
Abstract
The circadian rhythm is an evolutionarily conserved time-keeping system that comprises a wide variety of processes including sleep–wake cycles, eating–fasting cycles, and activity–rest cycles, coordinating the behavior and physiology of all organs for whole-body homeostasis. Acute disruption of circadian rhythm may lead to transient discomfort, whereas long-term irregular circadian rhythm will result in the dysfunction of the organism, therefore increasing the risks of numerous diseases especially cancers. Indeed, both epidemiological and experimental evidence has demonstrated the intrinsic link between dysregulated circadian rhythm and cancer. Accordingly, a rapidly increasing understanding of the molecular mechanisms of circadian rhythms is opening new options for cancer therapy, possibly by modulating the circadian clock. In this review, we first describe the general regulators of circadian rhythms and their functions on cancer. In addition, we provide insights into the mechanisms underlying how several types of disruption of the circadian rhythm (including sleep–wake, eating–fasting, and activity–rest) can drive cancer progression, which may expand our understanding of cancer development from the clock perspective. Moreover, we also summarize the potential applications of modulating circadian rhythms for cancer treatment, which may provide an optional therapeutic strategy for cancer patients.
Similar content being viewed by others

Background
It is well known that the rotation of the Earth is a near-24-h period which endows most organisms with an internal time-keeping system to adapt to the environmental changes. This evolutionarily conserved time-keeping mechanism termed circadian rhythm allows organisms to accommodate the changing environment, such as sleep and wake in animals and opening and closing of plant flowers [1]. As early as 1729, the French astronomer de Mairan firstly reported the endogenous rhythms associated with persistent movements of plant leaves even in constant darkness. He extended his observations to patients with sleep disorders in human beings [2]. However, since these initial observations, research on circadian rhythms was minimal until the discovery of circadian genes in Drosophila in the 1970s which opened the door for the in-depth understanding of circadian clocks [3, 4]. In the 1990s, an explosion of data in this field contributed to the canonical action model of the circadian clock in which the Clock/Bmal1 and Per/Cry complexes play the central role [5,6,7,8,9]. Although the components of circadian rhythms are not conserved between species, the basic mechanisms are universal in almost all models which comprise various biological processes including anabolism and catabolism, cell division and cell cycle, immune cell functions, apoptosis and DNA damage repair [10]. Perturbation of circadian rhythms by environmental disturbances (e.g., shift work, jet lag) has been implicated in multiple pathological conditions, such as cardiovascular diseases, sleep disorders, neurodegenerative disorders, and cancers [11,12,13].
Several key biological processes are regulated by circadian rhythms. Thus, the disruption of the circadian clock may contribute to abnormal cell proliferation, increased gene mutation, and resistance to apoptosis, which are important hallmarks of cancer [14, 15]. Based on the findings from epidemiology and laboratory studies, abnormal circadian rhythms have been listed as a potential carcinogen by the World Health Organization (WHO), which has increased the focus on defining the underlying mechanisms of circadian disruption-induced tumorigenesis [16, 17]. Indeed, accumulating evidence has demonstrated that altered circadian rhythms are closely related to tumorigenesis in breast cancer, prostate cancer, colorectal cancer (CRC), pancreatic adenocarcinoma, liver cancer, lung cancer, and others [18,19,20,21,22,23,24,25]. In addition, therapeutic efficiency in cancer treatment is also partially dependent on the time of drug administration, which could be a new therapeutic chronotherapy strategy in cancer management [26, 27]. The intrinsic links between circadian rhythms and tumorigenesis have therefore raised interest in manipulating these rhythms to prevent malignant transformation, to develop more efficacious therapies or novel adjuvant strategies, and ultimately improve the treatment outcome of cancer patients.
In this review, we will introduce the mechanism of disruptions of circadian rhythms-derived cancer progression and discuss the potential application of pharmacological modulation of the circadian clock and/or treating cancer using the clock as new therapeutic options for improved cancer management.
Regulation of circadian rhythms and their functions in cancer
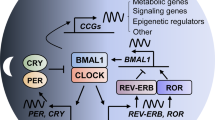
In almost all models, the signaling pathways regulating circadian rhythms are universal regardless of non-conservation of the clock components [28]. In mammals, the suprachiasmatic nucleus (SCN) of the hypothalamus holds the “master clock”, which clocks with the environmental light cycle by directly coordinating the autonomic nervous system efferent and neuroendocrine signals [29,30,31]. In detail, several proteins involved in the clockwork of the cell associate closely with the negative and positive transcriptional feedback loops (Fig. 1). For example, circadian locomotor output cycles kaput (CLOCK) and brain and muscle aryl hydrocarbon receptor nuclear translocator 1 (BMAL1), which contain two basic helix-loop-helix domains, can bind to the Cryptochrome (Cry) and Period (Per) genes through their E-boxes, thus positively regulating circadian transcription [32,33,34]. In contrast, the mammalian CRY and PER proteins, as a heterodimer interacting with casein kinase Iε (CKIε), perform a negative effect for CLOCK/BMAL1-driven transcription [35,36,37,38]. In addition, the expression of circadian genes is transcriptionally modulated by the RORs (subfamilies of nuclear hormone receptors) and REV-ERB, resulting in the activation or repression of gene transcription for several clock genes [39,40,41,42].

Regulation of circadian rhythms and their functions in cancer. a the master regulator of circadian clock is located in the suprachiasmatic nucleus (SCN) of the hypothalamus. The SCN coordinates several organ clocks in response to the environmental factors (including sleep/wake, eating/fasting, activity/rest, etc.), for controlling body homeostasis, such as heart rate, body temperature, and hormone levels. b at the molecular level, CLOCK and BMAL1 can bind to the CRY and PER genes through their E-boxes, thus positively regulating circadian transcription. But the mammalian CRY and PER proteins, as a heterodimer interacting with CKIε, perform a negative effect for CLOCK/BMAL1-driven transcription. In addition, the expression of circadian genes is transcriptionally modulated by the RORs and REV-ERB, resulting in the activation or repression of gene transcription for several clock genes. c, the mutation or deletion of core clock genes (including Per1/2, Clock, Bmal1, Cry1/2 and Rorc) can accelerate the development of various tumors, such as liver, ovarian, lung, and colorectal cancer, and lymphoma. CLOCK: circadian locomotor output cycles kaput; BMAL1: brain and muscle aryl hydrocarbon receptor nuclear translocator 1; CRY: cryptochrome; PER: period; CKIε: casein kinase Iε
Growing evidence from both epidemiological research and preclinical data based on animal models supports the relationship between chronic disruption of the circadian clock and the occurrence of cancer [43,44,45,46,47]. Indeed, circadian disruption was listed as a probable human carcinogen in 2007 by the International Agency for Research on Cancer (IARC), part of the WHO [48].
For example, several studies have revealed that shift workers have a higher risk of developing breast and prostate cancers, as evidenced by a strong correlation between a long period (> 20 years) of shiftwork and increased cancer risk [49,50,51,52]. Furthermore, previous studies have indicated that people in modern society have undergone a sub-health lifestyle change, including excessive calories intake at midnight or continuous caloric intake over the 24 h throughout days, which mimics aspects of shiftwork and potentially promotes prostate and breast cancer risk [53, 54].
Interestingly, rest/activity rhythm was also identified as a key factor due to the significant impact on the clinical treatment of metastatic CRC patients, who have a poor quality of life with erratic periods of rest/activity [55, 56]. In line with this, the disruption of the core clock Per2 gene further aggravated CRC cancer cell proliferation in ApcMin/+ genetic mice [57]. In addition, disruption of Per2 also accelerated KrasG12D and KrasG12D/p53−/− mutation-mediated lung cancer progression [23].
Additionally, mice with the core clock Per2 genes disrupted were more tumor-prone following treatment with the carcinogenic agent diethylnitrosamine (DEN) [58, 59]. Moreover, irradiation stimuli promoted the development and progression of liver cancer and lymphoma in Per1 and Per2 mutant mice and Bmal1−/+ mice [60]. Mice with deleted Cry1 and Cry2 or Rorc showed similar effects in lymphoma [60, 61]. There has also been a report on a link between Clock mutation ClockΔ19/+ and Tp53 mutation-induced liver cancer and lymphoma [62].
However, some studies demonstrated that clock gene mutations (including Clock, Per1−/− or Per2−/−, Cry1−/− and Cry2−/−) potentially led to an adverse influence on cancer progression [63, 64]. In a RAS mutation-triggered cutaneous squamous tumor model, Bmal1 deletion protects from tumorigenesis [65]. In addition, previous studies have revealed CLOCK and BMAL1 as key regulators for acute myeloid leukemia (AML) by contributing to proliferation and stemness [66, 67]. Moreover, deletions of Cry1 and Cry2 diminished tumor development in p53 null mice possibly due to triple mutant-induced genotoxic stress [68, 69]. Taken together, the dual function of clock genes, either as oncogenes or as tumor suppressors, may be attributed to tissue-specific mechanisms and implies the existence of intricate clock-dependent networks, which are involved in the maintenance of homeostasis during different tumor stages.
In summary, these rhythms mainly emerge from the interplay between circadian clocks and sleep–wake cycles, eating–fasting cycles, and activity–rest cycles. These circadian rhythms are able to modulate several key aspects of cellular and organ functions with profound implications in cancer management. In the following sections, we will discuss the interplay between these cycles with cancer and describe the links involved.
The interplay between sleep–wake cycles and cancer
Disruption of sleep–wake circadian rhythm has a major impact on the entire neuroendocrine-immune system, which modulates immune defense and energy metabolism, thus contributing to normal physiological activity by coordinating the biological response to everyday stresses, as well as cognitive and physical performance (Fig. 2).

The interplay between sleep–wake cycles and cancer. a sleep–wake cycles regulate the immune system. During normal sleep–wake cycles, SWS sustains the function of immune system by maintaining the balance of T-helper1 (Th1) and T-helper2 (Th2) cell-derived cytokines (Th1 cytokines: IL-2, IFNγ, and IL-12; Th2 cytokines: IL-4 and IL-10), which benefit the antigen presenting process. b sleep–wake cycles regulate the inflammatory response. Disturbance of sleep continuity (sleep time and efficiency) and architecture (SWS and REM sleep) may lead to body inflammatory response, including abnormal systemic inflammation, cellular inflammation and inflammation transcriptional activity, which are associated with development of chronic inflammation related disease, such as cancers. c Sleep–wake cycles and endocrine. Endocrine factors, including growth hormone, prolactin, thyroid hormone, cortisol, gonadal steroids, insulin, and so on, have been identified to be secreted during certain time periods. Disruption of sleep–wake cycles may break these balances and influence their secretions. d Sleep–wake cycles regulate DNA damage and repair. On the one hand, sleep disruption can reduce the levels of melatonin, an important antioxidant, which may lead to increased oxidative DNA damage. On the other hand, sleep deprivation downregulates the expression of several genes involved in DNA repair, such as ERCC6, PARP1, and RAD50, which may ultimately promote tumorigenesis. e, Feedback from cancer to sleep–wake cycles. IL-1β in the brain can regulate REM and NREM sleep by modulating various molecules and neurotransmitters, including COX-2, GABA, and nitric oxide (NO), while IL-6 enhances SWS and decreases REM sleep. SWS: slow-wave sleep; REM: rapid eyes movement; TSH: thyroid-stimulating hormone; PRL: prolactin; RAAS: renin–angiotensin–aldosterone systems; COX-2: cyclooxygenase-2.
Sleep–wake cycles regulate the immune system
Rhythmical biological regulation of the immune system is mediated by intricate crosstalk between the nervous and endocrine systems [70,71,72]. Dysregulation of sleep–wake cycles, like prolonged sleep deprivation, disrupts the immune system, resulting in increased rates of viral infection and weak antigen processing and antibody formation [70, 73, 74].
During normal sleep–wake cycles, the immune system displays a biphasic shift that is linked with maintaining the balance of T-helper1 (Th1) and T-helper2 (Th2) cell-derived cytokines (Th1 cytokines: IL-2, IFNγ, and IL-12; Th2 cytokines: IL-4 and IL-10) [75]. Mechanically, Th1 activity in the initial hours of nighttime sleep is enhanced and accompanied by moderate promotion of interferon (IFN)-γ/IL-4-producing Th cells, while Th2 activity dominates in the late hours of sleep or before wake up [76]. Once this pattern is broken, cytokines are excessively produced and directly induce immune disturbances, which subsequently lead to chronic inflammation and tissue damage. In sleep-deprived individuals, including stressed persons, insomniacs, and the elderly, a switch toward Th2 activity has been reported [76]. Sleep deprivation-induced production of growth hormone and prolactin turning cytokine balance into Type 1 dominance, as well as cortisol and norepinephrine leading to Type 2 dominance, are all involved in immune response alteration [70, 76, 77].
In addition, decreased cellular immunity is also attributed to the elevation of sympathetic tone induced by sleep deprivation. During sleep, the number of monocytes producing IL-12 or IL-10 is normally increased or decreased, respectively, thus resulting in a circadian rhythm. However, when chronic wakefulness occurs, rhythmic temporal variations of IL-12 and IL-10 produced from monocytes are lost, causing dysregulation of the immune system [77]. Furthermore, Irwin and colleagues revealed that the lytic activity of natural killer (NK) cell was diminished by an average of 28% in people with sleep deprivation, i.e., wakefulness between 03:00 to 07:00 h, compared with blood sampling between 07:00 and 09:00 h from healthy men [78]. They then demonstrated that partial sleep deprivation potentially attenuates the natural immune response, evidenced by decreased numbers and weak lytic activity of NK cells, lymphokine-activated killer (LAK), and its precursors [79]. Notably, a marked decrease of CD8+ T cells (cytotoxic and memory), which are derived from tumor necrosis factor (TNFα) stimuli, is the most pronounced effect resulting from sleep deprivation, solidly implying an impaired immune response if exposed to carcinogenic risk [76]. These findings suggest that even modest disruption of sleep–wake cycles resulting from working the night shift can significantly induce the dysregulation of natural immune responses, T cell-cytokine production, etc. These dysregulations need more than a single day of sleep to recover to baseline and potentially lead to an increased risk of cancer [80,81,82].
Sleep–wake cycles regulate inflammatory responses
Inflammatory responses are abnormally activated following dysregulation of the immune system caused by disruption of sleep–wake cycles, which is characterized by the activation of soluble intercellular adhesions molecule (sICAM)-modulated NF-κB inflammatory signaling and the subsequent induction of inflammatory markers TNF-γ, IL-6, and C-reactive protein (CRP) [83, 84]. In addition, sleep deprivation alters IL-6-mediated trans-signaling by changing the levels of soluble IL-6 (sIL-6) receptor in various types of brain cells and neighboring organs [85]. An analysis using a DNA microarray indicated that sleep-deprivation-induced upregulation of inflammatory genes in humans was a common response and might be not rapidly resolved [83]. Sleep restriction-triggered upregulation of proinflammatory cytokines (IL-6, IL-1β, IL-17, and CRP) were maintained at high expression levels even through two nights of recovery sleep [86]. In line with this, increased levels of CRP were also found in shift workers, indicating a progression of an inflammatory state and an increased risk of cancer [87].
Rigorous studies profiling inflammatory markers in the human sleep–wake cycle are limited to a few methodological studies that have looked at profiles of systemic and cellular inflammatory markers obtained repeatedly over the course of a regular sleep–wake cycle, compared with continuous wakefulness over 24 h [88]. These studies have shown a close association between the levels of several markers and sleep, the circadian oscillator, or both [70]. For example, IL-6 was reported to have a circadian profile whose levels peaked at 19:00 h and 05:00 h [89]. Furthermore, upregulation of IL-6 levels and production of TNF was attributed to nocturnal sleep [70]. A previous study indicated that experimental sleep deprivation during the nocturnal period resulted in an undersecretion of IL-6 and an attenuated secretion of TNF from Toll-like receptor 4 (TLR4)-induced monocytes [90]. Notably, sleep deprivation changed the nighttime pattern of IL-6 secretion into daytime, therefore resulting in an IL-6 oversecretion during the day [89]. Similarly, by investigating people with sleep disturbances, TLR4-mediated IL-6 production from monocytes was also found to be decreased at night and increased in the day [91]. Nevertheless, IL-6 levels were partially mediated by the circadian oscillator as indicated by the observation of transient (less than 1 h) peaks during nocturnal wakefulness [85].
Different stages of sleep, including slow-wave sleep (SWS) and rapid eyes movement (REM) sleep, differentially induced nocturnal changes of inflammatory cytokine activity. However, the levels of the inflammatory cytokines during the detailed period were not clear. The levels of inflammatory cytokines were reported to peak early in SWS, but higher levels of IL-6 were found during REM sleep compared with being awake [92]. Consistent with increased IL-6 during REM sleep, IL-6 and sIL-6R were upregulated later during the night, and the morning levels of TLR4-stimulated IL-6 production from monocytes were found to correlate with the amount of REM sleep [85, 91, 92].
In conclusion, sleep disruption may lead to abnormal inflammatory responses as evidenced by the upregulation of proinflammatory cytokines (especially IL-6 and CRP), which are potential risk factors for several cancers, including ovarian, brain, breast and colorectal cancers.
Sleep–wake cycles and endocrine factors
Endocrine factors, especially those governed by the hypothalamic-pituitary axis, have been identified to be regulated by circadian rhythms, therefore fluctuating over the day. These include growth hormone (GH), prolactin (PRL), thyroid hormone, cortisol, and gonadal steroids [93,94,95]. Insulin and adipokines, a form of nutrient-sensitive hormones, are also mediated by circadian rhythms (light/dark and feeding/fasting cycles) giving rise to varying circulating levels, partially controlled by time-of-day-dependent patterns [93].
Growth hormone and prolactin have been reported to change their secretory pattern due to exposure to acute sleep deprivation [96]. This treatment also mitigated nighttime peaks of growth hormone and prolactin, leading to dysregulation of the immune system [70]. Furthermore, a previous study demonstrated that the 24-h mean and amplitude of several endocrine factors modulated by the circadian clock, including thyroid-stimulating hormone, prolactin, and leptin, were suppressed by semi-chronic sleep deprivation with sleep debt during 6 days [93]. Sleep debt also influenced the circadian pattern of plasma cortisol, resulting in higher plasma levels in the afternoon and early evening [93].
Notably, hyperphagia and weight gain are closely associated with sleep deprivation. Mechanically, sleep deprivation triggers downregulation of the plasma concentration of the appetite-restraining adipokine leptin and upregulation of the plasma concentration of the appetite-stimulating peptide ghrelin, in which adipokine leptin and peptide ghrelin are produced in adipose tissue and the stomach, respectively [97]. Accordingly, this process leads to greater hunger and appetite, subsequently resulting in triglyceride metabolism and disturbed lipid levels [98]. Inhibition of appetite is required for the secretion and blood levels of leptin, which are positively regulated by daily sleep length and display significant reduction with sleep deprivation [97, 99]. Indeed, overweight or obesity risk in shift workers is found to have a potential relationship with sleep deprivation-induced changes of the hormonal milieu. Numerous studies have comprehensively revealed that weight gain or various levels of obesity, defined by increased body mass index and/or elevated waist-hip ratio, was potentially mediated by prolonged shift work-induced circadian disruption [100, 101]. In addition, marked weight gain was also confirmed, even 4–5 years after shift work exposure in prospective cohort studies [102]. However, weight modulation in shift work is a cumulative effect because it is also regulated by various factors, such as age, lifestyle as well as “night eating syndrome” [103]. Nevertheless, either overweight or obesity facilitated increased secretion of adipokines and release of macrophages from adipose tissue, thus triggering chronic inflammation and subsequent oxidative stress and DNA damage [104]. These events likely contribute to breast cancer promotion and poor prognosis at least in a subpopulation, for example, postmenopausal female shift workers [105].
Sleep–wake cycles regulate DNA damage and repair
Stress-induced DNA damage has long been recognized as an important risk factor for the development of several chronic diseases, including cancer [106,107,108]. Sleep disruption in shift workers may lead to increased oxidative DNA damage due to decreased secretion of melatonin (an important cellular antioxidant) caused by sleep disruption [109,110,111,112]. For example, urine samples from 217 dayshift workers and 223 nightshift workers were collected for analysis of 8-OH-dG (a marker of oxidative DNA damage) and aMT6s (a marker of circulating melatonin) levels by using high-performance liquid chromatography with electrochemical detection [113]. The results found that disruption of melatonin in nightshift workers is associated with impaired DNA repair machinery and increased cellular oxidative DNA damage, as evidenced by the positive correlation between aMT6s and urinary 8-OH-dG levels [113]. Another study also demonstrated that night work is associated with reduced DNA repair, and that this effect was probably due to melatonin suppression caused by night work, which raises the potential of melatonin supplementation for preventing DNA damage-induced chronic diseases [114]. Moreover, an observational study on 49 healthy doctors has found that nightshift doctors with sleep deprivation showed lower expression of DNA repair genes and increased DNA breaks compared with the dayshift colleagues, which was linked to the development of chronic disease [115]. In a recent controlled laboratory study, transcriptomic analysis of genes and associated pathways in circulating leukocytes obtained from night shift workers and healthy adults demonstrated a significantly impaired DNA repair pathway, which increased DNA damage and cancer risk in shift workers [116]. Taken together, these findings indicate that chronic disruption of sleep–wake cycles may cause increased DNA damage and decreased DNA repair, serving as potential risk factors for tumorigenesis [117,118,119].
