
Abstract
FLT3 mutations are one of the most common findings in acute myeloid leukemia (AML). FLT3 inhibitors have been in active clinical development. Midostaurin as the first-in-class FLT3 inhibitor has been approved for treatment of patients with FLT3-mutated AML. In this review, we summarized the preclinical and clinical studies on new FLT3 inhibitors, including sorafenib, lestaurtinib, sunitinib, tandutinib, quizartinib, midostaurin, gilteritinib, crenolanib, cabozantinib, Sel24-B489, G-749, AMG 925, TTT-3002, and FF-10101. New generation FLT3 inhibitors and combination therapies may overcome resistance to first-generation agents.
Similar content being viewed by others


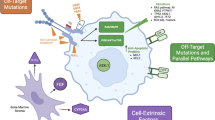
Introduction
Acute myeloid leukemia (AML) remains a highly resistant disease to conventional chemotherapy, with a median survival of only 4 months for relapsed and/or refractory disease [1]. Molecular profiling by PCR and next-generation sequencing has revealed a variety of recurrent gene mutations [2,3,4]. New agents are rapidly emerging as targeted therapy for high-risk AML [5, 6]. In 1996, FMS-like tyrosine kinase 3/internal tandem duplication (FLT3/ITD) was first recognized as a frequently mutated gene in AML [7]. According to 2017 ELN risk stratification, patients with FLT3/ITDhigh-positive AML are classified into adverse risk category. This mutation causes resistance to conventional chemotherapy. Although patients with AML can be cured with hematopoietic stem cell transplantation (HSCT), most of these patients are at high risk for relapse. Thus, the overall cure rate of AML is only 30–40% [1].
FLT3/ITD gene is found in approximately 30% of patients with AML with normal cytogenetics. FLT3/ITD belongs to the type III family of receptor tyrosine kinases [8]. The FLT3 gene is located on chromosome 13 .q12. It is expressed mainly in human hematopoietic progenitors and dendritic cells and plays key roles in leukemia cell proliferation, differentiation, and survival [9]. Constitutive activation of the FLT3/ITD gene triggers multiple downstream signaling cascades, such as STAT5, RAS, MEK, and PI3K/AKT pathways [10], and ultimately causes suppression of apoptosis and differentiation of leukemic cells, including dysregulation of leukemic cell proliferation [11].
Multiple FLT3 inhibitors are in clinical trials for treating patients with FLT3/ITD-mutated AML. In this review, we summarized the preclinical and clinical studies on new FLT3 inhibitors, including sorafenib, lestaurtinib, sunitinib, tandutinib, quizartinib, midostaurin, gilteritinib, crenolanib, cabozantinib, Sel24-B489, G-749, AMG 925, TTT-3002, and FF-10101.
First-generation FLT3 inhibitors
Sorafenib
Sorafenib is an oral multikinase inhibitor of RAF-1, VEGF, c-KIT, PDGFR, ERK, and FLT3. Currently, sorafenib is approved for treating hepatocellular carcinoma and renal cell carcinoma. Sorafenib also has a potent anti-leukemic effect on FLT3-mutated AML. It completely inhibits FLT3/ITD activity with an IC50 of 69.3 ng/ml [12].
Mechanisms of sorafenib effects on AML
There are several mechanisms that explain the efficacy of sorafenib for treating AML. First, sorafenib promotes secretion of IL-15 by FLT3/ITD-mutated leukemic cells and improves the survival time of patients with FLT3/ITD-positive AML [13]. Secondly, persistently low blast percentage, CD3+ cell invasion in the epidermis, high proportion of CD8+ lymphocytes in the bone marrow, and high expression levels of COL4A3, TLR9, FGF1, and IL-12 genes have been observed in patients treated with sorafenib [14]. Sorafenib has also been shown to block Src kinase-mediated STAT3 phosphorylation and reduces expression of apoptosis regulatory proteins such as Mcl-1 and Bcl-2 [15]. Finally, sorafenib decreases Smac mimetic-induced necroptosis in apoptosis-resistant leukemia cells [16].
Sorafenib plus chemotherapy for treating AML
Sorafenib combined with conventional chemotherapy is being explored for AML therapy. Sorafenib and decitabine have been used in preclinical and clinical trials to treat FLT3/ITD-mutated AML in vitro and in vivo [17]. Both drugs showed synergistic anti-tumor effects in a human FLT3/ITD-mutated AML cell line. In a clinical study, 5 out of 6 patients showed overall favorable responses, including 4 relapsed/refractory patients achieving complete remission with incomplete count recovery (CRi). The median survival time of these patients was 155 days, and the drugs were well tolerated. Ravandi et al. reported the effects of sorafenib combined with 5-azacytidine (AZA) in 43 patients with AML, including 40 patients with FLT3/ITD mutation [18]. All patients were intravenously administered AZA 75 mg/m2/d for 7 days and orally administrated sorafenib 400 mg continuously. Response rate (RR) was 46%, complete remission with incomplete count recovery (CRi) 27%, complete remission (CR) 16%, and partial remission (PR) 3%. Recently, Mahdi et al. used the same doses of azacytidine and sorafenib to successfully treat a pregnant patient with FLT3/ITD-mutated AML [19]. After 1 cycle of azacytidine and sorafenib treatment, the number of blasts in the bone marrow markedly decreased and FLT3/ITD was undetectable. The patient was also independent of transfusion, and her neutrophil count was almost normal after 4 cycles of treatment. Most importantly, the newborn was healthy. Sorafenib was also evaluated in a multicenter single-arm phase II study in patients aged ≥ 60 years with FLT3-mutated AML (Table 1) [20]. Sorafenib was added to induction, consolidation, and maintenance therapies. Fifty-four patients were enrolled in the study, 39 of which were FLT3/ITD-positive. The 1-year overall survival (OS) in FLT3/ITD patients was 62%, and disease-free survival (DFS) and OS were 12.2 and 15.0 months, respectively. In another multicenter, randomized, double-blind, placebo-controlled phase 2 trial from Germany, 267 patients with AML were treated with sorafenib and standard chemotherapy [21]. The result from this study showed that sorafenib had prolonged event-free survival (EFS), but the toxicity was also increased.
Application of sorafenib in HSCT
Sorafenib has shown encouraging results in HSCT for patients with FLT3/ITD-positive AML. In a retrospective analysis, 17 patients with FLT3/ITD-positive AML received sorafenib in combination with allo-HSCT [22]. Among the 17 patients, 10 patients started sorafenib only after transplantation. Fourteen of the 17 patients achieved CR, whereas 5 patients eventually progressed. Five patients showed pronounced signs of toxicity but remained in complete molecular remission when the dosage schedule was alternated. Sorafenib combined with allo-HSCT induced a lower relapse rate and longer leukemia-free survival (LFS) in patients with FLT3/ITD-mutated AML. In another study, 144 patients treated with the same regime were divided into 4 groups. The 3-year relapse rate of the four groups was 22.2%, 18.8%, 15.8%, and 46.1%, whereas OS and LFS rates were 74.9%, 78.1%, 84.6%, and 50.9% and 69.4%, 78.1%, 80.4%, and 34.8%, respectively [23]. Brunner et al. examined the effect of sorafenib as a maintenance drug for patients with FLT3/ITD-mutated AML in the first complete remission after HSCT [24]. The 2-year OS and PFS in the 26 sorafenib-treated patients were 81% and 82%, respectively. The 2-year cumulative incidence of relapse was 8.2%. However, there was no difference in 2-year non-relapse mortality or 1-year cGVHD between the sorafenib-treated patients and control. In another study of sorafenib as a maintenance drug after HSCT, 27 pediatric patients with FLT3/ITD-positive AML were enrolled [25]. Of these, 25 patients achieved complete molecular remission. The 1-year OS and PFS were 92 ± 6% and 92 ± 5%, respectively. Sorafenib was also used as a salvage therapy pre- and post-transplantation for 16 patients with refractory/relapsed FLT3-ITD-positive AML (Table 2) [26]. Out of the 16 patients, 13 achieved CR. The 2-year OS and DFS were 75.0 ± 10.8% and 50.5 ± 13.7%, respectively. Skin rash and gastrointestinal and cardiac toxicities were observed. In a report of a long-term follow-up of 29 patients with relapsed FLT3/ITD-positive AML after allo-SCT and sorafenib treatment [27], the median follow-up was 7.5 years. In this report, 6 patients survived, with 5 patients achieving sustained complete remission and 4 patients in treatment-free remission for a median of 4.4 years.
