
Abstract
Wnt/β-catenin signaling, a highly conserved pathway through evolution, regulates key cellular functions including proliferation, differentiation, migration, genetic stability, apoptosis, and stem cell renewal. The Wnt pathway mediates biological processes by a canonical or noncanonical pathway, depending on the involvement of β-catenin in signal transduction. β-catenin is a core component of the cadherin protein complex, whose stabilization is essential for the activation of Wnt/β-catenin signaling. As multiple aberrations in this pathway occur in numerous cancers, WNT-directed therapy represents an area of significant developmental therapeutics focus. The recently described role of Wnt/β-catenin pathway in regulating immune cell infiltration of the tumor microenvironment renewed the interest, given its potential impact on responses to immunotherapy treatments. This article summarizes the role of Wnt/β-catenin pathway in cancer and ongoing therapeutic strategies involving this pathway.
Similar content being viewed by others


Background
Nusse and Varmus discovered the components of the Wnt/β-catenin pathway in 1982 while studying oncogenic mechanisms of mouse mammary tumor virus (MMTV) [1]. Proviral insertion at the “integration site” was thought to be the mechanism of carcinogenesis, giving the name to the first gene discovered in this pathway as INT1. Simultaneous work in developmental biology and work in drosophila established INT1 gene to be the homologue of the Drosophila segment polarity gene, Wingless [2]. Subsequently, human INT1 was shown to be very similar to mouse INT1, thereby revealing the highly conserved nature of this pathway across various species [3]. Additional screens for MMTV proviral insertion sites in tumors yielded several other activated genes that are related to other developmental gene pathways, such as INT2, INT3, and INT4 [4,5,6]. For example, INT2 is a member of the fibroblast growth factor (FGF; INT2 is the same FGF-3 protein) family, and INT3 is related to the NOTCH gene family (INT3 protein is the same neurogenic locus notch homologue 4/NOTCH4) [7, 8]. With “INT” nomenclature turning out to be inadequate and confusing, consensus was reached to create the hybrid name “WNT” (for Wingless-related integration site) to denote genes belonging to the INT1/Wingless family. INT1—now called WNT1—became the founding member [9].
WNTs (translated products of WNT gene) are cysteine-rich glycoproteins, secreted by cells into the extracellular matrix, that activate receptor-mediated signaling with cells in immediate proximity [10]. The WNT protein family consists of at least 19 secreted glycoproteins (350–400 amino acids in length) highly conserved across species from invertebrates to mammals [11]. WNT binds to the N-terminal extra-cellular cysteine-rich domain of a Frizzled family receptor, a member of the superfamily of G-protein-coupled receptors. This disrupts the destruction complex of β-catenin (a tertiary complex formed by axin, adenomatous polyposis coli (APC), CK1α, and GSK3β) and triggers the cytoplasmic accumulation of β-catenin (Fig. 1).

Canonical Wnt/β-catenin pathway: “WNT ON state”: WNT proteins, by binding to frizzled receptors and the LRP co-receptor, act to suppress the activity of glycogen synthase kinase-3β (GSK-3β). ZNRF3 promotes degradation of WNT receptor functioning as tumor suppressors. This prevents phosphorylation of downstream molecules allowing β-catenin association with Tcf/Lef in the nucleus and subsequent increased cell proliferation. “WNT OFF state”: In the absence of WNT ligand, the destruction complex of β-catenin (marked by dotted line box), a tertiary complex formed by axin, APC, CK1α and GSK 3β, will phosphorylate β-catenin, which subsequently undergoes proteasomal degradation
T cell factor/lymphoid enhancer factor-1 (TCF/Lef1) is the transcription complex that mediates canonical WNT-triggered gene transcription [12, 13]. β-catenin translocates into the nucleus where it interacts with TCF/Lef1 and activates TCF/Lef1 transcription complex [14,15,16]. β-catenin also localizes to multiple subcellular locations including the cytoplasm where its levels are tightly controlled. β-catenin also promotes cell-to-cell adhesion by accumulating in cell–cell contact sites, namely the adherens junctions [17, 18]. Figure 1 illustrates the canonical pathway of Wnt/β-catenin signaling. In addition to classical canonical WNT-induced activation of β-catenin–TCF/Lef1 transcriptional complexes, WNT can elicit alternative responses through β-catenin independent mechanisms which are collectively known as noncanonical pathways [19]. In an alternative concept known as integrated Wnt pathway, the canonical and noncanonical pathways are combined and multiple inputs at the level of both Wnt-receptor binding and the downstream, intracellular response have been integrated [20].
