
Abstract
Gastrointestinal stromal tumor (GIST) is the most common mesenchymal tumor of the gastrointestinal tract. The majority of GISTs harbor gain of function mutations in either KIT or PDGFRα. Determination of the GIST molecular subtype upon diagnosis is important because this information informs therapeutic decisions in both the adjuvant and metastatic setting. The management of GIST was revolutionized by the introduction of imatinib, a KIT inhibitor, which has become the standard first line treatment for metastatic GIST. However, despite a clinical benefit rate of 80%, the majority of patients with GIST experience disease progression after 2–3 years of imatinib therapy. Second and third line options include sunitinib and regorafenib, respectively, and yield low response rates and limited clinical benefit. There have been recent FDA approvals for GIST including ripretinib in the fourth-line setting and avapritinib for PDGFRA exon 18-mutant GIST. This article aims to review the optimal treatment approach for the management of patients with advanced GIST. It examines the standard treatment options available but also explores the novel treatment approaches in the setting of imatinib refractory GIST.
Similar content being viewed by others

Background
Gastrointestinal stromal tumor (GIST) is the most common soft tissue sarcoma subtype. The incidence of GIST is between 10 and 15 cases per million worldwide and ~ 5000 in the USA [1, 2]. The median age at diagnosis is 66–69 years, with equal distribution of men and women [1, 2]. GIST originates from the interstitial cells of cajal (ICCs), and it can arise from any part of the gastrointestinal tract, most commonly in the stomach (55.6%), followed by the small bowel (31.8%), colorectum (6%), other locations (5.5%) and the esophagus (0.7%) [1].
The diagnosis of GIST relies on the combination of the clinical scenario, the tumor’s anatomic location, the immunohistochemistry (IHC) patterns, as well as molecular features. The majority of GIST are immunohistochemically positive for KIT (CD117) and DOG-1 [3]. Other IHC markers frequently expressed in GIST include CD34 antigen (70%), smooth muscle actin (SMA, 30–40%), S100 protein (10%) and desmin (< 5%) [6].
Molecular classification of GIST
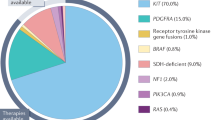
The majority of GIST (75–80%) harbor gain of function KIT mutations (Fig. 1). Exon 11 of KIT is the most frequently mutated region, affecting approximately two-thirds of GIST. In-frame deletions in KIT exon 11, particularly those involving codons 557 and 558, are associated with a worse prognosis compared to KIT exon 11 point mutations [4]. Mutations in KIT exon 9 occur in approximately 8–10% of GIST and are most commonly associated with small or large bowel tumors. Primary mutations in KIT exons 13, 17 and 18 are rare. Platelet-derived growth factor receptor alpha (PDGFRA)-mutant GIST represents the next most common molecular subtype, occurring in approximately 10% of GIST and generally arises in the stomach [5]. Exon 18 of PDGFRA is the most frequently mutated region, affecting approximately 8% of GIST. PDGFRA exon 18 D842V mutations account for 70% of PDGFRA mutant cases [7]. Rarely PDGFRA mutations occur within exon 12 or 14 [5].

Molecular sub-classification of GIST
Approximately 10–15% of GISTs are KIT and PDGFRA wild-type and are associated with genetic alterations in RAS-MAPK pathway (gain-of-function RAS/BRAF mutations or loss of function neurofibromatosis type 1 [NF1] mutations) or succinate dehydrogenase (SDHA/B/C/D) deficiency. SDHA/B/C/D deficiency GIST may be caused by a germline inactivating mutation in the suppressor genes encoding the SDH complex (SDHA, SDHB, SDHC and SDHD subunits) or by SDHC promotor-specific CpG island hypermethylation (SDHC epimutation) [5]. Other new molecular sub-classifications within GIST are oncogenic RTK translocation associated GIST or quadruple wild-type GISTs and quintuple wild-type GIST which refer to GISTs that are devoid of mutations in KIT/PDGFRA/RAS-MAPK pathway/SDH complex and oncogenic RTK translocations [5,6,7].
GISTs are generally resistant to conventional chemotherapy. The survival of metastatic GIST has greatly improved since 2002, when the US Food and Drug Administration (FDA) approved imatinib mesylate [8]. KIT exon 11 and 9 mutant GISTs are sensitive to imatinib. KIT exon 11 mutant GIST yields significantly higher response rates to imatinib and has longer overall survival than those with KIT exon 9 mutant or KIT/PDGFRA wild type GIST [9]. PDGFRA exon 18 D842V mutant GIST is resistant to imatinib. Avapritinib was recently FDA approved for this indication. Beyond this, a clinical trial should always be considered. Other PDGFRA mutant GISTs are generally responsive to imatinib. KIT and PDGFRA wild-type GIST have no standard/effective therapeutic alternative; therefore, clinical trial options should always be considered [5].
Knowledge of the molecular landscape in GIST is important because it provides prognostic information but also guides therapeutic selection. This review aims to discuss the management of advanced GIST focusing on the standard-of-care therapeutic options and novel therapeutics in clinical investigation.
Medical management of advanced GIST
Imatinib-sensitive GIST: first line therapy
Imatinib is a selective, small molecule inhibitor of three receptor tyrosine kinases: the transmembrane receptor KIT, the chimeric BCR-ABL fusion oncoprotein of chronic myeloid leukemia and PDGFRA. In 2002, the FDA approved imatinib for the management of patients with advanced GIST. This accelerated approval was based on the results of a multi-center, randomized study that evaluated the safety and efficacy of imatinib at two dose levels (either 400 mg and 600 mg daily) in 147 patients with advanced GIST [8]. The clinical benefit rate of imatinib was 81%; with 53.7% of patients achieving a RECIST partial response. The median time to an objective response was 3 months. Almost 14% of patients had early progressive disease despite imatinib therapy. No complete responses were observed. The majority of patients experienced mild to moderate adverse effects. The most common adverse events included edema (74%, frequently periorbital in location), nausea (52%), diarrhea (45%), myalgia or musculoskeletal pain (40%), fatigue (35%), dermatitis or rash (31%), headache (26%) and abdominal pain (26%).
The optimal imatinib dose (400 mg daily) to use in the first line setting was determined in two-phase III trials that randomized patients with advanced GIST to receive imatinib at 400 mg or 800 mg daily. The EORTC 62005 trial enrolled 946 patients. The higher imatinib dose arm yielded a statistically significant progression free survival (PFS) rate of 56% versus 50% for the lower dose arm, (estimated hazard ratio 0.82 [95% CI 0.69–0.98]; p = 0.026) [10]. The overall response rates were similar between the two treatment arms. The higher dose arm was associated with greater toxicity. The SWOG S0033/CALGB 15105 trial enrolled 746 patients. After a median follow-up of 4.5 years, no significant difference was observed with respect to median PFS (18 vs. 20 months), median OS (55 vs. 51 months) and objective response rate (40% vs. 42%), between the standard dose and the higher dose arms, respectively [11].
