces drug resistance in pancreatic adenocarcinoma. Can Res. 2018;78(18):5287–99.
Fugger K, Bajrami I, Silva Dos Santos M, Young SJ, Kunzelmann S, Kelly G, et al. Targeting the nucleotide salvage factor DNPH1 sensitizes BRCA-deficient cells to PARP inhibitors. Science. 2021;372(6538):156–65.
Gandhi M, Gross M, Holler JM, Coggins SA, Patil N, Leupold JH, et al. The lncRNA lincNMR regulates nucleotide metabolism via a YBX1—RRM2 axis in cancer. Nat Commun. 2020;11(1):3214.
Crook A, De Lima LA, Payne T, Bhinderwala F, Woods J, Singh VK, et al. Radiation exposure induces cross-species temporal metabolic changes that are mitigated in mice by amifostine. Sci Rep. 2021;11(1):14004.
Fu S, Li Z, Xiao L, Hu W, Zhang L, Xie B, et al. Glutamine synthetase promotes radiation resistance via facilitating nucleotide metabolism and subsequent DNA damage repair. Cell Rep. 2019;28(5):1136-43.e4.
Gunda V, Souchek J, Abrego J, Shukla SK, Goode GD, Vernucci E, et al. MUC1-mediated metabolic alterations regulate response to radiotherapy in pancreatic cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2017;23(19):5881–91.
Ponde NF, Zardavas D, Piccart M. Progress in adjuvant systemic therapy for breast cancer. Nat Rev Clin Oncol. 2019;16(1):27–44.
Shelton J, Lu X, Hollenbaugh JA, Cho JH, Amblard F, Schinazi RF. Metabolism, biochemical actions, and chemical synthesis of anticancer nucleosides, nucleotides, and base analogs. Chem Rev. 2016;116(23):14379–455.
Rich TA, Shepard RC, Mosley ST. Four decades of continuing innovation with fluorouracil: current and future approaches to fluorouracil chemoradiation therapy. J Clin Oncol Off J Am Soc Clin Oncol. 2004;22(11):2214–32.
Breugom AJ, Swets M, Bosset JF, Collette L, Sainato A, Cionini L, et al. Adjuvant chemotherapy after preoperative (chemo)radiotherapy and surgery for patients with rectal cancer: a systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015;16(2):200–7.
Lebwohl DE, Canetta R. New developments in chemotherapy of advanced breast cancer. Ann Oncol Off J Eur Soc Med Oncol. 1999;10(Suppl 6):139–46.
Parker WB. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem Rev. 2009;109(7):2880–93.
Brandalise SR, Pinheiro VR, Aguiar SS, Matsuda EI, Otubo R, Yunes JA, et al. Benefits of the intermittent use of 6-mercaptopurine and methotrexate in maintenance treatment for low-risk acute lymphoblastic leukemia in children: randomized trial from the Brazilian Childhood Cooperative Group–protocol ALL-99. J Clin Oncol Off J Am Soc Clin Oncol. 2010;28(11):1911–8.
Karran P, Attard N. Thiopurines in current medical practice: molecular mechanisms and contributions to therapy-related cancer. Nat Rev Cancer. 2008;8(1):24–36.
Sullivan RD, Miller E, Sikes MP. Antimetabolite-metabolite combination cancer chemotherapy. Effects of intraarterial methotrexate-intramuscular Citrovorum factor therapy in human cancer. Cancer. 1959;12:1248–62.
Vora A, Mitchell CD, Lennard L, Eden TO, Kinsey SE, Lilleyman J, et al. Toxicity and efficacy of 6-thioguanine versus 6-mercaptopurine in childhood lymphoblastic leukaemia: a randomised trial. Lancet (Lond Engl). 2006;368(9544):1339–48.
Mixed reviews for new AML drugs. Cancer Discov. 2019;9(2):OF1.
Ben-Josef E, Normolle D, Ensminger WD, Walker S, Tatro D, Ten Haken RK, et al. Phase II trial of high-dose conformal radiation therapy with concurrent hepatic artery floxuridine for unresectable intrahepatic malignancies. J Clin Oncol Off J Am Soc Clin Oncol. 2005;23(34):8739–47.