Wnt/β-catenin pathway is intricately involved in pathogenesis of several cancers. Recent findings of its role in regulating immunomodulation have renewed enthusiasm in the field.
Wnt/β-catenin pathway involvement in several malignancies
Colorectal cancers (CRCs)
The role of the Wnt/β-catenin pathway in carcinogenesis was first described in the setting of APC gene mutation. APC mutations, which typically are acquired early in the pathogenesis of most colon cancers (over 80%), lead to cytosolic accumulation of β-catenin that in combination with TCF/Lef1 shuttles to the nucleus where it functions as a transcription factor and promotes cellular proliferation [21, 22]. Nuclear expression of β-catenin has been associated with more aggressive cancer biology. In one study, nuclear expression of β-catenin was present in 18 out of 25 (72%) cases of ulcerative CRC while present in only 7 out of 26 (26.9%) cases of polypoid CRC (P < 0.001). This finding was independent of APC mutation and E-cadherin expression [23]. The Wnt/β-catenin pathway is also involved in cross talk with the Hippo/YAP pathway. Konsavage et al. showed that β-catenin/TCF4 complexes bind a DNA enhancer element within the first intron of the YAP gene to drive YAP expression in CRC cells, contributing to carcinogenesis [24]. The Hippo–YAP signaling pathway could be an effector pathway downstream from APC, independent from its involvement in the β-catenin destruction complex as well [25].
The WNT pathway has been implicated in the maintenance of cancer stem cells (CSC) in colorectal cancer. In vitro data suggest that chronic chemotherapeutic stress-induced “stemness” is associated with attenuated WNT signaling [26]. Vermeulen et al. showed that high activity of the WNT pathway was observed preferentially in tumor cells located close to stromal myofibroblasts, which are thought to secrete factors such as hepatocyte growth factor (HGF) that activate β-catenin-dependent transcription. This maintains CSC clonogenicity and restores the CSC phenotype in more differentiated tumor cells both in vitro and in vivo [27]. HGF additionally induces β-catenin nuclear translocation through Met/β-catenin dissociation, in a Wnt-independent pathway [28].
Noncolorectal gastrointestinal cancers
β-catenin mutations have been implicated in early steps of carcinogenesis by activating the WNT pathway in gastric and non-hepatitis-related hepatocellular cancers [29, 30]. Cholangiocarcinoma has also been shown to be a WNT-dependent cancer, which thrives on canonical WNT pathway upregulation. Activation of Wnt/β-catenin signaling, by the pluripotent mesenchymal stem cells recruited to the tumor site, seems to play a central role in tumor microenvironment modulation by promoting metastatic growth and resistance to chemotherapy in cholangiocarcinoma [31]. M2-polarized tumor-associated macrophages (TAMs) in the surrounding stroma are known to maintain a highly activated WNT pathway in the tumor [32].
Desmoid tumors
Crago et al. demonstrated that mutations affecting APC and CTNNB1 (gene encoding β-catenin) occur frequently in desmoid tumors (111 of 117; 95%). Even true CTNNB1 wild-type tumors (determined by next-generation sequencing) may have genomic alterations associated with WNT activation such as chromosome 6-loss/BMI1 mutation, supporting Wnt/β-catenin activation as an important pathway governing desmoid initiation [33].