In the EORTC 62005 trial, the strongest prognostic factor of risk for progression and death was the presence of a KIT exon 9 mutation. In patients with KIT exon 9 disease, the PFS was significantly longer in patients that received double-dose imatinib compared to those that received the standard dose. For patients that crossed over to double-dose imatinib, the response rates observed were significantly higher among patients with KIT exon 9 (57%) compared to KIT exon 11 (7%) mutant disease. The phase III SWOG S0033/CALGB 15105 trial failed to demonstrate a PFS benefit favoring the KIT exon 9 mutant cohort that received the higher dose imatinib. However, it did show improved response rates in the KIT exon 9 mutant cohort compared to the KIT exon 11 mutant cohort that crossed over to receive double-dose imatinib (67% vs. 17%, respectively). A preplanned meta-analysis of both the EORTC 62005 and SWOG S0033/CALGB 15105 studies confirmed a PFS advantage in favor of patients with KIT exon 9 mutant disease who received double-dose imatinib (400 mg, twice daily) compared to standard dose [12]. As a result, the standard dose for patients with KIT exon 9 mutant advanced GIST receiving first-line imatinib is 400 mg twice daily.
Imatinib refractory GIST
Sunitinib is another small molecule, receptor TKI. It targets KIT/PDGFRα, colony stimulating factor 1 receptor (CSF1R), Fms-related tyrosine kinase 3, RET and has anti-angiogenic activity by inhibiting vascular endothelial growth factor receptors (VEGFR1-3). An early phase study provided safety and promising efficacy data for sunitinib (50 mg, 4 weeks on, 2 weeks off schedule). This led to a randomized, double-blind, placebo-controlled, phase III trial of sunitinib in patients with advanced, imatinib refractory/intolerant GIST. The objective response rate with sunitinib was 7% (RECIST partial response). However, the study demonstrated a significantly longer time to progression, based on RECIST criteria, of 27.3 weeks versus 6.4 weeks in the placebo arm; p < 0.0001) [13]. FDA approved sunitinib in the management of advanced GIST in 2006. Another phase II study examined the benefit of an alternative dosing schedule of sunitinib delivered on a continuous, daily basis (37.5 mg once daily) to patients with advanced, imatinib refractory GIST. The clinical benefit rate was 53%, and median PFS was 34 weeks. Most adverse events encountered were ≤ grade 2 in severity and manageable. Morning or evening dosing was comparable with respect to efficacy and safety [14]. A continuous daily dosing schedule of sunitinib is commonly used to treat patients with advanced GIST.
The GIST molecular genotype has been shown to correlate with efficacy of sunitinib. Higher response rates were observed among GIST patients treated with sunitinib that harbored primary KIT exon 9 mutations compared to KIT exon 11 mutations (58% vs. 34%, respectively). Among patients with advanced GIST treated with sunitinib alone, longer PFS and OS were seen for those with exon 13 or 14 secondary KIT mutations compared to those with exon 17 or 18 secondary KIT mutations [15].
Regorafenib is another oral multi-targeted kinase inhibitor that targets a number of pathways involved in tumor growth including tumor angiogenesis (VEGFR-1, -2, and -3, and TIE2), oncogenesis (KIT, RET, RAF-1, BRAF and BRAFV600E), and the tumor microenvironment [PDGFR and fibroblast growth factor receptor (FGFR)]. The safety of regorafenib was first demonstrated in a phase I study that enrolled an unselected population of patients with solid tumors. This study confirmed the recommended phase II dose of 160 mg daily (3 weeks on, 1 week off schedule) [16]. A phase II GIST specific study investigated the efficacy of regorafenib among 34 patients with advanced GIST who had previously progressed on at least imatinib and sunitinib. The clinical benefit rate observed was 79% (4 patients achieved a partial response and 22 patients had stable disease at ≥ 16 weeks). The median PFS was 10 months [17]. Regorafenib was subsequently investigated in a phase III study that randomized 240 patients with advanced GIST to receive regorafenib versus placebo plus best supportive care in a 2:1 fashion. The study met its primary endpoint by demonstrating a significant improvement in median PFS (4.8 months and 0.9 months, respectively (hazard ratio [HR] 0.27, 95% confidence interval [CI] 0.19–0.39; p < 0·0001). A best response of partial response or stable disease was observed in 75.9% on regorafenib and 34.8% in the placebo arm. The disease control rate was also higher in the regorafenib arm compared to placebo, (52.6% and 9.1%, respectively) [18]. In 2013, the FDA approved regorafenib for use in the management of patients with advanced GIST refractory to imatinib and sunitinib.
A recent phase Ib trial tested the efficacy of rapid alternation of sunitinib and regorafenib: 3 days of sunitinib followed by 4 days of regorafenib, and repeated. Toxicity was encountered at the previously determined optimal doses of both agents. The recommended phase II dose was identified as sunitinib 37.5 mg and regorafenib 120 mg. Stable disease was the best response observed in 4 out of 14 participants. The median PFS was 1.9 months. Rapid alternation of tyrosine kinase inhibitors (TKIs) was an unprecedented investigational approach in GIST that may prove effective when drugs with complementary pharmacokinetics are combined in an effort to minimize toxicity while allowing optimal drug doses to be used [19].
Ripretinib is a potent switch pocket control inhibitor of KIT and PDGFR kinases and has activity against a broad range of mutations. The phase I study enrolled 142 patients with advanced GIST with KIT (95%) or PDGFRα (5%) mutations. The recommended phase II dose of ripretinib was 150 mg once daily. The ORR in second-, third- and fourth-line patients was 19.4%, 14.3% and 7.2%, respectively [20]. The INVICTUS, phase III, randomized, double-blind, placebo-controlled trial randomized 154 patients with advanced GIST in a 2:1 fashion to receive ripretinib or placebo. GIST patients had progressed on or were intolerant of imatinib, sunitinib and regorafenib. The study met its primary end point showing a significant improvement in median PFS favoring the ripretinib arm [6.3 months vs. 1·0 months (hazard ratio 0.15, 95% CI 0.09–0.25; p < 0.0001)]. The median OS was 15.1 months in the ripretinib group compared to 6·6 months in the placebo group (HR 0.36, 95% CI 0.21–0.62). Ripretinib yielded an objective response rate (ORR) of 9%. There was no response observed in the placebo arm. Ripretinib was generally well tolerated. The most common (≥ 20% of patients in the ripretinib group) treatment-related adverse events were alopecia, myalgia, nausea, fatigue, palmar–plantar erythrodysesthesia and diarrhea [21]. In May 2020, the US FDA approved ripretinib for patients with advanced GIST that had progressed on three or more receptor tyrosine kinase inhibitors including imatinib [22]. Intrigue, a phase III trial examining the efficacy of ripretinib compared to sunitinib in patients with advanced GIST who have progressed on or are intolerant of imatinib in the second line setting is currently ongoing [23].