Sunitinib
Sunitinib (SU11248) is a small-molecule FLT3 inhibitor with selectivity for PDGFR, VEGFR1, VEGFR2, KIT, and FLT3 [28]. It has both direct anti-tumor and antiangiogenic properties. The use of sunitinib is currently approved for treating renal cell carcinoma, gastrointestinal stromal tumor, and AML.
Mechanisms of sunitinib on AML
The mechanism of sunitinib’s effect against AML is similar to that of sorafenib [29]. One study found that STAT5 phosphorylation in patients with FLT3/ITD was also reduced [30]. Intriguingly, SU11248 shows synergistic effects with cytarabine or daunorubicin in inhibiting proliferation and survival of primary AML myeloblasts expressing mutant FLT3/ITD, FLT3/D835V, or FLT3/WT [31]. Furthermore, sunitinib induces G1 phase arrest, increases pro-apoptotic molecule expression, and decreases anti-apoptotic molecule expression in AML cells [32].
Sunitinib combined with chemotherapy for AML
In the past few years, more clinical trials of sunitinib with chemotherapy have been conducted. In a phase I/II clinical trial, sunitinib and intensive chemotherapy were chosen for 22 patients with FLT3/ITD-mutated AML aged over 60 years [33]. Thirteen patients, including 8 patients with FLT3/ITD mutation, achieved CR/CRi. The median overall, relapse-free, and event-free survival of the 17 patients were 1.6, 1.0, and 0.4 years, respectively. In another phase I study, 15 patients with refractory AML were treated with SU11248 [34]. Patients with FLT3 mutations showed morphologic or partial responses. No dose-limiting toxicity was observed in patients treated with SU11248 at 50 mg. The most common grade 2 toxicities were edema, fatigue, and oral ulcerations.
Lestaurtinib
Lestaurtinib (CEP-701) is an orally bioavailable indolocarbazole alkaloid compound derived from the bacterial fermentation product K-252a. It has activities against tropomyosin receptor kinases, neurotrophin receptors, FLT3, and JAK2 [35,36,37]. Different from other class III receptor tyrosine kinases, lestaurtinib has low IC50 against FLT3 phosphorylation. Interestingly, lestaurtinib is cytotoxic to human AML cell lines expressing both mutant and wild-type FLT3, and it prolongs the survival of FLT3/ITD leukemia in a mouse model [36].
Lestaurtinib for AML
In a phase II trial, lestaurtinib was used as a monotherapy in untreated older patients with AML [38]. Lestaurtinib was administered orally at doses of 60 mg and 80 mg twice daily for 8 weeks. Blast percentages in the bone marrow and peripheral blood in 3 out of 5 patients with mutated FLT3 were reduced transiently, and periods of transfusion independence were prolonged. In another phase I/II clinical trial, 14 patients with relapsed, refractory, or poor-risk FLT3/ITD-mutated AML received lestaurtinib as a single-agent salvage therapy at doses of 60 mg twice daily [39]. Five patients showed transient clinical responses. However, Levis et al. showed that lestaurtinib treatment after chemotherapy in the first relapse did not improve response rates nor prolong survival of patients with FLT3/ITD-mutated AML [40]. Furthermore, Knapper et al. also proved that lestaurtinib and chemotherapy as first-line therapy did not prolong 5-year overall or relapse-free survival of younger patients with untreated FLT3-mutated AML from the UK AML15 and AML17 trials (Table 3) [41].
Tandutinib
Tandutinib (MLN518, CT53518) is a novel quinazoline-based inhibitor of the type III receptor tyrosine kinases, FLT3, PDGFR, and KIT. Tandutinib, at a concentration that does not affect normal colony formation, was shown to inhibit blast growth in patients with FLT3/ITD-positive AML [42]. Tandutinib induces apoptosis and inhibits FLT3/ITD phosphorylation, cellular proliferation, and signaling of the MAPK and PI3K pathways [43].
The clinical effect of tandutinib in patients with AML was examined in a phase I trial. Tandutinib showed anti-leukemic activity and decreased the number of blasts in the peripheral blood as well as in the bone marrow in 40 patients with AML or high-risk MDS [44]. In addition, the combination of tandutinib with standard chemotherapy regimen exerts antiproliferative and pro-apoptotic effects on FLT3/ITD-positive blasts in AML [45]. Long-term effects of tandutinib remain to be determined.
Midostaurin
Midostaurin (CGP41251, PKC412) is a small-molecule tyrosine kinase inhibitor (TKI) and was approved by the US FDA in 2017 for the treatment of FLT3-mutated AML [46]. It has recently been approved for newly diagnosed patients with FLT3-mutated AML and advanced systemic mastocytosis.
The clinical activity of midostaurin has been investigated in multiple clinical trials. In a phase I trial, midostaurin was administered with bortezomib alone or in combination with mitoxantrone, etoposide, and cytarabine to patients with refractory or relapsed AML [47]. The overall response rate (ORR) and CR were 82.5 and 56.5%, respectively. Ramsingh et al. used various doses of midostaurin, all-trans retinoic acid, and CLAG chemotherapy to treat relapsed/refractory AML [48]. Among all patients, 22% achieved CR and 11% achieved CRi. However, Stone et al. reported that the efficacy of midostaurin improved significantly when administered in combination with a standard chemotherapy to newly diagnosed patients with AML (Table 4) [49]. The CR rate of patients treated with midostaurin at 50 mg twice daily was 80% (FLT3-mutant 92%, FLT3/WT 74%). However, the 1-year and 2-year OS of patients with FLT3-mutated AML were similar to those of patients with FLT3-WT. Furthermore, Stone recently reported that midostaurin in combination with a standard chemotherapy significantly prolonged the OS and EFS of patients with FLT3-mutated AML. The incidence of severe adverse events was not increased by the combined treatment [50].
Second-generation FLT3 inhibitors
Quizartinib
Quizartinib (AC220) is a selective and highly potent second-generation class III receptor TKI [51]. Quizartinib is a potent and selective FLT3 inhibitor for AML [52]. The quizartinib dosage with the highest efficacy is 1 mg/kg once a day.