Breast cancer
WNT/β-catenin signaling regulates the self-renewal and migration of CSCs, thereby promoting tumor growth and metastasis in breast cancer [34]. TAMs in the invasive front of primary mammary tumors have higher expression of molecules involved in WNT-signaling suggesting a role in tumor cell migration, invasiveness, and metastasis [35]. In triple negative breast cancers, β-catenin expression was associated with poor overall survival and disease-specific survival [36]. Upregulation of Wnt/β-catenin has been shown to be a mechanism of resistance to PI3K inhibitors, and the use of β-catenin inhibitors may sensitize PIK3CA mutant breast cancer to PI3K inhibition [37].
Adrenocortical tumors
Wnt/β-catenin activation has been shown to be an independent prognostic factor of overall and disease-free survival in patients with resected primary adrenocortical cancer. The presence of β-catenin nuclear staining was significantly associated with higher tumor stage and risk score, frequent necrosis, mitoses, and other associated mutations [38]. Results from animal models have suggested that β-catenin can function as an adrenal oncogene causing progressive sub-capsular cell hyperplasia, ectopic expansion of spongiocytes and sub-capsular cell with resulting dysplasia, and marked differentiation defects resulting in primary hyperaldosteronism and development of malignant characteristics such as uncontrolled neovascularization and local invasion when there is prolonged activation [39].
Melanoma
Wnt/β-catenin activation may have conflicting roles in the metastatic spread of melanoma [40]. β-catenin signaling decreased the migration of melanocytes and melanoma cell lines in vitro but promoted lung metastases in the NRAS-driven melanoma murine model. β-catenin seems to be a major driver of melanoma dissemination to lymph nodes and lungs in a mouse model based on melanocyte-specific PTEN loss and BRAF (V600E) mutation [41]. β-catenin level also controls tumor differentiation and regulates both MAPK/Erk and PI3K/Akt signaling. In fact, activation of Wnt/β-catenin and AKT pathways mediates chemo-resistance and increased invasion in melanoma cell lines [42].
Glioblastoma multiforme
Components of WNT pathway are usually overexpressed in glioblastoma multiforme (GBM) tumors. PLAG2 overexpression may play a role in inducing upregulation of WNT6, FZD9, and FZD2, ultimately leading to the maintenance of stemness features of GBM stem cells [43]. β-catenin also increases the expression DNA repair enzyme O6-methylguanine-DNA methyltransferase (MGMT) through Tcf/Lef binding located in the hmMGMT 5′-flanking regulatory region. Genetic or pharmacological inhibition of Wnt/β-catenin signaling reduces MGMT expression and increases the cytotoxic effects of temozolomide [44].
Renal cell carcinoma (RCC)
Higher levels of β-catenin are associated with poor prognosis, higher stage, node involvement, vascular invasion, and sarcomatoid differentiation in RCC [45]. Multilayer-omics analysis including methylome and transcriptome analyses also demonstrated a significant role of Wnt/β-catenin signaling pathway in RCC pathogenesis [46]. Mutations in GCN1L1, MED12, and CCNC, members of the CDK8 mediator complex that directly regulate β-catenin-driven transcription, were identified in 16% of the RCCs [46].
Osteosarcoma
Wnt receptor LRP-5 expression correlates with a worse event-free survival in patients with osteosarcoma [47]. Targeting LRP5 receptor signaling with a dominant-negative form of the receptor inhibited tumor growth and metastasis and reduced the expression of cancer cell invasiveness-associated markers in animal models of osteosarcoma.