Primary imatinib-resistant GIST/PDGFRA D842V-mutant GIST: first line therapy
PDGFRA-mutant GIST accounts for 5–10% of GIST and exhibits primary resistance to imatinib and sunitinib therapy. The median PFS of PDGFRα D842V mutant advanced GIST on placebo from retrospective analysis was reported to be 2.8 months [24]. Notably, the median PFS of unselected patients with GIST on placebo is less than 6 weeks [13, 18]. This highlights that PDGFRA D842V mutant GIST is clinically distinct from imatinib-sensitive disease. It often follows a more indolent disease course. Until January 2020, there was no standard of care systemic therapy available for this GIST molecular subtype and surgical resection was preferred in the setting of small volume disease progression. In a phase II trial, dasatinib yielded a response in one patient with PDGFRA D842V-mutant GIST. Based on this preliminary efficacy, dasatinib was added to NCCN compendium of approved agents for this molecular subtype [25].
Avapritinib is a potent, highly selective oral inhibitor of PDGFRα mutant kinases. The Navigator, phase I dose escalation/dose expansion study enrolled 231 patients with advanced GIST: PDGFRA exon 18 D842V mutant = 56 (24%), non-D842V mutant = 8 (4%) and KIT mutant disease = 167 (72%). In the PDGFRA D842V-mutant population, the ORR was 88%. There were five (9%) complete responses and 44 (79%) partial responses. The clinical benefit rate was 98%, which is remarkable in a GIST molecular subtype that was known to be refractory to all previously FDA-approved TKIs for GIST [26]. Avapritinib was generally tolerated. Cognitive effects emerged as an adverse event of special interest. Most cognitive effects, primarily driven by memory impairment, were grade 1 and generally manageable with dose modification. However, 8.3% of patients discontinued avapritinib for a treatment-related toxicity, of which 2% discontinued treatment because of intracranial bleeding [26]. The recommended phase II dose was determined to be 300 mg once daily and the FDA approved avapritinib in January 2020 for adults with inoperable GIST harboring a PDGFRA exon 18 mutations, including the D842V mutation.
Imatinib refractory GIST: alternative options
Enrollment in a clinical trial
Resistance to imatinib appears to confer a general resistance to non-selective TKIs. A number of novel therapeutic approaches to treat GIST are being investigated.
Next-generation selective tyrosine kinase inhibitors
Secondary KIT mutations are found in 50–67% of patients with secondary imatinib resistant, KIT mutant disease [27, 28]. They commonly arise in KIT exons 17, 13 and are less frequently observed in exons 18, 14 or within the PDGFRA gene. Next-generation TKIs targeting specific secondary KIT mutations are under investigation. Examples of selective TKIs include PLX3397, PLX9486 and AZD3229.
In preclinical models, PLX3397 appears to be a more potent KIT inhibitor than imatinib [29]. PLX9486 has selective activity against primary KIT mutations (exons 9 and 11) and activation loop mutations (exons 17 and 18). A phase I study of PLX9486 in GIST examined safety up to a dose of 1000 mg daily. The clinical benefit rate was 64%, and the median PFS was 6 months. Analysis of circulating tumor deoxyribonucleic acid (ctDNA) in this cohort confirmed the selectivity profile of PLX9486 with reductions in KIT ex 11, 17 and 18. PLX9486 (500 mg qd) was examined in combination with PLX3397 (600 mg qd). The median PFS for this combination was 6 months. Reduction in ctDNA levels of KIT exons 13 and 14 was not observed [30]. PLX9486 was also explored in combination with sunitinib creating synergistic potential against a broader range of KIT mutations by blocking active and inactive conformations. A total of 18 patients received this combination therapy. This was a heavily pre-treated cohort, 67% had received three or more prior lines of therapy. The maximum tolerated dose was not reached. The recommended phase II dose was PLX9486 1000 mg daily and sunitinib 37.5 mg once daily administered in 28 day cycles. The safety profile of the combination therapy was comparable to that of single agent sunitinib. The ORR was 20%. The median duration of response was 10 months. The median PFS for the combination therapy was 12 months [31, 32].
In preclinical models, avapritinib was highly active against imatinib-resistant patient derived GIST xenografts. The “Navigator,” phase I study of avapritinib demonstrated promising activity in ≥ 4th line patients (n = 109), 3rd line [regorafenib-naïve] (n = 23) and 2nd line non-D842V mutant GIST (n = 20) the ORR was 20%, 26% and 25%, respectively. The median duration of response was 7–10 months in the ≥ 4th line and 3rd line cohorts. Best response by mutational profile in the ≥ 4th line cohort failed to demonstrate activity in tumors harboring KIT V654A and T6701 mutations [33]. The “Voyager,” randomized, phase III study of avapritinib compared with regorafenib in 3rd or 4th line KIT/PDGRFA mutant GIST enrolled 476 patients [34]. In April 2020, a press release regarding the Voyager trial reported no statistically significant difference in median PFS between the avapritinib arm (4.2 months) and the regorafenib arm (5.6 months). The ORR was 17.1% in the avapritinib arm and 7.2% in the regorafenib arm.
AZD3229 is an oral, potent, pan-KIT/PDGFRα inhibitor. It has demonstrated activity in-vivo and in-vitro preclinical, GIST models across a broad range of primary and secondary KIT/PDGFRA mutations that confer resistance to standard of care agents [35]. This compound was designed to reduce activity against VEGFR-2 thereby, attempting to minimize off-target effects which often leads to sub-optimal dosing with standard of care agents used in GIST [36]. This agent demonstrates promising pre-clinical activity in GIST tumor models and merits investigation in the clinical setting.