The optimum dosages and efficacy of quizartinib alone and in combination with chemotherapy in patients with AML were investigated. A phase I open-label, sequential group dose-escalation trial was the first to evaluate the safety and tolerability of quizartinib in combination with chemotherapy in 19 patients newly diagnosed with AML [53]. Out of 16 patients who achieved good response, 14 achieved CR and 2 achieved a morphologic leukemia-free state. There were no apparent additional signs of toxicity. The most common grade 3 or 4 adverse events were febrile neutropenia, neutropenia, thrombocytopenia, and anemia. In another dose-escalation study, quizartinib was used as a maintenance therapy in 13 patients with FLT3/ITD-mutated AML after allo-HSCT [54]. Two patients treated with quizartinib at 40 and 60 mg/day interrupted treatment because of grade 3 gastric hemorrhage and anemia. One patient relapsed. However, there was no maximum tolerated dose (MTD), and 60 mg daily was the highest dose studied. Quizartinib has shown a strong activity in relapsed or refractory AML. Cortes et al. reported the results of a phase I trial of quizartinib in relapsed or refractory AML for the first time [55]. Out of 76 patients, 23 showed responses, with 10 achieving CR and 13 achieving PRs. The median duration of response was 13.3 weeks, and the median survival time was 14 weeks. The most common treatment-related adverse events were nausea, vomiting, and prolonged QT interval. The maximum tolerated dose (MTD) was 200 mg/day, and the dose-limiting toxicity was grade 3 QT prolongation. Cortes and Levis reported that the CR rate reached 44 to 54% in their phase II study of relapsed and refractory AML [56, 57]. Importantly, 30- or 60-mg/day quizartinib monotherapy was reported in 76 patients with relapsed/refractory FLT3/ITD-mutated AML. Composite complete remission (CRc) rates of both groups were similar to those who received higher quizartinib doses. The incidence of corrected QT interval (QTc) above 480 ms and 500 ms was also less common [58]. Quizartinib as a salvage chemotherapy has been administered to children with relapsed acute leukemia (Table 5) [59]. The responses were evaluated in 17 patients (2 CR, 1 CRp, 1 CRi, 10 SD, and 3 PD), 7 of which were FLT3/ITD-positive (1 CR, 1 CRp, 1 CRi, and 4 SD). FLT3 phosphorylation in all patients was completely inhibited with quizartinib at 60 mg/m2/day.
Crenolanib
Crenolanib is a potent and selective inhibitor of FLT3/WT, FLT3/ITD, FLT3-TKD, PDGFRα/β, KIT, and FLT3/D835 [60]. Crenolanib was less disruptive of erythroid colony growth, which may result in relatively less myelosuppression than that by quizartinib. Correlative data from an ongoing clinical trial showed that sufficient levels of crenolanib could inhibit both FLT3/ITD and resistant FLT3/D835 mutants in patients with AML [61]. In a phase II trial, the tolerability and efficacy of crenolanib combined with standard induction chemotherapy was examined in patients with newly diagnosed FLT3 mutant AML [62]. There were 26 patients including 19 patients with FLT3/ITD and 3 patients with FLT3/D835 mutations. Eighty-eight percent of patients achieved CR, and overall CR/CRi rate was 96%. During a median follow-up of 6 months, only 3 patients have relapsed. In the following year, the similar result was seen in crenolanib combined with 7+3 induction and high dose cytarabine consolidation in 29 patients < 60 years old with FLT3-mutated AML [63]. A head-to-head comparison with midostaurin in combination with 7+3 was planned to further evaluate the efficacy of crenolanib. In addition, crenolanib was also used in relapsed or refractory AML. Iyer et al. reported the result of 8 patients with first relapsed or primary refractory AML who received the treatment of high-dose ara-C/mitoxantrone (HAM) and crenolanib [64]. Four patients achieved CR/CRi after 1 cycle. Only 1 patient showed a transient elevation in total bilirubin. Maro et al. used salvage idarubicin and high-dose ara-C and crenolanib to treat patients with relapsed/refractory FLT3-positive AML [65]. The ORR was 36% and median OS was 259 days. No dose-limiting toxicities (DLT) were observed. Grade I GI toxicities including nausea, vomiting, diarrhea, and abdominal pain were the major non-hematological adverse events. Crenolanib was administered at 200 mg/m2/day 3 times a day in another single-center phase II study in 10 patients with relapsed/refractory AML who progressed after HSCT (Table 6) [66]. The ORR was 47%. Interestingly, crenolanib was recently shown to have synergistic antileukemia activity with FLT3-targeted CAR T cells [67].
Gilteritinib
Gilteritinib (ASP2215) is a novel dual FLT3/AXL inhibitor. Gilteritinib significantly reduced the colony-forming capacity of FLT3/ITD-positive leukemia cells [68]. Gilteritinib decreases the phosphorylation levels of FLT3 and its downstream targets in cell cultures as well as in animal models. No obvious toxicity was observed [69]. Gilteritinib was well tolerated in 252 relapsed/refractory AML patients. The ORR was 40%, whereas the RR was 52% in FLT3-mutated patients at doses ≥ 80 mg/day. More than 5% of the patients experienced serious adverse events such as fever, disease progression, neutropenia, sepsis, acute renal failure, pneumonia, pyrexia, bacteremia, and respiratory failure. Grade 3 diarrhea and transaminase elevation were limited in patients administrated at a dose of above 300 mg/day [70]. In another open-label, phase 1 study, gilteritinib was also shown to be well tolerated in Japanese patients with relapsed/refractory AML. The ORR in patients with mutated FLT3 and FLT/WT was 80% and 36.4%, respectively. The most common drug-related severe adverse events were thrombocytopenia and increased creatine phosphokinase. The recommended phase II dose was 120 mg/day and MTD was 200 mg/day (Table 7) [71]. A phase III clinical trial comparing guilteritinib to a salvage chemotherapy regimen in relapsed/refractory FLT3-mutated AML patients is currently being conducted.
Other FLT3 inhibitors
Cabozantinib
Cabozantinib is an oral inhibitor of multiple receptor tyrosine kinases VEGFR-1, VEGFR-2, VEGFR-3, Kit, MET, AXL, KIT, FLT3, and RET [72, 73]. It exhibits anti-tumor activity in several cancers, such as AML and renal cell carcinoma. Currently cabozantinib has been approved for treatment of advanced renal cell carcinoma. Cabozantinib exerts significant cytotoxicity to leukemia cell lines with FLT3/ITD. It induces apoptosis in leukemia cell by regulating the anti-apoptotic and pro-apoptotic proteins [74]. A phase I study of cabozantinib was done in 18 patients with AML [75]. Peripheral blast reductions were seen in 4 patients, 1 showed marrow blast reduction, and 1 had stable disease. The MTD of cabozantinib was 40 mg daily. The most common grade 2 or higher toxicities observed were fatigue, nausea, transaminitis, and electrolyte imbalance.
SEL24-B489
Sel24-B489 is a novel dual pan-PIM and FLT3/ITD inhibitor. SEL24-B489 suppresses the growth of AML cell lines. Unlike selective FLT3/ITD or PIM inhibitors, SEL24-B489 exhibits significantly broader on-target activity in AML cell lines, primary AML blasts, and FLT3-TKD-mutated cells [76].
G-749
G-749 is a novel FLT3 inhibitor against FLT3-ITD, D835Y, ITD/F691 L, and ITD/N676D. G-749 was shown to have sustained inhibition of FLT3 phosphorylation and downstream effectors in FLT3/ITD-positive as well as FLT3/WT- cell lines. It displayed potent anti-leukemic activity toward bone marrow blasts from patients with AML, including those with little or only minor responses to agents like AC220 or PKC412 [77]. G-749 was shown to induce complete elimination of leukemia cells and prolonged survival in animal models. G-749 appears to be a novel druggable candidate for the treatment of relapsed and refractory AML patients with various FLT3-ITD/FLT3-TKD mutants. G-749 may be a next-generation FLT3 inhibitor with the ability to overcome drug resistance.
AMG 925
AMG 925 is a highly bioavailable dual kinase inhibitor of cyclin-dependent kinase 4 (CDK4) and FLT3 and active against many FLT3 mutants reported to date [78]. AMG 925 suppresses the proliferation of tumor cell lines and exerts an anti-tumor activity by inhibiting STAT5 and RB phosphorylation. Furthermore, AMG 925 was also found to inhibit D835Y that are resistant to FLT3 inhibitors such as sorafenib and AC220 (quizartinib). In an animal model bearing AML xenograft, AMG 925 was shown to inhibit tumor growth by 96 to 99%. AMG 925 by targeting both FLT3 and CDK4 may improve clinical responses of patients with FLT3/ITD-mutated AML and overcome drug resistance [79].