Feedbacks from cancer to sleep–wake cycles
Various cytokines, such as TNF-α, TGF-β, IL-10, and especially IL-1β and IL-6, are well studied and have been shown to be involved in the complex crosstalk between cancer initiation/progression and sleep–wake cycles [120, 121].
Notably, IL-1β can not only act as a pleiotropic cytokine to promote cancer progression in the tumor microenvironment but can also gain access to the brain through passive diffusion, subsequently interacting with IL-1R1 expressed on endothelial cells at the blood–brain barrier or vagal afferents [122,123,124]. Accordingly, IL-1β in the brain has been identified as the key mediator to affect rhythmical behavior. A previous study reported that both duration and delta power (~ 0.5–4 Hz oscillations) of NREM (non-rapid eye movement) sleep were markedly enhanced by injecting central or systemic IL-1β, which might play a role in hypnosis [125]. In contrast, spontaneous NREM sleep could be inhibited by the administration of IL-1β receptor antagonists or IL-1β neutralizing antibodies [126, 127]. For REM sleep, IL-1β regulates it in a time and dose-dependent manner, shown by high levels of IL-1β inhibiting REM sleep, while low levels of IL-1β have no effect [121]. Moreover, IL-1β can influence sleep by modulating various molecules and neurotransmitters, including cyclooxygenase-2, NF-κB, GABA, nitric oxide (NO), prostaglandins, and adenosine [128]. For instance, inhibition of NO synthesis by an inhibitor, L-NAME, could decrease NO production and IL-1β-induced NREM sleep [129]. Notably, sleep deprivation could in turn stimulate the expression of IL-1β in the brain [130]. Overall, these studies suggest that IL-1β is under circadian and homeostatic control and has effects on multiple sleep nuclei.
IL-6 has been suggested to follow diurnal rhythms, but its detailed role is not yet totally clear [121, 131]. Like IL-1β, IL-6 during wakefulness is normally at low levels and peaks during sleep [89]. Sleep deprivation elevates plasma levels of circulating IL-6 [92]. A study in humans revealed that IL-6 enhanced SWS and decreased REM sleep following subcutaneous injection [132]. Paradoxically, human recombinant IL-6 injected intracerebroventricularly into rabbits seemed to be not somnogenic but pyrogenic [133]. However, recombinant rat IL-6 reduced NREM sleep after temporarily enhancing NREM sleep in rat models [134]. In fact, the relationship between IL-6 and sleep–wake cycles is intricate. For example, sleep enhanced the levels of sIL-6R to over 70% of the wake levels at the termination of sleep, and simultaneously induced IL-6 trans-signaling regardless of classical/membrane-bound IL-6 signaling [85]. This process further supports the role of sleep in immune defense [135, 136].
In summary, the above findings suggest that the disruption of sleep–wake cycles may serve as a potential risk factor for tumors mainly through modulating the neuroendocrine-immune system, the inflammatory responses, and the DNA damage repair systems. Moreover, increased levels of inflammatory cytokines secreted from tumors or tumor-associated leukocytes could in turn control the homeostasis of several sleep process, implying the complex crosstalk between cancers and sleep–wake cycles. Therefore, maintaining regular sleep pattern is highly encouraged in daily life for prevention of not only cancer but also other disorders.
The interplay between eating–fasting cycles and cancer
Dietary-related risk factors have been identified as one of the key determinants for cancer development [137,138,139]. In line with this notion, 14–20% of all cancer-related mortality in the USA is associated with obesity induced by dysregulation of eating–fasting cycles, and guidelines for nutrition administration that advocate maintaining eating–fasting cycles are thus suggested for reducing cancer incidence [138, 139]. In addition, preclinical data have revealed that cancer cells with hyperproliferation usually fail to adapt to fasting conditions, implying the possibility that calorie-limited diets based on eating–fasting cycles could become a potential strategy for the prevention and treatment of cancer [140, 141]. Notably, balancing eating–fasting cycles seems to improve acute and/or chronic side effects from treatments in cancer patients. Herein, we summarize the detailed mechanisms behind eating–fasting cycles-mediated signaling pathways for cancer initiation and development, and emphasis the important role of ordered eating–fasting cycles in cancer prevention or therapy (Fig. 3).

The interplay between eating–fasting cycles and cancer. a eating–fasting cycles regulate differential stress resistance and sensitization of cancer cells. Fasting triggers metabolic reprogramming of adipose tissue and muscle, which on the one hand sensitizes cancer cells to chemotherapy and other cancer therapies while on the other protects healthy cells from side effects from tumor therapy. b fasting or FMDs is able to decrease the levels of growth-promoting nutrients and factors, including glucose, IGF1 and insulin. Decreased glucose levels coupled with reduced glucose uptake via inhibition of GLUTs result in downregulated aerobic glycolysis and increased OXPHOS. This metabolic switch increases the accumulation of cellular ROS levels in cancer cells in response to chemotherapy, resultantly causing oxidative DNA damage and cell death. In addition, fasting or FMDs can also regulate IGF1R-mTOR-AMPK signaling to activate anticancer immunity by inducing autophagy. FMD: fasting-mimicking diets; GLUT: glucose transporter; OXPHOS: oxidative phosphorylation; ROS: reactive oxygen species
Effects of eating–fasting cycles on hormones and metabolites
The levels of metabolites and circulating hormones are changed in response to eating–fasting. This is characterized by downregulation of glucose, IGF1, leptin, and insulin, and upregulation of adiponectin, which contribute to either antitumor effects and/or protection from side effects [142, 143]. Increasingly strong evidence is demonstrating that fasting or fasting-mimicking diets (FMDs) inhibits the effect of IGF1 and glucose against cancer [144]. Mechanistically, fasting or an FMD compromises various onco-pathways, such as cAMP-PKA and IGF1R-AKT-mTOR-S6K signaling, activates anticancer immunity, and enhances the tolerance of normal cells in stress conditions by inducing autophagy [145,146,147]. In addition, fasting has been revealed to induce ketone body accumulation, leading to mitigation of tumor proliferation and promotion of differentiation by inhibiting histone deacetylases (HDACs) [148].
Eating–fasting cycles regulate differential stress resistance and sensitization of cancer cells
Fasting triggers metabolic reprogramming of adipose tissue and muscle, where carbon-related metabolic pathways are modulated to change the levels of certain metabolites and circulating hormones. On the one hand these processes sensitize cancer cells to chemotherapy and other cancer therapies, while on the other they protect healthy cells from side effects from tumor therapy [140, 141].
Several studies using animal models have demonstrated that short-term fasting (STF) promoted the anticancer effects of chemotherapeutic agents against numerous cancers, including pancreatic cancer, breast cancer, colorectal cancer, melanoma, and neuroblastoma, while simultaneously diminishing toxic effects of the treatments [149,150,151]. Furthermore, a 24–60-h fasting regime reinforced chemotoxicity tolerance of healthy cells, and further enhanced anticancer effects of chemotherapy compared to chemotherapy alone in different strains of mice with tumor xenografts [145, 152,153,154]. The distinct outcomes derived from STF in healthy or tumor cells are termed differential stress resistance (DSR) [155]. Similarly, healthy cells faced with nutrient deprivation utilize energy for tissue repair that facilitates resistance to chemotherapy, while tumor cells still maintain hyperproliferation thus resulting in increased DNA damage and apoptosis during chemotherapy [141, 152, 156]. Thus, differential stress sensitization (DSS) is defined as a phenomenon whereby STF protects healthy cells from chemotherapy-induced toxicity and enhances the antitumor effect of chemotherapy.
Differential stress resistance (DSR)
The hypothesis that starvation plays an adverse effect in cancer compared to normal cells is reasonable, especially when the organism simultaneously faces cell stressors such as chemotherapy. In detail, healthy cells during starvation reduce global ribosome biogenesis and growth-related gene transcription which facilitates entry into the self-maintenance mode to resist damage caused by various cancer treatments. By contrast, the self-maintenance mode is terminated due to onco-pathways in cancer cells, thus leading to the reduction of stress response signaling [141]. This study also demonstrated that low-glucose triggered further cell death of mouse or human glioma and neuroblastoma cancer in hydrogen peroxide or cyclophosphamide-treated models, while protecting healthy glia cells from treatment-induced toxicity [141]. In line with these results, etoposide treatment markedly promoted the survival rate of neuroblastoma allograft-bearing mice following 2-day fasting, while resulting in only moderate effects in non-fasted mice [141].
A subsequent study indicated that fasting-triggered inactivation of IGF1 signaling protected glia and neurons from the oxidative stress agent cyclophosphamide and reduced doxorubicin-induced mouse embryonic fibroblasts, but not glioma and neuroblastoma cells [144]. Furthermore, mice with a conditional liver Igf1 gene deletion, which mimics fasting characterized by decreased levels of circulating IGF1, showed a toxicity-resistant ability against chemotherapeutic drugs, such as doxorubicin [157]. Histopathology studies suggested that compromised cardiac myopathy induced by doxorubicin was present in control mice but not in limited ingredient diet (LID)-treated mice [144]. Taken together, these data demonstrate that fasting-induced IGF1 downregulation is involved in increased tolerability to chemotherapy.
Fasting also contributes to overcoming the bottleneck for clinical application of both mTOR inhibitors and dexamethasone, which are used as anti-emetics and anti-allergics or as anticancer agents in cancer therapy, but have non-negligible side effects, such as hyperglycemia [149]. Indeed, the high glucose-induced cAMP-PKA pathway, which was attributed to both dexamethasone and rapamycin treatment, was revealed to enhance the toxicity of mouse cardiomyocytes from doxorubicin [149]. These results could possibly be reversed by decreasing circulating glucose concentration via fasting or insulin injections. In addition, fasting and/or FMD-triggered autophagy is also involved in the reduction of doxorubicin-induced cardiomyopathy by removing damaged mitochondria and toxic aggregates, therefore resulting in the reduction of reactive oxygen species (ROS) with protective effects on cardiomyocytes [158].
Differential stress sensitization (DSS)
Dietary treatment alone (like fasting and FMDs) shows limited effects on cancer. However, based on the DSS-derived mechanism, dietary interventions combined with canonical cancer therapy potentially display promising anticancer effects [159,160,161]. Previous observations on glioma, melanoma, and breast cancer indicated that fasting induced unexpected upregulation of ribosome biogenesis and expression of assembly genes or proliferation-related genes, which contribute to the activation of AKT and S6K and subsequent ROS accumulation and DNA damage, further increasing the effect of DNA-damaging drugs [140]. In fact, periodic cycles of fasting or FMDs in mice were reported to inhibit cell growth in several solid tumors and lymphoid leukemia [147, 159]. Notably, periodic fasting or FMDs were also revealed to enhance the anticancer effects of chemo- or radiotherapy and tyrosine kinase inhibitors (TKIs) [162,163,164].
In addition to regulating glucose utilization and fatty acid β-oxidation in cancer, fasting or FMDs can also accelerate the conversion of energy metabolism mechanisms in cancer cells from aerobic glycolysis to mitochondrial OXPHOS for maintaining the growth of cancer cells in the nutrition-deprivation environment [152]. Subsequently, mitochondrial OXPHOS promotes ROS accumulation and decreases glutathione synthesis primarily derived by glycolysis and the pentose phosphate pathway, which leads to reduced ROS-mediated DNA damage. Strategies inhibiting glycolysis and glutaminolysis and promoting OXPHOS in order to delay tumor growth and overcome drug resistance are under investigation [152]. Of note, fasting or FMDs seem to promote metastasis, since several types of aggressive and metastatic cancer cells tend to be dependent on high-lactate production from high glycolytic activity [165].
Fasting or FMDs can trigger other alterations besides the metabolism changes that induce DSS in cancer cells. For example, fasting upregulated equilibrative nucleoside transporter 1 (ENT1) expression (gemcitabine transporter) to improve the anticancer activity of gemcitabine against pancreatic cancer [166]. In breast cancer and melanoma cells, fasting could induce SUMO modification of REV1 (a p53-binding protein and DNA polymerase) by the SUMO2 and/or SUMO3-dependent ways, consequently relieving the suppressive effect of REV1 on p53, which inhibited cancer cells by transcriptionally promoting expression of the pro-apoptotic genes [151]. In addition, a fasting-mimicking diet enhances the effect of the anti-estrogens tamoxifen and fulvestrant by reducing circulating IGF1 through EGR1 and PTEN-mediated inhibition of AKT/mTOR signaling [159]. In leukemia, a previous study reported that fasting could slow the progression of acute lymphoblastic leukemia (ALL) but not acute myeloid leukemia (AML) by activating the protein PR/SET domain 1 (PRDM1)-mediated leptin receptor and downstream signaling [167]. Furthermore, transcription factors PAX5 and IKZF1, commonly identified with more than 80% mutations in pre-B cell ALL, were indicated as tumor suppressors by exhibiting a persistent restriction on glucose uptake and energy supply [168]. Accordingly, energy crisis and cell death were observed in PAX5 and IKZF1 reconstituted pre-B-ALL cells, indicating a potential for developing the clinical application of fasting or FMDs in ALL [168].
Taken together, these observations demonstrate that although dysregulation of eating–fasting cycles have been identified as a key determinant for cancer development, fasting or FMDs can not only increase the sensitivity of cancer therapies but also reduce the side effects from chemotherapy, indicating a promising strategy for the prevention and treatment of cancer.
The interplay between activity–rest cycles and cancer
Based on epidemiological analyses, physical activity is associated with decreased risk of multiple cancers, including colorectal, breast, and prostate cancer [169,170,171,172,173]. Preclinical studies have also indicated that exercise has potential for cancer prevention (Fig. 4). Exercise training, such as treadmill running, voluntary wheel running, and swimming has been found to inhibit tumor initiation and progression in chemical, genetic or transplantable-triggered tumor mouse models [174,175,176,177]. However, exercise training has been shown to display different effects against a variety of cancers with different genetic backgrounds. For example, in the p53-deficient MMTV-Wnt mouse model bearing breast cancer, exercise training seemed to be ineffective against this cancer [178]. Nevertheless, strengthening physical exercise is still suggested for combating cancer regardless of cancer diagnosis. Notably, physical activity-driven antitumor effects can potentially be attributed to several mechanisms, which have different effects on different kinds of cancers.

The interplay between activity–rest cycles and cancer. a activity–rest cycles regulate immune function for tumor suppression. Exercise has been found to promote immune cell infiltration by epinephrine-driven NK cells circulation, resulting in a marked inhibition of cancer. Moreover, exercise also enhances naïve T cell populations and mitigates detrimental effects on T cells, leading to more efficient generation of immunological memory. b activity–rest cycles regulate the crosstalk between muscle and tumor. The crosstalk between muscle and tumor depends on skeletal muscle-secreted myokines. Exercise-induced myokines, including OSM, irisin, and SPARC, may play a role in cancer prevention and therapy. In addition, exercise-induced myokines can also induce the secretion of immune regulatory cytokines, including IL-6, IL-7, and IL-15, which indirectly regulate immune cell function. c, activity–rest cycles regulate tumor microenvironment. Exercise-mediated promotion of pro-angiogenic cytokines (such as VEGF) induces vascular remodeling to increase density and perfusion and reduce hypoxia in the tumor microenvironment. Furthermore, exercise was involved in the regulation of the Hippo signaling pathway, in which exercise-driven epinephrine markedly inhibited tumor formation by inducing Yap and Taz phosphorylation and subsequent degradation. NK cells: natural killer cells; OSM: oncostatin M; SPARC: secreted protein acidic and rich in cysteine; VEGF: vascular endothelial growth factor
Activity–rest cycles modulate immune functions
Exercise can modulate the circulation of immune cells by a mechanism mediated by adrenergic signaling and blood flow-induced shear stress, facilitating immune surveillance by eradicating identified malignant cells [179]. Indeed, an increasing number of studies have found exercise-induced immune cell mobilization as a common phenomenon independent of age, gender, or tumor type [180, 181]. A study focusing on patients with breast cancer convincingly demonstrated that NK cell mobilization in breast cancer survivors during exercise was comparable to that in age-matched healthy people [182]. However, a recent randomized controlled trial suggested that exercise displayed no significant impact on resting NK cell function and circulating myokines in women with high-risk breast cancer [183], indicating a potentially ambiguous role of exercise in regulating NK cell function for cancer immunotherapy.
In terms of preclinical studies, transplantable or genetic animal cancer models are usually used to investigate the association between exercise and cancer [184, 185]. For example, exercise was found to activate the immune system to suppress tumor progression in a genetic mouse model of mammary cancer following exercise training [186]. In addition, exercise-mediated immune system activation, such as NK cells and cytotoxic T cells infiltration, was also revealed in genetic tumor models, suggesting that additional immunological response was induced in the exercise-induced effect besides the initial immune response to the inoculated tumor cells [180, 187]. In a wheel-running mouse model, exercise was revealed to promote immune cell infiltration by epinephrine-driven NK cells circulation, resulting in a marked inhibition of cancer growth [185]. Exercise also enhances naïve T cell populations and mitigates detrimental effects on T cells, leading to more efficient generation of immunological memory [188, 189].
Increased body temperature is a major outcome of exercise, promoting the function and stimulus of immune cells [190]. A well-known observation is that hyperthermia can expand the diameter of intratumor blood vessels, therefore enhancing NK cells infiltration into the tumor and inhibiting cancer cell growth [191]. Furthermore, hyperthermia accelerates cytotoxic T cell recruitment into the tumor microenvironment by IL-6 trans-signaling-mediated tumor vasculature remodeling [184]. In fact, hyperthermia has been applied to clinical treatment for certain cancers, although the therapy temperature is generally higher than if exercise training-induced [192,193,194].
In summary, exercise could therefore be considered for cancer prevention or therapy due to its potential to promote an anticancer immunological response. However, exercise-mediated modulation of the immune system is complex, and the detailed mechanisms need to be further investigated [195,196,197].
Activity–rest cycles regulate the crosstalk between muscle and tumor
The crosstalk between muscle and tumor depends on skeletal muscle-secreted myokines produced during muscle contractions [198,199,200,201]. Recently, with the emergence of omics-based strategies, new exercise-induced myokines are gradually being identified in the muscle secretome [202,203,204,205,206]. Although the evidence, that exercise-driven myokines may play a role in cancer prevention or therapy, is still limited, several preclinical studies have indicated muscle-derived Oncostatin M (OSM) and Irisin may be effective against prostate and breast cancer [183, 207,208,209,210]. In addition, the myokine secreted protein acidic and rich in cysteine (SPARC) produced by exercise training has been identified as an anticancer factor, as evidenced by decreased tumorigenesis in trained mice bearing colon cancer [211,212,213]. Notably, myokines are divided into distinct classes, with the potential to modulate the proliferation and differentiation of cancer cells by either directly inducing cell growth or antagonizing certain ligands [208, 214,215,216]. Some myokines (like irisin) have been demonstrated to be modulators involved in the common cancer-related pathways, such as TGF-β or Wnt signaling [217,218,219].
Exercise-induced myokines can also induce the secretion of immune regulatory cytokines, including IL-6, IL-7, and IL-15, which indirectly regulate immune cell function [220,221,222,223]. For humans, IL-6 levels are controlled by exercise in a time and intensity-dependent way and are also related to the amount of muscle mass engaged in the exercise [224,225,226]. However, the importance of IL-6 produced during exercise in immune responses remains less well understood. A recent study indicated that exercise-mediated IL-6 seemed to improve immune cell infiltration. Anti-IL-6 antibody-mediated blockage of IL-6 signaling partially reversed the efficacy of exercise on inhibition of cancer cell proliferation [185]. Notably, direct IL-6 administration could not mimic exercise-related anticancer effects, implying that the role of IL-6 required a prior exercise-induced activation and recruitment of immune cells.
Activity–rest cycles regulate tumor microenvironment
Vascular remodeling attributed to abundant pro-angiogenic cytokines, such as VEGF (vascular endothelial growth factor) in the tumor microenvironment, is possibly the most well-known benefit of exercise on oncology indicated from pre-clinical investigations [227,228,229,230]. Numerous studies have revealed that vessel density and perfusion in tumors are involved in exercise-mediated VEGF production [231,232,233]. Furthermore, during exercise, endothelial cell recruitment is mediated by platelet derived growth factor receptor-beta (PDGFRβ) derived from platelets for tumor angiogenesis, dramatically reducing tumor hypoxia by upregulation of micro-vessel density and perfusion [232, 234, 235]. Hypoxia usually stimulates several stress response pathways to induce changes in the tumor microenvironment, especially immune microenvironment remodeling, therefore promoting tumor development [236,237,238]. For example, cytokines IL-4 and IL-10 triggered by hypoxia can induce differentiation of tumor-associated macrophages (TAMs) into an immunosuppressive M2 phenotype [239, 240]. In addition, the hypoxic environment stimulates dendritic cells to induce the expression of indoleamine 2,3-dioxygenase (IDO), which performs an immunomodulatory role in T cell suppression [241,242,243]. A previous study also found reduction of CD8+ tumor-infiltrating lymphocytes (TILs) presenting in tumor hypoxic regions [244, 245]. A hypoxic microenvironment has also been indicated to contribute to both dendritic cells-mediated inactivation of TILs and abnormal expression of PD-L1, resulting in immunotherapy resistance [246,247,248,249]. Interestingly, hyperoxia (60% oxygen) produced more than three-fold tumor infiltration CD8+ TILs compared to the control mice [244]. This finding provides the potential clinical implications of oxygen content regulation as every 10% increase in CD8 + TILs results in a 19% decrease in patient mortality [250]. However, sequestering chronic tumor patients in a 60% oxygen environment is not practical. Exercise, representing an available alternative, can reduce hypoxia in tumors, thus sensitizing cancer cells to immunotherapy by further increasing immune cell infiltration and reducing IDO-mediated immunosuppression.
