
Abstract
P-type ATPase inhibitors are among the most successful and widely prescribed therapeutics in modern pharmacology. Clinical transition has been safely achieved for H+/K+ ATPase inhibitors such as omeprazole and Na+/K+-ATPase inhibitors like digoxin. However, this is more challenging for Ca2+-ATPase modulators due to the physiological role of Ca2+ in cardiac dynamics. Over the past two decades, sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) modulators have been studied as potential chemotherapy agents because of their Ca2+-mediated pan-cancer lethal effects. Instead, recent evidence suggests that SERCA inhibition suppresses oncogenic Notch1 signaling emerging as an alternative to γ-secretase modulators that showed limited clinical activity due to severe side effects. In this review, we focus on how SERCA inhibitors alter Notch1 signaling and show that Notch on-target-mediated antileukemia properties of these molecules can be achieved without causing overt Ca2+ cellular overload.
Similar content being viewed by others

Background
NOTCH receptors are transmembrane cell-surface proteins that control cell to cell communication, embryogenesis, and tissue commitment [1]. In mammals, there are four NOTCH isoforms (I-IV) that share a similar basic structure organized in modules, including an extracellular domain (NECD) at the N‐terminus, a transmembrane domain (NTM), and a NOTCH intracellular domain (NICD) at the C‐terminus. After translation, pro-NOTCH proteins are proteolytically cleaved in the endoplasmic reticulum (ER)/Golgi compartment by a furin-like protease [2]. This cleavage (S1) releases non-covalent heterodimers that, once translocated on the surface of the cells [3, 4], are activated by the binding with ligands (Delta-like 1, 3, 4 and Jagged 1, 2) expressed on the neighboring cells [5]. This interaction generates a NOTCH conformational change that exposes site 2 (S2) to the proteolytic activity of a disintegrin and metalloprotease (ADAM-10 or TACE/ADAM-17). The resulting short-lived protein fragments are substrates of the γ-secretase complex (S3) that releases NICD to the cytoplasm [6, 7]. Then, NICD translocates into the nucleus, associates with the co-transcription factor of the Mastermind-like gene family (MAML) [8], and complex with the p300/CBF‐associated (PCAF) co-activators to bind the DNA-binding factor recombination signal binding protein for immunoglobulin kappa J region (RBPJ) and activate transcription [7].
Because Notch signaling controls the homeostasis of different physiological processes, its alteration may cause different pathological states or diseases including inherited syndromes or acquired malignancies [9, 10]. Notch operates in the context of the cellular microenvironment and the immune system [11, 12] to process either proliferative, pro-differentiation, or quiescence/stem cell maintenance signals [13,14,15]. Thus, it does not surprise that, depending on the cellular events, Notch may act as an oncogene or a tumor suppressor [9].
The majority of oncogenic alterations of NOTCH receptors in human cancers occur in the NOTCH1 gene. Activating NOTCH1 mutations are prevalent in hematological malignancies and sequenced in 40% to 70% of T cell acute lymphoblastic leukemia (T-ALL) [16], in 10–15% of chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL) [17, 18] and a subset of diffuse large B cell lymphoma (DLBCL) [19]. These mutations have been also described in solid cancers, such as breast cancer [20, 21], medulloblastoma [22], lung adenocarcinoma [23], melanoma [24], and colon cancer [25]. Most of these mutations occur in the juxta-membrane heterodimerization (HD) domain, which holds together the NECD with NTM, or in the PEST (rich in proline (P), glutamic acid (E), serine (S), and threonine (T)) degron domain, and result in a more stable and transcriptionally active NICD [16]. On the opposite, loss-of-function mutations occurring in the NECD module are detected in the skin squamous cell carcinoma (SCC) [26, 27], head and neck cancer [28, 29], and myeloid leukemia [30, 31].
Notch controls both cell-intrinsic and extrinsic circuits leading to tumor development, progression, and response to therapy. Several therapeutic efforts have historically focused on modulating Notch signaling by using small molecules such as γ-secretase inhibitors (GSI) or antibody-based strategies, albeit without achieving clinical translation [32, 33]. These approaches have a poor therapeutic window—wild-type versus mutant proteins—limiting their application in human diseases [34]. However, this is not the case for small molecules targeting SERCA. SERCA inhibition hijacks Notch1 trafficking and its activation emerging as a druggable approach for NOTCH1-dependent cancers [35]. Here we update on the current scientific advancements to impede NOTCH1 transfer to the cell surface by blocking SERCA activity as a strategy to target NOTCH1-mutated cancers.
SERCA
SERCA proteins belong to the superfamily of active transporters known as P-type ATPases (E1/E2-type) located in the ER. In 1993 Toyoshima and colleagues described the first complete structure of SERCA by cryo-electron microscopy [36]. Subsequently, novel high-content techniques shaped the resolution of several crystallography structures of SERCA [37]. These studies showed how ligands (e.g., vanadate, thapsigargin) bind SERCA, and what structural changes occur during the enzymatic catalytic cycle [38,39,40,41,42]. ATP2A1 (16p11.2), ATP2A2 (12q24.11), and ATP2A3 (17p13.2) genes encode for SERCA1, 2, and 3, respectively [43, 44]. Today, over 70 SERCA isoforms resulting from alternative splicing are deposited in the Protein Data Bank database [45]. While these transcripts share up to 85% of sequence homology, differences in tissue distribution, Ca2+ binding affinity in both normal and cancer tissue, are due to changes in the protein C-terminal region [46,47,48,49,50,51].
SERCA proteins maintain intracellular Ca2+ homeostasis by pumping Ca2+ from cytosol into the ER [52]. These 110 KDa pumps are organized in 10 transmembrane (TM) helices (M1-M10) along with two Ca2+ binding site (site I and II), a small luminal tail, three cytoplasmic domains (A, actuator; N, nucleotide binding; P, phosphorylation). These functional modules mediate ATP hydrolysis, hydron (H+), and calcium (Ca2+) binding and shuffling through the ER membrane [53]. Conformational changes during the catalytic cycle involving the N, A, and P domains along with the TM helices allow for a continued alternated access of Ca2+ from site I and II to the cytoplasmic and luminal sides [52]. The SERCA transport cycle is like the one described by Albers for other P-type ATPases [54] (Fig. 1a). The enzymatic reaction (E) alternates phases with high (2Ca2+E1) or low (E2) affinity to Ca2+ coupled with high (2Ca2+E1 ~ P) or low (E2 ~ P) energy phosphorylated states in the following sequence: E2 → 2Ca2+E1 → 2Ca2+E1 ~ P → 2Ca+E2 ~ P → E2 ~ P → E2. At the ground state (E2), SERCA presents two Ca2+ binding sites exposed to the luminal ER side [55]. In this orientation, ATP is located on the N domain far from the Asp351 residue of the P domain [56]. The exchange of 2/3 H+ causes a conformational change in the M1-M4 helices that enables a rotation of the A domain and the binding of cytosolic Ca2+ (2Ca2+E1). Next, the N domain shifts close to the Asp351 residue leading an energy-phosphorylated states 2Ca2+E1 ~ P [57, 58]. The following decay to a lower energy state, 2Ca2+E2 ~ P, sees a rotation of the N domain and A domain of 30° and 110°, respectively. Now the M1-M2 and M3-M4 helices pairs with the M5-M10 complex to release Ca2+ sites toward the luminal ER side [59, 60] in exchange of protons transferred to the cytosol. Dephosphorylation with the release of free phosphate, closes and reopens the cycle (E2) [61]. Thapsigargin and other SERCA inhibitors inhibit the enzyme in a Ca2+-free state (E2), avoiding the high-affinity binding of Ca2+ and the following activation of the catalytic cycle (Fig. 1b) [62,63,64].

SERCA catalytic cycle. a SERCA pumps Ca2+ from the cytoplasm to the endoplasmic reticulum (ER) to create an ion gradient of ~ 10,000-fold across the cell membranes. During the catalytic cycle the Ca2+-ATPase switches between diverse oriented structures (E1 and E2) characterized by a different affinity for Ca2+ ions. The enzymatic transition between conformations catalyzes ATP in a stoichiometric ratio with Ca2+ of 1:2. E1 binds two molecules of cytoplasmic Ca2+ (2Ca2+E1, top right) and hydrolyzes one molecule of ATP to induce the high energy state of SERCA (E1 ~ P). Following the decay in ADP, E2 ~ P releases Ca2+ in the ER lumen. The cycle is closed and reopened along with the dephosphorylation of SERCA. b Thapsigargin (or other SERCA inhibitors) locks SERCA in the ground E2 state (“dead-end”) preventing Ca2+ binding and ATP hydrolysis. Thapsigargin blockade causes a continuum leakage of Ca2+ from the ER to the cytosol reverting the physiological polarization of Ca2+ across these two compartments
SERCA, disease, and cancer
The specific tissue distribution of SERCA isoforms accounts for their non-redundant role in physiological processes and human diseases [65]. For example, while the fetal (1b, 1001 amino acid -AA-) and adult (1a, 994 AA) SERCA1 isoforms are functionally identical, the difference in 7 AA may explain the contribution of SERCA1b in muscle development [66]. Similarly, SERCA2 transcripts encode for four isoforms (a-d) [61] with the highest affinity for Ca2+ (KCa2+ 0.2 μM) [67] compared to SERCA1 and 3. The “a” isoform (997 AA) is expressed in slow-twitch skeletal and cardiac muscle, while the “b” (1042 AA) isoform is prevalent across different tissue [68]. The difference in size between the two variants is due to the presence in SERCA2b of an 11th transmembrane helix (TM11) and a luminal extension (LE) peptide which extends the C-terminal tail into the ER lumen [69, 70]. Biochemical and structural studies demonstrated that the TM11 tail is responsible for Ca2+ affinity, pump turnover, and the interaction with the ER redox and chaperon proteins [71, 72]. SERCA2c instead is expressed in cardiac muscle [73] and, at a lower extent, in hematopoietic and epithelial cells [74], while SERCA2d in skeletal muscle [75]. SERCA3 (a-f) is expressed in non-muscle tissues at variable levels and is frequently expressed with SERCA2b [76]. Compared to SERCA1 and 2, SERCA3 has the lowest affinity to Ca2+, suggesting that this pump is only active in the presence of higher cytoplasmic Ca2+ concentration [67].