TTT-3002
TTT-3002 is a novel FLT3 inhibitor with the most potent activity against a broad spectrum of FLT3-activating point mutations, including D835 and F691 L gatekeeper mutations. Compared with several other TKIs currently in clinical trials, TTT-3002 is only moderately protein-bound. TTT-3002 maintains its effect on cells isolated from patients with relapsed AML that are resistant to sorafenib and AC220. Tumor burden in an FLT3 TKI-resistant transplant mouse model was significantly reduced by oral administration of TTT-3002 [80]. TTT-3002 is cytotoxic to leukemic blasts isolated from FLT3/ITD-expressing AML patients while displaying minimal toxicity to normal hematopoietic stem/progenitor cells from healthy blood and bone marrow donors [81]. Therefore, these preclinical activities of TTT-3002 may suggest that it has the potential to become a promising new generation of FLT3 TKI for FLT3-mutated AML.
FF-10101
FF-10101 is a novel selective and irreversible FLT3 inhibitor with activities against FLT3/ITD, MOLM-13, MOLM-14, MV4-11, D835, Y842, and F691. FF-10101 covalently binds to the cysteine residue at 695 of FLT3 kinase and was shown to have high selectivity and inhibitory activity against FLT3 kinases. It significantly suppresses the growth of 32D cells with FLT3/ITD/D835Y-or FLT3/ITD/F691 L-expressing cells and primary AML cells with FLT3-ITD or FLT3-D835 mutations both in vitro and in vivo [82]. These evidences demonstrated that FF-10101 is a promising novel FLT3 inhibitor with activities against multiple FLT3 mutations including the activation loop mutations clinically identified as quizartinib-resistant mutations.
Overcoming resistance to FLT3 inhibitors
Many studies have shown that FLT3 inhibitors have favorable clinical activities for AML patients with FLT3/ITD, but response duration remains short because of the rapid development of resistance. Resistance to FLT3 inhibitors was attributed to the emergence of new mutations. The secondary FLT3 tyrosine kinase domain (TKD) mutation was one of the new mutations in the patients who showed resistance to FLT3 inhibitors [83, 84]. The constitutive activation of critical tyrosine residues in the FLT3 mutants and downstream signaling effectors was the common resistance mechanism of FLT3 TKIs [85].
Combining FLT3 inhibitors with other agents is the major drive in the clinical trials to overcome the resistance to current FLT3 TKIs. Dayal successfully used a collaborative FLT3 inhibitor, HSD 1169, to act against FLT3/ITD and sorafenib-resistant cell lines [86]. In a recent report, PI3K-delta inhibitor had synergistic anti-tumor activity with FLT3 inhibitors [87]. In addition, an autophagy inhibitor TAK-165 can induce cancer cell death through the activation of chaperone-mediated autophagy to enhance the efficacy of cancer therapies [88]. By integrating these novel inhibitors in combination with FLT3 inhibitors, their efficacy may be further improved in the near future. Bispecific antibodies, immune checkpoint inhibitors, and chimeric antigen receptor (CAR) T cells are major modalities of novel cancer immunotherapy [86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102]. Crenolanib was already shown to have synergistic activity with FLT3-targeted CAR T cells [67]. It is intriguing to consider integrating FLT3 inhibitors into cancer immunotherapy for enhancing activities and minimizing resistance.
Conclusion
FLT3 inhibitors have shown promising efficacies in aggressive AML. However, the duration of clinical response is short because of the rapid development of resistance. Novel next-generation FLT3 inhibitors are in active development to concur the resistance. Combining FLT3 inhibitors with other targeted agents are additional areas of investigation to minimize resistance to current FLT3 inhibitors.
Abbreviations
- allo-HSCT:
-
Allogeneic hematopoietic stem cell transplantation
- ATRA:
-
All-trans retinoic acid
- CR:
-
Complete remission
- CRi:
-
Complete remission with incomplete count recovery
- CRp:
-
Complete remission with incomplete platelet recovery
- DFS:
-
Disease-free survival
- EFS:
-
Event-free survival
- HSCT:
-
Hematopoietic stem cell transplantation
- LFS:
-
Leukemia-free survival
- OR:
-
Overall response
- OS:
-
Overall survival
- PD:
-
Progress disease
- PFS:
-
Progression-free survival
- RFS:
-
Relapse-free survival
- SD:
-
Stable disease
References
Rowe JM, Tallman MS. How I treat acute myeloid leukemia. Blood. 2010;116(17):3147–56.
Li YXQ, Lv N, Wang L, Zhao H, Wang X, Guo J, Chen C, Li Y, Yu L. Clinical implications of genome-wide DNA methylation studies in acute myeloid leukemia. J Hematol Oncol. 2017;10(1):41.
CY-C HY-C, Lin C-C, Kuo Y-Y, Hou H-A, Tzeng Y-S, Kao C-J, Chuang P-H, Tseng M-H, Hsiao T-H, Chou W-C, Tien H-F. The distinct biological implications of Asxl1 mutation and its roles in leukemogenesis revealed by a knock-in mouse model. J Hematol Oncol. 2017;10(1):139.
Hackl HAK, Wieser R. Molecular and genetic alterations associated with therapy resistance and relapse of acute myeloid leukemia. J Hematol Oncol. 2017;10(1):51.
Qin Y-ZWY, Xu L-P, Zhang X-H, Chen H, Han W, Chen Y-H, Wang F-R, Wang J-Z, Chen Y, Mo X-D, Zhao X-S, Chang Y-J, Liu K-Y, Huang X-J. The dynamics of RUNX1-RUNX1T1 transcript levels after allogeneic hematopoietic stem cell transplantation predict relapse in patients with t(8;21) acute myeloid leukemia. J Hematol Oncol. 2017;10(1):44.
Saygin CCH. Emerging therapies for acute myeloid leukemia. J Hematol Oncol. 2017;10(1):93.
Stone RMMP, Larson RA, Capdeville R. Midostaurin: its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018;2(4):444–53.
Hatcher JMWE, Sim T, Stone RM, Liu S, Griffin JD, Gray NS. Discovery of a highly potent and selective indenoindolone type 1 pan-FLT3 inhibitor. ACS Med Chem Lett. 2016;7(5):476–81.
Gallogly MMLH, Cooper BW. Midostaurin: a novel therapeutic agent for patients with FLT3-mutated acute myeloid leukemia and systemic mastocytosis. Adv Hematol. 2017;8(9):245–61.
Nguyen BWA, Young DJ, Ma H, Li L, Levis M, Brown P, Small D. FLT3 activating mutations display differential sensitivity to multiple tyrosine kinase inhibitors. Oncotarget. 2017;8(7):10931–44.
FR MM, Halfter H, Matsumura I, Schmidt R, Muller C, Gruning W, Kratz-Albers K, Serve S, Steur C, Buchner T, Kienast J, Kanakura Y, Berdel WE, Serve H. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood. 2000;96(12):3907–14.
Liu T IV, Sabato P, Gobburu JVS, Greer JM, Wright JJ, Smith BD, Pratz KW, Rudek MA. Sorafenib dose recommendation in acute myeloid leukemia based on exposure-FLT3 relationship. Clin Trans Sci. 2018;11(4):435–43.
Mathew NRBF, Braun L, O'Sullivan D, Thomas S, Waterhouse M, Muller TA, Hanke K, Taromi S, Apostolova P, Illert AL, Melchinger W, Duquesne S, Schmitt-Graeff A, Osswald L, Yan KL, Weber A, Tugues S, Spath S, Pfeifer D, Follo M, Claus R, Lubbert M, Rummelt C, Bertz H, Wasch R, Haag J, Schmidts A, Schultheiss M, Bettinger D, et al. Sorafenib promotes graft-versus-leukemia activity in mice and humans through IL-15 production in FLT3-ITD-mutant leukemia cells. Nat Med. 2018;24(3):282–91.