Hippo signaling is one of the most important pathways for tumor initiation and development [251, 252]. This pathway involves the growth and differentiation of tumor cells, and regulation of several signaling pathways related to the formation of the tumor microenvironment (including extracellular matrix remodeling) [253, 254]. Recently, a study has reported that exercise was involved in the regulation of the Hippo signaling pathway [255]. Mechanically, exercise-driven epinephrine markedly inhibited tumor formation by inducing Yap and Taz phosphorylation and subsequent degradation [255]. In line with this, serum samples from breast cancer patients who were scheduled to participate in exercise training were able to show a 50% decrease in breast cancer metastasis in an experimental tumor model, attributed to exercise-induced catecholamine release [255].
Additional mechanisms are currently being investigated in exercise-mediated reduction of cancer progression. Secretion of several systemic factors (such as catecholamines, myokines), upregulation of blood flow-related shear stress, sympathetic activation, as well as increased body temperature, display immediate stress on tumor metabolism and homeostasis during exercise training [256]. If people are undertaking long-term training, the effects mentioned above will result in steady intratumor changes, such as strong immunogenicity, metabolism adaptation, and improved blood perfusion, which should help mitigate tumor development [257, 258].
Potential application of clock-associated therapy in cancer management
The molecular understanding of circadian rhythms has raised new therapeutic frontiers for cancer which could put the circadian clock in an indispensable treatment role. Therefore, pharmacological modulation of the circadian clock and/or treating cancer in the clock may hold potential as new therapeutic options for better cancer management (Fig. 5).

Clock-based therapy in cancer management. a directly pharmacological targeting circadian clock for cancer therapy. Targeting the components of circadian clock has attracted much attention as a therapeutic approach to treat cancer. There are several pharmacologic agents targeting the components of circadian clock, including REV-ERBα/β, RORα/β/γ, CRY1/2, Casein Kinase family, and FBXL3. b, the effect of modulating circadian rhythms on conventional cancer therapy. Modulating the sleep–wake, eating–fasting, and activity–rest cycles can benefit the effect of chemotherapy, radiotherapy, and immunotherapy
Pharmacological targeting core components of the circadian clock for cancer therapy
Targeting the components of circadian clock has attracted much attention as a novel therapeutic approach to treat chronic diseases, such as chronic inflammatory diseases, metabolic syndrome, and cancer [259, 260]. Theoretically, there exist two drug approaches for targeting the circadian clock: either by directly modulating the core circadian genes, or targeting their regulators. However, as BMAL1 and CLOCK are transcriptional factors, it is notoriously challenging to directly target these circadian core genes [261, 262]. Hence, pharmacologic agents targeting proteins responsible for phosphorylation or degradation of clock components which negatively regulate BMAL1 and CLOCK have been developed as agonists or antagonists to disrupt the circadian network.
REV-ERBs
REV-ERBα/β are nuclear hormone receptors that can directly bind to the promoter of BMAL1 and CLOCK and thus negatively regulate their transcription [263,264,265]. The aberrant expression of REV-ERBs has been found in many cancer types in which they mainly regulate plasma glucose level, lipid, and energetic metabolism [266]. GSK4112, a small molecule molecular probe also known as SR6452, was the first synthetic agonist of REV-ERB obtained from a fluorescence resonance energy transfer biochemical screen, with an EC50 of 2.3 μM for inhibiting the transcriptional activity of Bmal1 [267, 268]. However, the unsatisfactory pharmacokinetic profile and specificity of GSK4112 limited its use as a chemical tool in vivo, which drove the development of pyrrole derivatives SR9009 and SR9011 [269, 270]. These two different agonists of REV-ERBs have been reported to display anticancer activity against different tumor types, including leukemia, brain, colon, breast, and melanoma, while exhibiting no obvious side effect on normal cells or tissues [271,272,273,274]. Further studies found that autophagy and de novo lipogenesis were identified as the key events in evoking the apoptotic response in malignant cells treated with SR9009 and SR9011 [274]. In addition, SR9011 and SR9009 also reduced glioblastoma stem cell (GSC) proliferation and were lethal to chemoresistant small-cell lung cancer (SCLC) cells by repressing the expression of the tricarboxylic acid (TCA) cycle enzymes [275] and suppression of autophagy [273], respectively. Other chemical agonists for REV-ERBα, including GSK2945, GSK0999, GSK5072, and GSK2667 have also been developed but their effects on cancer need further investigation [276].
RORs
Unlike REV-ERBs, RORs (including RORα, RORβ, and RORγ) can constitutively induce the transcription of Bmal1 through the ligand-independent recruitment of transcriptional co-activators [266, 277]. There is evidence showing that RORγ was upregulated in metastatic castrate-resistant prostate cancer and promoted the expression of the androgen receptor, thus raising the possibility of targeting RORγ for cancer treatment [278,279,280,281,282]. Notably, the ROR-γ-selective antagonists XY018 and SR2211 block the growth of prostate cancer cells with androgen receptor expression and restore sensitivity to enzalutamide treatment (a commonly used androgen inhibitor for prostate cancer), in which induction of apoptosis and decreased expression of key proliferation and survival proteins were identified as key events [278]. Moreover, RORγ was found to show important regulatory functions in pancreatic cancer stem cells, and pharmacological targeting of RORγ by SR2211 inhibits the tumor growth and prolongs survival in several in vivo pancreatic cancer models [283, 284]. In contrast, RORγ agonists can modulate multiple signaling pathways to enhance the antitumor immunity against leukemia, colon and breast cancer [285,286,287,288]. For example, LYC-54143 and LYC-53772 are two potent RORγ agonists which display cytotoxic activity by promoting cytokines/chemokines (i.e., IL-17A and GM-CSF) production and increasing co-stimulatory receptor expression (like CD226 and CD137) [288]. Based on this finding, another RORγ agonist, LYC-55716 (cintirorgon), is currently under Phase I clinical trial for treating patients with metastatic cancer alone [289] or in combination with pembrolizumab (clinical trial NCT03396497). Unlike RORγ, RORα was found to be downregulated in several cancer types, including breast, ovarian, and prostate cancer [290,291,292,293]. Treatment of RORα agonist SR1078 results in p53 stabilization and triggers apoptosis in human HepG2 cancer cells [294, 295]. Another study found that pevonedistat (MLN4924), a small molecule AMP mimetic, could stabilize RORα by inhibiting its ubiquitination and consequent degradation, which in turn attenuated cell proliferation of osteosarcoma, chondrosarcoma, and leukemia by inducing cell cycle arrest and apoptosis [296,297,298,299]. MLN4924 has been evaluated in 40 clinical trials related to cancer [300].
CRY1/2
Distinct from REV-ERBs, CRY1 and CRY2 are important for inhibiting clock-controlled gene transcription by directly interacting with BMAL1/CLOCK complexes [301,302,303,304]. KL001 was the first compound found to directly bind to and stabilize CRY by preventing FBXL3-mediated ubiquitin-dependent degradation [305]. A recent study has demonstrated that KL001 treatment decreased OLIG2 and SOX2 expression and inhibited GSC proliferation with reduced toxicity in normal brain cells [275, 306]. Interestingly, in vitro observations demonstrate that pharmacological targeting CRY by KS15 reduces proliferation of human breast cancer MCF-7 cells and increases the sensitivity of MCF-7 cells to treatment with doxorubicin and tamoxifen, but has no obvious cytotoxic effects on normal breast epithelial cells (MCF-10A) [307]. These contradictory findings indicate that the function of CRY in cancer treatment is context-dependent and that further research is needed to investigate its precise role.
Casein kinase (CK) family
The casein kinase 1 enzymes (CK1) are a family of serine/threonine kinases in which CK1δ and CK1ε phosphorylate PERs and CRYs and prime them for FBXL21 and β-TRCP mediated degradation [308,309,310,311]. The upregulation of CK1δ and CK1ε was observed in several cancer types, including melanoma, leukemia, pancreas, breast, and ovarian cancer, which highlights their potential as drug targets for anticancer therapy [312,313,314,315]. Indeed, breast cancer cells overexpressing CK1δ are sensitive to CK1δ/CK1ε inhibition caused by SR-3029 both in vitro and in vivo, while normal breast epithelial cells (MCF-10A) with low amounts of CK1δ are less sensitive to SR-3029 treatment [314, 316]. Another inhibitor, BTX-A51, was found to induce apoptosis in AML progenitor cells in vitro and suppress growth in vivo in both genetical-engineered AML mouse models and in patient-derived xenograft mouse models [317]. BTX-A51 is currently in Phase I clinical trials for the treatment of patients with relapsed or refractory AML (clinical trial NCT04243785). Similarly, CK2 (casein kinase II) has also been found to regulate cell growth and proliferation of several cancers [318]. For example, CK2α was upregulated in CRC cells and conferred resistance to 5-FU treatment by inhibiting ER stress-induced apoptosis, and treatment with a CK2α inhibitor may exert a synergistic effect with 5-FU against drug-resistant cancer cells [319]. Targeting CK2 with a pan-CK2 inhibitor, BMS-699, resulted in disruption of myeloid cell differentiation and increased efficacy of immunotherapy in mice with lung, colon and breast carcinoma and lymphoma [320]. Moreover, some potent CK2 inhibitors, such as CX-4945 (silmitasertib) and GO289, have been developed which inhibit the proliferation of several human tumors [321,322,323]. CX-4945 was the first CK2 inhibitor that entered into clinical trials for the treatment of human tumors [324, 325].
To sum up, several agonists or antagonists targeting the core components of circadian rhythm have been developed and show promising anticancer effect in various cancer types. However, few compounds are entering clinic trials to enable the evaluation of the true efficacy in patients, and the dependence of these anticancer effects on clock modulation still needs further investigation (Table 1).
Administrating drugs in clock: chronotherapy in cancer treatment
Chronotherapy has been defined as a strategy that utilizes the natural rhythms and cycles of physiological and biochemical processes to treat a disorder [326,327,328]. This type of therapy for cancer patients, aiming to reduce side effects and improve efficacy during cancer treatment, has been applied in the clinic even before more detailed mechanisms of the core circadian clock were known [329,330,331,332]. Subsequently, once the biological functions of circadian rhythms were revealed showing that drug PK, PD, and safety are highly depend on 24-h rhythm, the rationale for chronotherapy was supported [333,334,335]. Indeed, a suitable dosing time during the cycle can not only contribute to beneficial effects but also avoid adverse ones, especially for anticancer agents whose use may be limited due to their side effects on healthy host tissues [336,337,338]. For example, in cancer patients treated with constant-rate i.v. infusion of 5-fluorouracil (5-FU) for 5 days, the maximum plasma concentration (Cmax), and the best-tolerated time were found to be at 4:00 a.m [339, 340]. Recent reports have indicated circadian characteristics of inhibitors targeting the estrogen receptor and tyrosine kinases in both treated mice and patients, as evidenced by observed daily pharmacokinetic variations [341,342,343,344]. In addition, several Phase I to Phase III clinical trials have confirmed the efficacy of chrono-modulated treatment in various tumors [345, 346].
Importantly, chronotherapy potentially improves the survival rate and life quality of cancer patients by minimizing anticancer agents-driven cytotoxicity [347,348,349]. Mechanically, numerous anticancer drugs have consistently been demonstrated to display increased cytotoxicity to cells at specific phases of cell division, suggesting that optimization of dosing time for treatment by predicting circadian rhythms-related medicinal properties may translate into desired clinical outcomes [333, 338]. In a randomized trial, cancer patients with a sinusoidal chronotherapy schedule showed better tolerability and efficacy to drugs than with a constant-rate infusion [350]. Another study on 186 metastatic colorectal cancer patients in a randomized multicenter phase III trial also reported that oxaliplatin, 5-FU, and chronoFLO delivered by chrono-modulated infusion reduced ~ fivefold the rate of severe mucosal toxicity and ~ 50% functional impairment from peripheral sensitive neuropathy compared with constant drug delivery (14% vs. 76% and 16% vs. 31%, respectively) [329]. Furthermore, timed administration of irinotecan was performed in 31 cancer patients and demonstrated that chrono-mediated infusion of irinotecan from 2:00 a.m. to 8:00 a.m. induced less severe diarrhea and interpatient variability compared with the conventional 30-min infusion in the morning [351]. A very recent clinical study evaluated the effect of immunotherapy time-of-day infusion on the overall survival of patients with advanced melanoma. These results suggested that adaptive immune responses are less robust when infused in the evening than in the daytime [352, 353].
The application of a chronotherapeutic strategy for cancer treatment has stimulated developments in bioengineering, such as non-implantable multichannel time-programmable pumps for chrono-modulated drug delivery [354,355,356]. For example, in North America and the European Union, the IntelliJect™ device with four 30-mL reservoirs has been approved for treating cancer patients resulting in increased safety and efficacy of anticancer agents, based on chrono-mediated combination drug delivery of 5-FU, oxaliplatin, and leucovorin [328, 336, 357, 358]. The liver is a well-known rhythmic organ. However, liver having tumor metastases shows no marked circadian regulation. When anticancer drugs were infused in a chrono-modulated way directly into the hepatic artery of patients with liver metastases beneficial effects were observed [359, 360]. Indeed, this strategy has been confirmed as a safe and effective therapy by an international trial [361]. This study demonstrated that an automatic multichannel programmable pump, Mélodie®, could be used for treating colorectal cancer liver metastases by directly delivering anticancer agents (5-FU, oxaliplatin, irinotecan, and systemic cetuximab) into the hepatic artery, contributing to improvements in both toxic tolerability and drug effects [359, 361].
The emerging field of nanocarrier-mediated drug delivery on chronotherapy is now attracting increasing attention as it has been shown to increase cancer curability without added side effects and risks for the patients, as well as having potential cost savings [362,363,364]. An increasing body of data is showing that chronotherapy combined with nanotechnology offers important advantages for effective drug delivery, contributing to more efficient and safer cancer therapy [356, 365,366,367]. Recently, the synergistic anticancer effects of chrono-modulated delivery of paclitaxel (PTX)-loaded polymeric nanoparticles (PTX-NPs) were investigated on a human lung cancer-derived mouse xenograft model to identify the best time for drug delivery [368,369,370]. PTX in nanocarriers was initially rapidly released but subsequently turned into sustained release, showing a better anticancer effect than PTX single treatment [368]. Furthermore, the efficacy of PTX-NPs displayed a time-dependent effect and peaked at 15 h after light onset. Mechanically, PTX-NPs combined chronotherapy was verified to be associated with decreased Ki-67-mediated proliferation and CD31-mediated micro-vessel density, thereby resulting in inhibition of lung cancer by inducing cell apoptosis [368]. In addition to the above findings, a growing number of novel chrono-pharmaceutical delivery technologies are underway development and study. These strategies, by utilizing bedside or ambulatory pumps, facilitate more proficient cancer treatment by precisely delivering antitumor drugs in a circadian time-dependent manner.
In summary, chronotherapy is showing benefits not only for improving therapeutic efficiency but also for minimalizing the side effects of cancer treatment. However, the real application of chronotherapy in cancer treatment is still in its infancy, and more clinical trails are required to expand the clinical applications of chronotherapy in cancer treatment.
Fasting in cancer prevention and treatment
The fasting-feeding pattern is an important time cue that can also determine the robustness of daily circadian rhythms [371,372,373,374]. Although maintaining a robust fasting-feeding pattern is correlated with prevention of tumorigenesis, disruption of this circadian rhythm, either by time/calorie-restricted feeding or intermittent/periodic fasting, may benefit the outcome of cancer treatment [375, 376]. For example, chronic calorie restriction (CR) and periodic fasting is attracting increasing attention for potential application to clinical cancer prevention and therapy based on long-lasting epidemiological studies and preclinical reports [159, 377,378,379,380,381]. A diet that mimics the effects of fasting (FMD) has been developed and used in a pilot clinical study, aiming to maintain the cancer-preventive effects from CR or fasting while reducing adverse effects [162, 382].
To date, four feasibility studies regarding cancer patients undergoing chemotherapy treated with fasting have been published [383,384,385,386]. In one of these studies, 10 patients with various cancers, such as lung, breast, esophageal, prostate, uterus, and ovarian cancer, volunteered to fast for 140 h before chemotherapy or for 56 h following chemotherapy. The results indicated that fasting induced no obvious adverse effects except for hunger and lightheadedness. Notably, 6 fasting patients showed a marked decrease in gastrointestinal, fatigue, and weakness events following chemotherapy. Furthermore, fasting did not compromise the anticancer effects from chemotherapy in those patients, as evidenced by no distinct difference of tumor volume or malignant markers compared to patients without fasting [383]. In line with this, 13 HER2-negative breast cancer patients were scheduled for treatment with fasting (only water supply) for 24 h or with standard nutrition regimens before and after the treatment with neo-adjuvant taxotere, adriamycin, and cyclophosphamide chemotherapy. Good tolerance was displayed to the short-term fasting, while a significant reduction of the erythrocyte and thrombocyte counts was shown at 7 days after chemotherapy [384]. Interestingly, non-fasted patients showed the upregulated expression of γ-H2AX (which indicates the levels of DNA damage) in leukocytes in 30 min after chemotherapy rather than the fasted patients [384]. Another interesting study evaluated the influence of fasting time with cancer patients. In this study, 20 patients primarily diagnosed with breast or ovarian cancer were randomized to treatment with 24- or 48-h fasting before platinum-derived chemotherapy or another 24-h fasting after platinum-derived chemotherapy (total 72-h fasting) [385]. During the fasting, three or more out of six patients in each cohort needed to consume less than 200 kcal per day. Fasting-related toxicities were only relatively minor, including dizziness, fatigue, and headache with some evidence of neutropenia in the 48- and 72-h cohorts [385]. Recently, a randomized clinical trial on 34 patients with ovarian or breast cancer was conducted to assess the effects of an FMD on life quality and side effects of chemotherapy [386]. In this study, a 36 ~ 48-h limited intake of 400 kcal per day was required before the treatment of chemotherapy. Subsequently, the same calorie limitation was performed for a further 24 h following chemotherapy [386]. From these data we can conclude that FMD does not lead to serious side effects per se, while improving chemotherapy-induced adverse reaction, like fatigue.
Therefore, fasting, either periodic or short-term, is attracting increasing attention for clinical cancer prevention and therapy and indeed displays visible effect on cancer treatment in some clinic trails, suggesting a potential adjuvant therapy for cancer in the future.
Exercise benefits cancer therapy
Timing exercise has long been utilized as a Zeitgeber for regulating circadian clock of some major body systems, such as skeletal muscle and the blood vessels [387,388,389,390,391]. Maintaining this clock might be an efficacious strategy for preventing and combating metabolic disease, including obesity, diabetes, cardiovascular disease, and cancer [392,393,394].
Accumulating literature has shown that exercise training can not only relieve treatment side effects but may also hold the potential to increase the potency and efficacy of cancer therapies [256, 395,396,397]. Emerging evidence from pre-clinical studies has indicated that exercise regulates intratumoral vascular perfusion, systemic inflammation, sex hormone levels, and immune cell function during cancer treatment, implying that exercise may be not only healthy but also therapeutic [398,399,400].
The first line of treatment for most solid tumors is surgery, and radical tumor resection is usually considered as the most efficient therapeutic strategy, especially if the tumor is detected before it has metastasized [401]. Exercise training has been proved to be beneficial for the management of operable tumors at several distinct time points along the surgical treatment period, including the preoperative period, short-term immediately after surgery, and longer-term follow up [402,403,404,405] and is becoming increasingly popular. For example, in patients with early-stage lung cancer, preoperative exercise was found to improve walking endurance, peak exercise capacity, dyspnea and postoperative pulmonary complications, which are associated with the time of intensive care admissions, hospital stays, and hospital readmissions, as well as mortality in these patients [406]. Indeed, data from systematic review and meta-analysis have demonstrated that lung cancer patients undergoing an exercise regime had a lower risk of postoperative pulmonary complication, fewer days of intercostal catheter use, and a shorter length of hospitalization when compared with non-exercise patients [407,408,409]. In addition, exercise training immediately after cancer surgery is also beneficial to the outcome of cancer patients. Studies have been conducted to evaluate the effectiveness of an early onset exercise program on shoulder mobility, physical performance, and postoperative complications of breast cancer patients and found that shoulder-arm mobility of surgical breast cancer patients demonstrated significant improvement compared with the control group [410, 411]. Moreover, longer-term exercise following surgery has been established as a feasible, safe, and effective method of decreasing postoperative side effects and improving life quality in lung cancer patients [412]. Nevertheless, the mechanisms behind the effects of exercise on surgical outcomes are still poorly understood, and further studies are needed to clarify the potential of exercise training for better cancer management in the perioperative period.