Given their role in controlling cellular Ca2+ homeostasis, SERCA proteins have been involved in several human diseases from inherited syndrome to heart failure [77]. Loss-of-function mutations of ATP2A1 cause Brody myopathy, a rare autosomal dominant genetic condition, characterized by painless muscles cramping and stiffening after exercise or cold temperatures [78, 79]. Similarly, ATP2A2 mutations account for the development of the Darier disease (keratosis follicularis), a severe skin disorder characterized by skin wart-like blemishes, due to loss of adhesion between epidermal cells [80, 81].
The potential involvement of SERCA in cancer progression has been an active area of investigation given its role in Ca2+ homeostasis and its effect on cell survival and ER stress pathway [46]. For example, SERCA2 overexpression protects from apoptosis [77], while aberrant SERCA3 expression co-occurs in differentiating cells [46]. Park and colleagues showed that the overexpression of SERCA2 and Bcl-2 is a consequence of the repositioning of the nuclear factor kappa B (NF-kB) secondary to calcium/calmodulin-dependent protein kinase 2 alpha (CaMK2α) activation in metabolic stress-resistant breast cancer cell lines MDA-MB-231 and MCF-7 [82, 83]. They next showed that combined treatment with thapsigargin, a general SERCA inhibitor, and 2-deoxy-d-glucose (2-DG, acting as an NF-kB inhibitor) reduces the tumor burden, while 2-DG alone, that mimics glucose starvation, had a lower effect in a breast cancer xenograft model [83]. Overall, these data suggest that SERCA2 overexpression is a general mechanism to evade apoptosis and may result from the activation of metabolic stress [84].
A second observation is that SERCA expression may vary during epithelial differentiation and carcinogenesis playing different roles depending on the tissue of origin. For example, in colorectal cancer (CRC) increased SERCA2 transcripts correlate with tumor node metastasis or higher tumor grade [85], while decreased ATP2A2 mRNA abundance is measurable in transformed thyroid cells [86]. Similarly, SERCA3 protein levels seem to correlate with the grade of epithelial differentiation. In CRC, SERCA3 expression increases in colonic mucosa and hyperplastic polyps, while it reduces in dysplastic, moderately, or poorly differentiated carcinoma cells [87]. In lung adenocarcinoma SERCA3 mRNA is reduced compared to non-transformed cells, but after pharmacologic-induced differentiation, the physiologic expression levels of the enzyme are restored [88].
Interestingly, the expression of SERCA isoforms is not mutually exclusive in cancer cells. Cancer cells may simultaneously have multiple SERCA isoforms, and the net contribution to the cancer phenotype is a balance between the expression of different transcripts [89]. For example, SERCA2 and SERCA3 are oppositely regulated in acute promyelocytic leukemia (APL) cell lines HL-60 and NB4 and freshly isolated APL cells treated with the pro-differentiating agent all-trans-retinoic acid (ATRA) in vitro [90]. A similar effect is seen during the differentiation of MEG 01, UT7, M-07e, and CHRF 288-11 erythro-megakaryoblastic leukemia cell lines treated with 10–8 M of the phorbol ester PMA [91]. Collectively these studies suggest that the downregulation of SERCA3 and overexpression of SERCA2 are key processes in leukemia stem cell and cancer maintenance [46].
While SERCA mutations are rare in cancer, some studies have demonstrated the involvement of genetic alterations of ATP2A genes in tumor development, mainly for lung and colon cancer [92]. Liu and colleagues showed that heterozygous mutant ATP2A2 mice develop late-onset squamous cell tumors in the gut and in the skin where the expression of SERCA2 protein is reduced due to haploinsufficient loss-of-function mutations [93]. Toki and colleagues confirmed these initial observations and showed that the onset of ATP2A2-deficient tumor depends on the level of SERCA residual activity [94], suggesting that SERCA haploinsufficiency may predispose to multistage carcinogenesis by altering Ca2+ homeostasis [95].
SERCA and Notch
In 1999, Goran Periz and Mark E. Fortini described that the trafficking events leading to a correct NOTCH activation may be disrupted in the presence of a defective Ca2+-ATPase function in a Drosophila model [96]. In this work, the authors demonstrated that loss-of-function alleles of the Drosophila SERCA homologous gene Ca-P60A alter proper synthesis, folding, and trafficking of the NOTCH receptor in the ER/Golgi compartments. Consistently, in Drosophila S2 cultured lines, the treatment with general SERCA inhibitors such as thapsigargin and cyclopiazonic acid (CPA) primarily reduces the amount of NOTCH proteins that reach the cell surface [96]. While extremely interesting, these observations were not confirmed in mammalian cells until the Stegmaier’s laboratory embarked on a large gene expression-based screening (GE-HTS) effort to identify inhibitors of oncogenic NOTCH1 signatures or enhancer of NOTCH1 HD mutant L1601P∆P activity in T-ALL. Among the top hits were the genes ATP2A2 and ATP2A3, and SERCA inhibitors such as thapsigaricin (an analog of thapsigargin) and CPA. Together with other ion flux modulators, SERCA emerged as a novel potential therapeutic target in NOTCH1-associated cancers [35, 97]. Furthermore, these data suggest the hypothesis that Notch signaling could be dysfunctional in several genetic disorders associated with loss of function ATP2A1-3 mutations.
Thapsigargin and derivatives
Thapsigargin
The rise of SERCA inhibitors for cancer therapeutics dates back to 1960 when the National Cancer Institute (NCI) launched a program to identify compounds with antitumor activity from 35,000 plant extracts [98]. Sesquiterpene lactone (SL) derivatives demonstrated anti-inflammatory and antitumor activity in several tumor types, like laryngeal carcinomas, uveal melanomas, pituitary macroadenomas, kidney, prostate cancer, and hematological malignancies [99,100,101,102,103,104]. Among others, thapsigargin, parthenolide, and artemisinin were selected for their potency and initially used as tool compounds in different cancer models. Several SERCA inhibitors which differed in their source, chemical structure, potency and binding affinity to SERCA isoforms were subsequently developed [84].
Thapsigargin is an SL phyto-derivative compound isolated from the umbelliferous Mediterranean Thapsia Garganica [105]. Thapsigargin binds SERCA in its E2 Ca2+-free conformation through an irreversible lipophilic interaction, by stabilizing SERCA in a so-called dead-end inactive state with low Ca2+ affinity, preventing both ATPase and Ca2+ transport activity [42, 106]. Thapsigargin binds to all SERCA isoforms with different specificity within the transmembrane helices M3 (at Phe256), M5 (at Ile765), and M7 (at Tyr837) [42, 107]. SERCA1 appears to be the more sensitive to thapsigargin inhibition (Ki ~ 0.2 nM), while the affinity for SERCA2 (Ki ~ 1 nM) and SERCA3 (Ki ~ 12 nM) decreases of 20 and 60 times, respectively (Table 1) [50]. Following SERCA inhibition, the Ca2+ depletion from ER produces a modification in the plasma membrane permeability to extracellular Ca2+, leading to a rise of intracellular Ca2+ level that occurs as early as one-two minutes following thapsigargin treatment [103]. The initial increase of intracellular Ca2+ is followed by a second cytosolic peak in Ca2+ concentration that precedes apoptosis occurring within 24–48 h [103]. These on-target Ca2+ effects can be seen at sub-nanomolar range (Ki: 10–10 M) in several cell types including normal tissue [108].
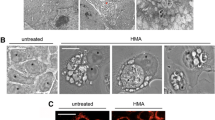
In the initial study in T-ALL, Roti and colleagues demonstrated that SERCA inhibition disrupts the trafficking of mutated NOTCH1 receptors that consequently accumulate in the ER/Golgi compartments upon drug treatment (Fig. 2a, b). This defective processing of newly synthesized NOTCH1 peptides ultimately results in a net reduction of NTM1 on the cell surface, the substrate of the γ-secretase complex. Hence, the consequent decrement on NICD1 level causes a Notch1 on-target inhibitory effect on leukemia growth in vitro, in T-ALL xenografts, and in a Drosophila intestinal stem cell model in which Notch1 inhibition perturbs differentiation of midgut pluripotent stem cells [35, 96]. Interestingly, mice treated with thapsigargin did not develop gastrointestinal toxicity, in sharp contrast with previous evidence of preclinical and clinical studies assessing the role of GSI [133]. These results suggested that mutated NOTCH1 receptors were more sensitive to the effects of thapsigargin than wild-type NOTCH1/NOTCH2 proteins expressed in normal cells providing a clinical therapeutic window for SERCA inhibitors.

SERCA and the Notch trafficking. a Schematic representation of the effects of SERCA inhibition on the maturation and trafficking of NOTCH1. In physiologic conditions, SERCA pumps Ca2+ into the ER required for the proper folding of NOTCH1 proteins. A furin-like protease (S1) releases from the ER/Golgi the non-covalent heterodimer NFL1 that migrates through the cytosol toward the cell membrane. Following the interaction of the extracellular NECD1 with the Notch ligands, NOTCH1 is cleaved sequentially by metalloproteases (S2) and γ-secretases [GS (S3)]. The final cleaved protein NICD1 migrates to the nucleus to complex with co-activators and activates transcription. b SERCA blockade by SERCA inhibitors (e.g., thapsigargin) induces a leak of ER and the accumulation of the full-length isoform of NOTCH1 at the ER/Golgi level. As a consequence, no substrate for metalloprotease or γ-secretase is available with the result of a reduced level of nuclear NICD1 proteins
Sharma and colleagues confirmed this original observation and demonstrated that thapsigargin alone or in combination with the monoclonal anti-NOTCH1 antibody mAb 604.107 inhibited “gain of function” mutants associated with T-ALL such as L1594P, R1599P, and I168N [134]. An important question is whether the lack of NOTCH1 Ca2+ binding modules EGF-like and LNR domains circumvents the requirement of the Ca2+-ATPase function. NOTCH1 peptides that are similar to the membrane-bound furin-processed forms ΔEGFΔLNR or NICD1 are insensitive to thapsigargin inhibition, and they properly localize to the cell surface, suggesting that the Ca2+-ATPase activity is required for the furin-mediated cleavage of NOTCH1 precursors in T-ALL cells [35]. These results differ from studies in flies where ΔECN (equivalent of ΔEGFΔLNR) proteins fail to correctly localize on apical eye disc membranes [96]. Collectively these data suggest that Ca2+ binding motives are required for thapsigargin effects and that Ca2+-ATPase activity interferes with general trafficking and secretion of NOTCH1. However, Roti and colleagues reported that ~ 25–30% of thapsigargin-treated mice (~ 0.4 mg/kg/injections/day) died during the study with no prior weight loss or clinical signs of illness, suggesting that at these doses thapsigargin may cause a sudden cardiac lethal event [35]. A different thapsigargin schedule, 0.4 mg/kg/injections 3 times per week, is tolerated and effective in reducing Notch1 signaling in TNF-induced synovial M1 macrophages in a Hes1-GFP/TNF transgenic mouse model of rheumatoid arthritis [135]. Nevertheless, despite the emerging role of SERCA as a Notch1 druggable target, the transition of naive thapsigargin into the clinic is worrisome because of the effect of this drug on cardiac SERCA2a [136].