Lange AJE, Lange J, Dworacki G, Nowak D, Simiczyjew A, Mordak-Domagala M, Sedzimirska M. The sorafenib anti-relapse effect after alloHSCT is associated with heightened alloreactivity and accumulation of CD8+PD-1+ (CD279+) lymphocytes in marrow. PLoS One. 2018;13(1):e0190525.
Zhao WZT, Qu B, Wu X, Zhu X, Meng F, Gu Y, Shu Y, Shen Y, Sun Y, Xu Q. Sorafenib induces apoptosis in HL60 cells by inhibiting Src kinase-mediated STAT3 phosphorylation. Anti-Cancer Drugs. 2011;22(1):79–88.
Feldmann FSB, Martens S, Vandenabeele P, Fulda S. Sorafenib inhibits therapeutic induction of necroptosis in acute leukemia cells. Oncotarget. 2017;8(40):68208–20.
Muppidi MRPS, Griffiths EA, Thompson JE, Ford LA, Freyer CW, Wetzler M, Wang ES. Decitabine and sorafenib therapy in FLT-3 ITD-mutant acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2015;15(Suppl):S73–9.
Ravandi FAM, Grunwald MR, Rudek MA, Rajkhowa T, Richie MA, Pierce S, Daver N, Garcia-Manero G, Faderl S, Nazha A, Konopleva M, Borthakur G, Burger J, Kadia T, Dellasala S, Andreeff M, Cortes J, Kantarjian H, Levis M. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. 2013;121(23):4655–62.
Mahdi AJGD, Chakraborty M, Rees A, Conner C, Wilson K, Rayment R, Alvares C. Successful molecular targeted treatment of AML in pregnancy with Azacitidine and Sorafenib with no adverse fetal outcomes. Br J Haematol. 2018;180(4):603–4.
Uy GLMS, Laumann K, Marcucci G, Zhao W, Levis MJ, Klepin HD, Baer MR, Powell BL, Westervelt P, DeAngelo DJ, Stock W, Sanford B, Blum WG, Bloomfield CD, Stone RM, Larson RA. A phase 2 study incorporating sorafenib into the chemotherapy for older adults with FLT3-mutated acute myeloid leukemia: CALGB 11001. Blood Adv. 2017;1(5):331–40.
Röllig CSH, Hüttmann A, Noppeney R, Müller-Tidow C, Krug U, Baldus CD, Brandts CHKV, Einsele H, Krämer A, Schäfer-Eckart K, Neubauer A, Burchert A, Giagounidis A, Krause SW, Mackensen A, Aulitzky W, Herbst R, Hänel M, Kiani A, Frickhofen N, Kullmer J, Kaiser U, Link H, Geer T, Reichle A, Junghanß C, Repp R, Heits F, Dürk H, Hase J, Klut IM, Illmer T, Bornhäuser M, Schaich M, Parmentier S, Görner M, Thiede C, von Bonin M, Schetelig J, Kramer M, Berdel WE, Ehninger G. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015;16(16):1691–9.
Tschan-Plessl AHJ, Heim D, Medinger M, Passweg JR, Gerull S. Synergistic effect of sorafenib and cGvHD in patients with high-risk FLT3-ITD+AML allows long-term disease control after allogeneic transplantation. Ann Hematol. 2015;94(11):1899–905.
Xuan LWY, Huang F, Jiang E, Deng L, Wu B, Fan Z, Liang X, Xu N, Ye J, Lin R, Yin C, Zhang Y, Sun J, Han M, Huang X, Liu Q. Effect of sorafenib on the outcomes of patients with FLT3-ITD acute myeloid leukemia undergoing allogeneic hematopoietic stem cell transplantation. Cancer. 2018;124(9):1954–63.
Brunner AMLS, Fathi AT, Wadleigh M, Ho VT, Collier K, Connolly C, Ballen KK, Cutler CS, Dey BR, El-Jawahri A, Nikiforow S, McAfee SL, Koreth J, Deangelo DJ, Alyea EP, Antin JH, Spitzer TR, Stone RM, Soiffer RJ, Chen YB. Haematopoietic cell transplantation with and without sorafenib maintenance for patients with FLT3-ITD acute myeloid leukaemia in first complete remission. Br J Haematol. 2016;175(3):496–504.
Battipaglia GRA, Massoud R, El Cheikh J, Jestin M, Antar A, Ahmed SO, Rasheed W, Shaheen M, Belhocine R, Brissot E, Dulery R, Eder S, Giannotti F, Isnard F, Lapusan S, Rubio MT, Vekhoff A, Aljurf M, Legrand O, Mohty M, Bazarbachi A. Efficacy and feasibility of sorafenib as a maintenance agent after allogeneic hematopoietic stem cell transplantation for Fms-like tyrosine kinase 3-mutated acute myeloid leukemia. Cancer. 2017;123(15):2867–74.
Ruella MKS, Shestova O, Fraietta JA, Qayyum S, Zhang Q, Maus MV, Liu X, Nunez-Cruz S, Klichinsky M, Kawalekar OU, Milone M, Lacey SF, Mato A, Schuster SJ, Kalos M, June CH, Gill S, Wasik MA. The addition of the BTK inhibitor ibrutinib to anti-CD19 chimeric antigen receptor T cells (CART19) improves responses against mantle cell lymphoma. Clin Cancer Res. 2016;22(11):2684–96.
Metzelder SKST, Lubbert M, Ditschkowski M, Gotze K, Scholl S, Meyer RG, Dreger P, Basara N, Fey MF, Salih HR, Finck A, Pabst T, Giagounidis A, Kobbe G, Wollmer E, Finke J, Neubauer A, Burchert A. Long-term survival of sorafenib-treated FLT3-ITD-positive acute myeloid leukaemia patients relapsing after allogeneic stem cell transplantation. Eur J Cancer. 2017;8(6):233–9.
Mendel DBLA, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan E, Moss KG, Haznedar JO, Sukbuntherng J, Blake RA, Sun L, Tang C, Miller T, Shirazian S, McMahon G, Cherrington JM. In vivo anti-tumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9(1):327–37.
O'Farrell AMAT, Yuen HA, Ngai TJ, Louie SG, Yee KW, Wong LM, Hong W, Lee LB, Town A, Smolich BD, Manning WC, Murray LJ, Heinrich MC, Cherrington JM. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. 2003;101(9):3597–605.
O'Farrell AMFJ, Fiedler W, Serve H, Paquette RL, Cooper MA, Yuen HA, Louie SG, Kim H, Nicholas S, Heinrich MC, Berdel WE, Bello C, Jacobs M, Scigalla P, Manning WC, Kelsey S, Cherrington JM. An innovative phase I clinical study demonstrates inhibition of FLT3 phosphorylation by SU11248 in acute myeloid leukemia patients. Clin Cancer Res. 2003;9(15):5465–76.
Yee KWSM, O'Farrell AM, Town AR, McGreevey L, Bainbridge T, Cherrington JM, Heinrich MC. Synergistic effect of SU11248 with cytarabine or daunorubicin on FLT3 ITD-positive leukemic cells. Blood. 2004;104(1):4202–9.
Teng CLYC, Hwang WL, Tsai JR, Liu HC, Hwang GY, Hsu SL. Effector mechanisms of sunitinib-induced G1 cell cycle arrest, differentiation, and apoptosis in human acute myeloid leukaemia HL60 and KG-1 cells. Ann Hematol. 2013;92(3):301–13.
Fiedler WKS, Kebenko M, Janning M, Krauter J, Schittenhelm M, Gotze K, Weber D, Gohring G, Teleanu V, Thol F, Heuser M, Dohner K, Ganser A, Dohner H, Schlenk RF. A phase I/II study of sunitinib and intensive chemotherapy in patients over 60 years of age with acute myeloid leukaemia and activating FLT3 mutations. Br J Haematol. 2015;169(5):694–700.