Radiotherapy is a cornerstone for cancer management, the effectiveness of which is dependent on sufficient delivery of oxygen to the tumor sites to generate enough ROS for eliminating cancer cells [413, 414]. Exercise training is a promising adjunct therapy for cancer patients receiving radiotherapy, mainly by affecting blood circulation and oxygen delivery to the tumor location [415]. Several randomized controlled trials have reported that exercise training could mitigate fatigue in cancer patients receiving radiotherapy and generate longer-term improvements and additional benefits for quality of life [416, 417], as well as reducing the severity of rectal toxicity during radiotherapy of prostate cancer [418]. Moreover, evidence from a preclinical mouse model of prostate cancer also demonstrated that exercise can improve radiotherapy efficiency by increasing natural killer cell infiltration and activity, resulting in increased tumor cell apoptosis [419]. However, very few clinical studies have been conducted to investigate the effect of either acute or prolonged exercise training on radiotherapy treatment response, thus delaying the application of exercise training in clinical cancer treatment.
Likewise, the efficacies of chemotherapy and immunotherapy are partially dependent on adequate intratumoral blood perfusion which can be enhanced by appropriate exercise training to assist in delivery of drugs or immune cells to the tumor sites [420]. Several preclinical studies using mouse models have found that soft aerobic exercise promotes tumor vascular function to enhance drug delivery to breast, prostate, and pancreatic tumors, thus increasing chemotherapy efficacy [421,422,423,424,425]. Moreover, exercise has been demonstrated to remodel the tumor vasculature in a PDX mouse model, which improves tumor regression and inhibits the recurrence of pancreatic ductal adenocarcinoma (PDAC) under gemcitabine treatment [426]. A previous study found that NK cell infiltration was significantly increased in tumors in wheel-running treated mice. Mechanistic analyses demonstrated that the growth suppression of tumors was attributed to an epinephrine-IL6-dependent mobilization and activation of NK cells [185]. Another recent study found that the combination of anti-PD-1 and exercise increased the percentage of CD8+ T cells and enhanced antitumor immune responses in mice with EO771 breast tumors [249]. However, much remains to be understood about the efficiency of exercise on immunotherapy in clinical cancer management.
Overall, the above findings indicate the potential therapeutic role of exercise training in cancer treatment. Nevertheless, current insights into the mechanistic effects of exercise on cancer therapy mainly stem from pre-clinical studies. Additional studies, especially a further understanding of the mechanisms involved and how they are controlled together with targeted clinical trials, are essential for a deeper understanding of the synergistic effects of exercise and conventional anticancer therapies.
Conclusions and perspectives
As discussed above, circadian control of biological processes is involved in almost all aspects of daily life, and tightly regulates multiple human behaviors, including sleep–wake, eating–fasting and activity–rest cycles. Chronic disruption of these biorhythms induced by external environmental or physiological changes may contribute to a wide range of human diseases, including sleep disorders, psychiatric and neurodegenerative diseases, cardiovascular disease, and especially cancers [11, 326, 427,428,429,430]. Indeed, several epidemiologic and animal studies have demonstrated the important role of the circadian clock in tumorigenesis and progression [427]. Although gene expression, cell division, and DNA repair are modulated by the clock, the concept that clock genes are general tumor suppressors remains unproven [326]. Moreover, tumor development will clearly cause clock disruptions, such as sleep deprivation and insufficient food intake, affecting bodily functions. This Janus effect is therefore attracting considerable interest worldwide for developing novel therapeutic strategies by modulating circadian rhythms. Table 2 summarizes the clinical studies related to clock-associated cancer therapy.
There are, however, some concerns that need to be addressed before the successful application of combining conventional treatment with targeting of the circadian clock for clinical cancer management. Firstly, targeting one of the clock components may prove no more effective than combinational therapeutic strategies using two or more targeted drugs. Secondly, the circadian clock can coordinate disparate cellular pathways. This may result in unwanted side effects or even opposite outcomes during treatment. Therefore, determining the appropriate dosage to successfully target circadian rhythm to obtain transient repression rather than chronic treatment may be required to avoid detrimental effects. Most importantly, the preclinical efficacy of manipulating circadian rhythms needs to be extended to larger study cohorts [327, 431], as results from a previous study with a small sample size claiming a substantial effect of chronotherapy on ovarian cancer were not reproduced in subsequent larger studies [432,433,434].
In summary, circadian rhythms participate in diverse biological pathways. Their manipulation holds the promise for beneficial treatment of not only cancer, but also other serious diseases. Based on significant advances in this area, which was honored by the 2017 Nobel Prize for Physiology or Medicine awarded jointly to Jeffrey C. Hall, Michael Rosbash and Michael W. Young, we believe that it will have excellent clinical potential in the near future.
Availability of data and materials
Not applicable.
Abbreviations
- AML:
-
Acute myeloid leukemia
- BMAL1:
-
Brain and muscle aryl hydrocarbon receptor nuclear translocator 1
- CKIε:
-
Casein kinase Iε
- CLOCK:
-
Circadian locomotor output cycles kaput
- CR:
-
Calorie restriction
- CRC:
-
Colorectal cancer
- CRP:
-
C-reactive protein
- DEN:
-
Diethylnitrosamine
- DSR:
-
Differential stress resistance
- DSS:
-
Differential stress sensitization
- ENT1:
-
Equilibrative nucleoside transporter 1
- FMDs:
-
Fasting-mimicking diets
- GH:
-
Growth hormone
- GSC:
-
Glioblastoma stem cell
- HDACs:
-
Histone deacetylases
- IARC:
-
International Agency for Research on Cancer
- IDO:
-
Indoleamine 2,3-dioxygenase
- IFN:
-
Interferon
- LAK:
-
Lymphokine-activated killer
- LID:
-
Limited ingredient diet
- NK:
-
Natural killer
- NO:
-
Nitric oxide
- NREM:
-
Non-rapid eye movement
- OSM:
-
Oncostatin M
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PDGFRβ:
-
Platelet derived growth factor receptor-beta
- PRDM1:
-
Protein PR/SET domain 1
- PRL:
-
Prolactin
- REM:
-
Rapid eyes movement
- RORs:
-
Subfamilies of nuclear hormone receptors
- ROS:
-
Reactive oxygen species
- SCLC:
-
Small-cell lung cancer
- SCN:
-
Suprachiasmatic nucleus
- sICAM:
-
Soluble intercellular adhesions molecule
- SPARC:
-
Secreted protein acidic and rich in cysteine
- STF:
-
Short-term fasting
- SWS:
-
Slow-wave sleep
- TAMs:
-
Tumor-associated macrophages
- TCA:
-
Tricarboxylic acid
- TILs:
-
Tumor-infiltrating lymphocytes
- TKIs:
-
Tyrosine kinase inhibitors
- TLR4:
-
Toll-like receptor 4
- VEGF:
-
Vascular endothelial growth factor
- WHO:
-
World Health Organization
References
Patke A, Young MW, Axelrod S. Molecular mechanisms and physiological importance of circadian rhythms. Nat Rev Mol Cell Biol. 2020;21:67–84.
Roenneberg T, Merrow M. Circadian clocks—the fall and rise of physiology. Nat Rev Mol Cell Biol. 2005;6:965–71.
Winfree AT. Acute temperature sensitivity of the circadian rhythm in Drosophila. J Insect Physiol. 1972;18:181–5.
Zimmerman WF, Pittendrigh CS, Pavlidis T. Temperature compensation of the circadian oscillation in Drosophila pseudoobscura and its entrainment by temperature cycles. J Insect Physiol. 1968;14:669–84.
Dunlap JC. Genetic and molecular analysis of circadian rhythms. Annu Rev Genet. 1996;30:579–601.
Dunlap JC, Loros JJ, Liu Y, Crosthwaite SK. Eukaryotic circadian systems: cycles in common. Genes Cells. 1999;4:01–10.
Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, Takahashi JS, Weitz CJ. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280:1564–9.
Honma S, Ikeda M, Abe H, Tanahashi Y, Namihira M, Honma K-I, Nomura M. Circadian oscillation ofBMAL1, a partner of a mammalian clock GeneClock, in rat suprachiasmatic nucleus. Biochem Biophys Res Commun. 1998;250:83–7.
Liu C, Weaver DR, Strogatz SH, Reppert SM. Cellular construction of a circadian clock: period determination in the suprachiasmatic nuclei. Cell. 1997;91:855–60.
Reinke H, Asher G. Crosstalk between metabolism and circadian clocks. Nat Rev Mol Cell Biol. 2019;20:227–41.
Musiek ES, Holtzman DM. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science. 2016;354:1004–8.
Richards J, Gumz ML. Advances in understanding the peripheral circadian clocks. FASEB J. 2012;26:3602–13.
Durgan DJ, Young ME. The cardiomyocyte circadian clock: emerging roles in health and disease. Circ Res. 2010;106:647–58.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70.
Straif K, Baan R, Grosse Y, Secretan B, El Ghissassi F, Bouvard V, Altieri A, Benbrahim-Tallaa L, Cogliano V, Group WIAfRoCMW. Carcinogenicity of shift-work, painting, and fire-fighting. Elsevier; 2007.
Stevens RG, Hansen J, Costa G, Haus E, Kauppinen T, Aronson KJ, Castaño-Vinyals G, Davis S, Frings-Dresen MH, Fritschi L. Considerations of circadian impact for defining ‘shift work’in cancer studies: IARC working group report. Occup Environ Med. 2011;68:154–62.
Lin H-H, Farkas ME. Altered circadian rhythms and breast cancer: from the human to the molecular level. Front Endocrinol (Lausanne). 2018;9:219.
Wendeu-Foyet MG, Menegaux F. Circadian disruption and prostate cancer risk: an updated review of epidemiological evidences. Cancer Epidemiol Biomark Prev. 2017;26:985–91.
Innominato PF, Focan C, Gorlia T, Moreau T, Garufi C, Waterhouse J, Giacchetti S, Coudert B, Iacobelli S, Genet D. Circadian rhythm in rest and activity: a biological correlate of quality of life and a predictor of survival in patients with metastatic colorectal cancer. Cancer Res. 2009;69:4700–7.
Bishehsari F, Levi F, Turek FW, Keshavarzian A. Circadian rhythms in GI health and diseases. Gastroenterology. 2016;151:e1.
Tahara Y, Shibata S. Circadian rhythms of liver physiology and disease: experimental and clinical evidence. Nat Rev Gastroenterol Hepatol. 2016;13:217.
Papagiannakopoulos T, Bauer MR, Davidson SM, Heimann M, Subbaraj L, Bhutkar A, Bartlebaugh J, Vander Heiden MG, Jacks T. Circadian rhythm disruption promotes lung tumorigenesis. Cell Metab. 2016;24:324–31.
Voigt RM, Forsyth CB, Green SJ, Engen PA, Keshavarzian A. Circadian rhythm and the gut microbiome. Int Rev Neurobiol. 2016;131:193–205.
Gery S, Koeffler HP. Circadian rhythms and cancer. Cell Cycle. 2010;9:1097–103.
Lévi F. Circadian chronotherapy for human cancers. Lancet Oncol. 2001;2:307–15.
Kelleher FC, Rao A, Maguire A. Circadian molecular clocks and cancer. Cancer Lett. 2014;342:9–18.
Schibler U, Sassone-Corsi P. A web of circadian pacemakers. Cell. 2002;111:919–22.
Ozturk N, Ozturk D, Kavakli IH, Okyar A. Molecular aspects of circadian pharmacology and relevance for cancer chronotherapy. Int J Mol Sci. 2017;18:2168.
Rosenwasser AM, Turek FW. Neurobiology of circadian rhythm regulation. Sleep Med Clin. 2015;10:403–12.
Hastings MH, Maywood ES, Brancaccio M. Generation of circadian rhythms in the suprachiasmatic nucleus. Nat Rev Neurosci. 2018;19:453–69.
Shearman LP, Sriram S, Weaver DR, Maywood ES, Chaves I, Zheng B, Kume K, Lee CC, van der Horst GT, Hastings MH, et al. Interacting molecular loops in the mammalian circadian clock. Science. 2000;288:1013–9.
Haque SN, Booreddy SR, Welsh DK. Effects of BMAL1 manipulation on the brain’s master circadian clock and behavior. Yale J Biol Med. 2019;92:251–8.
Kondratov RV, Shamanna RK, Kondratova AA, Gorbacheva VY, Antoch MP. Dual role of the CLOCK/BMAL1 circadian complex in transcriptional regulation. FASEB J. 2006;20:530–2.
Vielhaber EL, Duricka D, Ullman KS, Virshup DM. Nuclear export of mammalian PERIOD proteins. J Biol Chem. 2001;276:45921–7.
Akashi M, Okamoto A, Tsuchiya Y, Todo T, Nishida E, Node K. A positive role for PERIOD in mammalian circadian gene expression. Cell Rep. 2014;7:1056–64.
Schmalen I, Reischl S, Wallach T, Klemz R, Grudziecki A, Prabu JR, Benda C, Kramer A, Wolf E. Interaction of circadian clock proteins CRY1 and PER2 is modulated by zinc binding and disulfide bond formation. Cell. 2014;157:1203–15.
Lee H, Lee JW. The roles of CKI in circadian rhythm. Future Med Chem. 2019;11:2621–4.
Ukai-Tadenuma M, Kasukawa T, Ueda HR. Proof-by-synthesis of the transcriptional logic of mammalian circadian clocks. Nat Cell Biol. 2008;10:1154–63.
Duez H, Staels B. Rev-erb-alpha: an integrator of circadian rhythms and metabolism. J Appl Physiol. 1985;2009(107):1972–80.
Ikeda R, Tsuchiya Y, Koike N, Umemura Y, Inokawa H, Ono R, Inoue M, Sasawaki Y, Grieten T, Okubo N, et al. REV-ERBalpha and REV-ERBbeta function as key factors regulating Mammalian Circadian Output. Sci Rep. 2019;9:10171.
Jetten AM. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl Recept Signal. 2009;7:e003.
Reszka E, Przybek M, Muurlink O, Peplonska B. Circadian gene variants and breast cancer. Cancer Lett. 2017;390:137–45.
Morgan MN, Dvuchbabny S, Martinez CA, Kerr B, Cistulli PA, Cook KM. The cancer clock is (not) ticking: links between circadian rhythms and cancer. Clocks Sleep. 2019;1:435–58.
Chu LW, Till C, Yang B, Tangen CM, Goodman PJ, Yu K, Zhu Y, Han S, Hoque AM, Ambrosone C, et al. Circadian genes and risk of prostate cancer in the prostate cancer prevention trial. Mol Carcinog. 2018;57:462–6.
Arafa K, Emara M. Insights about circadian clock and molecular pathogenesis in gliomas. Front Oncol. 2020;10:199.
Sulli G, Lam MTY, Panda S. Interplay between circadian clock and cancer: new frontiers for cancer treatment. Trends Cancer. 2019;5:475–94.
Straif K, Baan R, Grosse Y, Secretan B, El Ghissassi F, Bouvard V, Altieri A, Benbrahim-Tallaa L, Cogliano V. Carcinogenicity of shift-work, painting, and fire-fighting. Lancet Oncol. 2007;8:1065–6.
Salamanca-Fernández E, Rodríguez-Barranco M, Guevara M, Ardanaz E, Olry de Labry Lima A, Sánchez MJ. Night-shift work and breast and prostate cancer risk: updating the evidence from epidemiological studies. An Sist Sanit Navar. 2018;41:211–26.
Gehlert S, Clanton M, On Behalf Of The Shift W, Breast Cancer Strategic Advisory G. Shift work and breast cancer. Int J Environ Res Public Health. 2020;17:9544.
Hansen J. Night shift work and risk of breast cancer. Curr Environ Health Rep. 2017;4:325–39.
Travis RC, Balkwill A, Fensom GK, Appleby PN, Reeves GK, Wang XS, Roddam AW, Gathani T, Peto R, Green J, et al. Night shift work and breast cancer incidence: three prospective studies and meta-analysis of published studies. J Natl Cancer Inst. 2016;108:djw169.
Kogevinas M, Espinosa A, Castelló A, Gómez-Acebo I, Guevara M, Martin V, Amiano P, Alguacil J, Peiro R, Moreno V, et al. Effect of mistimed eating patterns on breast and prostate cancer risk (MCC-Spain Study). Int J Cancer. 2018;143:2380–9.
Marinac CR, Natarajan L, Sears DD, Gallo LC, Hartman SJ, Arredondo E, Patterson RE. Prolonged nightly fasting and breast cancer risk: findings from NHANES (2009–2010). Cancer Epidemiol Biomark Prev. 2015;24:783–9.
Mormont MC, Waterhouse J, Bleuzen P, Giacchetti S, Jami A, Bogdan A, Lellouch J, Misset JL, Touitou Y, Lévi F. Marked 24-h rest/activity rhythms are associated with better quality of life, better response, and longer survival in patients with metastatic colorectal cancer and good performance status. Clin Cancer Res. 2000;6:3038–45.
Innominato PF, Komarzynski S, Palesh OG, Dallmann R, Bjarnason GA, Giacchetti S, Ulusakarya A, Bouchahda M, Haydar M, Ballesta A, et al. Circadian rest-activity rhythm as an objective biomarker of patient-reported outcomes in patients with advanced cancer. Cancer Med. 2018;7:4396–405.
Wood PA, Yang X, Taber A, Oh EY, Ansell C, Ayers SE, Al-Assaad Z, Carnevale K, Berger FG, Peña MM, et al. Period 2 mutation accelerates ApcMin/+ tumorigenesis. Mol Cancer Res. 2008;6:1786–93.
Fu L, Pelicano H, Liu J, Huang P, Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell. 2002;111:41–50.
Mteyrek A, Filipski E, Guettier C, Okyar A, Lévi F. Clock gene Per2 as a controller of liver carcinogenesis. Oncotarget. 2016;7:85832–47.
Lee S, Donehower LA, Herron AJ, Moore DD, Fu L. Disrupting circadian homeostasis of sympathetic signaling promotes tumor development in mice. PLoS ONE. 2010;5:e10995.
Ueda E, Kurebayashi S, Sakaue M, Backlund M, Koller B, Jetten AM. High incidence of T-cell lymphomas in mice deficient in the retinoid-related orphan receptor RORgamma. Cancer Res. 2002;62:901–9.
Gu X, Xing L, Shi G, Liu Z, Wang X, Qu Z, Wu X, Dong Z, Gao X, Liu G, et al. The circadian mutation PER2(S662G) is linked to cell cycle progression and tumorigenesis. Cell Death Differ. 2012;19:397–405.
Antoch MP, Gorbacheva VY, Vykhovanets O, Toshkov IA, Kondratov RV, Kondratova AA, Lee C, Nikitin AY. Disruption of the circadian clock due to the Clock mutation has discrete effects on aging and carcinogenesis. Cell Cycle. 2008;7:1197–204.
Antoch MP, Toshkov I, Kuropatwinski KK, Jackson M. Deficiency in PER proteins has no effect on the rate of spontaneous and radiation-induced carcinogenesis. Cell Cycle. 2013;12:3673–80.
Janich P, Pascual G, Merlos-Suárez A, Batlle E, Ripperger J, Albrecht U, Cheng HY, Obrietan K, Di Croce L, Benitah SA. The circadian molecular clock creates epidermal stem cell heterogeneity. Nature. 2011;480:209–14.
Puram RV, Kowalczyk MS, de Boer CG, Schneider RK, Miller PG, McConkey M, Tothova Z, Tejero H, Heckl D, Järås M, et al. Core circadian clock genes regulate leukemia stem cells in AML. Cell. 2016;165:303–16.
Disruption of core circadian clock genes reduces leukemia growth. Cancer Discov. 2016;6:569.
Lee JH, Gaddameedhi S, Ozturk N, Ye R, Sancar A. DNA damage-specific control of cell death by cryptochrome in p53-mutant ras-transformed cells. Cancer Res. 2013;73:785–91.
Chan AB, Parico GCG, Fribourgh JL, Ibrahim LH, Bollong MJ, Partch CL, Lamia KA. CRY2 missense mutations suppress P53 and enhance cell growth. Proc Natl Acad Sci USA. 2021;118:4077.
Lange T, Dimitrov S, Born J. Effects of sleep and circadian rhythm on the human immune system. Ann N Y Acad Sci. 2010;1193:48–59.
Petrovsky N. Towards a unified model of neuroendocrine-immune interaction. Immunol Cell Biol. 2001;79:350–7.
Haus E, Smolensky MH. Biologic rhythms in the immune system. Chronobiol Int. 1999;16:581–622.
Liu X, Chen B, Huang Z, Duan R, Li H, Xie L, Wang R, Li Z, Gao Y, Zheng Y, et al. Effects of poor sleep on the immune cell landscape as assessed by single-cell analysis. Commun Biol. 2021;4:1325.
Brooks JF 2nd, Behrendt CL, Ruhn KA, Lee S, Raj P, Takahashi JS, Hooper LV. The microbiota coordinates diurnal rhythms in innate immunity with the circadian clock. Cell. 2021;184:4154-67.e12.
Dimitrov S, Lange T, Fehm HL, Born J. A regulatory role of prolactin, growth hormone, and corticosteroids for human T-cell production of cytokines. Brain Behav Immun. 2004;18:368–74.