Targeting ETV1
ETS variant transcription factor 1 (ETV1) is a lineage specific transcription survival factor that is highly expressed by GIST cells and their precursor cells the interstitial cells of cajal. ETV1 promotes the growth and survival of both the ICC and GIST. KIT mutant GIST results in constitutive activation of the MAP kinase pathway. This stabilizes the ETV1 transcriptional output, which promotes GIST tumorigenesis. ETV1 may also be critical for the survival and maintenance of the intrinsically imatinib-resistant, KIT-independent stem cell/progenitors in GIST, suggesting that this may be a therapeutic target to consider in KIT wild-type GIST [32]. Combination inhibition of MAP kinase and KIT signaling represents a promising therapeutic approach. In preclinical studies, the combination of imatinib and binimetinib destabilized the ETV1 protein resulting in greater suppression of GIST cell growth compared to either drug alone and also induced apoptosis [37].
The combination of imatinib and binimetinib was investigated in patients with advanced GIST of any genotype. The phase Ib/II trial demonstrated safety and identified the recommended phase II dose (imatinib 400 mg daily and binimetinib 30 mg twice daily). The phase II study met its primary endpoint by demonstrating an ORR of 66.7% (n = 26/39) in the first-line management of advanced GIST. The median duration of response was 30 months. Notably, the rate of conversion to resection was 88.9% (95% CI, 52–100%)] among 9 patients who had initially inoperable disease. The median PFS for the combination therapy was 29.9 months, and the median overall survival had not yet been reached [38]. These results compare very favorably with imatinib alone and warrant further evaluation in a larger, randomized, controlled, phase III trial (Table 1).
Immunotherapy
The role of the immune system has been explored in preclinical KIT mutant GIST mouse models. Imatinib treatment induces a dramatic increase in CD8 + T cell number and proliferation and produces an increased ratio of CD8 + effector T cell: CD4 + regulatory T cells. Imatinib-sensitive KIT mutant tumors contained greater frequencies of CD3 + and CD8 + T cells, but a lower percentage of CD4 + T cells and T regs compared to imatinib-resistant KIT mutant GIST tumor mouse models. The anti-tumor effects of imatinib are reduced in the setting of depleted CD8 + but not CD4 + T cells, natural killer (NK) cells or myeloid cells. This underlines the contribution of CD8 + effector T cells to the anti-tumor effect of imatinib. Imatinib has been shown to regulate the intratumoral T cells and immune microenvironment through inhibition Indolamine 2,3-dioxygenase 1 (IDO1) [39]. Given the role of CD8 + effector T cells in imatinib response, concurrent administration of imatinib and an immune checkpoint inhibitor (ipilimumab) were tested in KIT mutant GIST mouse models. The combination was shown to be synergistic and significantly reduced tumor size compared to either treatment alone [39].
This preclinical work led to a phase Ib study that examined the combination of dasatinib, a multi-TKI and ipilimumab, an anti-CTLA4 antibody, in patients with advanced sarcomas. Twenty patients had advanced GIST. The combination was shown to be safe, however, not synergistic in its effect. Many patients progressed prior to the first disease assessment at 3 months (median PFS was 2.8 months). The ORR was 0%. The study population was heavily pre-treated (median number of prior therapies = 3) [40]. Dasatinib monotherapy was previously shown to have limited benefit in this setting producing a median PFS of less than 2 months [41]. Preclinical studies previously discussed highlighted that imatinib’s immunostimulatory effect on the tumor immune microenvironment is greatest in the setting of imatinib-sensitive disease. Hence, dasatinib was recognized as a poor TKI choice for the first immunotherapy-based trial in GIST. Combination therapy with an ICI and an approved TKI in a TKI sensitive population warrants exploration.
Two-phase II trials have examined nivolumab with or without ipilimumab in advanced GIST. The first study enrolled 29 patients and demonstrated a clinical benefit rate of 50% (n = 8/16) in the nivolumab alone arm compared to 23% (n = 3/13) in the combination arm. A partial response was observed in the combination arm. The median PFS was 12.1 weeks in the nivolumab alone arm and 8.3 weeks in the doublet checkpoint inhibition arm [42]. The Alliance A091401 included an expansion cohort in GIST. This cohort demonstrated poor activity for immune checkpoint inhibition among 21 heavily pre-treated patients. The clinical benefit rate was ~ 10% in each arm. No objective responses were seen in either arm. The median PFS was 2.9 months in the doublet arm versus 1.5 month in the monotherapy arm [43].
Targeting actionable genomic alterations in GIST beyond known oncogenic drivers
Oncogenic activation of the Phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) pathway represents a downstream effector in the KIT signaling pathway. In preclinical GIST cell line studies, cell growth arrest resulted from PI3K inhibition, and to a lesser degree from MEK/MAPK and mTOR inhibition. In imatinib-resistant GIST cell lines, only PI3K inhibition was shown to halt GIST cell growth and produce apoptosis. Hence, this work postulated that targeting critical downstream signaling proteins in imatinib-resistant disease, such as PI3K, represented a promising therapeutic avenue to explore [44].` Knowledge of the genomic landscape of GIST continues to evolve. Next-generation sequencing is increasingly being utilized in cancer patients in an effort to identify genomic alterations that may represent potential therapeutic targets [45, 46]. Studies examining the molecular spectrum of GIST have identified actionable alterations in the PI3K kinase and mTOR pathways, albeit at relatively low frequencies [47].
A phase II study investigated the efficacy of the combination imatinib (600 mg daily)/everolimus (2.5 mg daily) in patients with advanced GIST. A total of 75 patients were enrolled into one of 2 cohorts stratified by line of treatment [36]. There was a high proportion of grade 3/4 adverse events observed in both cohorts [54–75%]. The ORR for this combination was low at 0% and 2% in cohort 1 and 2, respectively. The median PFS was 1.9 months and 3.5 months in cohort 1 and 2, respectively [48]. Early studies using mTOR inhibitors have shown limited success, which may be due to the activation of AKT that occurs following mammalian target of rapamycin complex 1 (mTORC1) inhibition. Therefore, targeting PI3K or AKT, which lie upstream of mTORC1, may translate into more complete pathway inhibition.
GIST xenograft preclinical studies have demonstrated enhanced activity for combination of a PI3K inhibitors with imatinib compared to either drug alone [49, 50]. A phase Ib trial of Imatinib and an oral PI3K inhibitor, buparlisib enrolled 60 patients with advanced GIST refractory to imatinib and sunitinib. The combination failed to generate an objective response and was not developed further [51].
The most frequently observed gene specific copy number alteration in GIST is within the cyclin-dependent kinase inhibitor 2A (CDKN2A) locus that encompasses the tumor suppressors p16 and p14 [47, 52]. French investigators evaluated the efficacy of palbociclib, a CDK4/6 inhibitor, in 71 patients with advanced imatinib- and sunitinib-refractory GIST, 29 had confirmed CDKN2A gene loss. An interim analysis revealed that 86% of evaluable patients experienced progression of disease at 4 months. CDKN2A status was not shown to correlate with survival or outcome to prior therapy [53].