Fiedler WSH, Dohner H, Schwittay M, Ottmann OG, O'Farrell AM, Bello CL, Allred R, Manning WC, Cherrington JM, Louie SG, Hong W, Brega NM, Massimini G, Scigalla P, Berdel WE, Hossfeld DK. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2005;105(3):986–93.
Marshall JLKH, Deeken J, Bhargava P, Vogelzang NJ, Rizvi N, Luhtala T, Boylan S, Dordal M, Robertson P, Hawkins MJ, Ratain MJ. Phase I trial of orally administered CEP-701, a novel neurotrophin receptor-linked tyrosine kinase inhibitor. Investig New Drugs. 2005;23(1):31–7.
Levis MAJ, Tse KF, Zheng R, Baldwin BR, Smith BD, Jones-Bolin S, Ruggeri B, Dionne C, Small D. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood. 2002;99(11):3885–91.
Hexner EOMJ, Prchal J, Roboz GJ, Baer MR, Ritchie EK, Leibowitz D, Demakos EP, Miller C, Siuty J, Kleczko J, Price L, Jeschke G, Weinberg R, Basu T, Pahl HL, Orazi A, Najfeld V, Marchioli R, Goldberg JD, Silverman LR, Hoffman R. Phase I dose escalation study of lestaurtinib in patients with myelofibrosis. Leuk Lymphoma. 2015;56(9):2543–51.
Knapper SBA, Littlewood T, Kell WJ, Agrawal S, Chopra R, Clark R, Levis MJ, Small D. A phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood. 2006;108(10):3262–70.
Smith BDLM, Beran M, Giles F, Kantarjian H, Berg K, Murphy KM, Dauses T, Allebach J, Small D. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103(10):3669–76.
Levis MRF, Wang ES, Baer MR, Perl A, Coutre S, Erba H, Stuart RK, Baccarani M, Cripe LD, Tallman MS, Meloni G, Godley LA, Langston AA, Amadori S, Lewis ID, Nagler A, Stone R, Yee K, Advani A, Douer D, Wiktor-Jedrzejczak W, Juliusson G, Litzow MR, Petersdorf S, Sanz M, Kantarjian HM, Sato T, Tremmel L, Bensen-Kennedy DM, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood. 2011;117(12):3294–301.
Knapper SRN, Gilkes A, Hills RK, Gale RE, Cavenagh JD, Jones G, Kjeldsen L, Grunwald MR, Thomas I, Konig H, Levis MJ, Burnett AK. A randomized assessment of adding the kinase inhibitor lestaurtinib to first-line chemotherapy for FLT3-mutated AML. Blood. 2017;129(9):1143–54.
Griswold IJSL, La Rosee P, Demehri S, Heinrich MC, Braziel RM, McGreevey L, Haley AD, Giese N, Druker BJ, Deininger MW. Effects of MLN518, a dual FLT3 and KIT inhibitor on normal and malignant hematopoiesis. Blood. 2004;104(9):2912–8.
Kelly LMYJ, Boulton CL, Apatira M, Li J, Sullivan CM, Williams I, Amaral SM, Curley DP, Duclos N, Neuberg D, Scarborough RM, Pandey A, Hollenbach S, Abe K, Lokker NA, Gilliland DG, Giese NA. CT53518, a novel selective FLT3 antagonist for the treatment of acute myelogenous leukemia (AML). Cancer Cell. 2002;1(5):421–32.
DeAngelo DJSR, Heaney ML, Nimer SD, Paquette RL, Klisovic RB, Caligiuri MA, Cooper MR, Lecerf JM, Karol MD, Sheng S, Holford N, Curtin PT, Druker BJ, Heinrich MC. Phase 1 clinical results with tandutinib (MLN518), a novel FLT3 antagonist, in patients with acute myelogenous leukemia or high-risk myelodysplastic syndrome: safety, pharmacokinetics, and pharmacodynamics. Blood. 2006;108(12):3674–81.
Schittenhelm MMKK, Yee KW, Heinrich MC. The FLT3 inhibitor tandutinib (formerly MLN518) has sequence-independent synergistic effects with cytarabine and daunorubicin. Cell Cycle. 2009;8(16):2621–30.
Levis M. Midostaurin approved for FLT3-mutated AML. Blood. 2017;129(26):3403–6.
Walker ARWH, Walsh K, Bhatnagar B, Vasu S, Garzon R, Canning R, Geyer S, Wu YZ, Devine SM, Klisovic R, Blum W, Marcucci G. Midostaurin, bortezomib and MEC in relapsed/refractory acute myeloid leukemia. Leuk Lymphoma. 2016;57(9):2100–8.
Ramsingh G, Westervelt P, McBride A, Stockerl-Goldstein K, Vij R, Fiala M, Uy G, Cashen A, Dipersio JF, Abboud CN. Phase I study of cladribine, cytarabine, granulocyte colony stimulating factor (CLAG regimen) and midostaurin and all-trans retinoic acid in relapsed/refractory AML. Int J Hematol. 2014;99(3):272–8.
Stone RMFT, Paquette R, Schiller G, Schiffer CA, Ehninger G, Cortes J, Kantarjian HM, DeAngelo DJ, Huntsman-Labed A, Dutreix C, del Corral A, Giles F. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia. 2012;26(9):2061–8.
Stone RMMS, Sanford BL, Laumann K, Geyer S, Bloomfield CD, Thiede C, Prior TW, Dohner K, Marcucci G, Lo-Coco F, Klisovic RB, Wei A, Sierra J, Sanz MA, Brandwein JM, de Witte T, Niederwieser D, Appelbaum FR, Medeiros BC, Tallman MS, Krauter J, Schlenk RF, Ganser A, Serve H, Ehninger G, Amadori S, Larson RA, Dohner H. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454–64.
Kampa-Schittenhelm KMHM, Akmut F, Dohner H, Dohner K, Schittenhelm MM. Quizartinib (AC220) is a potent second generation class III tyrosine kinase inhibitor that displays a distinct inhibition profile against mutant-FLT3, -PDGFRA and -KIT isoforms. Mol Cancer. 2013;12:19.
Zarrinkar PPGR, Cramer MD, Gardner MF, Brigham D, Belli B, Karaman MW, Pratz KW, Pallares G, Chao Q, Sprankle KG, Patel HK, Levis M, Armstrong RC, James J, Bhagwat SS. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood. 2009;114(14):2984–92.
Altman JKFJ, Pratz KW, Trone D, Cortes JE, Tallman MS. Phase 1 study of quizartinib in combination with induction and consolidation chemotherapy in patients with newly diagnosed acute myeloid leukemia. Am J Hematol. 2018;93(2):213–21.
Sandmaier BMKS, Oran B, Gammon G, Trone D, Frankfurt O. Results of a phase 1 study of quizartinib as maintenance therapy in subjects with acute myeloid leukemia in remission following allogeneic hematopoietic stem cell transplant. Am J Hematol. 2018;93(2):222–31.
Cortes JEKH, Foran JM, Ghirdaladze D, Zodelava M, Borthakur G, Gammon G, Trone D, Armstrong RC, James J, Levis M. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol. 2013;31(29):3681–7.
Cortes JE, DH ea PA. Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in patients ≥ 60 years of age with FLT3 ITD positive or negative relapsed/refractory acute myeloid leukemia. ASH Annual Meeting Abstracts. 2012;48:2012.
Levis MJ, DH, EA PA. Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in patients with FLT3-ITD positive or negative relapsed/refractory acute myeloid leukemia after second-line chemotherapy or hematopoietic stem cell transplantation. ASH Annual Meeting Abstracts. 2012;673.
TM CJE, Schiller GJ, Trone D, Gammon G, Goldberg SL, Perl AE, MarieJP MG, Kantarjian HM, Levis MJ. Phase 2b study of 2 dosing regimens of quizartinib monotherapy in FLT3-ITD-mutated, relapsed or refractory AML. Blood. 2018;132(6):598–607.