Dimitrov S, Lange T, Tieken S, Fehm HL, Born J. Sleep associated regulation of T helper 1/T helper 2 cytokine balance in humans. Brain Behav Immun. 2004;18:341–8.
Lange T, Dimitrov S, Fehm HL, Westermann J, Born J. Shift of monocyte function toward cellular immunity during sleep. Arch Intern Med. 2006;166:1695–700.
Irwin M, Mascovich A, Gillin JC, Willoughby R, Pike J, Smith TL. Partial sleep deprivation reduces natural killer cell activity in humans. Psychosom Med. 1994;56:493–8.
Irwin M, McClintick J, Costlow C, Fortner M, White J, Gillin JC. Partial night sleep deprivation reduces natural killer and cellular immune responses in humans. FASEB J. 1996;10:643–53.
Schernhammer ES, Laden F, Speizer FE, Willett WC, Hunter DJ, Kawachi I, Fuchs CS, Colditz GA. Night-shift work and risk of colorectal cancer in the nurses’ health study. J Natl Cancer Inst. 2003;95:825–8.
Schernhammer ES, Razavi P, Li TY, Qureshi AA, Han J. Rotating night shifts and risk of skin cancer in the nurses’ health study. J Natl Cancer Inst. 2011;103:602–6.
Papantoniou K, Devore EE, Massa J, Strohmaier S, Vetter C, Yang L, Shi Y, Giovannucci E, Speizer F, Schernhammer ES. Rotating night shift work and colorectal cancer risk in the nurses’ health studies. Int J Cancer. 2018;143:2709–17.
Irwin MR, Wang M, Campomayor CO, Collado-Hidalgo A, Cole S. Sleep deprivation and activation of morning levels of cellular and genomic markers of inflammation. Arch Intern Med. 2006;166:1756–62.
Irwin MR, Wang M, Ribeiro D, Cho HJ, Olmstead R, Breen EC, Martinez-Maza O, Cole S. Sleep loss activates cellular inflammatory signaling. Biol Psychiatry. 2008;64:538–40.
Dimitrov S, Lange T, Benedict C, Nowell MA, Jones SA, Scheller J, Rose-John S, Born J. Sleep enhances IL-6 trans-signaling in humans. FASEB J. 2006;20:2174–6.
van Leeuwen WM, Lehto M, Karisola P, Lindholm H, Luukkonen R, Sallinen M, Härmä M, Porkka-Heiskanen T, Alenius H. Sleep restriction increases the risk of developing cardiovascular diseases by augmenting proinflammatory responses through IL-17 and CRP. PLoS ONE. 2009;4:e4589.
Puttonen S, Kivimäki M, Elovainio M, Pulkki-Råback L, Hintsanen M, Vahtera J, Telama R, Juonala M, Viikari JS, Raitakari OT, et al. Shift work in young adults and carotid artery intima-media thickness: The Cardiovascular Risk in Young Finns study. Atherosclerosis. 2009;205:608–13.
Irwin MR. Sleep and inflammation: partners in sickness and in health. Nat Rev Immunol. 2019;19:702–15.
Vgontzas AN, Papanicolaou DA, Bixler EO, Lotsikas A, Zachman K, Kales A, Prolo P, Wong ML, Licinio J, Gold PW, et al. Circadian interleukin-6 secretion and quantity and depth of sleep. J Clin Endocrinol Metab. 1999;84:2603–7.
Dimitrov S, Besedovsky L, Born J, Lange T. Differential acute effects of sleep on spontaneous and stimulated production of tumor necrosis factor in men. Brain Behav Immun. 2015;47:201–10.
Redwine L, Dang J, Hall M, Irwin M. Disordered sleep, nocturnal cytokines, and immunity in alcoholics. Psychosom Med. 2003;65:75–85.
Redwine L, Hauger RL, Gillin JC, Irwin M. Effects of sleep and sleep deprivation on interleukin-6, growth hormone, cortisol, and melatonin levels in humans. J Clin Endocrinol Metab. 2000;85:3597–603.
Knutson KL, Spiegel K, Penev P, Van Cauter E. The metabolic consequences of sleep deprivation. Sleep Med Rev. 2007;11:163–78.
Mullington JM, Haack M, Toth M, Serrador JM, Meier-Ewert HK. Cardiovascular, inflammatory, and metabolic consequences of sleep deprivation. Prog Cardiovasc Dis. 2009;51:294–302.
Aldabal L, Bahammam AS. Metabolic, endocrine, and immune consequences of sleep deprivation. Open Respir Med J. 2011;5:31–43.
Kuhs H, Farber D, Tolle R. Serum prolactin, growth hormone, total corticoids, thyroid hormones and thyrotropine during serial therapeutic sleep deprivation. Biol Psychiatry. 1996;39:857–64.
Spiegel K, Leproult R, L’Hermite-Balériaux M, Copinschi G, Penev PD, Van Cauter E. Leptin levels are dependent on sleep duration: relationships with sympathovagal balance, carbohydrate regulation, cortisol, and thyrotropin. J Clin Endocrinol Metab. 2004;89:5762–71.
Morgan L, Hampton S, Gibbs M, Arendt J. Circadian aspects of postprandial metabolism. Chronobiol Int. 2003;20:795–808.
Knutsson A, Hammar N, Karlsson B. Shift workers’ mortality scrutinized. Chronobiol Int. 2004;21:1049–53.
Tanaka K, Sakata K, Oishi M, Morimoto H, Nakada S, Uetani M, Nogawa K, Suwazono Y. Estimation of the benchmark duration of shiftwork associated with weight gain in male Japanese workers. Chronobiol Int. 2010;27:1895–910.
Karlsson B, Knutsson A, Lindahl B. Is there an association between shift work and having a metabolic syndrome? Results from a population based study of 27,485 people. Occup Environ Med. 2001;58:747–52.
van Amelsvoort LG, Schouten EG, Kok FJ. Duration of shiftwork related to body mass index and waist to hip ratio. Int J Obes Relat Metab Disord. 1999;23:973–8.
Yoshida J, Eguchi E, Nagaoka K, Ito T, Ogino K. Association of night eating habits with metabolic syndrome and its components: a longitudinal study. BMC Public Health. 2018;18:1366.
Al-Aubaidy HA, Jelinek HF. Oxidative DNA damage and obesity in type 2 diabetes mellitus. Eur J Endocrinol. 2011;164:899–904.
Ligibel J. Obesity and breast cancer. Oncology (Williston Park). 2011;25:994–1000.
O’Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015;60:547–60.
Srinivas US, Tan BWQ, Vellayappan BA, Jeyasekharan AD. ROS and the DNA damage response in cancer. Redox Biol. 2019;25:101084.
Ribezzo F, Shiloh Y, Schumacher B. Systemic DNA damage responses in aging and diseases. Semin Cancer Biol. 2016;37–38:26–35.
Zisapel N. New perspectives on the role of melatonin in human sleep, circadian rhythms and their regulation. Br J Pharmacol. 2018;175:3190–9.
Galano A, Tan DX, Reiter RJ. Melatonin: a versatile protector against oxidative DNA damage. Molecules. 2018;23:530.
Majidinia M, Sadeghpour A, Mehrzadi S, Reiter RJ, Khatami N, Yousefi B. Melatonin: A pleiotropic molecule that modulates DNA damage response and repair pathways. J Pineal Res. 2017;63:e12416.
Wei T, Li C, Heng Y, Gao X, Zhang G, Wang H, Zhao X, Meng Z, Zhang Y, Hou H. Association between night-shift work and level of melatonin: systematic review and meta-analysis. Sleep Med. 2020;75:502–9.
Bhatti P, Mirick DK, Randolph TW, Gong J, Buchanan DT, Zhang JJ, Davis S. Oxidative DNA damage during sleep periods among nightshift workers. Occup Environ Med. 2016;73:537–44.
Bhatti P, Mirick DK, Randolph TW, Gong J, Buchanan DT, Zhang JJ, Davis S. Oxidative DNA damage during night shift work. Occup Environ Med. 2017;74:680–3.
Cheung V, Yuen VM, Wong GTC, Choi SW. The effect of sleep deprivation and disruption on DNA damage and health of doctors. Anaesthesia. 2019;74:434–40.
Koritala BSC, Porter KI, Arshad OA, Gajula RP, Mitchell HD, Arman T, Manjanatha MG, Teeguarden J, Van Dongen HPA, McDermott JE, et al. Night shift schedule causes circadian dysregulation of DNA repair genes and elevated DNA damage in humans. J Pineal Res. 2021;70:e12726.
Ashok Kumar PV, Dakup PP, Sarkar S, Modasia JB, Motzner MS, Gaddameedhi S. It’s about time: advances in understanding the circadian regulation of DNA damage and repair in carcinogenesis and cancer treatment outcomes. Yale J Biol Med. 2019;92:305–16.
Zada D, Sela Y, Matosevich N, Monsonego A, Lerer-Goldshtein T, Nir Y, Appelbaum L. Parp1 promotes sleep, which enhances DNA repair in neurons. Mol Cell. 2021;81:4979-4993.e7.
Vaccaro A, Kaplan Dor Y, Nambara K, Pollina EA, Lin C, Greenberg ME, Rogulja D. Sleep loss can cause death through accumulation of reactive oxygen species in the gut. Cell. 2020;181:1307-28.e15.
Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E, de Gramont A. Targeting the TGFβ pathway for cancer therapy. Pharmacol Ther. 2015;147:22–31.
Opp MR. Cytokines and sleep. Sleep Med Rev. 2005;9:355–64.
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56.
Voronov E, Shouval DS, Krelin Y, Cagnano E, Benharroch D, Iwakura Y, Dinarello CA, Apte RN. IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci USA. 2003;100:2645–50.
Liu X, Nemeth DP, McKim DB, Zhu L, DiSabato DJ, Berdysz O, Gorantla G, Oliver B, Witcher KG, Wang Y, et al. Cell-type-specific interleukin 1 receptor 1 signaling in the brain regulates distinct neuroimmune activities. Immunity. 2019;50:317-33.e6.
Walker WH II, Borniger JC. Molecular mechanisms of cancer-induced sleep disruption. Int J Mol Sci. 2019;20:2780.
Imeri L, Opp MR, Krueger JM. An IL-1 receptor and an IL-1 receptor antagonist attenuate muramyl dipeptide- and IL-1-induced sleep and fever. Am J Physiol. 1993;265:R907–13.
Opp MR, Krueger JM. Anti-interleukin-1 beta reduces sleep and sleep rebound after sleep deprivation in rats. Am J Physiol. 1994;266:R688–95.
Alam MN, McGinty D, Bashir T, Kumar S, Imeri L, Opp MR, Szymusiak R. Interleukin-1beta modulates state-dependent discharge activity of preoptic area and basal forebrain neurons: role in sleep regulation. Eur J Neurosci. 2004;20:207–16.
Kapás L, Shibata M, Kimura M, Krueger JM. Inhibition of nitric oxide synthesis suppresses sleep in rabbits. Am J Physiol. 1994;266:R151–7.
Takahashi S, Kapás L, Fang J, Krueger JM. Somnogenic relationships between tumor necrosis factor and interleukin-1. Am J Physiol. 1999;276:R1132–40.
Bauer J, Hohagen F, Ebert T, Timmer J, Ganter U, Krieger S, Lis S, Postler E, Voderholzer U, Berger M. Interleukin-6 serum levels in healthy persons correspond to the sleep-wake cycle. Clin Investig. 1994;72:315.
Späth-Schwalbe E, Hansen K, Schmidt F, Schrezenmeier H, Marshall L, Burger K, Fehm HL, Born J. Acute effects of recombinant human interleukin-6 on endocrine and central nervous sleep functions in healthy men. J Clin Endocrinol Metab. 1998;83:1573–9.
Opp M, Obal F Jr, Cady AB, Johannsen L, Krueger JM. Interleukin-6 is pyrogenic but not somnogenic. Physiol Behav. 1989;45:1069–72.
Hogan D, Morrow JD, Smith EM, Opp MR. Interleukin-6 alters sleep of rats. J Neuroimmunol. 2003;137:59–66.
Bryant PA, Trinder J, Curtis N. Sick and tired: does sleep have a vital role in the immune system? Nat Rev Immunol. 2004;4:457–67.
Irwin M. Effects of sleep and sleep loss on immunity and cytokines. Brain Behav Immun. 2002;16:503–12.
Kerr J, Anderson C, Lippman SM. Physical activity, sedentary behaviour, diet, and cancer: an update and emerging new evidence. Lancet Oncol. 2017;18:e457–71.
Kushi LH, Doyle C, McCullough M, Rock CL, Demark-Wahnefried W, Bandera EV, Gapstur S, Patel AV, Andrews K, Gansler T. American Cancer Society Guidelines on nutrition and physical activity for cancer prevention: reducing the risk of cancer with healthy food choices and physical activity. CA Cancer J Clin. 2012;62:30–67.
Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–38.
Lee C, Raffaghello L, Brandhorst S, Safdie FM, Bianchi G, Martin-Montalvo A, Pistoia V, Wei M, Hwang S, Merlino A, et al. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci Transl Med. 2012;4:124ra27.
Raffaghello L, Lee C, Safdie FM, Wei M, Madia F, Bianchi G, Longo VD. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc Natl Acad Sci USA. 2008;105:8215–20.
Jardé T, Perrier S, Vasson MP, Caldefie-Chézet F. Molecular mechanisms of leptin and adiponectin in breast cancer. Eur J Cancer. 2011;47:33–43.
Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12:159–69.
Lee C, Safdie FM, Raffaghello L, Wei M, Madia F, Parrella E, Hwang D, Cohen P, Bianchi G, Longo VD. Reduced levels of IGF-I mediate differential protection of normal and cancer cells in response to fasting and improve chemotherapeutic index. Cancer Res. 2010;70:1564–72.
Cheng CW, Adams GB, Perin L, Wei M, Zhou X, Lam BS, Da Sacco S, Mirisola M, Quinn DI, Dorff TB, et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell-based regeneration and reverse immunosuppression. Cell Stem Cell. 2014;14:810–23.
Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, Levesque S, Castoldi F, Jacquelot N, Yamazaki T, et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell. 2016;30:147–60.
Di Biase S, Lee C, Brandhorst S, Manes B, Buono R, Cheng CW, Cacciottolo M, Martin-Montalvo A, de Cabo R, Wei M, et al. Fasting-mimicking diet reduces HO-1 to promote T cell-mediated tumor cytotoxicity. Cancer Cell. 2016;30:136–46.
Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. 2014;25:42–52.
Di Biase S, Shim HS, Kim KH, Vinciguerra M, Rappa F, Wei M, Brandhorst S, Cappello F, Mirzaei H, Lee C, et al. Fasting regulates EGR1 and protects from glucose- and dexamethasone-dependent sensitization to chemotherapy. PLoS Biol. 2017;15:e2001951.
Safdie F, Brandhorst S, Wei M, Wang W, Lee C, Hwang S, Conti PS, Chen TC, Longo VD. Fasting enhances the response of glioma to chemo- and radiotherapy. PLoS ONE. 2012;7:e44603.
Shim HS, Wei M, Brandhorst S, Longo VD. Starvation promotes REV1 SUMOylation and p53-dependent sensitization of melanoma and breast cancer cells. Cancer Res. 2015;75:1056–67.
Bianchi G, Martella R, Ravera S, Marini C, Capitanio S, Orengo A, Emionite L, Lavarello C, Amaro A, Petretto A, et al. Fasting induces anti-Warburg effect that increases respiration but reduces ATP-synthesis to promote apoptosis in colon cancer models. Oncotarget. 2015;6:11806–19.
Di Biase S, Longo VD. Fasting-induced differential stress sensitization in cancer treatment. Mol Cell Oncol. 2016;3:e1117701.
Tinkum KL, Stemler KM, White LS, Loza AJ, Jeter-Jones S, Michalski BM, Kuzmicki C, Pless R, Stappenbeck TS, Piwnica-Worms D, et al. Fasting protects mice from lethal DNA damage by promoting small intestinal epithelial stem cell survival. Proc Natl Acad Sci U S A. 2015;112:E7148–54.
de Groot S, Pijl H, van der Hoeven JJM, Kroep JR. Effects of short-term fasting on cancer treatment. J Exp Clin Cancer Res. 2019;38:209.
Schumacher B, Garinis GA, Hoeijmakers JH. Age to survive: DNA damage and aging. Trends Genet. 2008;24:77–85.
Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A. 1999;96:7324–9.
Antunes F, Erustes AG, Costa AJ, Nascimento AC, Bincoletto C, Ureshino RP, Pereira GJS, Smaili SS. Autophagy and intermittent fasting: the connection for cancer therapy? Clinics (Sao Paulo). 2018;73:e814s.
Caffa I, Spagnolo V, Vernieri C, Valdemarin F, Becherini P, Wei M, Brandhorst S, Zucal C, Driehuis E, Ferrando L, et al. Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature. 2020;583:620–4.
Kanarek N, Petrova B, Sabatini DM. Dietary modifications for enhanced cancer therapy. Nature. 2020;579:507–17.
Cathcart P, Craddock C, Stebbing J. Fasting: starving cancer. Lancet Oncol. 2017;18:431.
Brandhorst S, Choi IY, Wei M, Cheng CW, Sedrakyan S, Navarrete G, Dubeau L, Yap LP, Park R, Vinciguerra M, et al. A Periodic diet that mimics fasting promotes multi-system regeneration, enhanced cognitive performance, and healthspan. Cell Metab. 2015;22:86–99.
Lo Re O, Panebianco C, Porto S, Cervi C, Rappa F, Di Biase S, Caraglia M, Pazienza V, Vinciguerra M. Fasting inhibits hepatic stellate cells activation and potentiates anti-cancer activity of Sorafenib in hepatocellular cancer cells. J Cell Physiol. 2018;233:1202–12.
Shi Y, Felley-Bosco E, Marti TM, Orlowski K, Pruschy M, Stahel RA. Starvation-induced activation of ATM/Chk2/p53 signaling sensitizes cancer cells to cisplatin. BMC Cancer. 2012;12:571.
Strickaert A, Saiselet M, Dom G, De Deken X, Dumont JE, Feron O, Sonveaux P, Maenhaut C. Cancer heterogeneity is not compatible with one unique cancer cell metabolic map. Oncogene. 2017;36:2637–42.
D’Aronzo M, Vinciguerra M, Mazza T, Panebianco C, Saracino C, Pereira SP, Graziano P, Pazienza V. Fasting cycles potentiate the efficacy of gemcitabine treatment in in vitro and in vivo pancreatic cancer models. Oncotarget. 2015;6:18545–57.
Lu Z, Xie J, Wu G, Shen J, Collins R, Chen W, Kang X, Luo M, Zou Y, Huang LJ, et al. Fasting selectively blocks development of acute lymphoblastic leukemia via leptin-receptor upregulation. Nat Med. 2017;23:79–90.
Chan LN, Chen Z, Braas D, Lee JW, Xiao G, Geng H, Cosgun KN, Hurtz C, Shojaee S, Cazzaniga V, et al. Metabolic gatekeeper function of B-lymphoid transcription factors. Nature. 2017;542:479–83.
Moore SC, Lee IM, Weiderpass E, Campbell PT, Sampson JN, Kitahara CM, Keadle SK, Arem H, Berrington de Gonzalez A, Hartge P, et al. Association of leisure-time physical activity with risk of 26 types of cancer in 1.44 million adults. JAMA Intern Med. 2016;176:816–25.
Kenfield SA, Stampfer MJ, Giovannucci E, Chan JM. Physical activity and survival after prostate cancer diagnosis in the health professionals follow-up study. J Clin Oncol. 2011;29:726–32.
Van Blarigan EL, Meyerhardt JA. Role of physical activity and diet after colorectal cancer diagnosis. J Clin Oncol. 2015;33:1825–34.
Van Blarigan EL, Fuchs CS, Niedzwiecki D, Zhang S, Saltz LB, Mayer RJ, Mowat RB, Whittom R, Hantel A, Benson A, et al. Association of survival with adherence to the American Cancer Society nutrition and physical activity guidelines for cancer survivors after colon cancer diagnosis: the CALGB 89803/alliance trial. JAMA Oncol. 2018;4:783–90.
Cannioto RA, Hutson A, Dighe S, McCann W, McCann SE, Zirpoli GR, Barlow W, Kelly KM, DeNysschen CA, Hershman DL, et al. Physical activity before, during, and after chemotherapy for high-risk breast cancer: relationships with survival. J Natl Cancer Inst. 2021;113:54–63.
Ashcraft KA, Peace RM, Betof AS, Dewhirst MW, Jones LW. Efficacy and mechanisms of aerobic exercise on cancer initiation, progression, and metastasis: a critical systematic review of in vivo preclinical data. Cancer Res. 2016;76:4032–50.
Koelwyn GJ, Quail DF, Zhang X, White RM, Jones LW. Exercise-dependent regulation of the tumour microenvironment. Nat Rev Cancer. 2017;17:620–32.
Arfianti A, Pok S, Barn V, Haigh WG, Yeh MM, Ioannou GN, Teoh NC, Farrell GC. Exercise retards hepatocarcinogenesis in obese mice independently of weight control. J Hepatol. 2020;73:140–8.