Kemeny N, Jarnagin W, Gonen M, Stockman J, Blumgart L, Sperber D, et al. Phase I/II study of hepatic arterial therapy with floxuridine and dexamethasone in combination with intravenous irinotecan as adjuvant treatment after resection of hepatic metastases from colorectal cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2003;21(17):3303–9.
Rozencweig M, von Hoff DD, Slavik M, Muggia FM. Cis-diamminedichloroplatinum (II). A new anticancer drug. Ann Intern Med. 1977;86(6):803–12.
Wiltshaw E, Evans BD, Jones AC, Baker JW, Calvert AH. JMS, successor to cisplatin in advanced ovarian carcinoma? Lancet (Lond Engl). 1983;1(8324):587.
Herling CD, Coombes KR, Benner A, Bloehdorn J, Barron LL, Abrams ZB, et al. Time-to-progression after front-line fludarabine, cyclophosphamide, and rituximab chemoimmunotherapy for chronic lymphocytic leukaemia: a retrospective, multicohort study. Lancet Oncol. 2019;20(11):1576–86.
Holowiecki J, Grosicki S, Giebel S, Robak T, Kyrcz-Krzemien S, Kuliczkowski K, et al. Cladribine, but not fludarabine, added to daunorubicin and cytarabine during induction prolongs survival of patients with acute myeloid leukemia: a multicenter, randomized phase III study. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30(20):2441–8.
Juliusson G, Christiansen I, Hansen MM, Johnson S, Kimby E, Elmhorn-Rosenborg A, et al. Oral cladribine as primary therapy for patients with B-cell chronic lymphocytic leukemia. J Clin Oncol Off J Am Soc Clin Oncol. 1996;14(7):2160–6.
Amadori S, Suciu S, Selleslag D, Aversa F, Gaidano G, Musso M, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncolo Off J Am Soc Clin Oncol. 2016;34(9):972–9.
Earl HM, Hiller L, Howard HC, Dunn JA, Young J, Bowden SJ, et al. Addition of gemcitabine to paclitaxel, epirubicin, and cyclophosphamide adjuvant chemotherapy for women with early-stage breast cancer (tAnGo): final 10-year follow-up of an open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18(6):755–69.
Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N Engl J Med. 2018;379(25):2395–406.
Shanbhag S, Ambinder RF. Hodgkin lymphoma: a review and update on recent progress. CA Cancer J Clin. 2018;68(2):116–32.
Baena-Canada JM, Martinez MJ, Garcia-Olmedo O, Jimenez-Barcenas R, Muriel-Cueto P. Interaction between capecitabine and brivudin in a patient with breast cancer. Nat Rev Clin Oncol. 2010;7(1):55–8.
Faderl S, Wetzler M, Rizzieri D, Schiller G, Jagasia M, Stuart R, et al. Clofarabine plus cytarabine compared with cytarabine alone in older patients with relapsed or refractory acute myelogenous leukemia: results from the CLASSIC I Trial. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30(20):2492–9.
Bonate PL, Arthaud L, Cantrell WR Jr, Stephenson K, Secrist JA 3rd, Weitman S. Discovery and development of clofarabine: a nucleoside analogue for treating cancer. Nat Rev Drug Discov. 2006;5(10):855–63.
Wei AH, Dohner H, Pocock C, Montesinos P, Afanasyev B, Dombret H, et al. Oral azacitidine maintenance therapy for acute myeloid leukemia in first remission. N Engl J Med. 2020;383(26):2526–37.
Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther J Am Soc Gene Therapy. 2017;25(9):2214–24.
Gandhi V, Keating MJ, Bate G, Kirkpatrick P. Nelarabine. Nat Rev Drug Discov. 2006;5(1):17–8.
Elion GB. The purine path to chemotherapy. Science. 1989;244(4900):41–7.