Mipsagargin and JQ-FT
Applying lessons from the experience of monoclonal antibody-based strategies in cancer, one way to overcome limitations associated with thapsigargin clinical translation is by developing pro-drugs that selectively target the desired cell type [103]. This is, for example, the mode of action of mipsagargin/G202, a thapsigargin derivative coupled with a peptide cleaved by the carboxypeptidase prostate-specific membrane antigen (PSMA) [137] (Fig. 3a, Table 2). PSMA is overexpressed in the neovasculature of several tumors including hepatocellular carcinoma, mesothelioma, ovarian, bladder, renal, and breast cancer [138]. In biochemical assays, G202 showed a lower SERCA inhibitory capacity and potency confirming previous studies that most of the derivatives [139] are less potent compared to thapsigargin, hence potentially less effective in vivo [140]. Nevertheless, three days of mipsagargin treatment at 56 mg/kg led to a reduction of more than 50% in tumor size in prostate cancer cell xenograft models expressing PSMA [137]. These proof of concept preclinical data justified phase II study clinical trial (NCT01777594) in patients with advanced sorafenib-refractory hepatocellular carcinoma (HCC) [141]. Patients treated with mipsagargin at 40 mg/m2 on days 1–3 (dose level -1) or 40 mg/m2 on day 1 and 66.8 mg/m2 on day 2–3 (dose level 1) experienced a disease stabilization in 63% of cases, a decrease in tumor blood flow, a median time to progression (TTP) of 134 days (P < 0.001), compared to the historic median TTP of 63 days, a progression-free survival (PFS) of 129 days, and overall survival (OS) of 205 days [141, 142]. Patients treated with mipsagargin at dose level -1 experienced grade 1 and 2 adverse events (AE) such as increased blood creatinine (68% of patients), fatigue (56%), and nausea (44%) [141]. These AE were seen also in patients with advanced/recurrent glioblastoma multiforme, where mipsagargin treatment led to disease stabilization in 22% of patients [143].

Strategy to overcome thapsigargin’s toxicity. a Mipsagargin is a thapsigargin derivative coupled with a masking peptide that is a substrate for the carboxypeptidase prostate-specific membrane antigen (PSMA). This peptide reduces the affinity of these molecules toward SERCA in non-neoplastic cells. However, if mipsagargin intercepts PSMA on tumor cells or neoplastic neoangiogenic vessels are cleaved into a cytotoxic analog of thapsigargin (12-ADT-Asp) and diffuse into the cancer cells. b JQ-FT is a derivative of thapsigargin, 8-O-debutanoyl-thapsigargin, linked with folic acid. In tumor cells expressing folate (FA) receptor, JQ-FT enters through endocytosis and proteases release 8-O-debutanoyl-thapsigargin from FA directly into the cytosol of targeted cells. c Identification of SERCA inhibitors by high-throughput screening, ATPase activity assays (Ca2+-ATPase vs. Na+/K+-ATPase vs. H+/K+-ATPase) or in silico prediction
Instead of an enzymatic pro-drug-based approach such as the one described for prostate-specific antigen [144] and PSMA [137], Roti and Qi leveraged the dependency of ALL on folic acid (FA) metabolism. They first demonstrated that folate receptor 2 (FR2) is aberrantly expressed in T-ALL and that the endocytic trafficking of this receptor can be used as a carrier for folate-conjugated probes. They next showed that the alcohol derivative of thapsigargin suitable for conjugation, 8-O-debutanoyl-thapsigargin, similarly to the parental drug inhibits NOTCH1 and it is preferentially active against mutant T-ALL. They then connected the carboxylate of folic acid to the C8-alcohol of 8-O-debutanoyl-thapsigargin via a cleavable ester linkage to generate JQ-FT (Table 2) [145]. In a series of preclinical validation studies, the authors demonstrated that JQ-FT is stable in cell culture conditions and that the cleavage of JQ-FT occurs through an endocytic FR-mediated process (Fig. 3b). Moreover, JQ-FT is 150-fold more tolerable in mice compared to unconjugated thapsigargin, without killing the antileukemic effect in a preclinical NOTCH1 mutated T-ALL model in vivo. This approach enhanced the therapeutic window of thapsigargin as a NOTCH1 inhibitor and provided dual selectivity: leukemia over the normal cell and NOTCH1 mutated over wild-type receptors [145].
Cyclopiazonic acid and 2,5-Di-(tert-butyl)-1,4-benzohydroquinone
An alternative strategy to reduce the potential toxicity of thapsigargin or to avoid complicated chemical synthesis routes is through the identification of SERCA inhibitors that retain the anti-Notch properties but lack Ca2+-related toxicities. For example, CPA and 2,5-Di-(tert-butyl)-1,4-benzohydroquinone (DBHQ) have been recognized for their ability to lock SERCA in a pocket different from the one of thapsigargin [41, 50, 146]. In fact, the CPA binding pocket is located between the SERCA transmembrane helices M1, M2, M3, and M4 helices, with polar interaction at the Gln56, Asp59, and Asn101 residues [41], while DBHQ at Asp59 on and Pro308 [146]. Both compounds, similar to thapsigargin, reversibly stabilize SERCA in the E2 ~ P conformational state (Table 1) [84, 146]. CPA and DBHQ are less potent compared to thapsigargin; in fact, CPA inhibits SERCA1 with a Ki is 120 nM while DBHQ with a Ki of 0,4 μM [50]. Because of their poor pharmacokinetic properties and lack of potency, DBHQ and CPA had a limited application in tumor models, with only few studies exploring the feasibility of their application as anticancer or antimalarial compounds [40, 50, 147,148,149,150,151]. However, similarly to thapsigargin, CPA induces a NOTCH1 off transcriptional program and triggers a NOTCH1 trafficking defect, suggesting that a thapsigargin-like binding mode of action is not an absolute requirement to achieve the suppression of the Notch1 signaling [35].
Emerging SERCA inhibitors
Dual target compounds
Because P-type ATPases are compelling therapeutic targets in several human diseases [152], several efforts are ongoing to develop tolerable SERCA inhibitors for cancer therapeutics (Table 1). Works from Xing’s laboratory demonstrated that a stable analog of a putative Bcl-2 inhibitor HA 14-1, sHA 14-1, moderately inhibits SERCA1a and 2b with IC50 values of 29.2 ± 4.9 and 23.5 ± 4.2 μM, respectively [128, 153]. Mechanistically, sHA 14-1 induces a rapid ER Ca2+ release that triggered the expression of ER stress-associated transcription factor ATF4/CREB-2 and apoptosis in B cell acute lymphoblastic leukemia (B-ALL) cell lines [128]. The same group pursued a structure–activity relationship (SAR) optimization of sHA 14-1 that led to the identification of the ethyl 2-amino-6-(3,5-dimethoxyphenyl)-4-(2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate (CXL017) [125, 154]. Similar to the parent compound, CXL017 simultaneously inhibits SERCA and Bcl-2 [155] while induces apoptosis in hematopoietic multi-drug-resistant cancer cell lines including T-ALL [126]. Additional studies demonstrated that the active isomer (−) CXL017 synergizes with other SERCA inhibitors including thapsigargin, CPA, and DBHQ, indicating that CXL017 may bind SERCA at a unique allosteric site [126].
Casearin J
Using in silico approaches and docking simulation, De Ford and colleagues demonstrated that tricyclic clerodane diterpenes (TCD) may target SERCA [116]. In the following study, they showed that TCD isolated from Casearia sylvestris casearin J (CJ) [115], affects the Notch1 pathway in human T-ALL cells. CJ reduces the cell surface expression of NOTCH1 receptors and prevents the formation of the cleaved NICD1 molecules, which results in the transcriptional suppression of MYC and HES1. The authors also demonstrated that CJ is more active against HD-mutated T-ALL cells compared to a cell line carrying NOTCH1 juxtamembrane mutations such as Jurkat. While this study does not rule out the activity of CJ on NOTCH1 wild-type cells or mechanistically on NOTCH1 full length (NFL1) proteins, it confirms that T-ALL is sensitive to Ca2+-ATPase suppression further supporting the need to explore SERCA inhibitors with binding sites different from the one of thapsigargin in search of small molecules with tolerable off-target effects.
Curcumin
Among other SERCA inhibitors, curcumin has been extensively tested in several health conditions due to its antioxidant, anti-inflammatory, and anticancer properties [156, 157]. Curcumin (1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione), also known as diferuloylmethane, is the main natural polyphenol extracted from the rhizome of Curcuma longa (turmeric) and in others Curcuma spp. [158]. Curcumin binds SERCA in a hydrophobic task between M3 and M5 [159] and stabilizes the enzyme in its 2Ca2+E1 conformational state preventing ATP binding [117]. Curcumin shows Ki values of 5.8 ± 1.6, 8.6 ± 2.5, and 53 ± 6 μM for SERCA1b, SERCA3a, and SERCA2b, respectively [50], although its poor bioavailability has been tested in vivo [124] and in clinical trials (NCT00094445, NCT01490996) [160, 161] without clear evidence of therapeutic benefits. Furthermore, curcumin (CU) was combined with standard chemotherapy [162], FOLFOX (5-fluorouracil, folinic acid, and oxaliplatin), in advanced metastatic CRC. CUFOX was well tolerated, and adverse events (AEs), quality of life, and neurotoxicity were comparable to those seen in standard chemotherapy [161]. While the study was too small (28 patients) and the cohorts potentially biased for tumor staging and number of metastatic sites, the authors reported a significant increase in OS and PFS in patients treated with CUFOX compared to FOLFOX alone [161].