Pedersen L, Christensen JF, Hojman P. Effects of exercise on tumor physiology and metabolism. Cancer J. 2015;21:111–6.
Colbert LH, Westerlind KC, Perkins SN, Haines DC, Berrigan D, Donehower LA, Fuchs-Young R, Hursting SD. Exercise effects on tumorigenesis in a p53-deficient mouse model of breast cancer. Med Sci Sports Exerc. 2009;41:1597–605.
Gustafson MP, Wheatley-Guy CM, Rosenthal AC, Gastineau DA, Katsanis E, Johnson BD, Simpson RJ. Exercise and the immune system: taking steps to improve responses to cancer immunotherapy. J Immunother Cancer. 2021;9:e001872.
Idorn M, Hojman P. Exercise-dependent regulation of NK cells in cancer protection. Trends Mol Med. 2016;22:565–77.
Song M, Chan AT. The potential role of exercise and nutrition in harnessing the immune system to improve colorectal cancer survival. Gastroenterology. 2018;155:596–600.
Evans ES, Hackney AC, McMurray RG, Randell SH, Muss HB, Deal AM, Battaglini CL. Impact of acute intermittent exercise on natural killer cells in breast cancer survivors. Integr Cancer Ther. 2015;14:436–45.
Coletta AM, Agha NH, Baker FL, Niemiro GM, Mylabathula PL, Brewster AM, Bevers TB, Fuentes-Mattei E, Basen-Engquist K, Gilchrist SC, et al. The impact of high-intensity interval exercise training on NK-cell function and circulating myokines for breast cancer prevention among women at high risk for breast cancer. Breast Cancer Res Treat. 2021;187:407–16.
Fisher DT, Chen Q, Skitzki JJ, Muhitch JB, Zhou L, Appenheimer MM, Vardam TD, Weis EL, Passanese J, Wang WC, et al. IL-6 trans-signaling licenses mouse and human tumor microvascular gateways for trafficking of cytotoxic T cells. J Clin Invest. 2011;121:3846–59.
Pedersen L, Idorn M, Olofsson GH, Lauenborg B, Nookaew I, Hansen RH, Johannesen HH, Becker JC, Pedersen KS, Dethlefsen C, et al. Voluntary running suppresses tumor growth through epinephrine- and IL-6-dependent NK cell mobilization and redistribution. Cell Metab. 2016;23:554–62.
Rundqvist H, Velica P, Barbieri L, Gameiro PA, Bargiela D, Gojkovic M, Mijwel S, Reitzner SM, Wulliman D, Ahlstedt E, et al. Cytotoxic T-cells mediate exercise-induced reductions in tumor growth. Elife. 2020;9:e59996.
Gross E, Sunwoo JB, Bui JD. Cancer immunosurveillance and immunoediting by natural killer cells. Cancer J. 2013;19:483–9.
Bartlett DB, Duggal NA. Moderate physical activity associated with a higher naive/memory T-cell ratio in healthy old individuals: potential role of IL15. Age Ageing. 2020;49:368–73.
Duggal NA, Pollock RD, Lazarus NR, Harridge S, Lord JM. Major features of immunesenescence, including reduced thymic output, are ameliorated by high levels of physical activity in adulthood. Aging Cell. 2018;17:e12750.
Evans SS, Repasky EA, Fisher DT. Fever and the thermal regulation of immunity: the immune system feels the heat. Nat Rev Immunol. 2015;15:335–49.
Hurwitz MD. Hyperthermia and immunotherapy: clinical opportunities. Int J Hyperthermia. 2019;36:4–9.
Mahmood J, Shukla HD, Soman S, Samanta S, Singh P, Kamlapurkar S, Saeed A, Amin NP, Vujaskovic Z. Immunotherapy, radiotherapy, and hyperthermia: a combined therapeutic approach in pancreatic cancer treatment. Cancers (Basel). 2018;10:469.
Chang M, Hou Z, Wang M, Li C, Lin J. Recent advances in hyperthermia therapy-based synergistic immunotherapy. Adv Mater. 2021;33:e2004788.
Toraya-Brown S, Fiering S. Local tumour hyperthermia as immunotherapy for metastatic cancer. Int J Hyperthermia. 2014;30:531–9.
Wang J, Liu S, Li G, Xiao J. Exercise regulates the immune system. Adv Exp Med Biol. 2020;1228:395–408.
Scheffer DDL, Latini A. Exercise-induced immune system response: Anti-inflammatory status on peripheral and central organs. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165823.
Ticinesi A, Lauretani F, Tana C, Nouvenne A, Ridolo E, Meschi T. Exercise and immune system as modulators of intestinal microbiome: implications for the gut-muscle axis hypothesis. Exerc Immunol Rev. 2019;25:84–95.
Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol. 2012;8:457–65.
Hoffmann C, Weigert C. Skeletal muscle as an endocrine organ: the role of myokines in exercise adaptations. Cold Spring Harb Perspect Med. 2017;7:a029793.
Ruiz-Casado A, Martin-Ruiz A, Perez LM, Provencio M, Fiuza-Luces C, Lucia A. Exercise and the Hallmarks of Cancer. Trends Cancer. 2017;3:423–41.
Miao Y, Hu B, Wang Q, Yang Q, Zhou S. Myokines related to leukocyte recruitment are down-regulated in osteosarcoma. Int J Med Sci. 2018;15:859–66.
Whitham M, Febbraio MA. The ever-expanding myokinome: discovery challenges and therapeutic implications. Nat Rev Drug Discov. 2016;15:719–29.
Catoire M, Mensink M, Kalkhoven E, Schrauwen P, Kersten S. Identification of human exercise-induced myokines using secretome analysis. Physiol Genomics. 2014;46:256–67.
Weigert C, Lehmann R, Hartwig S, Lehr S. The secretome of the working human skeletal muscle—a promising opportunity to combat the metabolic disaster? Proteomics Clin Appl. 2014;8:5–18.
Catoire M, Kersten S. The search for exercise factors in humans. FASEB J. 2015;29:1615–28.
Pourteymour S, Eckardt K, Holen T, Langleite T, Lee S, Jensen J, Birkeland KI, Drevon CA, Hjorth M. Global mRNA sequencing of human skeletal muscle: search for novel exercise-regulated myokines. Mol Metab. 2017;6:352–65.
Rocha-Rodrigues S, Matos A, Afonso J, Mendes-Ferreira M, Abade E, Teixeira E, Silva B, Murawska-Cialowicz E, Oliveira MJ, Ribeiro R. Skeletal muscle-adipose tissue-tumor axis: molecular mechanisms linking exercise training in prostate cancer. Int J Mol Sci. 2021;22:4469.
Kim JS, Galvao DA, Newton RU, Gray E, Taaffe DR. Exercise-induced myokines and their effect on prostate cancer. Nat Rev Urol. 2021;18:519–42.
Gannon NP, Vaughan RA, Garcia-Smith R, Bisoffi M, Trujillo KA. Effects of the exercise-inducible myokine irisin on malignant and non-malignant breast epithelial cell behavior in vitro. Int J Cancer. 2015;136:E197-202.
Kim JS, Wilson RL, Taaffe DR, Galvao DA, Gray E, Newton RU. Myokine expression and tumor-suppressive effect of serum following 12 weeks of exercise in prostate cancer patients on ADT. Med Sci Sports Exerc. 2021;54:197–205.
Aoi W, Naito Y, Takagi T, Tanimura Y, Takanami Y, Kawai Y, Sakuma K, Hang LP, Mizushima K, Hirai Y, et al. A novel myokine, secreted protein acidic and rich in cysteine (SPARC), suppresses colon tumorigenesis via regular exercise. Gut. 2013;62:882–9.
Liu YP, Hsiao M. Exercise-induced SPARC prevents tumorigenesis of colon cancer. Gut. 2013;62:810–1.
Matsuo K, Sato K, Suemoto K, Miyamoto-Mikami E, Fuku N, Higashida K, Tsuji K, Xu Y, Liu X, Iemitsu M, et al. A mechanism underlying preventive effect of high-intensity training on colon cancer. Med Sci Sports Exerc. 2017;49:1805–16.
Severinsen MCK, Pedersen BK. Muscle-organ crosstalk: the emerging roles of myokines. Endocr Rev. 2020;41:594–609.
Kim JS, Galvao DA, Newton RU, Gray E, Taaffe DR. Publisher correction: exercise-induced myokines and their effect on prostate cancer. Nat Rev Urol. 2021;19:64.
Severinsen MCK, Pedersen BK. Corrigendum to: Muscle-organ crosstalk: the emerging roles of myokines. Endocr Rev. 2021;42:97–9.
Li X, Liu Y, Liu Q, Wang S, Ma Y, Jin Q. Recombinant human irisin regulated collagen II, matrix metalloproteinase-13 and the Wnt/beta-catenin and NF-kappaB signaling pathways in interleukin-1beta-induced human SW1353 cells. Exp Ther Med. 2020;19:2879–86.
Chen X, Sun K, Zhao S, Geng T, Fan X, Sun S, Zheng M, Jin Q. Irisin promotes osteogenic differentiation of bone marrow mesenchymal stem cells by activating autophagy via the Wnt//beta-catenin signal pathway. Cytokine. 2020;136:155292.
Dong HN, Park SY, Le CT, Choi DH, Cho EH. Irisin regulates the functions of hepatic stellate cells. Endocrinol Metab (Seoul). 2020;35:647–55.
Bay ML, Pedersen BK. Muscle-organ crosstalk: focus on immunometabolism. Front Physiol. 2020;11:567881.
Roy P, Chowdhury S, Roy HK. Exercise-induced myokines as emerging therapeutic agents in colorectal cancer prevention and treatment. Future Oncol. 2018;14:309–12.
Khan SU, Ghafoor S. Myokines: discovery challenges and therapeutic impediments. J Pak Med Assoc. 2019;69:1014–7.
Leal LG, Lopes MA, Batista ML Jr. Physical exercise-induced myokines and muscle-adipose tissue crosstalk: a review of current knowledge and the implications for health and metabolic diseases. Front Physiol. 2018;9:1307.
Hennigar SR, McClung JP, Pasiakos SM. Nutritional interventions and the IL-6 response to exercise. FASEB J. 2017;31:3719–28.
Pedersen BK, Febbraio MA. Muscle as an endocrine organ: focus on muscle-derived interleukin-6. Physiol Rev. 2008;88:1379–406.
Pedersen BK, Febbraio M. Muscle-derived interleukin-6—a possible link between skeletal muscle, adipose tissue, liver, and brain. Brain Behav Immun. 2005;19:371–6.
Tsai MS, Kuo ML, Chang CC, Wu YT. The effects of exercise training on levels of vascular endothelial growth factor in tumor-bearing mice. Cancer Biomark. 2013;13:307–13.
Vital TM, Stein AM, de Melo Coelho FG, Arantes FJ, Teodorov E, Santos-Galduroz RF. Physical exercise and vascular endothelial growth factor (VEGF) in elderly: a systematic review. Arch Gerontol Geriatr. 2014;59:234–9.
Shalamzari SA, Agha-Alinejad H, Alizadeh S, Shahbazi S, Khatib ZK, Kazemi A, Saei MA, Minayi N. The effect of exercise training on the level of tissue IL-6 and vascular endothelial growth factor in breast cancer bearing mice. Iran J Basic Med Sci. 2014;17:231–58.
Buss LA, Dachs GU. Effects of exercise on the tumour microenvironment. Adv Exp Med Biol. 2020;1225:31–51.
Buss LA, Ang AD, Hock B, Robinson BA, Currie MJ, Dachs GU. Effect of post-implant exercise on tumour growth rate, perfusion and hypoxia in mice. PLoS ONE. 2020;15:e0229290.
Betof AS, Lascola CD, Weitzel D, Landon C, Scarbrough PM, Devi GR, Palmer G, Jones LW, Dewhirst MW. Modulation of murine breast tumor vascularity, hypoxia and chemotherapeutic response by exercise. J Natl Cancer Inst. 2015;107:div040.
Goel S, Wong AH, Jain RK. Vascular normalization as a therapeutic strategy for malignant and nonmalignant disease. Cold Spring Harb Perspect Med. 2012;2:a006486.
Buss LA, Dachs GU. Voluntary exercise slows breast tumor establishment and reduces tumor hypoxia in ApoE(-/-) mice. J Appl Physiol. 1985;2018(124):938–49.
Lin B, Song X, Yang D, Bai D, Yao Y, Lu N. Anlotinib inhibits angiogenesis via suppressing the activation of VEGFR2, PDGFRbeta and FGFR1. Gene. 2018;654:77–86.
Jones LW, Antonelli J, Masko EM, Broadwater G, Lascola CD, Fels D, Dewhirst MW, Dyck JR, Nagendran J, Flores CT, et al. Exercise modulation of the host-tumor interaction in an orthotopic model of murine prostate cancer. J Appl Physiol. 1985;2012(113):263–72.
Jones LW, Viglianti BL, Tashjian JA, Kothadia SM, Keir ST, Freedland SJ, Potter MQ, Moon EJ, Schroeder T, Herndon JE 2nd, et al. Effect of aerobic exercise on tumor physiology in an animal model of human breast cancer. J Appl Physiol. 1985;2010(108):343–8.
Patel DI, Abuchowski K, Bedolla R, Rivas P, Musi N, Reddick R, Kumar AP. Nexrutine and exercise similarly prevent high grade prostate tumors in transgenic mouse model. PLoS ONE. 2019;14:e0226187.
Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104:2224–34.
Murdoch C, Lewis CE. Macrophage migration and gene expression in response to tumor hypoxia. Int J Cancer. 2005;117:701–8.
Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–74.
Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon KS, Auffinger B, Tobias AL, Han Y, Lesniak MS. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res. 2012;18:6110–21.
Kiyozumi Y, Baba Y, Okadome K, Yagi T, Ishimoto T, Iwatsuki M, Miyamoto Y, Yoshida N, Watanabe M, Komohara Y, et al. IDO1 expression is associated with immune tolerance and poor prognosis in patients with surgically resected esophageal cancer. Ann Surg. 2019;269:1101–8.
Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, Sethumadhavan S, Philbrook P, Ko K, Cannici R, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med. 2015;7:277ra30.
Leone RD, Horton MR, Powell JD. Something in the air: hyperoxic conditioning of the tumor microenvironment for enhanced immunotherapy. Cancer Cell. 2015;27:435–6.
Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014;74:665–74.
Barsoum IB, Hamilton TK, Li X, Cotechini T, Miles EA, Siemens DR, Graham CH. Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: role of nitric oxide. Cancer Res. 2011;71:7433–41.
Ruf M, Moch H, Schraml P. PD-L1 expression is regulated by hypoxia inducible factor in clear cell renal cell carcinoma. Int J Cancer. 2016;139:396–403.
Buss LA, Williams T, Hock B, Ang AD, Robinson BA, Currie MJ, Dachs GU. Effects of exercise and anti-PD-1 on the tumour microenvironment. Immunol Lett. 2021;239:60–71.
Ashcraft KA, Warner AB, Jones LW, Dewhirst MW. Exercise as adjunct therapy in cancer. Semin Radiat Oncol. 2019;29:16–24.
Dey A, Varelas X, Guan KL. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat Rev Drug Discov. 2020;19:480–94.
Moya IM, Halder G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat Rev Mol Cell Biol. 2019;20:211–26.
Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. 2016;29:783–803.
Totaro A, Panciera T, Piccolo S. YAP/TAZ upstream signals and downstream responses. Nat Cell Biol. 2018;20:888–99.
Dethlefsen C, Hansen LS, Lillelund C, Andersen C, Gehl J, Christensen JF, Pedersen BK, Hojman P. Exercise-induced catecholamines activate the hippo tumor suppressor pathway to reduce risks of breast cancer development. Cancer Res. 2017;77:4894–904.
Hojman P, Gehl J, Christensen JF, Pedersen BK. Molecular mechanisms linking exercise to cancer prevention and treatment. Cell Metab. 2018;27:10–21.
Hojman P. Exercise protects from cancer through regulation of immune function and inflammation. Biochem Soc Trans. 2017;45:905–11.
Bigley AB, Spielmann G, LaVoy EC, Simpson RJ. Can exercise-related improvements in immunity influence cancer prevention and prognosis in the elderly? Maturitas. 2013;76:51–6.
Roenneberg T, Merrow M. The circadian clock and human health. Curr Biol. 2016;26:R432–43.
Rijo-Ferreira F, Takahashi JS. Genomics of circadian rhythms in health and disease. Genome Med. 2019;11:82.
Trott AJ, Menet JS. Regulation of circadian clock transcriptional output by CLOCK:BMAL1. PLoS Genet. 2018;14:e1007156.
Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet. 2017;18:164–79.
Cho H, Zhao X, Hatori M, Yu RT, Barish GD, Lam MT, Chong LW, DiTacchio L, Atkins AR, Glass CK, et al. Regulation of circadian behaviour and metabolism by REV-ERB-alpha and REV-ERB-beta. Nature. 2012;485:123–7.
Solt LA, Wang Y, Banerjee S, Hughes T, Kojetin DJ, Lundasen T, Shin Y, Liu J, Cameron MD, Noel R, et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature. 2012;485:62–8.
Uriz-Huarte A, Date A, Ang H, Ali S, Brady HJM, Fuchter MJ. The transcriptional repressor REV-ERB as a novel target for disease. Bioorg Med Chem Lett. 2020;30:127395.
Kojetin DJ, Burris TP. REV-ERB and ROR nuclear receptors as drug targets. Nat Rev Drug Discov. 2014;13:197–216.
Grant D, Yin L, Collins JL, Parks DJ, Orband-Miller LA, Wisely GB, Joshi S, Lazar MA, Willson TM, Zuercher WJ. GSK4112, a small molecule chemical probe for the cell biology of the nuclear heme receptor Rev-erbalpha. ACS Chem Biol. 2010;5:925–32.
Chen H, Chu G, Zhao L, Yamauchi N, Shigeyoshi Y, Hashimoto S, Hattori MA. Rev-erbalpha regulates circadian rhythms and StAR expression in rat granulosa cells as identified by the agonist GSK4112. Biochem Biophys Res Commun. 2012;420:374–9.
Geldof L, Deventer K, Roels K, Tudela E, Van Eeno P. In vitro metabolic studies of REV-ERB Agonists SR9009 and SR9011. Int J Mol Sci. 2016;17:1676.
Mazzarino M, Rizzato N, Stacchini C, de la Torre X, Botre F. A further insight into the metabolic profile of the nuclear receptor Rev-erb agonist, SR9009. Drug Test Anal. 2018;10:1670–81.
Wagner PM, Monjes NM, Guido ME. Chemotherapeutic effect of SR9009, a REV-ERB agonist, on the human glioblastoma T98G cells. ASN Neuro. 2019;11:1759091419892713.
Wang Y, Kojetin D, Burris TP. Anti-proliferative actions of a synthetic REV-ERBalpha/beta agonist in breast cancer cells. Biochem Pharmacol. 2015;96:315–22.
Shen W, Zhang W, Ye W, Wang H, Zhang Q, Shen J, Hong Q, Li X, Wen G, Wei T, et al. SR9009 induces a REV-ERB dependent anti-small-cell lung cancer effect through inhibition of autophagy. Theranostics. 2020;10:4466–80.
Sulli G, Rommel A, Wang X, Kolar MJ, Puca F, Saghatelian A, Plikus MV, Verma IM, Panda S. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature. 2018;553:351–5.
Dong Z, Zhang G, Qu M, Gimple RC, Wu Q, Qiu Z, Prager BC, Wang X, Kim LJY, Morton AR, et al. Targeting glioblastoma stem cells through disruption of the circadian clock. Cancer Discov. 2019;9:1556–73.
Trump RP, Bresciani S, Cooper AW, Tellam JP, Wojno J, Blaikley J, Orband-Miller LA, Kashatus JA, Boudjelal M, Dawson HC, et al. Optimized chemical probes for REV-ERBalpha. J Med Chem. 2013;56:4729–37.
Guillaumond F, Dardente H, Giguere V, Cermakian N. Differential control of Bmal1 circadian transcription by REV-ERB and ROR nuclear receptors. J Biol Rhythms. 2005;20:391–403.
Wang J, Zou JX, Xue X, Cai D, Zhang Y, Duan Z, Xiang Q, Yang JC, Louie MC, Borowsky AD, et al. ROR-gamma drives androgen receptor expression and represents a therapeutic target in castration-resistant prostate cancer. Nat Med. 2016;22:488–96.
Wang J, Zou JX, Xue X, Cai D, Zhang Y, Duan Z, Xiang Q, Yang JC, Louie MC, Borowsky AD, et al. Corrigendum: ROR-gamma drives androgen receptor expression and represents a therapeutic target in castration-resistant prostate cancer. Nat Med. 2016;22:692.
Stone L. Prostate cancer: ROR-gamma drives androgen receptor expression. Nat Rev Urol. 2016;13:237.
Zheng J, Wang J, Wang Q, Zou H, Wang H, Zhang Z, Chen J, Wang Q, Wang P, Zhao Y, et al. Targeting castration-resistant prostate cancer with a novel RORgamma antagonist elaiophylin. Acta Pharm Sin B. 2020;10:2313–22.