Absence of an appropriate clinical trial
In the absence of an appropriate clinical trial, other therapeutic options include (1) an alternative TKI (off-label use), (2) re-challenge with imatinib or other TKI (3) the addition of an mTOR inhibitor to imatinib or sorafenib.
Alternative TKIs previously investigated in patients with advanced GIST
A number of other multi-tyrosine kinase inhibitors have been examined in the setting of imatinib refractory advanced disease. These studies were small phase II trials. The drugs investigated include sorafenib, pazopanib, cabozantinib, nilotinib, dasatinib, masitinib, linsitinib, dovitinib, vatalanib and ponatinib [34, 41, 54,55,56,57,58,59,60,61,62,63]. The response and survival outcomes reported in these studies are conveyed in Table 2. Some of these drugs are National Comprehensive Cancer Network (NCCN) compendium listed for the management of imatinib refractory advanced GIST. In the absence of a clinical trial, it is reasonable to consider one of these agents where available.
Re-challenge with Imatinib or other TKI
In the setting of limited progression, when standard and investigational therapies fail to control disease, consideration can be made to re-challenge with a TKI that was previously tolerated and effective. A retrospective analysis of 71 patients with advanced GIST refractory to imatinib, sunitinib and regorafenib, re-challenged with imatinib (400 mg daily) demonstrated a median time to progression of 5.4 months and overall survival of 10.6 months [64].
Notably, in the setting of progression on TKI therapy, discontinuation of therapy may result in accelerated tumor growth due to withdrawal of treatment for sensitive clones of the disease. Therefore, in the absence of a standard, investigational or clinical trial option, TKI therapy should be continued as an element of best supportive care [2].
Addition of an mTOR inhibitor to imatinib or other TKI.
As previously highlighted activation of the mTOR pathway is a downstream effector of KIT signaling. Hence, in the setting of imatinib refractory disease in the absence of standard or clinical trial options the addition of sirolimus to a TKI can be considered. Safety has been confirmed for the addition of sirolimus to sorafenib or sunitinib in patients with advanced cancer [65]. However, we do not have efficacy data for this treatment approach.
PDGFRA D842V-mutant GIST: investigational options
Crenolanib is a selective inhibitor of PDGFRα and FLT3 with nanomolar activity against PDGFRα D842V mutant GIST. A phase I study confirmed an objective response rate of 12.5% (2/16) and a clinical benefit rate of 31% (partial response, n = 2, stable disease, n = 3) in heavily pretreated patients with PDGFRα D842V mutant GIST. An international, randomized phase III trial of crenolanib versus placebo in 120 patients with PDGFRα D842V mutant GIST continues [66].
Biomarker development in GIST
Molecular classification of GIST is imperative to inform optimal therapeutic selection in the management of GIST. Testing GIST tumor tissue obtained from a surgical or biopsy procedure is the standard diagnostic approach used to determine the molecular landscape of GIST. Intra-tumoral heterogeneity within GIST limits the ability to detect the complete genomic spectrum from testing isolated tumor tissue samples. As previously outlined, the molecular spectrum of GIST may change following exposure to tyrosine kinase inhibition. Many novel therapies under investigation are selective for specific KIT/PDGFRα mutations. Hence, there is an emergent need to develop a noninvasive approach to molecularly characterize GIST in a serial manner. CtDNA extracted from plasma may overcome the limitations of tumor tissue. Sequenced ctDNA has detected primary and secondary mutations in patients with advanced GIST [67, 68]. The detection of ctDNA has been shown to correlate with tumor burden and response to treatment in GIST and other tumor types [69,70,71].
To date, studies examining sequenced ctDNA in GIST have been small and used older sequencing technologies. Sequenced ctDNA represents a candidate blood biomarker of GIST molecular behavior and treatment response that warrants further investigation and validation in a prospective manner. Several clinical trials are incorporating sequencing ctDNA material in an effort to add to this evolving area of research.
An exploratory analysis of ctDNA extracted from patients with PDGFRA D842V-mutant GIST enrolled in the phase 1 study of avapritinib was presented at the European Society of Medical Oncology annual meeting in 2018. They found that baseline ctDNA levels were the strongest independent baseline predictive indicator of PFS over established predictive indicators including Eastern Cooperative Oncology Group (ECOG) performance status, lesion size and age. Moreover, in the majority of patients with PDGFRα D842V-mutant GIST treated with avapritinib, ctDNA levels fell below the limit of detection (0.05%) by two months on treatment and large declines in on-treatment ctDNA levels were associated with high baseline ctDNA, an independent risk factor for progression. These data indicate that baseline ctDNA levels may have utility as a predictive biomarker in patients with advanced GIST; however, further research is warranted to understand the role and utility of sequenced ctDNA in the management of advanced GIST [72].
Conclusion and future directions
The management of GIST was revolutionized in 2001 following the introduction of imatinib. Since then the landscape of advanced KIT- and PDGFRA-mutant GIST management has evolved to include second-, third- and fourth-line TKIs, sunitinib, regorafenib and ripretinib, as well as avapritinib for advanced PDGFRA D842V mutant GIST. The obvious clinical challenges that we face now are how to treat patients who have progressed on ripretinib fourth-line therapy, and how to leverage molecular data to optimize the treatment paradigm.
While there has been tremendous progress in the clinical management of advanced GIST, some fundamental challenges remain: (1) imatinib resistance. For the majority (90%+) of patients, they derive clinical benefit to front-line imatinib therapy, but this is not indefinite. Once their disease develops resistance to imatinib, subsequent lines of TKI therapies, including newer generations of TKIs, have limited clinical benefit. Hence, the next phase of clinical investigations should focus on intervening early and developing therapeutic strategies to transform TKI-mediated cytostatic effects to cytotoxic effects. Some of these strategies will involve novel combination therapeutic strategies of targeting different survival pathways for synergistic cell killing effects, and novel therapeutic targets, such as the tumor microenvironment including immune and non-immune stroma and vasculature compartments.
Furthermore, there are significant unmet needs for the subset of patients with KIT/PDGFRA-wild type GISTs. These include SDH-deficient GIST mainly affecting pediatric and young adult population, NF1-mutant GIST both sporadic and syndromic (associated with type I neurofibromatosis), RAS-mutant, FGFR mutant GISTs and others. These groups of patients shall be considered differently; and therapeutics development shall be tailored to their particular genetic defects and potential molecular vulnerabilities.
Lastly, a multidisciplinary approach to the management of GIST within a dedicated sarcoma center is the ideal. The addition of surgical resection, radiation therapy and local radiological interventional options to TKI therapy may be appropriate in certain situations but requires careful selection. Clinical trials exploring novel investigational therapies should always been considered.