Hematological malignancies
Wnt/β-catenin signaling pathway is required for self-renewal of CSCs. Yeung et al. demonstrated that β-catenin was activated during development of mixed-lineage leukemia (MLL) CSCs. Suppression of β-catenin reversed CSCs to a pre-CSC-like stage and reduced the growth of human MLL leukemic cells [48]. CSCs rely on Wnt/β-catenin pathway in addition to other important pathways like PI3K/Akt/mTOR and JAK/STAT for acquiring therapy resistance [49]. In chronic myelogenous leukemia, deletion of β-catenin synergized with imatinib resulted in a delay of disease recurrence after imatinib discontinuation [50]. Wnt pathway has been shown to be upregulated in mantle cell lymphoma-initiating cells (MCL-IL) [51]. In addition, MCL-ILs were shown to be sensitive when targeted downstream at β-catenin-mediated transcription complex with inhibitors such as CCT036477, iCRT14, and PKF118-310. β-catenin mRNA or protein expression was shown to be upregulated in the bone marrow aspirates of patients with myelofibrosis and in other Philadelphia-negative myeloproliferative neoplasms [52, 53]. The Wnt/β-catenin pathway is also involved in the pathogenesis of multiple myeloma (MM), with silencing of the pathway resulting in autophagy and apoptosis in MM cells [54]. Loss of the deubiquitinating enzyme CYLD has been shown to enhance MM aggressiveness via Wnt pathway activation [55].
WNT inhibitors in clinical development
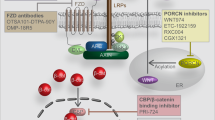
Several therapeutic strategies have been developed with the aim to inhibit WNT-pathway, and many agents are undergoing early phase clinical trials (Table 1 and Fig. 2). Membrane bound O-acyl transferase porcupine (PORCN) has emerged as a molecular target of interest to treat WNT-driven cancers [56]. Addition of palmitoyl groups to WNT proteins is catalyzed by PORCN, a biochemical process known as palmitoylation or S-acylation in the endoplasmic reticulum, which enhances WNT secretion into the cytoplasm [57]. WNT974, a PORCN inhibitor, produced cytostatic effects in ovarian cancer cells in vitro [58] and decreased tumor growth and metastatic spread in head and neck squamous cell carcinoma models in vivo [59]. A phase I/II trial evaluating WNT974 in combination with LGX818, a specific BRAF inhibitor, and cetuximab in patients with metastatic colorectal cancer bearing WNT and BRAF mutations is ongoing (NCT02278133). Another trial is evaluating WNT974 in patients with metastatic head and neck squamous cell carcinoma (NCT02649530).

Therapeutic targets in Wnt/β-catenin pathway and developmental therapeutics. Multiple strategies have been under investigation to counteract the canonical pathway of Wnt signaling. a, b Wnt soluble receptors and antibodies directed to Frizzled receptors impair the interaction ligand/receptor and its conveyed signal. c COX inhibitors reduces β-catenin cytoplasmic levels through different ways. PGE2, the main product of COX2 enzyme, is thought to mediate β-catenin transcription. Also, COX inhibitors such as aspirin were related to increase β-catenin ubiquitination and proteasomal destruction. d Tankyrase activates axin through induction of PARsylation and proteasomal degradation; tankyrase inhibitors increase the levels of axin, facilitating the formation of the β-catenin destruction complex and reducing β-catenin availability. e CBP inhibitors reduce the interaction between CBP and Tcf/Lef, reducing Tcf/Lef activity. f. PORCN inhibitors reduce the essential palmitoylation of Wnt, precluding its release to the extracellular space. g SAM68 is a regulator of alternative splicing of Tcf and impairs β-catenin/Tcf/Lef interaction
WNT agonists, specifically WNT-5a activation, have shown to inhibit metastases. Increased WNT-5a signaling suppressed endothelial tumor cell migration and invasion and inhibited metastasis in model of breast cancer in vivo. Foxy-5 is a WNT-5a mimic hexapeptide that binds and activates the WNT-5a receptors, Frizzled-2 and Frizzled-5 [60]. Two phase I trials are evaluating Foxy-5 in advanced solid tumors (breast, colorectal, and prostate) with loss or reduced expression of WNT-5a protein (NCT02020291 and NCT02655952).
CWP232291 is a novel small molecule that binds Src associated with mitosis 68K protein (Sam68). Preclinical data showed selective inhibitory activity on a WNT gene reporter and decreased expression of the β-catenin target genes, cyclin D1 and survivin. CWP232291 is being tested in phase I clinical trials in patients with acute myeloid leukemia (NCT01398462) and relapsed or refractory myeloma (NCT02426723) [61].