Because curcumin has been shown to target multiple signaling molecules, several investigators pointed to the role of this drug in the suppression of the Notch signaling, albeit without a unifying mechanism. For example, Liu and colleagues showed that curcumin (10–90 μM) inhibits the proliferation of the SMMC-7721 hepatoma cancer cell line and suppresses NOTCH1 mRNA and protein expression [163] in a way that was similarly described in osteosarcoma cells [164]. Subramanian and colleagues showed that 30 μM curcumin represses NOTCH1, Jagged-1, and HES1 transcript and consequently diminishes NICD1 level in TE-7 esophageal cancer cells. The authors also demonstrated that curcumin reduces the transcription and the expression of proteins of the γ-secretase complex such as presenilin-1 and 2 (PSEN-1, 2), nicastrin (NCSTN), anterior pharynx-defective-1 (APH-1), and presenilin enhancer-2 (PEN-2). These results suggest a general mechanism of transcriptional inhibition or protein stability rather than a direct effect of curcumin on the γ-secretase complex as claimed in the manuscript [165]. An alternative hypothesis is that Notch1 signaling is decreased upon curcumin treatment as a consequence of the inhibition of a NOTCH1 transcriptional regulator [166]. Curcumin analogs with enhanced activity toward SERCA [85] or with a novel binding site between M3, M4 helices and the L78 loop [167] have been recently developed; however, their role as potential Notch modulator is far to be addressed [168].
CAD204520
Despite the potential risk of Ca2+-related toxicity, SERCA inhibitors are used for clinical applications indicating that for some of these molecules the leak of ER Ca2+ toward the cytosol is moderate or compensated. This is probably the case for cisplatin, a widely used platinum-containing compound that among other effects inhibits SERCA and Na+/K+-ATPase simultaneously [169] or the SERCA antimalarial drug artemisinin and its derivatives (artesunate and di-hydro-artemisinin) that are tolerable with minimal side effects [170, 171]. As such the effect of a given SERCA inhibitor on cytosolic and ER Ca2+ levels may depend on its molecular mechanism of interaction with the ATPase [172].
From a secondary analysis of a small molecule screening of 191,000 compounds, Marchesini and Gherli identified one of such molecules (Fig. 3c) [114], CAD204520, with favorable pharmacodynamic properties. CAD204520, (4-[2-[2-[3-propyl-6-(trifluoromethoxy)-1H-indol-2-yl]-1-piperidyl]ethyl] morpholine) dihydrochloride, binds SERCA between the transmembrane helices M1, M2, M3, and M4 with two polar interactions to Asp59 on M1 (2.9 Å) and Asn101 on M2 (2.7 Å) and with several hydrophobic interactions involving Leu61, Val62, Ile307, Pro308, and Pro312. This binding groove is similar to the binding of CPA and DBHQ but different from that of thapsigargin. Interestingly, in preclinical studies, CAD204520 suppresses mutated Notch1 signaling without causing overt cardiac toxicity. We demonstrated that in cardiomyocytes isolated from Wistar rats CAD204520 treatment reduces contractile efficiency by ~ 25%, a cardiomechanics impairment that is tolerated at a therapeutic concentration in a T-ALL orthotope model in vivo. These results are consistent with the transient effects on cytosolic Ca2+ shifts and the lack of unfolded protein response (UPR) activation in cardiomyocytes upon CAD204520 treatment. Collectively these data also support the work from Sehgal and colleagues that showed that inhibition of SERCA ATPase activity and apoptosis can be efficiently achieved without triggering measurable changes in Ca2+ pools [172]. This effect is a consequence of how ligands lock the Ca2+-ATPase, kinetics, and the number of rotating bonds, suggesting that the development of new SERCA inhibitors requires careful consideration of substrate binding [173]. A second question concerning how SERCA inhibitors bind to the Ca2+-ATPase is whether different binding sites may cause a different mechanism of resistance. Our team and others have demonstrated the rapid cell adaptation to thapsigargin treatment due to mutations occurring in the M3 segment spanning between Asp254 and Leu260 [114, 174, 175]. However, in these cells, we showed the lack of cross-resistance with CAD204520, suggesting that early characterization of the binding mechanism may overcome potential relapse from mutant clones.
Conclusions
Modulation of intracellular Ca2+ homeostasis plays critical roles in key processes that regulate cellular survival, growth, differentiation, metabolism, and death in normal and cancer cells. Thus, it is not surprising that several anticancer agents suppress pro-survival and activate pro-apoptotic pathways through modulation of Ca2+ signaling-dependent mechanisms. This is, for example, the case for chemotherapeutics such as cytotoxic alkylating agents [169] or antimetabolites that rely on a Ca2+ signaling component to induce cancer cell death [176]. Similarly, natural compounds including alkaloids, flavonoids, diterpenoids, and polyphenolics have been extensively investigated for their ability to modulate intracellular Ca2+ concentration and to participate in apoptotic signaling pathways. Among them, SL, such as thapsigargin, has been long regarded as target compounds for drug development. Thapsigargin has a broad spectrum of growth-suppressing activity in several tumor types including poorly dividing cells [177]. However, we have demonstrated the SERCA inhibition may efficiently control the trafficking of NOTCH1 and that this blockade can be achieved without causing overt cardiac toxicities in preclinical leukemia models [114]. Importantly the effects of SERCA suppression can be rescued by the overexpression of unprocessed NOTCH1 peptides such as NICD1, indicating that the antileukemia effect is on target for Notch1 inhibition rather than Ca2+ overload [35]. An important standing question is why mutated NOTCH1 appears more sensitive to SERCA suppression compared to wild-type isoform or other proteins more broadly. One hypothesis to explain NOTCH1 and SERCA functional dependency is by mechanisms of co-regulation. It has been previously shown that PSEN and SERCA co-localize in the ER [178]. Since PSEN-1 is a key regulator of NOTCH1 maturation and preferentially binds NFL1 polypeptides processed through the ER, it is possible that NOTCH1-PSEN-1-SERCA is part of a co-functional protein complex. Interestingly, treatment of T-ALL cell lines with the selective PSEN-1 inhibitor MRK-560 inhibited mutant NOTCH1 processing and led to cell cycle arrest. MRK-560 treatment decreases leukemia burden and increased OS with no associated gut toxicity in T-ALL patient-derived xenografts in vivo, suggesting that, similar to SERCA inhibition, disruption of PSEN-1 may preferentially affect mutated proteins [179]. The second hypothesis is a Ca2+ mediated one. In fact, Malecki and colleagues previously demonstrated that clinically relevant activating NOTCH1 HD mutations destabilize the NOTCH1-negative regulatory region and have deleterious effects on NOTCH1 folding and maturation. Because EGF and LNR repeats of NOTCH1 rely on Ca2+ for folding and activation, it may be possible that changes in ER Ca2+ may preferentially impair unstable NOTCH1 mutant proteins [180] compared to wild-type providing a therapeutic window for SERCA inhibitors. Finally, a hypothesis not yet explored to explain NFL1 accumulation at concentrations not sufficient to trigger the general mechanism of UPR is through a Ca2+-mediated transcriptional activation of inhibitors of furin-like proteases. This would explain for example why CAD204520 efficiently targets cancers with isolated PEST deletions that would not be predicted to be unstable given a normal LNR and HD protein sequence.
In conclusion, the transient disruption of SERCA activity can be leveraged for targeting Notch1 in cancers. Legitimate concerns associated with SERCA inhibition can be overcome by the identification of druggable modalities to lock ATPase function without causing dose-limiting ER Ca2+ toxicities.
Availability of data and materials
Not applicable.
Abbreviations
- 2-DG:
-
2-Deoxy-d-glucose
- AA:
-
Amino acid
- Adomain:
-
Actuator domain
- AE:
-
Adverse event
- APL:
-
Acute promyelocytic leukemia
- APH:
-
Anterior-pharynx-defective
- ATRA:
-
All-trans-retinoic acid
- B-ALL:
-
B cell acute lymphoblastic leukemia
- Ca2 + :
-
Calcium
- CAD204520:
-
(4-[2-[2-[3-Propyl-6-(trifluoromethoxy)-1H-indol-2-yl]-1-piperidyl]ethyl] morpholine) dihydrochloride
- CaMK2α:
-
Calcium/calmodulin-dependent protein kinase 2 alpha
- CJ:
-
Casearin J
- CLL:
-
Chronic lymphocytic leukemia
- CPA:
-
Cyclopiazonic acid
- CRC:
-
Colorectal cancer
- CU:
-
Curcumin
- CXL017:
-
Ethyl 2-Amino-6-(3,5-dimethoxyphenyl)-4-(2-ethoxy-2-oxoethyl)-4H- chromene-3-carboxylate
- DBHQ:
-
2,5-Di-(tert-butyl)-1,4-benzohydroquinone
- DLBCL:
-
Diffuse large B cell lymphoma
- DLL:
-
Delta-like ligand
- EGF:
-
Epidermal growth factor
- E:
-
Enzymatic reaction
- EFS:
-
Event-free survival
- ER:
-
Endoplasmic reticulum
- FA:
-
Folic acid
- FR:
-
Folate receptor
- GE-HTS:
-
Gene expression-based high-throughput screening
- GSI:
-
γ-Secretase inhibitors
- H+ :
-
Hydron
- HD:
-
Heterodimerization domain
- HCC:
-
Hepatocellular carcinoma
- JAG:
-
Jagged ligand
- Ki :
-
Constant of inhibition
- LNR:
-
LIN-12/NOTCH repeats
- MAML:
-
Mastermind-like
- MCL:
-
Mantle cell lymphoma
- MRD:
-
Minimal residual disease
- N domain:
-
Nucleotide binding domain
- NCI:
-
National Cancer Institute
- NCSTN:
-
Nicastrin
- NECD:
-
NOTCH extracellular domain
- NICD:
-
NOTCH intracellular domain
- NF-kB:
-
Nuclear factor kappa B
- NFL:
-
NOTCH full length
- NTM:
-
NOTCH transmembrane domain
- OS:
-
Overall survival
- P domain:
-
Phosphorylation domain
- PEN:
-
Presenilin enhancer
- PEST:
-
Proline, glutamic acid, serine, threonine
- PFS:
-
Progression-free survival
- PSEN:
-
Presenilin
- PSMA:
-
Prostate-specific membrane antigen
- RBPJ:
-
Recombination signal binding protein for immunoglobulin kappa J region
- SAR:
-
Structure–activity relationship
- SERCA:
-
Sarco-endoplasmic reticulum Ca2+-ATPase
- sHA 14–1:
-
Stable analog of HA 14–1
- SL:
-
Sesquiterpene lactone
- T-ALL:
-
T cell acute lymphoblastic leukemia
- TCD:
-
Tricyclic clerodane diterpenes
- TM:
-
Transmembrane
- TTP:
-
Time to progression
- UPR:
-
Unfolded protein responses
References
Bray SJ. Notch signalling in context. Nat Rev Mol Cell Biol. 2016;17(11):722–35.