Availability of data and materials
Not applicable.
Abbreviations
- GIST:
-
Gastrointestinal stromal tumors
- ICCs:
-
Interstitial cells of cajal
- IHC:
-
Immunohistochemistry
- CD117:
-
Cluster of differentiation 117
- DOG-1:
-
Discovered on GIST-1
- PDGFRA:
-
Platelet-derived growth factor receptor alpha
- NF1:
-
Neurofibromatosis type 1
- SDH:
-
Succinate dehydrogenase
- RTK:
-
Receptor tyrosine kinase
- FDA:
-
Food and Drug Administration
- EORTC:
-
European Organization for Research and Treatment of Cancer
- SWOG:
-
Southwest Oncology Group
- CALGB:
-
Cancer and Leukemia Group B
- PFS:
-
Progression free survival
- CSF1R:
-
Colony stimulating factor 1 receptor
- RECIST:
-
Response evaluation criteria in solid tumors
- TKIs:
-
Tyrosine kinase inhibitors
- ORR:
-
Objective response rate
- ctDNA:
-
Circulating tumor deoxyribonucleic acid
- ETV1:
-
ETS variant transcription factor 1
- NK:
-
Natural killer
- IDO1:
-
Indolamine 2:3-dioxygenase 1
- CTLA4:
-
Cytotoxic T-lymphocyte-associated protein 4
- PI3K:
-
Phosphoinositide 3-kinase
- AKT:
-
Protein kinase B
- mTOR:
-
Mammalian target of rapamycin
- mTORC1:
-
Mammalian target of rapamycin complex 1
- CDKN2A:
-
Cyclin-dependent kinase inhibitor 2A
- NCCN :
-
National Comprehensive Cancer Network
- ECOG:
-
Eastern Cooperative Oncology Group
References
Soreide K, Sandvik OM, Soreide JA, Giljaca V, Jureckova A, Bulusu VR. Global epidemiology of gastrointestinal stromal tumours (GIST): a systematic review of population-based cohort studies. Cancer Epidemiol. 2016;40:39–46.
Demetri GD, von Mehren M, Antonescu CR, DeMatteo RP, Ganjoo KN, Maki RG, et al. NCCN Task Force report: update on the management of patients with gastrointestinal stromal tumors. J Natl Comprehens Cancer Netw JNCCN. 2010;8 Suppl 2:S1–41; quiz S2–4.
Miettinen M, Wang ZF, Lasota J. DOG1 antibody in the differential diagnosis of gastrointestinal stromal tumors: a study of 1840 cases. Am J Surg Pathol. 2009;33(9):1401–8.
Martín J, Poveda A, Llombart-Bosch A, Ramos R, López-Guerrero JA, García del Muro J, et al. Deletions affecting codons 557–558 of the c-KIT gene indicate a poor prognosis in patients with completely resected gastrointestinal stromal tumors: a study by the Spanish Group for Sarcoma Research (GEIS). J Clin Oncol. 2005;23(25):6190–8.
Bannon AE, Klug LR, Corless CL, Heinrich MC. Using molecular diagnostic testing to personalize the treatment of patients with gastrointestinal stromal tumors. Expert Rev Mol Diagnost. 2017;17(5):445–57.
Shi E, Chmielecki J, Tang CM, Wang K, Heinrich MC, Kang G, et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J Transl Med. 2016;14(1):339.
Brenca M, Rossi S, Polano M, Gasparotto D, Zanatta L, Racanelli D, et al. Transcriptome sequencing identifies ETV6-NTRK3 as a gene fusion involved in GIST. J Pathol. 2016;238(4):543–9.
Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–80.
Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342–9.
Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet (London). 2004;364(9440):1127–34.
Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626–32.
Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28(7):1247–53.
Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet (London). 2006;368(9544):1329–38.
George S, Blay JY, Casali PG, Le Cesne A, Stephenson P, Deprimo SE, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer (Oxford, England: 1990). 2009;45(11):1959–68.
Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352–9.
Mross K, Frost A, Steinbild S, Hedbom S, Buchert M, Fasol U, et al. A phase I dose-escalation study of regorafenib (BAY 73–4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res. 2012;18(9):2658–67.
George S, Wang Q, Heinrich MC, Corless CL, Zhu M, Butrynski JE, et al. Efficacy and safety of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of imatinib and sunitinib: a multicenter phase II trial. J Clin Oncol. 2012;30(19):2401–7.
Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet (London). 2013;381(9863):295–302.
Serrano C, Leal A, Kuang Y, Morgan JA, Barysauskas CM, Phallen J, et al. Phase I study of rapid alternation of sunitinib and regorafenib for the treatment of tyrosine kinase inhibitor refractory gastrointestinal stromal tumors. Clin Cancer Res. 2019;25(24):7287–93.
George S HM, Chi P, et al., editor Initial results of phase 1 study of DCC-2618, a broad-spectrum KIT and PDGFRα inhibitor, in patients (pts) with gastrointestinal stromal tumor (GIST) by number of prior regimens. Eur Soc Med Oncol Cong; Oct 19–23, 2018 (abstr 1603O); Munich, Germany.
Blay JY, Serrano C, Heinrich MC, Zalcberg J, Bauer S, Gelderblom H, et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21(7):923–34.
Administration UFaD. QINLOCK prescribing information. 2020. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213973s000lbl.pdf.
Clinicaltrials.gov. A Study of DCC-2618 vs. sunitinib in advanced gist patients after treatment with imatinib (intrigue) 2020. Available from: https://clinicaltrials.gov/ct2/show/NCT03673501.
Cassier PA, Fumagalli E, Rutkowski P, Schöffski P, Van Glabbeke M, Debiec-Rychter M, et al. Outcome of patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors in the tyrosine kinase inhibitor era. Clin Cancer Res. 2012;18(16):4458–64.
Network NCC. NCCN Guidelines Version 2.2017 Soft Tissue Sarcoma 2017. Available from: https://www.nccn.org/professionals/physician_gls/pdf/sarcoma.pdf.
Heinrich MC, Jones RL, von Mehren M, Schöffski P, Serrano C, Kang YK, et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): a multicentre, open-label, phase 1 trial. Lancet Oncol. 2020;21(7):935–46.
Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11(11):4182–90.
Liegl B, Kepten I, Le C, Zhu M, Demetri GD, Heinrich MC, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216(1):64–74.
Kim TS, Cavnar MJ, Cohen NA, Sorenson EC, Greer JB, Seifert AM, et al. Increased KIT inhibition enhances therapeutic efficacy in gastrointestinal stromal tumor. Clin Cancer Res. 2014;20(9):2350–62.