Cooper TMCJ, Eckroth E, Malvar J, Sposto R, Gaynon P, Chang BH, Gore L, August K, Pollard JA, DuBois SG, Silverman LB, Oesterheld J, Gammon G, Magoon D, Annesley C, Brown PA. A phase I study of quizartinib combined with chemotherapy in relapsed childhood leukemia: a therapeutic advances in childhood leukemia & lymphoma (TACL) study. Clin Cancer Res. 2016;22(16):4014–22.
Kampa-Schittenhelm KMFJ, Haeusser LA, Illing B, Pavlovsky AA, Blumenstock G, Schittenhelm MM. Crenolanib is a type I tyrosine kinase inhibitor that inhibits mutant KIT D816 isoforms prevalent in systemic mastocytosis and core binding factor leukemia. Oncotarget. 2017;8(47):82897–909.
Zimmerman EITD, Buaboonnam J, Hu S, Orwick S, Roberts MS, Janke LJ, Ramachandran A, Stewart CF, Inaba H, Baker SD. Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood. 2013;122(22):3607–15.
Wang ES RMS, Tallman MS, Walter RB, Eckardt JR, Collins R. Crenolanib, a type I FLT3 TKI, can be safely combined with cytarabine and anthracycline induction chemotherapy and results in high response rates in patients with newly diagnosed FLT3 mutant acute myeloid leukemia (AML). Blood. 2016;128(22):1071.
Wang ES, MST RMS, Walter RB, Karanes C, Jain V, Collins RH. Low relapse rate in younger patients ≤ 60 years old with newly diagnosed FLT3-mutated acute myeloid leukemia (AML) treated with crenolanib and cytarabine/anthracycline chemotherapy. Blood. 2017;130:566.
Iyer SP, Jethava Y, Karanes C, Eckardt JR, Collins R. Safety study of salvage chemotherapy high-dose Ara-C/mitoxantrone (HAM) and type I FLT3-TKI crenolanib in first relapsed/primary refractory AML. Blood. 2016;128(22):3983.
Maro Ohanian HMK, Borthakur G, Kadia TM, Konopleva M, Garcia-Manero G, Estrov Z, Ferrajoli A, Takahashi K, Jabbour EJ, Daver N, Kornblau SM, Wierda WG, Burger JA, Naqvi K, Benton CB, Bose P, Eckardt JR, Ravandi F, Cortes JE. Efficacy of a type I FLT3 inhibitor, crenolanib, with idarubicin and high-dose Ara-C in multiply relapsed/refractory FLT3+ AML. Blood. 2016;128:2744.
Jasleen K, Randhawa HMK, Borthakur G, Thompson PA, Konopleva M, Daver N, Pemmaraju N, Jabbour E, Kadia TM, Estrov Z, Ramachandran A, Paradela J, Andreef M, Levis M, Ravandi F, Cortes JE. Results of a phase II study of crenolanib in relapsed/refractory acute myeloid leukemia patients (Pts) with activating FLT3 mutations. Blood. 2014;124:389.
Jetani H, Garcia-Cadenas I, Nerreter T, Thomas S, Rydzek J, Meijide JB, Bonig H, Herr W, Sierra J, Einsele H, Hudecek M. CAR T-cells targeting FLT3 have potent activity against FLT3(−)ITD(+) AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia. 2018;32(5):1168–79.
Cucchi DGJDB, Kaspers GJL, Janssen J, Ossenkoppele GJ, de Haas V, Zwaan CM, van den Heuvel-Eibrink MM, Philippe J, Csikos T, Kwidama Z, de Moerloose B, de Bont E, Lissenberg-Witte BI, Zweegman S, Verwer F, Vandepoele K, Schuurhuis GJ, Sonneveld E, Cloos J. RNA-based FLT3-ITD allelic ratio is associated with outcome and ex vivo response to FLT3 inhibitors in pediatric AML. Blood. 2018;131(22):2485–9.
Mori MKN, Ueno Y, Yamada M, Tanaka R, Saito R, Shimada I, Mori K, Kuromitsu S. Gilteritinib, a FLT3/AXL inhibitor, shows anti-leukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Investig New Drugs. 2017;35(5):556–65.
Perl AEAJ, Cortes J, Smith C, Litzow M, Baer MR, Claxton D, Erba HP, S G, Goldberg S, Jurcic JG, Larson RA, Liu C, Ritchie E, Schiller G, Spira AI, Strickland SA, Tibes R, Ustun C, Wang ES, Stuart R, Röllig C, Neubauer A, Martinelli G, Bahceci E, Levis M. Selective inhibition of FLT3 by gilteritinib in relapsed/refractory acute myeloid leukemia: a multicenter, first-in-human, open-label, phase 1/2 study. Lancet Oncol. 2017;18(8):1061–75.
Usuki KST, Kobayashi Y, Miyamoto T, Iida H, Morita S, Bahceci E, Kaneko M, Kusano M, Yamada S, Takeshita S, Miyawaki S, Naoe T. Clinical profile of gilteritinib in Japanese patients with relapsed/refractory AML an open-label phase 1 study. Cancer Sci. 2018;109(10):3235–44.
Tarabadkar ESTH, Blom A, Parvathaneni U, Olencki T, Nghiem P, Bhatia S. Clinical benefit from tyrosine kinase inhibitors in metastatic Merkel cell carcinoma: a case series of 5 patients. Am J Case Rep. 2018;19:505–11.
vdHT OS. Cabozantinib in the treatment of advanced renal cell carcinoma in adults following prior vascular endothelial growth factor targeted therapy: clinical trial evidence and experience. Ther Adv Urol. 2018;10(3):109–23.
Lu JWWA, Liao HA, Chen CY, Hou HA, Hu CY, Tien HF, Ou DL, Lin LI. Cabozantinib is selectively cytotoxic in acute myeloid leukemia cells with FLT3-internal tandem duplication (FLT3-ITD). Cancer Lett. 2016;376(2):218–25.
Fathi ATBT, Hernandez D, Amrein PC, Ballen KK, McMasters M, Avigan DE, Joyce R, Logan EK, Hobbs G, Brunner AM, Joseph C, Perry AM, Burke M, Behnan T, Foster J, Bergeron MK, Moran JA, Ramos AY, Som TT, Rae J, Fishman KM, McGregor KL, Connolly C, Neuberg DS, Levis MJ. Cabozantinib is well tolerated in acute myeloid leukemia and effectively inhibits the resistance-conferring FLT3/tyrosine kinase domain/F691 mutation. Cancer. 2018;124(2):306–14.
Czardybon WWR, Golas A, Galezowski M, Sabiniarz A, Dolata I, Salwinska M, Guzik P, Zawadzka M, Gabor-Worwa E, Winnik B, Zurawska M, Kolasinska E, Wincza E, Bugaj M, Danielewicz M, Majewska E, Mazan M, Dubin G, Noyszewska-Kania M, Jablonska E, Szydlowski M, Sewastianik T, Pula B, Szumera-Cieckiewicz A, Prochorec-Sobieszek M, Madro E, Lech-Maranda E, Warzocha K, Tamburini J, et al. A novel, dual pan-PIM/FLT3 inhibitor SEL24 exhibits broad therapeutic potential in acute myeloid leukemia. Oncotarget. 2018;9(24):16917–31.
Lee HKKH, Lee IY, Lee J, Lee J, Jung DS, Lee SY, Park SH, Hwang H, Choi JS, Kim JH, Kim SW, Kim JK, Cools J, Koh JS, Song HJ. G-749, a novel FLT3 kinase inhibitor, can overcome drug resistance for the treatment of acute myeloid leukemia. Blood. 2014;123(14):2209–19.