Genistein, a dietary compound present in soy-based foods, is thought to mediate anticancer activity through pleiotropic mechanisms that may include the inhibition of WNT pathway [62]. Phase I and II studies investigating the clinical activity of genistein and other soy isoflavones compounds in cancer treatment and chemoprevention have been published with disappointing results [63,64,65].
OMP-54F28 (Ipafricept) is a fusion protein that combines the immunoglobulin Fc domain with the cysteine-rich domain of frizzled family receptor 8 (Fzd8) competing with the native Fzd8 receptor for its ligands and antagonizes WNT signaling [66]. A dose-escalation phase I showed that OMP-54F28 was well tolerated and dose escalation cohorts with standard therapy in advanced hepatocellular cancers (NCT02069145), pancreatic cancers (NCT02050178), and recurrent platinum-sensitive ovarian cancer (NCT02092363) are under way [67]. Another monoclonal antibody against frizzled receptors OMP-18R5 is in phase 1 clinical trials for patients with metastatic breast cancer in combination with paclitaxel. DKN-01 is a humanized monoclonal antibody (Mab) with neutralizing activity against Dickkopf-1 (Dkk-1). Strong preclinical evidence of synergistic activity with chemotherapy agents is the basis for clinical testing of these agents in combination.
Preclinical studies have addressed several other strategies to counteract Wnt/β-catenin-mediated carcinogenesis, although they have not been yet tested in clinical trials. Inducing or stabilizing the “destruction complex” of β-catenin, therefore reducing its intracellular levels and precluding its transcriptional activity, is a promising strategy. Axin is a concentration-limiting component of the β-catenin destruction complex, and its stability is regulated by tankyrase, the key regulator enzyme that is responsible for poly(ADP-ribosyl)ation (PARsylation) of axin and induces its proteasomal degradation [68, 69]. Waaler et al. demonstrated the antitumoral effect of inhibiting tankyrase in a model of colorectal adenoma and adenocarcinoma [70]. Potential adverse events of this therapy include diarrhea and intestinal toxicity [71]. Tankyrase inhibition has been shown to revert resistance to PI3K and AKT inhibitors in colorectal cancer patient-derived sphere cultures and mouse tumor xenografts [72]. High nuclear β-catenin expression predicted resistance to PI3K and AKT inhibitors. Combined treatment with a WNT/tankyrase inhibitor reduced nuclear β-catenin, reverted resistance to PI3K and AKT inhibitors, and repressed tumor growth.
Another possible strategy to reduce signaling through the canonical pathway of Wnt/β-catenin consists of antagonizing β-catenin/TCF-mediated transcription. Because transcriptional regulation of the β-catenin/TCF complex needs some coactivators, such as the cAMP response element-binding protein (CREB)-binding protein (CBP), this molecule was considered a potential target for inhibition [73]. INT-001, a specific antagonist of CBP, showed antitumoral activity in preclinical models of pancreatic, colon, and tamoxifen-resistant breast cancers [74,75,76].
Multiple observational studies and randomized controlled trials have demonstrated a chemo-preventative role for aspirin, particularly in the development of colorectal neoplasia. Given the critical importance of WNT dysregulation in colorectal carcinogenesis, the interplay between aspirin and canonical WNT signaling has become a focus of investigation [77]. Through inhibiting cyclooxygenase-2 (COX-2), aspirin decreases the availability of prostaglandin E2 (PGE2), therefore reducing WNT levels [78]. Interestingly, cancer cells when exposed to aspirin in vitro led to the inhibition of WNT pathway through increasing β-catenin ubiquitination and consequent degradation, independent on COX inhibition as well [79]. Figure 2 summarizes the developmental strategies under investigation in Wnt/β-catenin pathway.