Wagner AJ TW, Shields AF, et al, editor A phase I pharmacokinetic (PK) and pharmacodynamic (PD) study of PLX9486 alone and in combination (combo) with the KIT inhibitors pexidartinib (pexi) or sunitinib (su) in patients (Pts) with advanced solid tumors and gastrointestinal stromal tumor (GIST). Annual Society of Clinical Oncology; 2018; Chicago, USA.
Clinicaltrials.gov. PLX9486 as a Single Agent and in Combination With PLX3397 or PLX9486 With Sunitinib in Patients With Advanced Solid Tumors 2020. Available from: https://clinicaltrials.gov/ct2/show/NCT02401815?term=NCT02401815&draw=2&rank=1.
J Trent WT, R Chugh, AC Tsiatis, P Severson, K Inokuchi, C Zhang, G Michelson, WJ Wagner. The potent and selective KIT inhibitor PLX9846 dosed in combination with sunitinib demonstrates promising PFS in patients with advanced GIST: final results of phase I/II study. 2020 CTOS Virtual Annual Meeting.
Heinrich M vMM, Jones RL, et al., editor Avapritinib is highly active and well-tolerated in patients (pts) with advanced GIST driven by diverse variety of oncogenic mutations in KIT and PDGFRA. CTOS Annual Meeting; 2018; Rome, Italy.
(VOYAGER) Study of Avapritinib vs Regorafenib in Patients With Locally Advanced Unresectable or Metastatic GIST. Available from: https://clinicaltrials.gov/ct2/show/NCT03465722.
Kettle JG, Anjum R, Barry E, Bhavsar D, Brown C, Boyd S, et al. Discovery of N-(4-{[5-Fluoro-7-(2-methoxyethoxy)quinazolin-4-yl]amino}phenyl)-2-[4-(propan-2-yl)-1 H-1,2,3-triazol-1-yl]acetamide (AZD3229), a Potent Pan-KIT Mutant Inhibitor for the Treatment of Gastrointestinal Stromal Tumors. J Med Chem. 2018;61(19):8797–810.
Pilla Reddy V, Anjum R, Grondine M, Smith A, Bhavsar D, Barry E, et al. The Pharmacokinetic-Pharmacodynamic (PKPD) Relationships of AZD3229, a Novel and Selective Inhibitor of KIT, in a Range of Mouse Xenograft Models of GIST. Clin Cancer Res. 2020;26(14):3751–9.
Ran L, Sirota I, Cao Z, Murphy D, Chen Y, Shukla S, et al. Combined inhibition of MAP kinase and KIT signaling synergistically destabilizes ETV1 and suppresses GIST tumor growth. Cancer Discov. 2015;5(3):304–15.
Chi P, Qin L-X, Kelly CM, D'Angelo SP, Dickson MA, Gounder MM, et al. A phase II study of MEK162 (binimetinib [BINI]) in combination with imatinib in patients with untreated advanced gastrointestinal stromal tumor (GIST). J Clin Oncol. 2020;38(15_suppl):11508.
Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011;17(9):1094–100.
D’Angelo SP, Shoushtari AN, Keohan ML, Dickson MA, Gounder MM, Chi P, et al. Combined KIT and CTLA-4 blockade in patients with refractory GIST and other advanced sarcomas: a phase Ib study of dasatinib plus ipilimumab. Clin Cancer Res. 2017;23(12):2972–80.
Trent JC, Wathen K, Mehren Mv, Samuels BL, Staddon AP, Choy E, et al. A phase II study of dasatinib for patients with imatinib-resistant gastrointestinal stromal tumor (GIST). J Clin Oncol. 2011;29(15_suppl):10006.
Singh AS, Chmielowski B, Hecht JR, Rosen LS, Chow WA, Wang X, et al. A randomized phase II study of nivolumab monotherapy versus nivolumab combined with ipilimumab in advanced gastrointestinal stromal tumor (GIST). J Clin Oncol. 2019;37(15_suppl):11017.
Chen JL, Mahoney MR, George S, Antonescu CR, Liebner DA, Tine BAV, et al. A multicenter phase II study of nivolumab +/− ipilimumab for patients with metastatic sarcoma (Alliance A091401): Results of expansion cohorts. J Clin Oncol. 2020;38(15_suppl):11511.
Bauer S, Duensing A, Demetri GD, Fletcher JA. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene. 2007;26(54):7560–8.
Hyman DM, Solit DB, Arcila ME, Cheng DT, Sabbatini P, Baselga J, et al. Precision medicine at Memorial Sloan Kettering Cancer Center: clinical next-generation sequencing enabling next-generation targeted therapy trials. Drug Discovery Today. 2015;20(12):1422–8.
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703–13.
Kelly CM BT DAS, Dickson MA, Gounder M, Keohan ML, Vonya T, Yoon SS, Singer s, DeMatteo RP, Antonescu CR, Zehir A, Ladanyi M, Solit DB, Ping Chi, William D. Tap, editor The clinical impact of performing routine next generation sequencing in gastrointestinal stromal tumors (GIST). American Society of Clinical Oncology; 2017; Chicago, USA.
Schöffski P, Reichardt P, Blay JY, Dumez H, Morgan JA, Ray-Coquard I, et al. A phase I-II study of everolimus (RAD001) in combination with imatinib in patients with imatinib-resistant gastrointestinal stromal tumors. Ann Oncol. 2010;21(10):1990–8.
Van Looy T, Wozniak A, Floris G, Sciot R, Li H, Wellens J, et al. Phosphoinositide 3-kinase inhibitors combined with imatinib in patient-derived xenograft models of gastrointestinal stromal tumors: rationale and efficacy. Clin Cancer Res. 2014;20(23):6071–82.
Floris G, Wozniak A, Sciot R, Li H, Friedman L, Van Looy T, et al. A potent combination of the novel PI3K Inhibitor, GDC-0941, with imatinib in gastrointestinal stromal tumor xenografts: long-lasting responses after treatment withdrawal. Clin Cancer Res. 2013;19(3):620–30.
Gelderblom H, Jones RL, George S, Valverde Morales C, Benson C, Jean-Yves B, et al. Imatinib in combination with phosphoinositol kinase inhibitor buparlisib in patients with gastrointestinal stromal tumour who failed prior therapy with imatinib and sunitinib: a Phase 1b, multicentre study. Br J Cancer. 2020;122(8):1158–65.
Merten L, Agaimy A, Moskalev EA, Giedl J, Kayser C, Geddert H, et al. Inactivating Mutations of RB1 and TP53 Correlate With Sarcomatous Histomorphology and Metastasis/Recurrence in Gastrointestinal Stromal Tumors. Am J Clin Pathol. 2016;146(6):718–26.