Rand MD, Grimm LM, Artavanis-Tsakonas S, Patriub V, Blacklow SC, Sklar J, et al. Calcium depletion dissociates and activates heterodimeric notch receptors. Mol Cell Biol. 2000;20(5):1825–35.
Pagliaro L, Sorrentino C, Roti G. Targeting Notch trafficking and processing in cancers. Cells. 2020;9(10):2212.
Blaumueller CM, Qi H, Zagouras P, Artavanis-Tsakonas S. Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell. 1997;90(2):281–91.
Lai EC. Notch signaling: control of cell communication and cell fate. Development. 2004;131(5):965–73.
Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, et al. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol Cell. 2000;5(2):197–206.
Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, et al. A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell. 2000;5(2):207–16.
Wu L, Aster JC, Blacklow SC, Lake R, Artavanis-Tsakonas S, Griffin JD. MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for NOTCH receptors. Nat Genet. 2000;26(4):484–9.
Aster JC, Pear WS, Blacklow SC. The varied roles of Notch in cancer. Annu Rev Pathol. 2017;12:245–75.
Gridley T. Notch signaling and inherited disease syndromes. Hum Mol Genet. 2003;12(suppl_1):R9–13.
Khosla R, Vyas AK, Trehanpati N. Dichotomy of Notch signalling in regulating tumour immune surveillance. Scand J Immunol. 2019;89(3):e12744.
Meurette O, Mehlen P. Notch signaling in the tumor microenvironment. Cancer Cell. 2018;34(4):536–48.
Artavanis-Tsakonas S. The molecular biology of the Notch locus and the fine tuning of differentiation in Drosophila. Trends Genet. 1988;4(4):95–100.
Gering M, Patient R. Notch signalling and haematopoietic stem cell formation during embryogenesis. J Cell Physiol. 2010;222(1):11–6.
Bugeon L, Taylor HB, Progatzky F, Lin MI, Ellis CD, Welsh N, et al. The NOTCH pathway contributes to cell fate decision in myelopoiesis. Haematologica. 2011;96(12):1753–60.
Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269–71.
Kridel R, Meissner B, Rogic S, Boyle M, Telenius A, Woolcock B, et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood. 2012;119(9):1963–71.
Arruga F, Gizdic B, Serra S, Vaisitti T, Ciardullo C, Coscia M, et al. Functional impact of NOTCH1 mutations in chronic lymphocytic leukemia. Leukemia. 2014;28(5):1060–70.
Fabbri G, Holmes AB, Viganotti M, Scuoppo C, Belver L, Herranz D, et al. Common nonmutational NOTCH1 activation in chronic lymphocytic leukemia. Proc Natl Acad Sci. 2017;114(14):E2911–9.
Robinson DR, Kalyana-Sundaram S, Wu Y-M, Shankar S, Cao X, Ateeq B, et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat Med. 2011;17(12):1646.
Stoeck A, Lejnine S, Truong A, Pan L, Wang H, Zang C, et al. Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma. Cancer Discov. 2014;4(10):1154–67.
Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547(7663):311–7.
Westhoff B, Colaluca IN, D’Ario G, Donzelli M, Tosoni D, Volorio S, et al. Alterations of the Notch pathway in lung cancer. Proc Natl Acad Sci. 2009;106(52):22293–8.
Golan T, Messer AR, Amitai-Lange A, Melamed ZE, Ohana R, Bell RE, et al. Interactions of melanoma cells with distal keratinocytes trigger metastasis via notch signaling inhibition of MITF. Mol Cell. 2015;59(4):664–76.
Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11(5):338–51.
Pickering CR, Zhou JH, Lee JJ, Drummond JA, Peng SA, Saade RE, et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin Cancer Res. 2014;20(24):6582–92.
South AP, Purdie KJ, Watt SA, Haldenby S, Den Breems NY, Dimon M, et al. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J Invest Dermatol. 2014;134(10):2630–8.
Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333(6046):1154–7.
Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157–60.
Klinakis A, Lobry C, Abdel-Wahab O, Oh P, Haeno H, Buonamici S, et al. A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature. 2011;473(7346):230.
Lobry C, Ntziachristos P, Ndiaye-Lobry D, Oh P, Cimmino L, Zhu N, et al. Notch pathway activation targets AML-initiating cell homeostasis and differentiation. J Exp Med. 2013;210(2):301–19.
Roti G, Stegmaier K. Targeting NOTCH1 in hematopoietic malignancy. Crit Rev Oncog. 2011;16(1–2):103–15.
Sorrentino C, Cuneo A, Roti G. Therapeutic targeting of Notch signaling pathway in hematological malignancies. Mediterr J Hematol Infect Dis. 2019;11(1):e2019037.
Riccio O, Van Gijn ME, Bezdek AC, Pellegrinet L, Van Es JH, Zimber-Strobl U, et al. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. EMBO Rep. 2008;9(4):377–83.
Roti G, Carlton A, Ross KN, Markstein M, Pajcini K, Su AH, et al. Complementary genomic screens identify SERCA as a therapeutic target in NOTCH1 mutated cancer. Cancer Cell. 2013;23(3):390–405.
Toyoshima C, Sasabe H, Stokes DL. Three-dimensional cryo-electron microscopy of the calcium ion pump in the sarcoplasmic reticulum membrane. Nature. 1993;362(6419):469.
Aguayo-Ortiz R, Espinoza-Fonseca LM. Linking biochemical and structural states of SERCA: achievements, challenges, and new opportunities. Int J Mol Sci. 2020;21(11):4146.
Zhang P, Toyoshima C, Yonekura K, Green NM, Stokes DL. Structure of the calcium pump from sarcoplasmic reticulum at 8-Å resolution. Nature. 1998;392(6678):835.
Toyoshima C, Nakasako M, Nomura H, Ogawa H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature. 2000;405(6787):647.
Moncoq K, Trieber CA, Young HS. The molecular basis for cyclopiazonic acid inhibition of the sarcoplasmic reticulum calcium pump. J Biol Chem. 2007;282(13):9748–57.
Laursen M, Bublitz M, Moncoq K, Olesen C, Moller JV, Young HS, et al. Cyclopiazonic acid is complexed to a divalent metal ion when bound to the sarcoplasmic reticulum Ca2+-ATPase. J Biol Chem. 2009;284(20):13513–8.
Toyoshima C, Nomura H. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature. 2002;418(6898):605–11.
Lipskaia L, Hulot J-S, Lompré A-M. Role of sarco/endoplasmic reticulum calcium content and calcium ATPase activity in the control of cell growth and proliferation. Pflügers Archiv. 2009;457(3):673–85.
Bobe R, Bredoux R, Corvazier E, Lacabaratz-Porret C, Martin V, Kovacs T, et al. How many Ca2+ ATPase isoforms are expressed in a cell type? A growing family of membrane proteins illustrated by studies in platelets. Platelets. 2005;16(3–4):133–50.
Research Collaboratory for Structural Bioinformatics Protein Data Bank. https://www.rcsb.org/. Accessed 29 July 2020
Papp B, Brouland JP, Arbabian A, Gelebart P, Kovacs T, Bobe R, et al. Endoplasmic reticulum calcium pumps and cancer cell differentiation. Biomolecules. 2012;2(1):165–86.
Vandecaetsbeek I, Vangheluwe P, Raeymaekers L, Wuytack F, Vanoevelen J. The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb Perspect Biol. 2011;3(5):a004184.
Wuytack F, Raeymaekers L, Missiaen L. Molecular physiology of the SERCA and SPCA pumps. Cell Calcium. 2002;32(5–6):279–305.
Baba-Aissa F, Raeymaekers L, Wuytack F, Dode L, Casteels R. Distribution and isoform diversity of the organellar Ca2+ pumps in the brain. Mol Chem Neuropathol. 1998;33(3):199–208.
Wootton LL, Michelangeli F. The effects of the phenylalanine 256 to valine mutation on the sensitivity of sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) Ca2+ pump isoforms 1, 2, and 3 to thapsigargin and other inhibitors. J Biol Chem. 2006;281(11):6970–6.
Brini M, Calì T, Ottolini D, Carafoli E. Calcium pumps: why so many? Compr Physiol. 2011;2(2):1045–60.
Primeau JO, Armanious GP, M’Lynn EF, Young HS. The sarcoendoplasmic reticulum calcium ATPase. Membrane protein complexes: structure and function. Berlin: Springer; 2018. p. 229–58.
Bublitz M, Musgaard M, Poulsen H, Thøgersen L, Olesen C, Schiøtt B, et al. Ion pathways in the sarcoplasmic reticulum Ca2+-ATPase. J Biol Chem. 2013;288(15):10759–65.
Albers R. Biochemical aspects of active transport. Annu Rev Biochem. 1967;36(1):727–56.
Olesen C, Picard M, Winther A-ML, Gyrup C, Morth JP, Oxvig C, et al. The structural basis of calcium transport by the calcium pump. Nature. 2007;450(7172):1036–42.
Jensen AML, Sørensen TLM, Olesen C, Møller JV, Nissen P. Modulatory and catalytic modes of ATP binding by the calcium pump. EMBO J. 2006;25(11):2305–14.
Sørensen TL-M, Møller JV, Nissen P. Phosphoryl transfer and calcium ion occlusion in the calcium pump. Science. 2004;304(5677):1672–5.
Møller JV, Nissen P, Sørensen TL, le Maire M. Transport mechanism of the sarcoplasmic reticulum Ca2+-ATPase pump. Curr Opin Struct Biol. 2005;15(4):387–93.
Toyoshima C. Ion pumping by calcium ATPase of sarcoplasmic reticulum. Regulatory mechanisms of striated muscle contraction. Berlin: Springer; 2007. p. 295–303.
Toyoshima C. How Ca2+-ATPase pumps ions across the sarcoplasmic reticulum membrane. Biochim Biophys Acta. 2009;1793(6):941–6.