Li Z, Wang X, Eksterowicz J, Gribble MW Jr, Alba GQ, Ayres M, Carlson TJ, Chen A, Chen X, Cho R, Connors RV, DeGraffenreid M, Deignan JT, Duquette J, Fan P, Fisher B, Fu J, Huard JN, Kaizerman J, Keegan KS, Li C, Li K, Li Y, Liang L, Liu W, Lively SE, Lo MC, Ma J, McMinn DL, Mihalic JT, et al. Discovery of AMG 925, a FLT3 and CDK4 dual kinase inhibitor with preferential affinity for the activated state of FLT3. J Med Chem. 2014;57(8):3430–49.
Keegan KLC, Li Z, Ma J, Ragains M, Coberly S, Hollenback D, Eksterowicz J, Liang L, Weidner M, Huard J, Wang X, Alba G, Orf J, Lo MC, Zhao S, Ngo R, Chen A, Liu L, Carlson T, Queva C, McGee LR, Medina J, Kamb A, Wickramasinghe D, Dai K. Preclinical evaluation of AMG 925, a FLT3/CDK4 dual kinase inhibitor for treating acute myeloid leukemia. Mol Cancer Ther. 2014;13(4):880–9.
Ma HSNB, Duffield AS, Li L, Galanis A, Williams AB, Brown PA, Levis MJ, Leahy DJ, Small D. FLT3 kinase inhibitor TTT-3002 overcomes both activating and drug resistance mutations in FLT3 in acute myeloid leukemia. Cancer Res. 2014;74(18):5206–17.
Ma HNB, Li L, Greenblatt S, Williams A, Zhao M, Levis M, Rudek M, Duffield A, Small D. TTT-3002 is a novel FLT3 tyrosine kinase inhibitor with activity against FLT3-associated leukemias in vitro and in vivo. Blood. 2014;123(10):1525–34.
Yamaura TNT, Uda K, Ogura H, Shin W, Kurokawa N, Saito K, Fujikawa N, Date T, Takasaki M, Terada D, Hirai A, Akashi A, Chen F, Adachi Y, Ishikawa Y, Hayakawa F, Hagiwara S, Naoe T, Kiyoi H. A novel irreversible FLT3 inhibitor, FF-10101, shows excellent efficacy against AML cells with FLT3 mutations. Blood. 2018;131(4):426–38.
Alvarado YKH, Luthra R, Ravandi F, Borthakur G, Garcia-Manero G, Konopleva M, Estrov Z, Andreeff M, Cortes JE. Treatment with FLT3 inhibitor in patients with FLT3-mutated acute myeloid leukemia is associated with development of secondary FLT3-tyrosine kinase domain mutations. Cancer. 2014;120(14):2142–9.
Baker SDZE, Wang YD, Orwick S, Zatechka DS, Buaboonnam J, Neale GA, Olsen SR, Enemark EJ, Shurtleff S, Rubnitz JE, Mullighan CG, Inaba H. Emergence of polyclonal FLT3 tyrosine kinase domain mutations during sequential therapy with sorafenib and sunitinib in FLT3-ITD-positive acute myeloid leukemia. Clin Cancer Res. 2013;19(20):5758–68.
Ishiko JMM, Matsumura I, Shibayama H, Sugahara H, Scholz G, Serve H, Kanakura Y. Roles of tyrosine residues 845, 892 and 922 in constitutive activation of murine FLT3 kinase domain mutant. Oncogene. 2005;24(55):8144–53.
Dayal NO-TC, Hernandez DE, Sooreshjani MA, Carter-Cooper BA, Lapidus RG, Sintim HO. Dual FLT3/TOPK inhibitor with activity against FLT3-ITD secondary mutations potently inhibits acute myeloid leukemia cell lines. Future Med Chem. 2018;10(7):823–35.
He YSL, Xu Y, Fu L, Li Y, Bao X, Fu H, Xie C, Lou L. Combined inhibition of PI3Kdelta and FLT3 signaling exerts synergistic anti-tumor activity and overcomes acquired drug resistance in FLT3-activated acute myeloid leukemia. Cancer Lett. 2018;420:49–59.
Ouchida ATLY, Geng J, Najafov A, Ofengeim D, Sun X, Yu Q, Yuan J. Synergistic effect of a novel autophagy inhibitor and Quizartinib enhances cancer cell death. Cell Death Dis. 2018;9(2):138.
G-CI JH, Nerreter T, Thomas S, Rydzek J, Meijide JB, Bonig H, Herr W, Sierra J, Einsele H, Hudecek M. CAR T-cells targeting FLT3 have potent activity against FLT3(−)ITD(+) AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia. 2018;32(5):1168–79.
Topp MSGN, Stein AS, Zugmaier G, O'Brien S, Bargou RC, Dombret H, Fielding AK, Heffner L, Larson RA, Neumann S, Foa R, Litzow M, Ribera JM, Rambaldi A, Schiller G, Bruggemann M, Horst HA, Holland C, Jia C, Maniar T, Huber B, Nagorsen D, Forman SJ, Kantarjian HM. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm phase 2 study. Lancet Oncol. 2014;16(1):57–66.
Liu BSY, Liu D. Recent development in clinical applications of PD-1 and PD-L1 antibodies for cancer immunotherapy. J Hematol Oncol. 2017;10(1):174.
Liu BSY, Liu D. Clinical trials of CAR-T cells in China. J Hematol Oncol. 2017;10(1):166.
Fan MLM, Gao L, Geng S, Wang J, Wang Y, Yan Z, Yu L. Chimeric antigen receptors for adoptive T cell therapy in acute myeloid leukemia. J Hematol Oncol. 2017;10(1):151.
Diggs LPHE. Utility of PD-L1 immunohistochemistry assays for predicting PD-1/PD-L1 inhibitor response. Biomark Res. 2017;5(1):12.
Zhang CLJ, Zhong JF, Zhang X. Engineering CAR-T cells. Biomark Res. 2017;5(1):22.
Qin LZR, Li P. Incorporation of functional elements enhances the anti-tumor capacity of CAR T cells. Exp Hematol Oncol. 2017;6(1):28.
Wei GDL, Wang J, Hu Y, Huang H. Advances of CD19-directed chimeric antigen receptor-modified T cells in refractory/relapsed acute lymphoblastic leukemia. Exp Hematol Oncol. 2017;6(1):10.
Zhang XYY, Fan D, Xiong D. The development of bispecific antibodies and their applications in tumor immune escape. Exp Hematol Oncol. 2017;6(1):12.
Kantarjian HSA, Gokbuget N, Fielding AK, Schuh AC, Ribera JM, Wei A, Dombret H, Foa R, Bassan R, Arslan O, Sanz MA, Bergeron J, Demirkan F, Lech-Maranda E, Rambaldi A, Thomas X, Horst HA, Bruggemann M, Klapper W, Wood BL, Fleishman A, Nagorsen D, Holland C, Zimmerman Z, Topp MS. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836–47.
Lichtenegger FSKC, Haubner S, Köhnke T, Subklewe M. Recent developments in immunotherapy of acute myeloid leukemia. J Hematol Oncol. 2017;10(1):142.
June CHOCR, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–5.
June CHSM. Chimeric antigen receptor therapy. N Engl J Med. 2018;379(1):64–73.
Acknowledgements
We gratefully acknowledge Professor Zhizhe Chen for editing the manuscript.
Funding
There is no funding for this study.
Availability of data and materials
The material supporting the conclusion of this review has been included within the article.
Author information
Authors and Affiliations
Contributions
XPZ designed the study. MW and CTL drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This is not applicable for this review.
Consent for publication
This is not applicable for this review.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wu, M., Li, C. & Zhu, X. FLT3 inhibitors in acute myeloid leukemia. J Hematol Oncol 11, 133 (2018). https://doi.org/10.1186/s13045-018-0675-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-018-0675-4