Gao M, Guo L, Wang H, Huang J, Han F, Xiang S, Wang J. Orphan nuclear receptor RORgamma confers doxorubicin resistance in prostate cancer. Cell Biol Int. 2020;44:2170–6.
Lytle NK, Ferguson LP, Rajbhandari N, Gilroy K, Fox RG, Deshpande A, Schurch CM, Hamilton M, Robertson N, Lin W, et al. A multiscale map of the stem cell state in pancreatic adenocarcinoma. Cell. 2019;177:572-86.e22.
Yao W, Maitra A. Hear pancreatic cancer stem cells ROR. Cell. 2019;177:516–8.
Jetten AM, Cook DN. (Inverse) Agonists of retinoic acid-related orphan receptor gamma: regulation of immune responses, inflammation, and autoimmune disease. Annu Rev Pharmacol Toxicol. 2020;60:371–90.
Liu X, Zawidzka EM, Li H, Lesch CA, Dunbar J, Bousley D, Zou W, Hu X, Carter LL. RORgamma agonists enhance the sustained antitumor activity through intrinsic Tc17 cytotoxicity and Tc1 recruitment. Cancer Immunol Res. 2019;7:1054–63.
Chang MR, Dharmarajan V, Doebelin C, Garcia-Ordonez RD, Novick SJ, Kuruvilla DS, Kamenecka TM, Griffin PR. Synthetic RORgammat agonists enhance protective immunity. ACS Chem Biol. 2016;11:1012–8.
Hu X, Liu X, Moisan J, Wang Y, Lesch CA, Spooner C, Morgan RW, Zawidzka EM, Mertz D, Bousley D, et al. Synthetic RORgamma agonists regulate multiple pathways to enhance antitumor immunity. Oncoimmunology. 2016;5:e1254854.
Mahalingam D, Wang JS, Hamilton EP, Sarantopoulos J, Nemunaitis J, Weems G, Carter L, Hu X, Schreeder M, Wilkins HJ. Phase 1 open-label, multicenter study of first-in-class RORgamma agonist LYC-55716 (cintirorgon): safety, tolerability, and preliminary evidence of antitumor activity. Clin Cancer Res. 2019;25:3508–16.
Zhu Y, McAvoy S, Kuhn R, Smith DI. RORA, a large common fragile site gene, is involved in cellular stress response. Oncogene. 2006;25:2901–8.
Mao W, Xiong G, Wu Y, Wang C, St Clair D, Li JD, Xu R. RORalpha suppresses cancer-associated inflammation by repressing respiratory complex I-dependent ROS generation. Int J Mol Sci. 2021;22:10665.
Park SC, Park IG, Kim H, Lee JM. N-terminal domain mediated regulation of RORalpha1 inhibits invasive growth in prostate cancer. Int J Mol Sci. 2019;20:1684.
Wang Z, Xiong F, Wang X, Qi Y, Yu H, Zhu Y, Zhu H. Nuclear receptor retinoid-related orphan receptor alpha promotes apoptosis but is reduced in human gastric cancer. Oncotarget. 2017;8:11105–13.
Wang Y, Solt LA, Kojetin DJ, Burris TP. Regulation of p53 stability and apoptosis by a ROR agonist. PLoS ONE. 2012;7:e34921.
Wang Y, Kumar N, Nuhant P, Cameron MD, Istrate MA, Roush WR, Griffin PR, Burris TP. Identification of SR1078, a synthetic agonist for the orphan nuclear receptors RORalpha and RORgamma. ACS Chem Biol. 2010;5:1029–34.
Zhang S, Zhang J, Deng Z, Liu H, Mao W, Jiang F, Xia Z, Li JD. Circadian clock components RORalpha and Bmal1 mediate the anti-proliferative effect of MLN4924 in osteosarcoma cells. Oncotarget. 2016;7:66087–99.
Zhang Y, Shi CC, Zhang HP, Li GQ, Li SS. MLN4924 suppresses neddylation and induces cell cycle arrest, senescence, and apoptosis in human osteosarcoma. Oncotarget. 2016;7:45263–74.
Wu MH, Lee CY, Huang TJ, Huang KY, Tang CH, Liu SH, Kuo KL, Kuan FC, Lin WC, Shi CS. MLN4924, a protein neddylation inhibitor, suppresses the growth of human chondrosarcoma through inhibiting cell proliferation and inducing endoplasmic reticulum stress-related apoptosis. Int J Mol Sci. 2018;20:72.
Han K, Wang Q, Cao H, Qiu G, Cao J, Li X, Wang J, Shen B, Zhang J. The NEDD8-activating enzyme inhibitor MLN4924 induces G2 arrest and apoptosis in T-cell acute lymphoblastic leukemia. Oncotarget. 2016;7:23812–24.
Rahman S, Wittine K, Sedic M, Markova-Car EP. Small molecules targeting biological clock; a novel prospective for anti-cancer drugs. Molecules. 2020;25:4937.
Cao X, Yang Y, Selby CP, Liu Z, Sancar A. Molecular mechanism of the repressive phase of the mammalian circadian clock. Proc Natl Acad Sci U S A. 2021;118:e2021174118.
Ye R, Selby CP, Chiou YY, Ozkan-Dagliyan I, Gaddameedhi S, Sancar A. Dual modes of CLOCK:BMAL1 inhibition mediated by cryptochrome and period proteins in the mammalian circadian clock. Genes Dev. 2014;28:1989–98.
Chiou YY, Yang Y, Rashid N, Ye R, Selby CP, Sancar A. Mammalian period represses and de-represses transcription by displacing CLOCK-BMAL1 from promoters in a Cryptochrome-dependent manner. Proc Natl Acad Sci U S A. 2016;113:E6072–9.
Parico GCG, Perez I, Fribourgh JL, Hernandez BN, Lee HW, Partch CL. The human CRY1 tail controls circadian timing by regulating its association with CLOCK:BMAL1. Proc Natl Acad Sci U S A. 2020;117:27971–9.
Hirota T, Lee JW, St John PC, Sawa M, Iwaisako K, Noguchi T, Pongsawakul PY, Sonntag T, Welsh DK, Brenner DA, et al. Identification of small molecule activators of cryptochrome. Science. 2012;337:1094–7.
Solanas G, Benitah SA. Alterations to the circadian clock make brain tumours vulnerable. Nature. 2019;574:337–8.
Chun SK, Chung S, Kim HD, Lee JH, Jang J, Kim J, Kim D, Son GH, Oh YJ, Suh YG, et al. A synthetic cryptochrome inhibitor induces anti-proliferative effects and increases chemosensitivity in human breast cancer cells. Biochem Biophys Res Commun. 2015;467:441–6.
Eide EJ, Woolf MF, Kang H, Woolf P, Hurst W, Camacho F, Vielhaber EL, Giovanni A, Virshup DM. Control of mammalian circadian rhythm by CKIepsilon-regulated proteasome-mediated PER2 degradation. Mol Cell Biol. 2005;25:2795–807.
Isojima Y, Nakajima M, Ukai H, Fujishima H, Yamada RG, Masumoto KH, Kiuchi R, Ishida M, Ukai-Tadenuma M, Minami Y, et al. CKIepsilon/delta-dependent phosphorylation is a temperature-insensitive, period-determining process in the mammalian circadian clock. Proc Natl Acad Sci U S A. 2009;106:15744–9.
Yang Y, Xu T, Zhang Y, Qin X. Molecular basis for the regulation of the circadian clock kinases CK1delta and CK1epsilon. Cell Signal. 2017;31:58–65.
Guo G, Wang K, Hu SS, Tian T, Liu P, Mori T, Chen P, Johnson CH, Qin X. Autokinase activity of casein kinase 1 delta/epsilon governs the period of mammalian circadian rhythms. J Biol Rhythms. 2019;34:482–96.
Janovska P, Normant E, Miskin H, Bryja V. Targeting casein kinase 1 (CK1) in hematological cancers. Int J Mol Sci. 2020;21:9026.
Sinnberg T, Wang J, Sauer B, Schittek B. Casein kinase 1alpha has a non-redundant and dominant role within the CK1 family in melanoma progression. BMC Cancer. 2016;16:594.
Rosenberg LH, Lafitte M, Quereda V, Grant W, Chen W, Bibian M, Noguchi Y, Fallahi M, Yang C, Chang JC, et al. Therapeutic targeting of casein kinase 1delta in breast cancer. Sci Transl Med. 2015;7:318ra202.
Shen C, Nayak A, Melendez RA, Wynn DT, Jackson J, Lee E, Ahmed Y, Robbins DJ. Casein kinase 1alpha as a regulator of Wnt-driven cancer. Int J Mol Sci. 2020;21:5940.
Bibian M, Rahaim RJ, Choi JY, Noguchi Y, Schurer S, Chen W, Nakanishi S, Licht K, Rosenberg LH, Li L, et al. Development of highly selective casein kinase 1delta/1epsilon (CK1delta/epsilon) inhibitors with potent antiproliferative properties. Bioorg Med Chem Lett. 2013;23:4374–80.
Minzel W, Venkatachalam A, Fink A, Hung E, Brachya G, Burstain I, Shaham M, Rivlin A, Omer I, Zinger A, et al. Small molecules co-targeting CKIalpha and the transcriptional kinases CDK7/9 control AML in preclinical models. Cell. 2018;175:171–85.
Hanif IM, Hanif IM, Shazib MA, Ahmad KA, Pervaiz S. Casein kinase II: an attractive target for anti-cancer drug design. Int J Biochem Cell Biol. 2010;42:1602–5.
Kim HJ, Han YS, Lee JH, Lee SH. Casein Kinase 2alpha Enhances 5-fluorouracil resistance in colorectal cancer cells by inhibiting endoplasmic reticulum stress. Anticancer Res. 2020;40:1419–26.
Hashimoto A, Gao C, Mastio J, Kossenkov A, Abrams SI, Purandare AV, Desilva H, Wee S, Hunt J, Jure-Kunkel M, et al. Inhibition of casein kinase 2 disrupts differentiation of myeloid cells in cancer and enhances the efficacy of immunotherapy in mice. Cancer Res. 2018;78:5644–55.
Kim HM, Jeong I, Kim HJ, Kang SK, Kwon WS, Kim TS, Park KH, Jung M, Soong J, Lin SC, et al. Casein kinase 2 inhibitor, CX-4945, as a potential targeted anticancer agent in gastric cancer. Anticancer Res. 2018;38:6171–80.
Oshima T, Niwa Y, Kuwata K, Srivastava A, Hyoda T, Tsuchiya Y, Kumagai M, Tsuyuguchi M, Tamaru T, Sugiyama A, et al. Cell-based screen identifies a new potent and highly selective CK2 inhibitor for modulation of circadian rhythms and cancer cell growth. Sci Adv. 2019;5:eaau9060.
Siddiqui-Jain A, Drygin D, Streiner N, Chua P, Pierre F, O’Brien SE, Bliesath J, Omori M, Huser N, Ho C, et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010;70:10288–98.
D’Amore C, Borgo C, Sarno S, Salvi M. Role of CK2 inhibitor CX-4945 in anti-cancer combination therapy—potential clinical relevance. Cell Oncol (Dordr). 2020;43:1003–16.
Pierre F, Chua PC, O’Brien SE, Siddiqui-Jain A, Bourbon P, Haddach M, Michaux J, Nagasawa J, Schwaebe MK, Stefan E, et al. Pre-clinical characterization of CX-4945, a potent and selective small molecule inhibitor of CK2 for the treatment of cancer. Mol Cell Biochem. 2011;356:37–43.
Sancar A, Van Gelder RN. Clocks, cancer, and chronochemotherapy. Science. 2021;371:eabb0738.
Yang Y, Lindsey-Boltz LA, Vaughn CM, Selby CP, Cao X, Liu Z, Hsu DS, Sancar A. Circadian clock, carcinogenesis, chronochemotherapy connections. J Biol Chem. 2021;297:101068.
Ballesta A, Innominato PF, Dallmann R, Rand DA, Levi FA. Systems chronotherapeutics. Pharmacol Rev. 2017;69:161–99.
Levi F, Zidani R, Misset JL. Randomised multicentre trial of chronotherapy with oxaliplatin, fluorouracil, and folinic acid in metastatic colorectal cancer. Int Organ Cancer Chronother Lancet. 1997;350:681–6.
Sandor V. Chronotherapy with 5-fluorouracil, oxaliplatin, and folinic acid in colorectal cancer. Lancet. 1997;350:1325–6.
Efficace F, Innominato PF, Bjarnason G, Coens C, Humblet Y, Tumolo S, Genet D, Tampellini M, Bottomley A, Garufi C, et al. Validation of patient’s self-reported social functioning as an independent prognostic factor for survival in metastatic colorectal cancer patients: results of an international study by the Chronotherapy Group of the European Organisation for Research and Treatment of Cancer. J Clin Oncol. 2008;26:2020–6.
Giacchetti S, Bjarnason G, Garufi C, Genet D, Iacobelli S, Tampellini M, Smaaland R, Focan C, Coudert B, Humblet Y, et al. Phase III trial comparing 4-day chronomodulated therapy versus 2-day conventional delivery of fluorouracil, leucovorin, and oxaliplatin as first-line chemotherapy of metastatic colorectal cancer: the European Organisation for Research and Treatment of Cancer Chronotherapy Group. J Clin Oncol. 2006;24:3562–9.
Dong D, Yang D, Lin L, Wang S, Wu B. Circadian rhythm in pharmacokinetics and its relevance to chronotherapy. Biochem Pharmacol. 2020;178:114045.
Hill RJW, Innominato PF, Levi F, Ballesta A. Optimizing circadian drug infusion schedules towards personalized cancer chronotherapy. PLoS Comput Biol. 2020;16:e1007218.
Bicker J, Alves G, Falcao A, Fortuna A. Timing in drug absorption and disposition: the past, present, and future of chronopharmacokinetics. Br J Pharmacol. 2020;177:2215–39.
Levi F, Okyar A, Dulong S, Innominato PF, Clairambault J. Circadian timing in cancer treatments. Annu Rev Pharmacol Toxicol. 2010;50:377–421.
Innominato PF, Roche VP, Palesh OG, Ulusakarya A, Spiegel D, Levi FA. The circadian timing system in clinical oncology. Ann Med. 2014;46:191–207.
Dallmann R, Okyar A, Levi F. Dosing-time makes the poison: circadian regulation and pharmacotherapy. Trends Mol Med. 2016;22:430–45.
Levi F, Schibler U. Circadian rhythms: mechanisms and therapeutic implications. Annu Rev Pharmacol Toxicol. 2007;47:593–628.
Altinok A, Levi F, Goldbeter A. Identifying mechanisms of chronotolerance and chronoefficacy for the anticancer drugs 5-fluorouracil and oxaliplatin by computational modeling. Eur J Pharm Sci. 2009;36:20–38.
Binkhorst L, Kloth JSL, de Wit AS, de Bruijn P, Lam MH, Chaves I, Burger H, van Alphen RJ, Hamberg P, van Schaik RHN, et al. Circadian variation in tamoxifen pharmacokinetics in mice and breast cancer patients. Breast Cancer Res Treat. 2015;152:119–28.
Kloth JS, Binkhorst L, de Wit AS, de Bruijn P, Hamberg P, Lam MH, Burger H, Chaves I, Wiemer EA, van der Horst GT, et al. Relationship between sunitinib pharmacokinetics and administration time: preclinical and clinical evidence. Clin Pharmacokinet. 2015;54:851–8.
Lankheet NA, Kloth JS, Gadellaa-van Hooijdonk CG, Cirkel GA, Mathijssen RH, Lolkema MP, Schellens JH, Voest EE, Sleijfer S, de Jonge MJ, et al. Pharmacokinetically guided sunitinib dosing: a feasibility study in patients with advanced solid tumours. Br J Cancer. 2014;110:2441–9.
Liu J, Wang CY, Ji SG, Xu X, Wang PP, Zhang B, Zhao LY, Liu L, Lin PP, Liu LK, et al. chronopharmacokinetics of erlotinib and circadian rhythms of related metabolic enzymes in Lewis tumor-bearing mice. Eur J Drug Metab Pharmacokinet. 2016;41:627–35.
Bajetta E, Pietrantonio F, Buzzoni R, Ferrario E, Valvo F, Mariani L, Dotti KF, Biondani P, Formisano B, Gevorgyan A, et al. Chronomodulated capecitabine and adjuvant radiation in intermediate-risk to high-risk rectal cancer: a phase II study. Am J Clin Oncol. 2014;37:545–9.
Akgun Z, Saglam S, Yucel S, Gural Z, Balik E, Cipe G, Yildiz S, Kilickap S, Okyar A, Kaytan-Saglam E. Neoadjuvant chronomodulated capecitabine with radiotherapy in rectal cancer: a phase II brunch regimen study. Cancer Chemother Pharmacol. 2014;74:751–6.
Bernard S, Cajavec Bernard B, Levi F, Herzel H. Tumor growth rate determines the timing of optimal chronomodulated treatment schedules. PLoS Comput Biol. 2010;6:e1000712.
Hesse J, Martinelli J, Aboumanify O, Ballesta A, Relogio A. A mathematical model of the circadian clock and drug pharmacology to optimize irinotecan administration timing in colorectal cancer. Comput Struct Biotechnol J. 2021;19:5170–83.
Dulong S, Ballesta A, Okyar A, Levi F. Identification of circadian determinants of cancer chronotherapy through in vitro chronopharmacology and mathematical modeling. Mol Cancer Ther. 2015;14:2154–64.
Ortiz-Tudela E, Iurisci I, Beau J, Karaboue A, Moreau T, Rol MA, Madrid JA, Levi F, Innominato PF. The circadian rest-activity rhythm, a potential safety pharmacology endpoint of cancer chemotherapy. Int J Cancer. 2014;134:2717–25.
Innominato PF, Karaboue A, Focan C, Chollet P, Giacchetti S, Bouchahda M, Ulusakarya A, Torsello A, Adam R, Levi FA, et al. Efficacy and safety of chronomodulated irinotecan, oxaliplatin, 5-fluorouracil and leucovorin combination as first- or second-line treatment against metastatic colorectal cancer: results from the International EORTC 05011 Trial. Int J Cancer. 2020;148:2512–21.
Qian DC, Kleber T, Brammer B, Xu KM, Switchenko JM, Janopaul-Naylor JR, Zhong J, Yushak ML, Harvey RD, Paulos CM, et al. Effect of immunotherapy time-of-day infusion on overall survival among patients with advanced melanoma in the USA (MEMOIR): a propensity score-matched analysis of a single-centre, longitudinal study. Lancet Oncol. 2021;22:1777–86.
Levi F. Daytime versus evening infusions of immune checkpoint inhibitors. Lancet Oncol. 2021;22:1648–50.
Khan Z, Pillay V, Choonara YE, du Toit LC. Drug delivery technologies for chronotherapeutic applications. Pharm Dev Technol. 2009;14:602–12.
Levi F, Okyar A. Circadian clocks and drug delivery systems: impact and opportunities in chronotherapeutics. Expert Opin Drug Deliv. 2011;8:1535–41.
Sunil SA, Srikanth MV, Rao NS, Uhumwangho MU, Latha K, Murthy KV. Chronotherapeutic drug delivery systems: an approach to circadian rhythms diseases. Curr Drug Deliv. 2011;8:622–33.
Popov I, Jelic S, Radosavljevic D. Nikolic-Tomasevic A [Chronotherapy with high dose carboplatin, 5-fluorouracil and leucovorin in advanced colorectal carcinoma]. Srp Arh Celok Lek. 1998;126:355–61.
Focan C, Denis B, Kreutz F, Focan-Henrard D, Levi F. Ambulatory chronotherapy with 5-fluorouracil, folinic acid, and carboplatin for advanced non-small cell lung cancer. A phase II feasibility trial. J Infus Chemother. 1995;5:148–52.
Levi FA, Boige V, Hebbar M, Smith D, Lepere C, Focan C, Karaboue A, Guimbaud R, Carvalho C, Tumolo S, et al. Conversion to resection of liver metastases from colorectal cancer with hepatic artery infusion of combined chemotherapy and systemic cetuximab in multicenter trial OPTILIV. Ann Oncol. 2016;27:267–74.
Bouchahda M, Adam R, Giacchetti S, Castaing D, Brezault-Bonnet C, Hauteville D, Innominato PF, Focan C, Machover D, Levi F. Rescue chemotherapy using multidrug chronomodulated hepatic arterial infusion for patients with heavily pretreated metastatic colorectal cancer. Cancer. 2009;115:4990–9.
Levi F, Karaboue A, Etienne-Grimaldi MC, Paintaud G, Focan C, Innominato P, Bouchahda M, Milano G, Chatelut E. Pharmacokinetics of irinotecan, oxaliplatin and 5-fluorouracil during hepatic artery chronomodulated infusion: a translational European OPTILIV Study. Clin Pharmacokinet. 2017;56:165–77.
Youan BB. Chronopharmaceutical drug delivery systems: Hurdles, hype or hope? Adv Drug Deliv Rev. 2010;62:898–903.
Ohdo S. Chrono-drug-delivery focused on biological clock: intra- and inter-individual variability of molecular clock. Adv Drug Deliv Rev. 2010;62:857–8.