Gorski PA, Ceholski DK, Young HS. Structure–function relationship of the SERCA pump and its regulation by phospholamban and sarcolipin. Membrane dynamics and calcium signaling. Berlin: Springer; 2017. p. 77–119.
Sagara Y, Fernandez-Belda F, de Meis L, Inesi G. Characterization of the inhibition of intracellular Ca2+ transport ATPases by thapsigargin. J Biol Chem. 1992;267(18):12606–13.
Nørregaard A, Vilsen B, Andersen JP. Transmembrane segment M3 is essential to thapsigargin sensitivity of the sarcoplasmic reticulum Ca(2+)-ATPase. J Biol Chem. 1994;269(43):26598–601.
Winther AML, Bublitz M, Karlsen JL, Møller JV, Hansen JB, Nissen P, et al. The sarcolipin-bound calcium pump stabilizes calcium sites exposed to the cytoplasm. Nature. 2013;495(7440):265–9.
Periasamy M, Kalyanasundaram A. SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve. 2007;35(4):430–42.
Kósa M, Brinyiczki K, Van Damme P, Goemans N, Hancsák K, Mendler L, et al. The neonatal sarcoplasmic reticulum Ca2+-ATPase gives a clue to development and pathology in human muscles. J Muscle Res Cell Motil. 2015;36(2):195–203.
Chandrasekera PC, Kargacin ME, Deans JP, Lytton J. Determination of apparent calcium affinity for endogenously expressed human sarco(endo)plasmic reticulum calcium-ATPase isoform SERCA3. Am J Physiol Cell Physiol. 2009;296(5):C1105–14.
Verboomen H, Wuytack F, De Smedt H, Himpens B, Casteels R. Functional difference between SERCA2a and SERCA2b Ca2+ pumps and their modulation by phospholamban. Biochem J. 1992;286(2):591–5.
Gunteski-Hamblin A-M, Greeb J, Shull G. A novel Ca2+ pump expressed in brain, kidney, and stomach is encoded by an alternative transcript of the slow-twitch muscle sarcoplasmic reticulum Ca-ATPase gene. Identification of cDNAs encoding Ca2+ and other cation-transporting ATPases using an oligonucleotide probe derived from the ATP-binding site. J Biol Chem. 1988;263(29):15032–40.
Campbell AM, Kessler PD, Fambrough DM. The alternative carboxyl termini of avian cardiac and brain sarcoplasmic reticulum/endoplasmic reticulum Ca (2+)-ATPases are on opposite sides of the membrane. J Biol Chem. 1992;267(13):9321–5.
Ushioda R, Miyamoto A, Inoue M, Watanabe S, Okumura M, Maegawa KI, et al. Redox-assisted regulation of Ca2+ homeostasis in the endoplasmic reticulum by disulfide reductase ERdj5. Proc Natl Acad Sci. 2016;113(41):E6055–63.
Inoue M, Sakuta N, Watanabe S, Zhang Y, Yoshikaie K, Tanaka Y, et al. Structural basis of sarco/endoplasmic reticulum Ca(2+)-ATPase 2b regulation via transmembrane helix interplay. Cell Rep. 2019;27(4):1221-1230e3.
Gelebart P, Martin V, Enouf J, Papp B. Identification of a new SERCA2 splice variant regulated during monocytic differentiation. Biochem Biophys Res Commun. 2003;303(2):676–84.
Dally S, Bredoux R, Corvazier E, Andersen JP, Clausen JD, Dode L, et al. Ca2+-ATPases in non-failing and failing heart: evidence for a novel cardiac sarco/endoplasmic reticulum Ca2+-ATPase 2 isoform (SERCA2c). Biochem J. 2006;395(2):249–58.
Kimura T, Nakamori M, Lueck JD, Pouliquin P, Aoike F, Fujimura H, et al. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum Mol Genet. 2005;14(15):2189–200.
Wuytack F, Papp B, Verboomen H, Raeymaekers L, Dode L, Bobe R, et al. A sarco/endoplasmic reticulum Ca(2+)-ATPase 3-type Ca2+ pump is expressed in platelets, in lymphoid cells, and in mast cells. J Biol Chem. 1994;269(2):1410–6.
Chemaly ER, Troncone L, Lebeche D. SERCA control of cell death and survival. Cell Calcium. 2018;69:46–61.
Novelli A, Valente EM, Bernardini L, Ceccarini C, Sinibaldi L, Caputo V, et al. Autosomal dominant Brody disease cosegregates with a chromosomal (2;7)(p11.2;p12.1) translocation in an Italian family. Eur J Hum Genet. 2004;12(7):579–83.
Odermatt A, Taschner PE, Khanna VK, Busch HF, Karpati G, Jablecki CK, et al. Mutations in the gene-encoding SERCA1, the fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+ ATPase, are associated with Brody disease. Nat Genet. 1996;14(2):191–4.
Foggia L, Hovnanian A. Calcium pump disorders of the skin. Am J Med Genet Sem Med Genet. 2004;131c(1):20–31.
Dhitavat J, Fairclough RJ, Hovnanian A, Burge SM. Calcium pumps and keratinocytes: lessons from Darier’s disease and Hailey-Hailey disease. Br J Dermatol. 2004;150(5):821–8.
Rodriguez-Mora O, LaHair MM, Howe CJ, McCubrey JA, Franklin RA. Calcium/calmodulin-dependent protein kinases as potential targets in cancer therapy. Expert Opin Ther Targets. 2005;9(4):791–808.
Park KC, Kim SW, Jeon JY, Jo AR, Choi HJ, Kim J, et al. Survival of cancer stem-like cells under metabolic stress via CaMK2alpha-mediated upregulation of sarco/endoplasmic reticulum calcium ATPase expression. Clin Cancer Res. 2018;24(7):1677–90.
Michelangeli F, East JM. A diversity of SERCA Ca2+ pump inhibitors. Biochem Soc Trans. 2011;39(3):789–97.
Fan L, Li A, Li W, Cai P, Yang B, Zhang M, et al. Novel role of Sarco/endoplasmic reticulum calcium ATPase 2 in development of colorectal cancer and its regulation by F36, a curcumin analog. Biomed Pharmacother. 2014;68(8):1141–8.
Pacifico F, Ulianich L, De Micheli S, Treglia S, Leonardi A, Vito P, et al. The expression of the sarco/endoplasmic reticulum Ca2+-ATPases in thyroid and its down-regulation following neoplastic transformation. J Mol Endocrinol. 2003;30(3):399–409.
Brouland J-P, Gélébart P, Kovacs T, Enouf J, Grossmann J, Papp B. The loss of sarco/endoplasmic reticulum calcium transport ATPase 3 expression is an early event during the multistep process of colon carcinogenesis. Am J Pathol. 2005;167(1):233–42.
Arbabian A, Brouland JP, Apáti Á, Pászty K, Hegedűs L, Enyedi Á, et al. Modulation of endoplasmic reticulum calcium pump expression during lung cancer cell differentiation. FEBS J. 2013;280(21):5408–18.
Lipskaia L, Keuylian Z, Blirando K, Mougenot N, Jacquet A, Rouxel C, et al. Expression of sarco (endo) plasmic reticulum calcium ATPase (SERCA) system in normal mouse cardiovascular tissues, heart failure and atherosclerosis. Biochim Biophys Acta. 2014;1843(11):2705–18.
Launay S, Gianni M, Kovacs T, Bredoux R, Bruel A, Gelebart P, et al. Lineage-specific modulation of calcium pump expression during myeloid differentiation. Blood. 1999;93(12):4395–405.
Lacabaratz-Porret C, Launay S, Corvazier E, Bredoux R, Papp B, Enouf J. Biogenesis of endoplasmic reticulum proteins involved in Ca2+ signalling during megakaryocytic differentiation: an in vitro study. Biochem J. 2000;350(Pt 3):723–34.
Korošec B, Glavač D, Rott T, Ravnik-Glavač M. Alterations in the ATP2A2 gene in correlation with colon and lung cancer. Cancer Genet Cytogenet. 2006;171(2):105–11.
Liu LH, Boivin GP, Prasad V, Periasamy M, Shull GE. Squamous cell tumors in mice heterozygous for a null allele of Atp2a2, encoding the sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2 Ca2+ pump. J Biol Chem. 2001;276(29):26737–40.
Toki H, Minowa O, Inoue M, Motegi H, Karashima Y, Ikeda A, et al. Novel allelic mutations in murine Serca2 induce differential development of squamous cell tumors. Biochem Biophys Res Commun. 2016;476(4):175–82.
Prasad V, Boivin GP, Miller ML, Liu LH, Erwin CR, Warner BW, et al. Haploinsufficiency of Atp2a2, encoding the sarco (endo) plasmic reticulum Ca2+-ATPase isoform 2 Ca2+ pump, predisposes mice to squamous cell tumors via a novel mode of cancer susceptibility. Can Res. 2005;65(19):8655–61.
Periz G, Fortini ME. Ca(2+)-ATPase function is required for intracellular trafficking of the Notch receptor in Drosophila. EMBO J. 1999;18(21):5983–93.
Baldoni S, Del Papa B, Dorillo E, Aureli P, De Falco F, Rompietti C, et al. Bepridil exhibits anti-leukemic activity associated with NOTCH1 pathway inhibition in chronic lymphocytic leukemia. Int J Cancer. 2018;143(4):958–70.
Ghantous A, Gali-Muhtasib H, Vuorela H, Saliba NA, Darwiche N. What made sesquiterpene lactones reach cancer clinical trials? Drug Discov Today. 2010;15(15–16):668–78.
Zhang Z, Yu S, Miao L, Huang X, Zhang X, Zhu Y, et al. Artesunate combined with vinorelbine plus cisplatin in treatment of advanced non-small cell lung cancer: a randomized controlled trial. Zhong Xi Yi Jie He Xue Bao. 2008;6(2):134–8.
Berger TG, Dieckmann D, Efferth T, Schultz ES, Funk J-O, Baur A, et al. Artesunate in the treatment of metastatic uveal melanoma-first experiences. Oncol Rep. 2005;14(6):1599–603.
Singh N, Verma K. Case report of a laryngeal squamous cell carcinoma treated with artesunate. Arch Oncol. 2002;10(4):279–80.
Singh NP, Panwar VK. Case report of a pituitary macroadenoma treated with artemether. Integr Cancer Ther. 2006;5(4):391–4.