Toulmonde M, Blay JY, Bouche O, Mir O, Penel N, Isambert N, et al. Activity and Safety of Palbociclib in Patients with Advanced Gastrointestinal Stromal Tumors Refractory to Imatinib and Sunitinib: A Biomarker-driven Phase II Study. Clin Cancer Res. 2019;25(15):4611–5.
Ganjoo KN, Villalobos VM, Kamaya A, Fisher GA, Butrynski JE, Morgan JA, et al. A multicenter phase II study of pazopanib in patients with advanced gastrointestinal stromal tumors (GIST) following failure of at least imatinib and sunitinib. Ann Oncol. 2014;25(1):236–40.
Mir O, Cropet C, Toulmonde M, Cesne AL, Molimard M, Bompas E, et al. Pazopanib plus best supportive care versus best supportive care alone in advanced gastrointestinal stromal tumours resistant to imatinib and sunitinib (PAZOGIST): a randomised, multicentre, open-label phase 2 trial. Lancet Oncol. 2016;17(5):632–41.
Schöffski P, Mir O, Kasper B, Papai Z, Blay JY, Italiano A, et al. Activity and safety of the multi-target tyrosine kinase inhibitor cabozantinib in patients with metastatic gastrointestinal stromal tumour after treatment with imatinib and sunitinib: European Organisation for Research and Treatment of Cancer phase II trial 1317 'CaboGIST'. Eur J Cancer (Oxford, England: 1990). 2020;134:62–74.
Park SH, Ryu MH, Ryoo BY, Im SA, Kwon HC, Lee SS, et al. Sorafenib in patients with metastatic gastrointestinal stromal tumors who failed two or more prior tyrosine kinase inhibitors: a phase II study of Korean gastrointestinal stromal tumors study group. Invest New Drugs. 2012;30(6):2377–83.
Kindler HL, Campbell NP, Wroblewski K, Maki RG, D'Adamo DR, Chow WA, et al. Sorafenib (SOR) in patients (pts) with imatinib (IM) and sunitinib (SU)-resistant (RES) gastrointestinal stromal tumors (GIST): Final results of a University of Chicago Phase II Consortium trial. J Clin Oncol. 2011;29(15_suppl):10009.
Sawaki A, Nishida T, Doi T, Yamada Y, Komatsu Y, Kanda T, et al. Phase 2 study of nilotinib as third-line therapy for patients with gastrointestinal stromal tumor. Cancer. 2011;117(20):4633–41.
Heinrich MC, vonMehren M, Demetri GD, Fletcher JA, Sun J, Hodgson JG, et al. A phase 2 study of ponatinib in patients (pts) with advanced gastrointestinal stromal tumors (GIST) after failure of tyrosine kinase inhibitor (TKI) therapy: Initial report. J Clin Oncol. 2014;32(15_suppl):10506.
Adenis A, Blay JY, Bui-Nguyen B, Bouché O, Bertucci F, Isambert N, et al. Masitinib in advanced gastrointestinal stromal tumor (GIST) after failure of imatinib: a randomized controlled open-label trial. Ann Oncol. 2014;25(9):1762–9.
Joensuu H, De Braud F, Grignagni G, De Pas T, Spitalieri G, Coco P, et al. Vatalanib for metastatic gastrointestinal stromal tumour (GIST) resistant to imatinib: final results of a phase II study. Br J Cancer. 2011;104(11):1686–90.
Kang YK, Yoo C, Ryoo BY, Lee JJ, Tan E, Park I, et al. Phase II study of dovitinib in patients with metastatic and/or unresectable gastrointestinal stromal tumours after failure of imatinib and sunitinib. Br J Cancer. 2013;109(9):2309–15.
Vincenzi B, Nannini M, Badalamenti G, Grignani G, Fumagalli E, Gasperoni S, et al. Imatinib rechallenge in patients with advanced gastrointestinal stromal tumors following progression with imatinib, sunitinib and regorafenib. Therap Adv Med Oncol. 2018;10:1758835918794623.
Gangadhar TC, Cohen EE, Wu K, Janisch L, Geary D, Kocherginsky M, et al. Two drug interaction studies of sirolimus in combination with sorafenib or sunitinib in patients with advanced malignancies. Clin Cancer Res. 2011;17(7):1956–63.
Clinicaltrials.gov. Randomized Trial of Crenolanib in Subjects With D842V Mutated GIST. Available from: https://clinicaltrials.gov/ct2/show/NCT02847429.
Bauer S HT, Mühlenberg T, et al., editor Plasma sequencing to detect a multitude of secondary KIT resistance mutations in metastatic gastrointestinal stromal tumors (GIST) (abstract 10518). ASCO Annual Meeting 2015; Chicago.
Maier J, Lange T, Kerle I, Specht K, Bruegel M, Wickenhauser C, et al. Detection of mutant free circulating tumor DNA in the plasma of patients with gastrointestinal stromal tumor harboring activating mutations of CKIT or PDGFRA. Clin Cancer Res. 2013;19(17):4854–67.
Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14(9):985–90.
Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11(6):426–37.
Heinrich MC, Hodgson JG, Mehren Mv, Demetri GD, Fletcher JA, Sun JG, et al. Detection of KIT mutants in circulating tumor DNA (ctDNA) and their association with ponatinib anti-tumor activity in patients (pts) with advanced gastrointestinal stromal tumors (GIST). J Clin Oncol. 2015;33(15_suppl):10517.
Suzanne George SB, Robin L. Jones, et al., editor Correlation of ctDNA and Response in Patients with PDGFR-D842 GIST Treated with Avapritinib. European society of medical oncology; 2018; Munich, Germany.
Acknowledgements
Not applicable.
Funding
None.
Author information
Authors and Affiliations
Contributions
All authors wrote, revised and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for Publication
Not applicable.
Competing interests
Ciara M. Kelly discloses the following relationships: Consulting Role: Exicure. Institutional Research Funding: Agios; Amgen; Exicure; Incyte; Kartos; Merck. Financial interest (employment of an immediate family member): Daiichi Sankyo. Laura Gutierrez Sainz has no disclosures to report. Ping Chi discloses the following relationships: Consulting or Advisory Role: Deciphera; Exelixis; Merck. Institutional Research Funding: Array BioPharma; Deciphera.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kelly, C.M., Gutierrez Sainz, L. & Chi, P. The management of metastatic GIST: current standard and investigational therapeutics. J Hematol Oncol 14, 2 (2021). https://doi.org/10.1186/s13045-020-01026-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-01026-6