Bird ST, Tian F, Flowers N, Przepiorka D, Wang R, Jung TH, et al. Idelalisib for treatment of relapsed follicular lymphoma and chronic lymphocytic leukemia: a comparison of treatment outcomes in clinical trial participants vs medicare beneficiaries. JAMA Oncol. 2020;6(2):248–54.
Burger JA, Okkenhaug K. Haematological cancer: idelalisib-targeting PI3Kdelta in patients with B-cell malignancies. Nat Rev Clin Oncol. 2014;11(4):184–6.
Pfeiffer P, Yilmaz M, Moller S, Zitnjak D, Krogh M, Petersen LN, et al. TAS-102 with or without bevacizumab in patients with chemorefractory metastatic colorectal cancer: an investigator-initiated, open-label, randomised, phase 2 trial. Lancet Oncol. 2020;21(3):412–20.
Zhou C, Chen G, Huang Y, Zhou J, Lin L, Feng J, et al. Camrelizumab plus carboplatin and pemetrexed versus chemotherapy alone in chemotherapy-naive patients with advanced non-squamous non-small-cell lung cancer (CameL): a randomised, open-label, multicentre, phase 3 trial. Lancet Respir Med. 2021;9(3):305–14.
Sahasranaman S, Howard D, Roy S. Clinical pharmacology and pharmacogenetics of thiopurines. Eur J Clin Pharmacol. 2008;64(8):753–67.
Petit E, Langouet S, Akhdar H, Nicolas-Nicolaz C, Guillouzo A, Morel F. Differential toxic effects of azathioprine, 6-mercaptopurine and 6-thioguanine on human hepatocytes. Toxicol In Vitro. 2008;22(3):632–42.
Carson DA, Wasson DB, Kaye J, Ullman B, Martin DW Jr, Robins RK, et al. Deoxycytidine kinase-mediated toxicity of deoxyadenosine analogs toward malignant human lymphoblasts in vitro and toward murine L1210 leukemia in vivo. Proc Natl Acad Sci USA. 1980;77(11):6865–9.
Lindemalm S, Liliemark J, Juliusson G, Larsson R, Albertioni F. Cytotoxicity and pharmacokinetics of cladribine metabolite, 2-chloroadenine in patients with leukemia. Cancer Lett. 2004;210(2):171–7.
Heidelberger C, Chaudhuri NK, Danneberg P, Mooren D, Griesbach L, Duschinsky R, et al. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature. 1957;179(4561):663–6.
Deren TL, Wilson WL. Use of 5-fluorouracil in treatment of bladder carcinomas. J Urol. 1960;83:390–3.
Olson KB, Greene JR. Evaluation of 5-fluorouracil in the treatment of cancer. J Natl Cancer Inst. 1960;25:133–40.
Vaitkevicius VK, Brennan MJ, Beckett VL, Kelly JE, Talley RW. Clinical evaluation of cancer chemotherapy with 5-fluorouracil. Cancer. 1961;14:131–52.
Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3(5):330–8.
Rutman RJ, Cantarow A, Paschkis KE. Studies in 2-acetylaminofluorene carcinogenesis. I. The intracellular distribution of nucleic acids and protein in rat liver. Cancer Res. 1954;14(2):111–4.
DiMasi JA, Paquette C. The economics of follow-on drug research and development: trends in entry rates and the timing of development. Pharmacoeconomics. 2004;22(2 Suppl 2):1–14.
Fukushima M, Suzuki N, Emura T, Yano S, Kazuno H, Tada Y, et al. Structure and activity of specific inhibitors of thymidine phosphorylase to potentiate the function of antitumor 2’-deoxyribonucleosides. Biochem Pharmacol. 2000;59(10):1227–36.
Ursem C, Van Loon K, Venook A. Adjuvant therapy trials. Cancer J. 2016;22(3):196–8.
Koeffler HP, Leong G. Preleukemia: one name, many meanings. Leukemia. 2017;31(3):534–42.
Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol Off J Eur Soc Med Oncol. 2006;17(Suppl 5):v7-12.