Denmeade SR, Isaacs JT. The SERCA pump as a therapeutic target: making a “smart bomb” for prostate cancer. Cancer Biol Ther. 2005;4(1):14–22.
Christensen SB, Skytte DM, Denmeade SR, Dionne C, Moller JV, Nissen P, et al. A Trojan horse in drug development: targeting of thapsigargins towards prostate cancer cells. Anticancer Agents Med Chem. 2009;9(3):276–94.
Doan NT, Paulsen ES, Sehgal P, Moller JV, Nissen P, Denmeade SR, et al. Targeting thapsigargin towards tumors. Steroids. 2015;97:2–7.
Sagara Y, Wade J, Inesi G. A conformational mechanism for formation of a dead-end complex by the sarcoplasmic reticulum ATPase with thapsigargin. J Biol Chem. 1992;267(2):1286–92.
Xu C, Ma H, Inesi G, Al-Shawi MK, Toyoshima C. Specific structural requirements for the inhibitory effect of thapsigargin on the Ca2+ ATPase SERCA. J Biol Chem. 2004;279(17):17973–9.
Sagara Y, Inesi G. Inhibition of the sarcoplasmic reticulum Ca2+ transport ATPase by thapsigargin at subnanomolar concentrations. J Biol Chem. 1991;266(21):13503–6.
Gravett AM, Liu WM, Krishna S, Chan W-C, Haynes RK, Wilson NL, et al. In vitro study of the anti-cancer effects of artemisone alone or in combination with other chemotherapeutic agents. Cancer Chemother Pharmacol. 2011;67(3):569–77.
Sun Q, Wang J, Li Y, Zhuang J, Zhang Q, Sun X, et al. Synthesis and evaluation of cytotoxic activities of artemisinin derivatives. Chem Biol Drug Des. 2017;90(5):1019–28.
Stockwin LH, Han B, Yu SX, Hollingshead MG, ElSohly MA, Gul W, et al. Artemisinin dimer anticancer activity correlates with heme-catalyzed reactive oxygen species generation and endoplasmic reticulum stress induction. Int J Cancer. 2009;125(6):1266–75.
Michaelis M, Kleinschmidt MC, Barth S, Rothweiler F, Geiler J, Breitling R, et al. Anti-cancer effects of artesunate in a panel of chemoresistant neuroblastoma cell lines. Biochem Pharmacol. 2010;79(2):130–6.
Liu Y, Wong VKW, Ko BC-B, Wong M-K, Che C-M. Synthesis and cytotoxicity studies of artemisinin derivatives containing lipophilic alkyl carbon chains. Organ Lett. 2005;7(8):1561–4.
Marchesini M, Gherli A, Montanaro A, Patrizi L, Sorrentino C, Pagliaro L, et al. Blockade of oncogenic NOTCH1 with the SERCA inhibitor CAD204520 in T cell acute lymphoblastic leukemia. Cell Chem Biol. 2020;27:678–97.
De Ford C, Heidersdorf B, Haun F, Murillo R, Friedrich T, Borner C, et al. The clerodane diterpene casearin J induces apoptosis of T-ALL cells through SERCA inhibition, oxidative stress, and interference with Notch1 signaling. Cell Death Dis. 2016;7:e2070.
De Ford C, Calderon C, Sehgal P, Fedosova NU, Murillo R, Olesen C, et al. Discovery of tricyclic clerodane diterpenes as sarco/endoplasmic reticulum Ca(2+)-ATPase inhibitors and structure–activity relationships. J Nat Prod. 2015;78(6):1262–70.
Bilmen JG, Khan SZ, Javed MH, Michelangeli F. Inhibition of the SERCA Ca2+ pumps by curcumin. Curcumin putatively stabilizes the interaction between the nucleotide-binding and phosphorylation domains in the absence of ATP. Eur J Biochem. 2001;268(23):6318–27.
Garrido-Armas M, Corona JC, Escobar ML, Torres L, Ordóñez-Romero F, Hernández-Hernández A, et al. Paraptosis in human glioblastoma cell line induced by curcumin. Toxicol In Vitro. 2018;51:63–73.
Calibasi-Kocal G, Pakdemirli A, Bayrak S, Ozupek NM, Sever T, Basbinar Y, et al. Curcumin effects on cell proliferation, angiogenesis and metastasis in colorectal cancer. J BUON. 2019;24(4):1482–7.
Mishra D, Singh S, Narayan G. Curcumin induces apoptosis in Pre-B acute lymphoblastic leukemia cell lines via PARP-1 cleavage. Asian Pac J Cancer Prev. 2016;17(8):3865–9.
Thacker PC, Karunagaran D. Curcumin and emodin down-regulate TGF-β signaling pathway in human cervical cancer cells. PLoS ONE. 2015;10(3):e120045.
Tabatabaei Mirakabad FS, Akbarzadeh A, Milani M, Zarghami N, Taheri-Anganeh M, Zeighamian V, et al. A comparison between the cytotoxic effects of pure curcumin and curcumin-loaded PLGA-PEG nanoparticles on the MCF-7 human breast cancer cell line. Artif Cells Nanomed Biotechnol. 2016;44(1):423–30.
Amin AR, Haque A, Rahman MA, Chen ZG, Khuri FR, Shin DM. Curcumin induces apoptosis of upper aerodigestive tract cancer cells by targeting multiple pathways. PLoS ONE. 2015;10(4):e0124218.
Wang L, Wang L, Song R, Shen Y, Sun Y, Gu Y, et al. Targeting sarcoplasmic/endoplasmic reticulum Ca2+-ATPase 2 by curcumin induces ER stress-associated apoptosis for treating human liposarcoma. Mol Cancer Ther. 2011;10(3):461–71.
Das SG, Srinivasan B, Hermanson DL, Bleeker NP, Doshi JM, Tang R, et al. Structure-activity relationship and molecular mechanisms of ethyl 2-amino-6-(3,5-dimethoxyphenyl)-4-(2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate (CXL017) and its analogues. J Med Chem. 2011;54(16):5937–48.
Bleeker NP, Cornea RL, Thomas DD, Xing C. A novel SERCA inhibitor demonstrates synergy with classic SERCA inhibitors and targets multidrug-resistant AML. Mol Pharm. 2013;10(11):4358–66.
Hymery N, Masson F, Barbier G, Coton E. Cytotoxicity and immunotoxicity of cyclopiazonic acid on human cells. Toxicol In Vitro. 2014;28(5):940–7.
Hermanson D, Addo SN, Bajer AA, Marchant JS, Das SG, Srinivasan B, et al. Dual mechanisms of sHA 14–1 in inducing cell death through endoplasmic reticulum and mitochondria. Mol Pharmacol. 2009;76(3):667–78.
Wu L, Huang X, Kuang Y, Xing Z, Deng X, Luo Z. Thapsigargin induces apoptosis in adrenocortical carcinoma by activating endoplasmic reticulum stress and the JNK signaling pathway: an in vitro and in vivo study. Drug Des Dev Therapy. 2019;13:2787.
Loukianov E, Ji Y, Grupp IL, Kirkpatrick DL, Baker DL, Loukianova T, et al. Enhanced myocardial contractility and increased Ca2+ transport function in transgenic hearts expressing the fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+-ATPase. Circ Res. 1998;83(9):889–97.
Jung M, Kim H, Nam KY, No KT. Three-dimensional structure of Plasmodium falciparum Ca2+-ATPase (PfATP6) and docking of artemisinin derivatives to PfATP6. Bioorg Med Chem Lett. 2005;15(12):2994–7.
Nagasundaram N, Chakraborty C, Karthick V, Balaji V, Siva R, Lu A, et al. Mechanism of artemisinin resistance for malaria PfATP6 L263 mutations and discovering potential antimalarials: an integrated computational approach. Sci Rep. 2016;6:30106.
DeAngelo DJ, Stone RM, Silverman LB, Aster JC. A phase I clinical trial of the Notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. J Clin Oncol. 2006;24:6585.
Sharma A, Gadkari RA, Ramakanth SV, Padmanabhan K, Madhumathi DS, Devi L, et al. A novel monoclonal antibody against Notch1 targets leukemia-associated mutant Notch1 and depletes therapy resistant cancer stem cells in solid tumors. Sci Rep. 2015;5:11012.
Sun W, Zhang H, Wang H, Chiu YG, Wang M, Ritchlin CT, et al. Targeting Notch-activated M1 macrophages attenuates joint tissue damage in a mouse model of inflammatory arthritis. J Bone Miner Res. 2017;32(7):1469–80.
Lytton J, Westlin M, Hanley MR. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem. 1991;266(26):17067–71.
Denmeade SR, Mhaka AM, Rosen DM, Brennen WN, Dalrymple S, Dach I, et al. Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci Transl Med. 2012;4(140):140.
Thi Quynh Doan N, Brogger Christensen S. Thapsigargin, origin, chemistry, structure-activity relationships and prodrug development. Curr Pharm Des. 2015;21(38):5501–17.
Christensen SB, Andersen A, Poulsen JC, Treiman M. Derivatives of thapsigargin as probes of its binding site on endoplasmic reticulum Ca2+ ATPase. Stereoselectivity and important functional groups. FEBS Lett. 1993;335(3):345–8.
Aloysius H, Hu L. Targeted prodrug approaches for hormone refractory prostate cancer. Med Res Rev. 2015;35(3):554–85.
Mahalingam D, Peguero J, Cen P, Arora SP, Sarantopoulos J, Rowe J, et al. A phase II, multicenter, single-arm study of Mipsagargin (G-202) as a second-line therapy following sorafenib for adult patients with progressive advanced hepatocellular carcinoma. Cancers. 2019;11(6):833.
Mahalingam D, Peguero J, Cen P, Allgood V, Shazer R, Campos L. Mipsagargin, a PSMA-directed prodrug, provides clinical benefit in patients with advanced sorafenib-refractory hepatocellular carcinoma. J Hepatol. 2017;66(1):S207.
Piccioni D, Juarez T, Brown B, Rose L, Allgood V, Kesari S. ATCT-18 phase II study of Mipsagargin (G-202), a PSMA-activated prodrug targeting the tumor endothelium, in adult patients with recurrent or progressive glioblastOMA. Neuro-oncology. 2015;17(Suppl 5):v5.
Denmeade SR, Jakobsen CM, Janssen S, Khan SR, Garrett ES, Lilja H, et al. Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. J Natl Cancer Inst. 2003;95(13):990–1000.