Cancer immunity and Wnt/β-catenin pathway
Overexpression of immune checkpoint molecules in the tumor microenvironment has a critical role in antitumor immunity evasion and cancer progression [80]. Currently, four immune checkpoint inhibitors (ICI) are approved for treatment of cancer, including ipilimumab (anti-cytotoxic T-lymphocyte-associated protein 4 (anti-CTLA4)), nivolumab, pembrolizumab (anti-programmed death-1 (anti-PD-1)), and atezolizumab (anti-programmed death ligand-1 (anti-PD-L1)). Anti-PD-1/PD-L1 antibodies have demonstrated clinical activity in more than 15 cancer types, but the majority of patients with advanced cancer still do not derive clinical benefit from these drugs suggesting that immunosuppressive mechanisms within the tumor microenvironment may play a role in de novo resistance to these therapies.
Clinical responses to multiple T cell-based therapies, including immune checkpoint inhibitors, have correlated with tumors with a T cell-inflamed microenvironment [81, 82]. These tumors are characterized by infiltration of CD8+ T cells, chemokines, and an interferon signature, which ultimately correlate with improved survival and responses to immunotherapies [83, 84]. On the other hand, it has been recognized that tumors without T cell infiltration have worse prognosis and do not benefit from immunotherapy [84]. Thus, understanding mechanisms driving T cell exclusion is critical to increase the number of patients that will benefit from immunotherapy.
Wnt/β-catenin pathway has been identified as one of the important oncogenic pathway signaling related to immune evasion (Fig. 3) [85, 86]. In a teratoma model, enhanced expression of Wnt correlated with impaired immune cell recruitment. Both T- and B cell infiltration was reduced which was independent of teratoma size and differentiation suggesting impaired immune surveillance [87]. Luke et al. evaluated gene expression data from The Cancer Genome Atlas (TCGA) to segregate tumors based on a T cell-inflamed gene expression signature and classified 8890 tumor samples into T cell-inflamed, non-T cell inflamed, and intermediate subtypes [88]. Approximately one third of the interrogated tumors were characterized as non-T cell inflamed. They further interrogated exomic sequencing to address the presence of mutations in Wnt/β-catenin pathway in this subtype. Activating mutations in CTNNB1 and inactivating mutations in negative regulators such as Axin1, Axin2, APC1, and APC2 were related to non-T cell inflamed gene signature, accounting for about 13% of the sequenced tumors. Most of the mutations were noted in Exon 3 of the CTNNB1 gene, with mis-sense mutation being the most common. Evidence of β-catenin pathway activation, in the presence of mutation or otherwise, was noted in 24 of 30 solid tumors of TCGA. Evidence for pathway activation without mutation was evidenced by increased expression of β-catenin pathway elements such as WNT ligands, Fzd receptors, or β-catenin itself. Immunohistochemistry demonstrated inverse correlations between β-catenin and CD8+ T cell infiltration.

Mechanisms of immune exclusion through Wnt/beta-catenin pathway: Activation of Wnt/beta-catenin pathway in tumor leads to noninflammatory milieu through multiple mechanisms. a By acting on Batf3-lineage CD103+ dendritic cells, decreasing CCL4 production by inducing the gene expression of the transcription repressor ATF3. This in turn reduces CD8+ T cell priming and infiltration. b By interacting with tumor-associated macrophages (TAM) through Snail (a soluble factor product of a Wnt-regulated gene) which can in turn increase beta-catenin activity by IL-1β. c β-catenin can enhance Treg survival through unknown mechanisms
These results suggest that increased expression of β-catenin in the tumor should be investigated and validated as a predictive tool to improve the selection of candidate patients for checkpoint inhibition therapy. In addition, one might hypothesize that inhibition of Wnt/β-catenin signaling could improve CD8+ T cell infiltration and priming, therefore possibly producing a more favorable scenario to immune checkpoint inhibition [89]. Thus, the investigation of the possible role of Wnt/β-catenin inhibitors as possible adjuvants to anti-PD1, anti-CTLA4, or anti-PD-L1 is an interesting strategy and should be evaluated in preclinical and clinical studies.