Cohen SS. Sponges, cancer chemotherapy, and cellular aging. Perspect Biol Med. 1963;6(2):215–27.
Turka LA, Dayton J, Sinclair G, Thompson CB, Mitchell BS. Guanine ribonucleotide depletion inhibits T cell activation. Mechanism of action of the immunosuppressive drug mizoribine. J Clin Investig. 1991;87(3):940–8.
Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–6.
Nawrocki ST, Griffin P, Kelly KR, Carew JS. MLN4924: a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin Investig Drugs. 2012;21(10):1563–73.
Madak JT, Bankhead A 3rd, Cuthbertson CR, Showalter HD, Neamati N. Revisiting the role of dihydroorotate dehydrogenase as a therapeutic target for cancer. Pharmacol Ther. 2019;195:111–31.
Baumann P, Mandl-Weber S, Volkl A, Adam C, Bumeder I, Oduncu F, et al. Dihydroorotate dehydrogenase inhibitor A771726 (leflunomide) induces apoptosis and diminishes proliferation of multiple myeloma cells. Mol Cancer Ther. 2009;8(2):366–75.
Chiarella AM, Ryu YK, Manji GA, Rustgi AK. Extracellular ATP and adenosine in cancer pathogenesis and treatment. Trends Cancer. 2021;7(8):731–50.
Overman MJ, LoRusso P, Strickler JH, Patel SP, Clarke SJ, Noonan AM, et al. Safety, efficacy and pharmacodynamics (PD) of MEDI9447 (oleclumab) alone or in combination with durvalumab in advanced colorectal cancer (CRC) or pancreatic cancer (panc). J Clin Oncol. 2018;36(15_suppl):4123.
Li XY, Moesta AK, Xiao C, Nakamura K, Casey M, Zhang H, et al. Targeting CD39 in cancer reveals an extracellular ATP- and inflammasome-driven tumor immunity. Cancer Discov. 2019;9(12):1754–73.
Konieczkowski DJ, Johannessen CM, Garraway LA. A convergence-based framework for cancer drug resistance. Cancer Cell. 2018;33(5):801–15.
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714–26.
Ariav Y, Ch’ng JH, Christofk HR, Ron-Harel N, Erez A. Targeting nucleotide metabolism as the nexus of viral infections, cancer, and the immune response. Sci Adv. 2021;7(21):eabg6165.
Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10(10):1073–80.
Motta V, Soares F, Sun T, Philpott DJ. NOD-like receptors: versatile cytosolic sentinels. Physiol Rev. 2015;95(1):149–78.
Giuliani AL, Sarti AC, Di Virgilio F. Extracellular nucleotides and nucleosides as signalling molecules. Immunol Lett. 2019;205:16–24.
Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol. 2016;16(3):177–92.
Fong L, Hotson A, Powderly JD, Sznol M, Heist RS, Choueiri TK, et al. Adenosine 2A receptor blockade as an immunotherapy for treatment-refractory renal cell cancer. Cancer Discov. 2020;10(1):40–53.
Cekic C, Sag D, Li Y, Theodorescu D, Strieter RM, Linden J. Adenosine A2B receptor blockade slows growth of bladder and breast tumors. J Immunol. 2012;188(1):198–205.
Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Blockade of A2b adenosine receptor reduces tumor growth and immune suppression mediated by myeloid-derived suppressor cells in a mouse model of melanoma. Neoplasia. 2013;15(12):1400–9.
Ohta A, Sitkovsky M. Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol. 2014;5:304.
Kinsey GR, Huang L, Jaworska K, Khutsishvili K, Becker DA, Ye H, et al. Autocrine adenosine signaling promotes regulatory T cell-mediated renal protection. J Am Soc Nephrol. 2012;23(9):1528–37.
Romio M, Reinbeck B, Bongardt S, Huls S, Burghoff S, Schrader J. Extracellular purine metabolism and signaling of CD73-derived adenosine in murine Treg and Teff cells. Am J Physiol Cell Physiol. 2011;301(2):C530–9.