Roti G, Qi J, Kitara S, Sanchez-Martin M, Saur Conway A, Varca AC, et al. Leukemia-specific delivery of mutant NOTCH1 targeted therapy. J Exp Med. 2018;215(1):197–216.
Obara K, Miyashita N, Xu C, Toyoshima I, Sugita Y, Inesi G, et al. Structural role of countertransport revealed in Ca(2+) pump crystal structure in the absence of Ca(2+). Proc Natl Acad Sci USA. 2005;102(41):14489–96.
Paula S, Abell J, Deye J, Elam C, Lape M, Purnell J, et al. Design, synthesis, and biological evaluation of hydroquinone derivatives as novel inhibitors of the sarco/endoplasmic reticulum calcium ATPase. Bioorg Med Chem. 2009;17(18):6613–9.
Elam C, Lape M, Deye J, Zultowsky J, Stanton DT, Paula S. Discovery of novel SERCA inhibitors by virtual screening of a large compound library. Eur J Med Chem. 2011;46(5):1512–23.
Lape M, Elam C, Paula S. Comparison of current docking tools for the simulation of inhibitor binding by the transmembrane domain of the sarco/endoplasmic reticulum calcium ATPase. Biophys Chem. 2010;150(1–3):88–97.
Cardi D, Pozza A, Arnou B, Marchal E, Clausen JD, Andersen JP, et al. Purified E255L mutant SERCA1a and purified PfATP6 are sensitive to SERCA-type inhibitors but insensitive to artemisinins. The Journal of biological chemistry. 2010;285(34):26406–16.
Tadini-Buoninsegni F, Smeazzetto S, Gualdani R, Moncelli MR. Drug interactions with the Ca(2+)-ATPase from sarco(endo)plasmic reticulum (SERCA). Front Mol Biosci. 2018;5:36.
Burghoorn HP, Soteropoulos P, Paderu P, Kashiwazaki R, Perlin DS. Molecular evaluation of the plasma membrane proton pump from Aspergillus fumigatus. Antimicrob Agents Chemother. 2002;46(3):615–24.
Tian D, Das SG, Doshi JM, Peng J, Lin J, Xing C. sHA 14–1, a stable and ROS-free antagonist against anti-apoptotic Bcl-2 proteins, bypasses drug resistances and synergizes cancer therapies in human leukemia cell. Cancer Lett. 2008;259(2):198–208.
Das SG, Doshi JM, Tian D, Addo SN, Srinivasan B, Hermanson DL, et al. Structure-activity relationship and molecular mechanisms of ethyl 2-amino-4-(2-ethoxy-2-oxoethyl)-6-phenyl-4h-chromene-3-carboxylate (sha 14–1) and its analogues. J Med Chem. 2009;52(19):5937–49.
Aridoss G, Zhou B, Hermanson DL, Bleeker NP, Xing C. Structure-activity relationship (SAR) study of ethyl 2-amino-6-(3,5-dimethoxyphenyl)-4-(2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate (CXL017) and the potential of the lead against multidrug resistance in cancer treatment. J Med Chem. 2012;55(11):5566–81.
Pae HO, Jeong SO, Jeong GS, Kim KM, Kim HS, Kim SA, et al. Curcumin induces pro-apoptotic endoplasmic reticulum stress in human leukemia HL-60 cells. Biochem Biophys Res Commun. 2007;353(4):1040–5.
Bakhshi J, Weinstein L, Poksay KS, Nishinaga B, Bredesen DE, Rao RV. Coupling endoplasmic reticulum stress to the cell death program in mouse melanoma cells: effect of curcumin. Apoptosis. 2008;13(7):904–14.
Xu XY, Meng X, Li S, Gan RY, Li Y, Li HB. Bioactivity, health benefits, and related molecular mechanisms of curcumin: current progress, challenges, and perspectives. Nutrients. 2018;10(10):1553.
Sviatenko LK. Molecular docking of curcumin analogues as serca inhibitory agents. Bicник Днiпpoпeтpoвcькoгo yнiвepcитeтy Cep: Xiмiя. 2013(21, Bип. 20):15–21.
Dhillon N, Aggarwal BB, Newman RA, Wolff RA, Kunnumakkara AB, Abbruzzese JL, et al. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res. 2008;14(14):4491–9.
Howells LM, Iwuji CO, Irving GR, Barber S, Walter H, Sidat Z, et al. Curcumin combined with FOLFOX chemotherapy is safe and tolerable in patients with metastatic colorectal cancer in a randomized phase IIa trial. J Nutr. 2019;149(7):1133–9.
Irving GR, Iwuji CO, Morgan B, Berry DP, Steward WP, Thomas A, et al. Combining curcumin (C3-complex, Sabinsa) with standard care FOLFOX chemotherapy in patients with inoperable colorectal cancer (CUFOX): study protocol for a randomised control trial. Trials. 2015;16:110.
Liu ZC, Yang ZX, Zhou JS, Zhang HT, Huang QK, Dang LL, et al. Curcumin regulates hepatoma cell proliferation and apoptosis through the Notch signaling pathway. Int J Clin Exp Med. 2014;7(3):714–8.
Li Y, Zhang J, Ma D, Zhang L, Si M, Yin H, et al. Curcumin inhibits proliferation and invasion of osteosarcoma cells through inactivation of Notch-1 signaling. FEBS J. 2012;279(12):2247–59.
Subramaniam D, Ponnurangam S, Ramamoorthy P, Standing D, Battafarano RJ, Anant S, et al. Curcumin induces cell death in esophageal cancer cells through modulating Notch signaling. PLoS ONE. 2012;7(2):e30590.
Zhou X, Su J, Feng S, Wang L, Yin X, Yan J, et al. Antitumor activity of curcumin is involved in down-regulation of YAP/TAZ expression in pancreatic cancer cells. Oncotarget. 2016;7(48):79076–88.
Yang B, Zhang M, Gao J, Li J, Fan L, Xiang G, et al. Small molecule RL71 targets SERCA2 at a novel site in the treatment of human colorectal cancer. Oncotarget. 2015;6(35):37613–25.
Hackler L Jr, Ozsvari B, Gyuris M, Sipos P, Fabian G, Molnar E, et al. The curcumin analog C-150, influencing NF-kappaB, UPR and Akt/Notch pathways has potent anticancer activity in vitro and in vivo. PLoS ONE. 2016;11(3):e0149832.
Tadini-Buoninsegni F, Sordi G, Smeazzetto S, Natile G, Arnesano F. Effect of cisplatin on the transport activity of PII-type ATPases. Metallomics. 2017;9(7):960–8.
Crespo-Ortiz MP, Wei MQ. Antitumor activity of artemisinin and its derivatives: from a well-known antimalarial agent to a potential anticancer drug. J Biomed Biotechnol. 2012;2012:247597.
Bhaw-Luximon A, Jhurry D. Artemisinin and its derivatives in cancer therapy: status of progress, mechanism of action, and future perspectives. Cancer Chemother Pharmacol. 2017;79(3):451–66.
Sehgal P, Szalai P, Olesen C, Praetorius HA, Nissen P, Christensen SB, et al. Inhibition of the sarco/endoplasmic reticulum (ER) Ca(2+)-ATPase by thapsigargin analogs induces cell death via ER Ca(2+) depletion and the unfolded protein response. J Biol Chem. 2017;292(48):19656–73.
Winther AM, Liu H, Sonntag Y, Olesen C, le Maire M, Soehoel H, et al. Critical roles of hydrophobicity and orientation of side chains for inactivation of sarcoplasmic reticulum Ca2+-ATPase with thapsigargin and thapsigargin analogs. J Biol Chem. 2010;285(37):28883–92.
Zhong L, Inesi G. Role of the S3 stalk segment in the thapsigargin concentration dependence of sarco-endoplasmic reticulum Ca2+ ATPase inhibition. J Biol Chem. 1998;273(21):12994–8.
Horn M, Kroef V, Allmeroth K, Schuller N, Miethe S, Peifer M, et al. Unbiased compound-protein interface mapping and prediction of chemoresistance loci through forward genetics in haploid stem cells. Oncotarget. 2018;9(11):9838–51.
Can G, Akpinar B, Baran Y, Zhivotovsky B, Olsson M. 5-Fluorouracil signaling through a calcium-calmodulin-dependent pathway is required for p53 activation and apoptosis in colon carcinoma cells. Oncogene. 2013;32(38):4529–38.
Baliakas P, Hadzidimitriou A, Sutton LA, Rossi D, Minga E, Villamor N, et al. Recurrent mutations refine prognosis in chronic lymphocytic leukemia. Leukemia. 2015;29(2):329–36.
Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, et al. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Cell Biol. 2008;181(7):1107–16.
Habets RA, de Bock CE, Serneels L, Lodewijckx I, Verbeke D, Nittner D, et al. Safe targeting of T cell acute lymphoblastic leukemia by pathology-specific NOTCH inhibition. Sci Transl Med. 2019;11(494):6246.
Malecki MJ, Sanchez-Irizarry C, Mitchell JL, Histen G, Xu ML, Aster JC, et al. Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Mol Cell Biol. 2006;26(12):4642–51.
Acknowledgements
Authors thank Anna Montanaro, Andrea Gherli, Elisa Cerretani, Anna D’Antuono for critical suggestions, Pamela Criscuoli for administrative support, and the physicians at the Hematology and BMT unit of the University of Parma. Francesco Marchesini draws the 3D Notch pathway and supervised the art and design work (francescomarchesini@gmail.com).
Funding
This work was supported by an AIRC Start-up Investigator Grant (n. 17107 G.R.), Fondazione Cariparma (3576/2017, 0180/2018 G.R.), Fondazione Grande Ale Onlus (G.R.), Leukemia Research Foundation (G.R.), Feliciani Ferretti Fellowship (L.P.), Associazione Italiana contro le Leucemie-Linfomi e Mieloma ONLUS (AIL) Parma chapter (G.R.). M.M is an EHA-ASH TRTH 2018 and a Beat-leukemia scholar.
Author information
Authors and Affiliations
Contributions
LP, MM, GR helped in conception and design. LP, MM, GR wrote, reviewed, and/or revised the manuscript. GR supervised the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pagliaro, L., Marchesini, M. & Roti, G. Targeting oncogenic Notch signaling with SERCA inhibitors. J Hematol Oncol 14, 8 (2021). https://doi.org/10.1186/s13045-020-01015-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-01015-9