Zhang Z, Qin S, Chen Y, Zhou L, Yang M, Tang Y, Zuo J, Zhang J, Mizokami A, Nice EC, et al. Inhibition of NPC1L1 disrupts adaptive responses of drug-tolerant persister cells to chemotherapy. EMBO Mol Med. 2022;14:e14903.
Patil SS, Shahiwala A. Patented pulsatile drug delivery technologies for chronotherapy. Expert Opin Ther Pat. 2014;24:845–56.
Dumpa NR, Sarabu S, Bandari S, Zhang F, Repka MA. Chronotherapeutic drug delivery of ketoprofen and ibuprofen for improved treatment of early morning stiffness in arthritis using hot-melt extrusion technology. AAPS PharmSciTech. 2018;19:2700–9.
Maqbool I, Akhtar M, Ahmad R, Sadaquat H, Noreen S, Batool A, Khan SU. Novel multiparticulate pH triggered delayed release chronotherapeutic drug delivery of celecoxib-beta-cyclodextrin inclusion complexes by using Box–Behnken design. Eur J Pharm Sci. 2020;146:105254.
Hu J, Fu S, Peng Q, Han Y, Xie J, Zan N, Chen Y, Fan J. Paclitaxel-loaded polymeric nanoparticles combined with chronomodulated chemotherapy on lung cancer: in vitro and in vivo evaluation. Int J Pharm. 2017;516:313–22.
Hu J, Fu S, Peng Q, Han Y, Xie J, Zan N, Chen Y, Fan J. Corrigendum to “Paclitaxel-loaded polymeric nanoparticles combined with chronomodulated chemotherapy on lung cancer: in vitro and in vivo evaluation” [Int. J. Pharmaceut. 516 (2017) 313-322]. Int J Pharm. 2020;573:118878.
Zhao X, Fan J, Wu P, Wei C, Chen Q, Ming Z, Yan J, Yang L. Chronic chemotherapy with paclitaxel nanoparticles induced apoptosis in lung cancer in vitro and in vivo. Int J Nanomed. 2019;14:1299–309.
Potter GD, Cade JE, Grant PJ, Hardie LJ. Nutrition and the circadian system. Br J Nutr. 2016;116:434–42.
Longo VD, Panda S. Fasting, circadian rhythms, and time-restricted feeding in healthy lifespan. Cell Metab. 2016;23:1048–59.
Manoogian ENC, Panda S. Circadian rhythms, time-restricted feeding, and healthy aging. Ageing Res Rev. 2017;39:59–67.
Parr EB, Heilbronn LK, Hawley JA. A time to eat and a time to exercise. Exerc Sport Sci Rev. 2020;48:4–10.
Nencioni A, Caffa I, Cortellino S, Longo VD. Fasting and cancer: molecular mechanisms and clinical application. Nat Rev Cancer. 2018;18:707–19.
Chaix A, Manoogian ENC, Melkani GC, Panda S. Time-restricted eating to prevent and manage chronic metabolic diseases. Annu Rev Nutr. 2019;39:291–315.
O’Flanagan CH, Smith LA, McDonell SB, Hursting SD. When less may be more: calorie restriction and response to cancer therapy. BMC Med. 2017;15:106.
Brandhorst S, Longo VD. Fasting and caloric restriction in cancer prevention and treatment. Recent Results Cancer Res. 2016;207:241–66.
Castejon M, Plaza A, Martinez-Romero J, Fernandez-Marcos PJ, Cabo R, Diaz-Ruiz A. Energy restriction and colorectal cancer: a call for additional research. Nutrients. 2020;12:114.
Zhang J, Deng Y, Khoo BL. Fasting to enhance cancer treatment in models: the next steps. J Biomed Sci. 2020;27:58.
Sadeghian M, Rahmani S, Khalesi S, Hejazi E. A review of fasting effects on the response of cancer to chemotherapy. Clin Nutr. 2021;40:1669–81.
Wei M, Brandhorst S, Shelehchi M, Mirzaei H, Cheng CW, Budniak J, Groshen S, Mack WJ, Guen E, Di Biase S, et al. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci Transl Med. 2017;9:eaai8700.
Safdie FM, Dorff T, Quinn D, Fontana L, Wei M, Lee C, Cohen P, Longo VD. Fasting and cancer treatment in humans: a case series report. Aging (Albany N Y). 2009;1:988–1007.
de Groot S, Vreeswijk MP, Welters MJ, Gravesteijn G, Boei JJ, Jochems A, Houtsma D, Putter H, van der Hoeven JJ, Nortier JW, et al. The effects of short-term fasting on tolerance to (neo) adjuvant chemotherapy in HER2-negative breast cancer patients: a randomized pilot study. BMC Cancer. 2015;15:652.
Dorff TB, Groshen S, Garcia A, Shah M, Tsao-Wei D, Pham H, Cheng CW, Brandhorst S, Cohen P, Wei M, et al. Safety and feasibility of fasting in combination with platinum-based chemotherapy. BMC Cancer. 2016;16:360.
Bauersfeld SP, Kessler CS, Wischnewsky M, Jaensch A, Steckhan N, Stange R, Kunz B, Bruckner B, Sehouli J, Michalsen A. The effects of short-term fasting on quality of life and tolerance to chemotherapy in patients with breast and ovarian cancer: a randomized cross-over pilot study. BMC Cancer. 2018;18:476.
Tahara Y, Aoyama S, Shibata S. The mammalian circadian clock and its entrainment by stress and exercise. J Physiol Sci. 2017;67:1–10.
Sato S, Basse AL, Schonke M, Chen S, Samad M, Altintas A, Laker RC, Dalbram E, Barres R, Baldi P, et al. Time of exercise specifies the impact on muscle metabolic pathways and systemic energy homeostasis. Cell Metab. 2019;30:92-110.e4.
Choi Y, Cho J, No MH, Heo JW, Cho EJ, Chang E, Park DH, Kang JH, Kwak HB. Re-setting the circadian clock using exercise against sarcopenia. Int J Mol Sci. 2020;21:3106.
Garcia-Ortiz L, Recio-Rodriguez JI, Martin-Cantera C, Cabrejas-Sanchez A, Gomez-Arranz A, Gonzalez-Viejo N, Iturregui-San Nicolas E, Patino-Alonso MC, Gomez-Marcos MA, Group E. Physical exercise, fitness and dietary pattern and their relationship with circadian blood pressure pattern, augmentation index and endothelial dysfunction biological markers: EVIDENT study protocol. BMC Public Health. 2010;10:233.
Matteucci E, Consani C, Masoni MC, Giampietro O. Circadian blood pressure variability in type 1 diabetes subjects and their nondiabetic siblings—influence of erythrocyte electron transfer. Cardiovasc Diabetol. 2010;9:61.
Chen HM, Cheung DST, Lin YY, Wu YC, Liu CY, Lin KC, Lin YJ, Lin CC. Relationships of exercise timing with sleep, fatigue and rest-activity rhythms of lung cancer patients in Taiwan: an exploratory study. Eur J Cancer Care (Engl). 2020;29:e13233.
Gabriel BM, Zierath JR. Circadian rhythms and exercise—re-setting the clock in metabolic disease. Nat Rev Endocrinol. 2019;15:197–206.
Yamanaka Y, Hashimoto S, Takasu NN, Tanahashi Y, Nishide SY, Honma S, Honma K. Morning and evening physical exercise differentially regulate the autonomic nervous system during nocturnal sleep in humans. Am J Physiol Regul Integr Comp Physiol. 2015;309:R1112–21.
Tong CKW, Lau B, Davis MK. Exercise training for cancer survivors. Curr Treat Options Oncol. 2020;21:53.
Cavalheri V, Granger CL. Exercise training as part of lung cancer therapy. Respirology. 2020;25(Suppl 2):80–7.
Cavalheri V, Tahirah F, Nonoyama M, Jenkins S, Hill K. Exercise training undertaken by people within 12 months of lung resection for non-small cell lung cancer. Cochrane Database Syst Rev. 2013;6:CD009955.
Idorn M, Thor SP. Exercise and cancer: from “healthy” to “therapeutic”? Cancer Immunol Immunother. 2017;66:667–71.
Holmen Olofsson G, Jensen AWP, Idorn M, Thor Straten P. Exercise oncology and immuno-oncology; a (future) dynamic duo. Int J Mol Sci. 2020;21:3816.
Saner NJ, Lee MJ, Kuang J, Pitchford NW, Roach GD, Garnham A, Genders AJ, Stokes T, Schroder EA, Huo Z, et al. Exercise mitigates sleep-loss-induced changes in glucose tolerance, mitochondrial function, sarcoplasmic protein synthesis, and diurnal rhythms. Mol Metab. 2021;43:101110.
Wyld L, Audisio RA, Poston GJ. The evolution of cancer surgery and future perspectives. Nat Rev Clin Oncol. 2015;12:115–24.
Wilson DJ. Exercise for the patient after breast cancer surgery. Semin Oncol Nurs. 2017;33:98–105.
Avancini A, Sartori G, Gkountakos A, Casali M, Trestini I, Tregnago D, Bria E, Jones LW, Milella M, Lanza M, et al. Physical activity and exercise in lung cancer care: will promises be fulfilled? Oncologist. 2020;25:e555–69.
Bourke L, Smith D, Steed L, Hooper R, Carter A, Catto J, Albertsen PC, Tombal B, Payne HA, Rosario DJ. Exercise for men with prostate cancer: a systematic review and meta-analysis. Eur Urol. 2016;69:693–703.
Yang A, Sokolof J, Gulati A. The effect of preoperative exercise on upper extremity recovery following breast cancer surgery: a systematic review. Int J Rehabil Res. 2018;41:189–96.
Rosero ID, Ramirez-Velez R, Lucia A, Martinez-Velilla N, Santos-Lozano A, Valenzuela PL, Morilla I, Izquierdo M. Systematic review and meta-analysis of randomized, controlled trials on preoperative physical exercise interventions in patients with non-small-cell lung cancer. Cancers (Basel). 2019;11:944.
Cavalheri V, Granger C. Preoperative exercise training for patients with non-small cell lung cancer. Cochrane Database Syst Rev. 2017;6:CD012020.
Ni HJ, Pudasaini B, Yuan XT, Li HF, Shi L, Yuan P. Exercise training for patients pre- and postsurgically treated for non-small cell lung cancer: a systematic review and meta-analysis. Integr Cancer Ther. 2017;16:63–73.
Cavalheri V, Tahirah F, Nonoyama M, Jenkins S, Hill K. Exercise training for people following lung resection for non-small cell lung cancer—a Cochrane systematic review. Cancer Treat Rev. 2014;40:585–94.
Kilgour RD, Jones DH, Keyserlingk JR. Effectiveness of a self-administered, home-based exercise rehabilitation program for women following a modified radical mastectomy and axillary node dissection: a preliminary study. Breast Cancer Res Treat. 2008;109:285–95.
Scaffidi M, Vulpiani MC, Vetrano M, Conforti F, Marchetti MR, Bonifacino A, Marchetti P, Saraceni VM, Ferretti A. Early rehabilitation reduces the onset of complications in the upper limb following breast cancer surgery. Eur J Phys Rehabil Med. 2012;48:601–11.
Cavalheri V, Burtin C, Formico VR, Nonoyama ML, Jenkins S, Spruit MA, Hill K. Exercise training undertaken by people within 12 months of lung resection for non-small cell lung cancer. Cochrane Database Syst Rev. 2019;6:CD009955.
Allen C, Her S, Jaffray DA. Radiotherapy for cancer: present and future. Adv Drug Deliv Rev. 2017;109:1–2.
Citrin DE. Recent developments in radiotherapy. N Engl J Med. 2017;377:1065–75.
Schumacher O, Galvao DA, Taaffe DR, Chee R, Spry N, Newton RU. Exercise modulation of tumour perfusion and hypoxia to improve radiotherapy response in prostate cancer. Prostate Cancer Prostatic Dis. 2021;24:1–14.
Segal RJ, Reid RD, Courneya KS, Sigal RJ, Kenny GP, Prud’Homme DG, Malone SC, Wells GA, Scott CG, Slovinec D’Angelo ME. Randomized controlled trial of resistance or aerobic exercise in men receiving radiation therapy for prostate cancer. J Clin Oncol. 2009;27:344–51.
Lipsett A, Barrett S, Haruna F, Mustian K, O’Donovan A. The impact of exercise during adjuvant radiotherapy for breast cancer on fatigue and quality of life: a systematic review and meta-analysis. Breast. 2017;32:144–55.
Kapur G, Windsor PM, McCowan C. The effect of aerobic exercise on treatment-related acute toxicity in men receiving radical external beam radiotherapy for localised prostate cancer. Eur J Cancer Care (Engl). 2010;19:643–7.
Dufresne S, Gueritat J, Chiavassa S, Noblet C, Assi M, Rioux-Leclercq N, Rannou-Bekono F, Lefeuvre-Orfila L, Paris F, Rebillard A. Exercise training improves radiotherapy efficiency in a murine model of prostate cancer. FASEB J. 2020;34:4984–96.
Esteves M, Monteiro MP, Duarte JA. Role of regular physical exercise in tumor vasculature: favorable modulator of tumor milieu. Int J Sports Med. 2021;42:389–406.
McCullough DJ, Stabley JN, Siemann DW, Behnke BJ. Modulation of blood flow, hypoxia, and vascular function in orthotopic prostate tumors during exercise. J Natl Cancer Inst. 2014;106:dju036.
Schadler KL, Thomas NJ, Galie PA, Bhang DH, Roby KC, Addai P, Till JE, Sturgeon K, Zaslavsky A, Chen CS, et al. Tumor vessel normalization after aerobic exercise enhances chemotherapeutic efficacy. Oncotarget. 2016;7:65429–40.
Garcia E, Becker VG, McCullough DJ, Stabley JN, Gittemeier EM, Opoku-Acheampong AB, Sieman DW, Behnke BJ. Blood flow responses to mild-intensity exercise in ectopic vs. orthotopic prostate tumors; dependence upon host tissue hemodynamics and vascular reactivity. J Appl Physiol. 2016;121:15–24.
Jones LW, Dewhirst MW. Therapeutic properties of aerobic training after a cancer diagnosis: more than a one-trick pony? J Natl Cancer Inst. 2014;106:dju042.
Jones LW, Eves ND, Courneya KS, Chiu BK, Baracos VE, Hanson J, Johnson L, Mackey JR. Effects of exercise training on antitumor efficacy of doxorubicin in MDA-MB-231 breast cancer xenografts. Clin Cancer Res. 2005;11:6695–8.
Florez Bedoya CA, Cardoso ACF, Parker N, Ngo-Huang A, Petzel MQ, Kim MP, Fogelman D, Romero SG, Wang H, Park M, et al. Exercise during preoperative therapy increases tumor vascularity in pancreatic tumor patients. Sci Rep. 2019;9:13966.
Ruan W, Yuan X, Eltzschig HK. Circadian rhythm as a therapeutic target. Nat Rev Drug Discov. 2021;20:287–307.
Crnko S, Du Pre BC, Sluijter JPG, Van Laake LW. Circadian rhythms and the molecular clock in cardiovascular biology and disease. Nat Rev Cardiol. 2019;16:437–47.
Allada R, Bass J. Circadian Mechanisms in Medicine. N Engl J Med. 2021;384:550–61.
Koronowski KB, Sassone-Corsi P. Communicating clocks shape circadian homeostasis. Science. 2021;371:eabd0951.
Sancar A, Lindsey-Boltz LA, Gaddameedhi S, Selby CP, Ye R, Chiou YY, Kemp MG, Hu J, Lee JH, Ozturk N. Circadian clock, cancer, and chemotherapy. Biochemistry. 2015;54:110–23.
Hrushesky WJ. Circadian timing of cancer chemotherapy. Science. 1985;228:73–5.
Hrushesky WJ, Bjarnason GA. Circadian cancer therapy. J Clin Oncol. 1993;11:1403–17.
Levi F, Benavides M, Chevelle C, Le Saunier F, Bailleul F, Misset JL, Regensberg C, Vannetzel JM, Reinberg A, Mathe G. Chemotherapy of advanced ovarian cancer with 4’-O-tetrahydropyranyl doxorubicin and cisplatin: a randomized phase II trial with an evaluation of circadian timing and dose-intensity. J Clin Oncol. 1990;8:705–14.
Lockhart AC, Bauer TM, Aggarwal C, Lee CB, Harvey RD, Cohen RB, Sedarati F, Nip TK, Faessel H, Dash AB, et al. Phase Ib study of pevonedistat, a NEDD8-activating enzyme inhibitor, in combination with docetaxel, carboplatin and paclitaxel, or gemcitabine, in patients with advanced solid tumors. Invest New Drugs. 2019;37:87–97.
Faessel HM, Mould DR, Zhou X, Faller DV, Sedarati F, Venkatakrishnan K. Population pharmacokinetics of pevonedistat alone or in combination with standard of care in patients with solid tumours or haematological malignancies. Br J Clin Pharmacol. 2019;85:2568–79.
Focan C, Levi F, Kreutz F, Focan-Henrard D, Lobelle JP, Adam R, Dallemagne B, Jehaes C, Markiewicz S, Weerts J, et al. Continuous delivery of venous 5-fluorouracil and arterial 5-fluorodeoxyuridine for hepatic metastases from colorectal cancer: feasibility and tolerance in a randomized phase II trial comparing flat versus chronomodulated infusion. Anticancer Drugs. 1999;10:385–92.
Price TJ, Ross PJ, Hickish T, Tait D, Norman AR, Ford HE, Middleton G, Sumpter K, Hill M, Oates J, et al. Phase III study of mitomycin-C with protracted venous infusion or circadian-timed infusion of 5-fluorouracil in advanced colorectal carcinoma. Clin Colorectal Cancer. 2004;3:235–42.
Coudert B, Focan C, Genet D, Giacchetti S, Cvickovic F, Zambelli A, Fillet G, Chollet P, Amoroso D, Van Der Auwera J, et al. A randomized multicenter study of optimal circadian time of vinorelbine combined with chronomodulated 5-fluorouracil in pretreated metastatic breast cancer patients: EORTC trial 05971. Chronobiol Int. 2008;25:680–96.
Escudier B, Roigas J, Gillessen S, Harmenberg U, Srinivas S, Mulder SF, Fountzilas G, Peschel C, Flodgren P, Maneval EC, et al. Phase II study of sunitinib administered in a continuous once-daily dosing regimen in patients with cytokine-refractory metastatic renal cell carcinoma. J Clin Oncol. 2009;27:4068–75.
Qvortrup C, Jensen BV, Fokstuen T, Nielsen SE, Keldsen N, Glimelius B, Bjerregaard B, Mejer J, Larsen FO, Pfeiffer P. A randomized study comparing short-time infusion of oxaliplatin in combination with capecitabine XELOX(30) and chronomodulated XELOX(30) as first-line therapy in patients with advanced colorectal cancer. Ann Oncol. 2010;21:87–91.
Li J, Chen R, Ji M, Zou SL, Zhu LN. Cisplatin-based chronotherapy for advanced non-small cell lung cancer patients: a randomized controlled study and its pharmacokinetics analysis. Cancer Chemother Pharmacol. 2015;76:651–5.
Re GL, Santeufemia DA, Re FL, Bortolus R, Doretto P, Marus W, Buttazzi L, Lenardon O, Falda A, Piazza R, et al. Interleukin-2 chronotherapy for metastatic renal cell carcinoma: Results of a phase I–II study. Cytokine. 2020;128:154984.
Roosendaal J, Jacobs BAW, Pluim D, Rosing H, de Vries N, van Werkhoven E, Nuijen B, Beijnen JH, Huitema ADR, Schellens JHM, et al. Phase I pharmacological study of continuous chronomodulated capecitabine treatment. Pharm Res. 2020;37:89.
Levi F, Karaboue A, Gorden L, Innominato PF, Saffroy R, Giacchetti S, Hauteville D, Guettier C, Adam R, Bouchahda M. Cetuximab and circadian chronomodulated chemotherapy as salvage treatment for metastatic colorectal cancer (mCRC): safety, efficacy and improved secondary surgical resectability. Cancer Chemother Pharmacol. 2011;67:339–48.
Farker K, Merkel U, Wedding U, Hippius M, Hoffken K, Hoffmann A. Chronomodulated chemotherapy with oxaliplatin, 5-FU and sodium folinate in metastatic gastrointestinal cancer patients: original analysis of non-hematological toxicity and patient characteristics in a pilot investigation. Int J Clin Pharmacol Ther. 2006;44:31–7.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from National Key Research and Development Program of China (2020YFA0509400, 2019YFC1709101), Guangdong Basic and Applied Basic Research Foundation (2019B030302012), 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYJC21004), National Natural Science Foundation of China (81821002, 82130082, 81790251, 82003098 and 82103168), the Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (ZYYCXTD-D-202003), and Sichuan Science and Technology Program (2019YFH0108, 22GJHZ0007).
Author information
Authors and Affiliations
Contributions
YT, WZ, and CH designed this manuscript. LZ and ZZ drafted the manuscript and drew the figures. EN revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have read and approved the final manuscript.
Competing interests
The authors declared that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, L., Zhang, Z., Nice, E. et al. Circadian rhythms and cancers: the intrinsic links and therapeutic potentials. J Hematol Oncol 15, 21 (2022). https://doi.org/10.1186/s13045-022-01238-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-022-01238-y