Ludwig N, Gillespie DG, Reichert TE, Jackson EK, Whiteside TL. Purine metabolites in tumor-derived exosomes may facilitate immune escape of head and neck squamous cell carcinoma. Cancers. 2020;12(6):1602.
Hausler SF, Montalban del Barrio I, Strohschein J, Chandran PA, Engel JB, Honig A, et al. Ectonucleotidases CD39 and CD73 on OvCA cells are potent adenosine-generating enzymes responsible for adenosine receptor 2A-dependent suppression of T cell function and NK cell cytotoxicity. Cancer Immunol Immunother CII. 2011;60(10):1405–18.
Gorini S, Callegari G, Romagnoli G, Mammi C, Mavilio D, Rosano G, et al. ATP secreted by endothelial cells blocks CX(3)CL 1-elicited natural killer cell chemotaxis and cytotoxicity via P2Y(1)(1) receptor activation. Blood. 2010;116(22):4492–500.
Nowak M, Lynch L, Yue S, Ohta A, Sitkovsky M, Balk SP, et al. The A2aR adenosine receptor controls cytokine production in iNKT cells. Eur J Immunol. 2010;40(3):682–7.
Young A, Mittal D, Stagg J, Smyth MJ. Targeting cancer-derived adenosine: new therapeutic approaches. Cancer Discov. 2014;4(8):879–88.
Airas L, Hellman J, Salmi M, Bono P, Puurunen T, Smith DJ, et al. CD73 is involved in lymphocyte binding to the endothelium: characterization of lymphocyte-vascular adhesion protein 2 identifies it as CD73. J Exp Med. 1995;182(5):1603–8.
Allard B, Turcotte M, Spring K, Pommey S, Royal I, Stagg J. Anti-CD73 therapy impairs tumor angiogenesis. Int J Cancer. 2014;134(6):1466–73.
Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol. 2017;18(12):1332–41.
Duan S, Guo W, Xu Z, He Y, Liang C, Mo Y, et al. Natural killer group 2D receptor and its ligands in cancer immune escape. Mol Cancer. 2019;18(1):29.
Vecchio E, Caiazza C, Mimmi S, Avagliano A, Iaccino E, Brusco T, et al. Metabolites profiling of melanoma interstitial fluids reveals uridine diphosphate as potent immune modulator capable of limiting tumor growth. Front Cell Dev Biol. 2021;9:730726.
Lee JS, Adler L, Karathia H, Carmel N, Rabinovich S, Auslander N, et al. Urea cycle dysregulation generates clinically relevant genomic and biochemical signatures. Cell. 2018;174(6):1559-70.e22.
Keshet R, Lee JS, Adler L, Iraqi M, Ariav Y, Lim LQJ, et al. Targeting purine synthesis in ASS1-expressing tumors enhances the response to immune checkpoint inhibitors. Nat Cancer. 2020;1(9):894–908.
Canale FP, Basso C, Antonini G, Perotti M, Li N, Sokolovska A, et al. Metabolic modulation of tumours with engineered bacteria for immunotherapy. Nature. 2021;598(7882):662–6.
Lam KC, Araya RE, Huang A, Chen Q, Di Modica M, Rodrigues RR, et al. Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironment. Cell. 2021;184(21):5338–56.
Giuffrida L, Sek K, Henderson MA, Lai J, Chen AXY, Meyran D, et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat Commun. 2021;12(1):3236.
Mastelic-Gavillet B, Navarro Rodrigo B, Decombaz L, Wang H, Ercolano G, Ahmed R, et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8(+) T cells. J Immunother Cancer. 2019;7(1):257.
Quemeneur L, Gerland LM, Flacher M, Ffrench M, Revillard JP, Genestier L. Differential control of cell cycle, proliferation, and survival of primary T lymphocytes by purine and pyrimidine nucleotides. J Immunol. 2003;170(10):4986–95.
Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378(24):2288–301.
Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol Off J Am Soc Clin Oncol. 2015;33(25):2780–8.
Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. 2017;377(26):2545–54.
Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515(7528):558–62.
Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21(1):44–59.
Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16(4):375–84.
Mavligit GM, Gutterman JU, Burgess MA, Khankhanian N, Seibert GB, Speer JF, et al. Prolongation of postoperative disease-free interval and survival in human colorectal cancer by B.C.G. or B.C.G. plus 5-fluorouracil. Lancet (Lond Engl). 1976;1(7965):871–6.
Newlands ES, Oon CJ, Roberts JT, Elliott P, Mould RF, Topham C, et al. Clinical trial of combination chemotherapy and specific active immunotherapy in disseminated melanoma. Br J Cancer. 1976;34(2):174–9.
McCracken JD, Chen T, White J, Samson M, Stephens R, Coltman CA Jr, et al. Combination chemotherapy, radiotherapy, and BCG immunotherapy in limited small-cell carcinoma of the lung: a Southwest Oncology Group Study. Cancer. 1982;49(11):2252–8.
Repp R, van Ojik HH, Valerius T, Groenewegen G, Wieland G, Oetzel C, et al. Phase I clinical trial of the bispecific antibody MDX-H210 (anti-FcgammaRI x anti-HER-2/neu) in combination with Filgrastim (G-CSF) for treatment of advanced breast cancer. Br J Cancer. 2003;89(12):2234–43.
Pusztai L, Yau C, Wolf DM, Han HS, Du L, Wallace AM, et al. Durvalumab with olaparib and paclitaxel for high-risk HER2-negative stage II/III breast cancer: results from the adaptively randomized I-SPY2 trial. Cancer Cell. 2021;39(7):989-98.e5.
Theelen W, Chen D, Verma V, Hobbs BP, Peulen HMU, Aerts J, et al. Pembrolizumab with or without radiotherapy for metastatic non-small-cell lung cancer: a pooled analysis of two randomised trials. Lancet Respir Med. 2021;9(5):467–75.
Meric-Bernstam F, Larkin J, Tabernero J, Bonini C. Enhancing anti-tumour efficacy with immunotherapy combinations. Lancet (Lond Engl). 2021;397(10278):1010–22.
Nowak AK, Robinson BW, Lake RA. Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Can Res. 2003;63(15):4490–6.
Prehn RT, Lappe MA. An immunostimulation theory of tumor development. Transplant Rev. 1971;7:26–54.
Macpherson I. Letter: Cancer immunochemotherapy: the role of immunostimulation. Lancet (Lond Engl). 1974;1(7865):1058.
Stewart TH, Hollinshead AC, Harris JE, Belanger R, Crepeau A, Hooper GD, et al. Immunochemotherapy of lung cancer. Ann N Y Acad Sci. 1976;277(00):436–66.
Veronesi U, Adamus J, Aubert C, Bajetta E, Beretta G, Bonadonna G, et al. A randomized trial of adjuvant chemotherapy and immunotherapy in cutaneous melanoma. N Engl J Med. 1982;307(15):913–6.
Nowak AK, Lake RA, Marzo AL, Scott B, Heath WR, Collins EJ, et al. Induction of tumor cell apoptosis in vivo increases tumor antigen cross-presentation, cross-priming rather than cross-tolerizing host tumor-specific CD8 T cells. J Immunol. 2003;170(10):4905–13.
Li L, Ng SR, Colon CI, Drapkin BJ, Hsu PP, Li Z, et al. Identification of DHODH as a therapeutic target in small cell lung cancer. Sci Transl Med. 2019;11(517):eaaw7852.
Hayek S, Pietrancosta N, Hovhannisyan AA, Alves de Sousa R, Bekaddour N, Ermellino L, et al. Cerpegin-derived furo[3,4-c]pyridine-3,4(1H,5H)-diones enhance cellular response to interferons by de novo pyrimidine biosynthesis inhibition. Eur J Med Chem. 2020;186:111855.
Tu HF, Ko CJ, Lee CT, Lee CF, Lan SW, Lin HH, et al. Afatinib exerts immunomodulatory effects by targeting the pyrimidine biosynthesis enzyme CAD. Cancer Res. 2021;81(12):3270–82.
Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Can Res. 2010;70(8):3052–61.
Peereboom DM, Alban TJ, Grabowski MM, Alvarado AG, Otvos B, Bayik D, et al. Metronomic capecitabine as an immune modulator in glioblastoma patients reduces myeloid-derived suppressor cells. JCI Insight. 2019;4(22):655688.
Loo Yau H, Bell E, Ettayebi I, de Almeida FC, Boukhaled GM, Shen SY, et al. DNA hypomethylating agents increase activation and cytolytic activity of CD8(+) T cells. Mol Cell. 2021;81(7):1469-83.e8.
Weng RR, Lu HH, Lin CT, Fan CC, Lin RS, Huang TC, et al. Epigenetic modulation of immune synaptic-cytoskeletal networks potentiates gammadelta T cell-mediated cytotoxicity in lung cancer. Nat Commun. 2021;12(1):2163.
Stone ML, Chiappinelli KB, Li H, Murphy LM, Travers ME, Topper MJ, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc Natl Acad Sci USA. 2017;114(51):E10981–90.
Yang R, Elsaadi S, Misund K, Abdollahi P, Vandsemb EN, Moen SH, et al. Conversion of ATP to adenosine by CD39 and CD73 in multiple myeloma can be successfully targeted together with adenosine receptor A2A blockade. J Immunother Cancer. 2020;8(1):e000610.
Kamai T, Kijima T, Tsuzuki T, Nukui A, Abe H, Arai K, et al. Increased expression of adenosine 2A receptors in metastatic renal cell carcinoma is associated with poorer response to anti-vascular endothelial growth factor agents and anti-PD-1/anti-CTLA4 antibodies and shorter survival. Cancer Immunol Immunother CII. 2021;70(7):2009–21.
Borea PA, Gessi S, Merighi S, Vincenzi F, Varani K. Pharmacology of adenosine receptors: the state of the art. Physiol Rev. 2018;98(3):1591–625.
Le X, Negrao MV, Reuben A, Federico L, Diao L, McGrail D, et al. Characterization of the immune landscape of EGFR-mutant NSCLC identifies CD73/adenosine pathway as a potential therapeutic target. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer. 2021;16(4):583–600.
Sidders B, Zhang P, Goodwin K, O’Connor G, Russell DL, Borodovsky A, et al. Adenosine signaling is prognostic for cancer outcome and has predictive utility for immunotherapeutic response. Clin Cancer Res Off J Am Assoc Cancer Res. 2020;26(9):2176–87.
Liu H, Kuang X, Zhang Y, Ye Y, Li J, Liang L, et al. ADORA1 inhibition promotes tumor immune evasion by regulating the ATF3-PD-L1 axis. Cancer Cell. 2020;37(3):324-39.e8.
Willingham SB, Ho PY, Hotson A, Hill C, Piccione EC, Hsieh J, et al. A2AR antagonism with CPI-444 induces antitumor responses and augments efficacy to anti-PD-(L)1 and anti-CTLA-4 in preclinical models. Cancer Immunol Res. 2018;6(10):1136–49.
Giuffrida L, Sek K, Henderson MA, Lai J, Chen AXY, Meyran D, et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nature Commun. 2021;12(1):3236.
Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Investig. 2017;127(3):929–41.
Schneider G, Glaser T, Lameu C, Abdelbaset-Ismail A, Sellers ZP, Moniuszko M, et al. Extracellular nucleotides as novel, underappreciated pro-metastatic factors that stimulate purinergic signaling in human lung cancer cells. Mol Cancer. 2015;14:201.
Bastid J, Regairaz A, Bonnefoy N, Dejou C, Giustiniani J, Laheurte C, et al. Inhibition of CD39 enzymatic function at the surface of tumor cells alleviates their immunosuppressive activity. Cancer Immunol Res. 2015;3(3):254–65.