While tumor infiltration with CD8+ effector T cell is favorable, presence of regulatory T cells (Treg) has been associated with poor antitumor immunity. Tregs are known to suppress adaptive responses mainly by reducing CD8+ T cell proliferation, activation, and effector function [90, 91]. Some studies have shown that expression of β-catenin has been associated with Treg infiltration, survival, and activity [92, 93]. In an adoptive transfer mouse colorectal cancer model, β-catenin expression was enforced in intratumoral CD4+ T cells using lentiviral transduction. This led to increased IL-17a expression, enhanced proliferation, and inhibited apoptosis of colorectal cancer cells [87]. Active WNT signaling has been shown to disrupt Foxp3 transcriptional activity, a crucial step for both the development and function of regulatory T cells [94]. Hence, it has to be recognized that blocking WNT for immunomodulation is still controversial at this time. On the other hand, induction of Wnt signaling has shown to be important in maintenance of stemness of memory CD8+ T cells by blocking T cell differentiation [95]. This strategy may be used in generating memory T cell populations, thereby improving effectiveness of cancer vaccines, and may find applications in programming antitumor T cells for adoptive immunotherapy [96].
Wnt/β-catenin pathway has also been associated with modulation of innate immunity, such as dendritic cells [97]. Spranger et al. detected a correlation between activation of the Wnt/β-catenin signaling pathway and absence of a T cell gene expression signature in human metastatic melanoma [98]. Active β-catenin signaling was observed in 48% of non-T cell-infiltrated melanomas, which was thought to be the important pathway mediating immune exclusion. In mouse models, β-catenin induced expression of the transcription repressor ATF3, which suppressed the transcription of CCL4. The defective production of CCL4 led to impaired infiltration and activation of Batf3-lineage CD103+ dendritic cells, reduced CD8+ T cell priming and infiltration, and consequent non-response to immune check point blockade. In the absence of active β-catenin signaling, the normal production of CCL4 was restored, leading to CD103+ dendritic cell activation and infiltration and proficient priming of CD8+ T cell. Wnt/β-catenin signaling in intestinal dendritic cells has shown to regulate the balance between inflammatory versus regulatory responses in the gut [99]. β-catenin when expressed in intestinal dendritic cells was associated with expression of anti-inflammatory mediators such as retinoic acid-metabolizing enzymes, interleukin-10, and transforming growth factor-β, and the stimulation of Treg induction [100].
Wnt/Beta-catenin signaling has been shown to be involved in cross talk between cancer cells and tumor-associated macrophages. Kaler et al. demonstrated a possible role of interleukin-1β secreted by TAMs in the increased availability of β-catenin through phosphorylation of GSK3β in colon cancer cells, disrupting the function of β-catenin destruction complex [101]. Colorectal cancer cells stimulate macrophage production of IL-1β through Snail, a soluble factor product of a Wnt-regulated gene. These data demonstrated a novel Wnt-dependent cross talk mechanism between tumor cells and macrophages [102].
Conclusions
The Wnt/β-catenin pathway is increasingly recognized as a potentially important target for anticancer therapies, with several relevant inhibitors at various stages of clinical development. The expanding role of immunotherapies in cancer and recent insight into the role of Wnt-pathway in cancer-related immune-regulation may give a new dimension to this field of developmental therapeutics. Only tumors with upregulated Wnt/β-catenin signaling such as colorectal cancer have been explored as targets for Wnt inhibition. However, with its role in immunomodulation, Wnt inhibitors may have a broader role in cancers such as melanoma, lung, and renal cancers where immunotherapy has come to the forefront. There are some nuances to be addressed in the exact role of Wnt signaling in immunomodulation before this could be adopted in clinical practice.