Hilchey SP, Kobie JJ, Cochran MR, Secor-Socha S, Wang JC, Hyrien O, et al. Human follicular lymphoma CD39+-infiltrating T cells contribute to adenosine-mediated T cell hyporesponsiveness. J Immunol. 2009;183(10):6157–66.
Zhang H, Vijayan D, Li XY, Robson SC, Geetha N, Teng MWL, et al. The role of NK cells and CD39 in the immunological control of tumor metastases. Oncoimmunology. 2019;8(6):e1593809.
Kashyap AS, Thelemann T, Klar R, Kallert SM, Festag J, Buchi M, et al. Antisense oligonucleotide targeting CD39 improves anti-tumor T cell immunity. J Immunother Cancer. 2019;7(1):67.
Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, et al. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc Natl Acad Sci USA. 2013;110(36):14711–6.
Buisseret L, Pommey S, Allard B, Garaud S, Bergeron M, Cousineau I, et al. Clinical significance of CD73 in triple-negative breast cancer: multiplex analysis of a phase III clinical trial. Ann Oncol Off J Eur Soc Med Oncol. 2018;29(4):1056–62.
Tripathi A, Lin E, Xie W, Flaifel A, Steinharter JA, Stern Gatof EN, et al. Prognostic significance and immune correlates of CD73 expression in renal cell carcinoma. J Immunother Cancer. 2020;8(2):e001467.
Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology. 2016;5(8):e1208875.
Goswami S, Walle T, Cornish AE, Basu S, Anandhan S, Fernandez I, et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat Med. 2020;26(1):39–46.
Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Adenosine limits the therapeutic effectiveness of anti-CTLA4 mAb in a mouse melanoma model. Am J Cancer Res. 2014;4(2):172–81.
Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res Off J Am Assoc Cancer Res. 2013;19(20):5626–35.
Sitkovsky MV, Hatfield S, Abbott R, Belikoff B, Lukashev D, Ohta A. Hostile, hypoxia-A2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer Immunol Res. 2014;2(7):598–605.
Shevchenko I, Mathes A, Groth C, Karakhanova S, Muller V, Utikal J, et al. Enhanced expression of CD39 and CD73 on T cells in the regulation of anti-tumor immune responses. Oncoimmunology. 2020;9(1):1744946.
Thompson EA, Powell JD. Inhibition of the adenosine pathway to potentiate cancer immunotherapy: potential for combinatorial approaches. Annu Rev Med. 2021;72:331–48.
Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359(6371):104–8.
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359(6371):91–7.
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359(6371):97–103.
Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science. 2021;371(6529):602–9.
Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science. 2021;371(6529):595–602.
Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377(25):2500–1.
Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51(2):202–6.
Arbour KC, Riely GJ. Systemic therapy for locally advanced and metastatic non-small cell lung cancer: a review. JAMA. 2019;322(8):764–74.
Yap TA, Parkes EE, Peng W, Moyers JT, Curran MA, Tawbi HA. Development of immunotherapy combination strategies in cancer. Cancer Discov. 2021;11(6):1368–97.
D’Angelo SP, Melchiori L, Merchant MS, Bernstein D, Glod J, Kaplan R, et al. Antitumor activity associated with prolonged persistence of adoptively transferred NY-ESO-1 (c259)T cells in synovial sarcoma. Cancer Discov. 2018;8(8):944–57.
Ramachandran I, Lowther DE, Dryer-Minnerly R, Wang R, Fayngerts S, Nunez D, et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J Immunother Cancer. 2019;7(1):276.
Funding
This work was supported by grants from the Shanghai Science and Technology Commission (19411966700). The funder had no role in the study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
G.-Y.L., Y.-Z.J., H.-L.W., and Y.G. designed and finalized the study. H.-L.W. and Y.G. wrote the paper. H.-L.W., Y.G., P.J., and Y.-F.X. revised the paper. All authors approved the final version submitted. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, Hl., Gong, Y., Ji, P. et al. Targeting nucleotide metabolism: a promising approach to enhance cancer immunotherapy. J Hematol Oncol 15, 45 (2022). https://doi.org/10.1186/s13045-022-01263-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-022-01263-x