Abbreviations
- anti-CTLA4:
-
Anti-cytotoxic T-lymphocyte-associated protein 4
- anti-PD-1:
-
Anti-programmed death-1
- anti-PD-L1:
-
Anti-programmed death ligand-1
- APC:
-
Adenomatous polyposis coli
- CBP:
-
cAMP response element-binding (CREB)-binding protein
- COX-2:
-
Cyclooxygenase-2
- CRC:
-
Colorectal cancer
- CSC:
-
Cancer stem cells
- FGF:
-
Fibroblast growth factor
- Fzd8:
-
Frizzled family receptor 8
- GBM:
-
Glioblastoma multiforme
- MGMT:
-
O6-methylguanine-DNA methyltransferase
- MLL:
-
Mixed-lineage leukemia
- MM:
-
Multiple myeloma
- MMTV:
-
Mouse mammary tumor virus
- PARsylation:
-
Poly(adenosine diphosphate-ribosyl)ation
- PGE2:
-
Prostaglandin E2
- PORCN:
-
O-acyl transferase porcupine
- RCC:
-
Renal cell carcinoma
- TAM:
-
Tumor-associated macrophage
- TCGA:
-
The Cancer Genome Atlas
- Treg:
-
Regulatory T cells
References
Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. 1982;31:99–109.
Nusse R, van Ooyen A, Cox D, Fung YK, Varmus H. Mode of proviral activation of a putative mammary oncogene (int-1) on mouse chromosome 15. Nature. 1984;307:131–6.
van Ooyen A, Kwee V, Nusse R. The nucleotide sequence of the human int-1 mammary oncogene; evolutionary conservation of coding and non-coding sequences. Embo J. 1985;4:2905–9.
Dickson C, Smith R, Brookes S, Peters G. Tumorigenesis by mouse mammary tumor virus: proviral activation of a cellular gene in the common integration region int-2. Cell. 1984;37:529–36.
Gallahan D, Callahan R. Mammary tumorigenesis in feral mice: identification of a new int locus in mouse mammary tumor virus (Czech II)-induced mammary tumors. J Virol. 1987;61:66–74.
Roelink H, Wagenaar E, Lopes da Silva S, Nusse R. Wnt-3, a gene activated by proviral insertion in mouse mammary tumors, is homologous to int-1/Wnt-1 and is normally expressed in mouse embryos and adult brain. Proc Natl Acad Sci U S A. 1990;87:4519–23.
Dickson C, Peters G. Potential oncogene product related to growth factors. Nature. 1987;326:833.
Gallahan D, Callahan R. The mouse mammary tumor associated gene INT3 is a unique member of the NOTCH gene family (NOTCH4). Oncogene. 1997;14:1883–90.
Nusse R, Brown A, Papkoff J, Scambler P, Shackleford G, McMahon A, Moon R, Varmus H. A new nomenclature for int-1 and related genes: the Wnt gene family. Cell. 1991;64:231.
Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2012;13:767–79.
Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997;11:3286–305.
Riese J, Yu X, Munnerlyn A, Eresh S, Hsu SC, Grosschedl R, Bienz M. LEF-1, a nuclear factor coordinating signaling inputs from wingless and decapentaplegic. Cell. 1997;88:777–87.
Brunner E, Peter O, Schweizer L, Basler K. Pangolin encodes a Lef-1 homologue that acts downstream of Armadillo to transduce the Wingless signal in Drosophila. Nature. 1997;385:829–33.
Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destree O, Clevers H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391–9.
Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, Kuhl M, Wedlich D, Birchmeier W. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998;280:596–9.
Huber O, Korn R, McLaughlin J, Ohsugi M, Herrmann BG, Kemler R. Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech Dev. 1996;59:3–10.
Fagotto F. Looking beyond the Wnt pathway for the deep nature of beta-catenin. EMBO Rep. 2013;14:422–33.
Miller RK, Hong JY, Munoz WA, McCrea PD. Beta-catenin versus the other armadillo catenins: assessing our current view of canonical Wnt signaling. Prog Mol Biol Transl Sci. 2013;116:387–407.
Veeman MT, Axelrod JD, Moon RT. A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Dev Cell. 2003;5:367–77.
van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009;136:3205–14.
Morin PJ. Beta-catenin signaling and cancer. Bioessays. 1999;21:1021–30.
Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–42.
Chiang JM, Chou YH, Chen TC, Ng KF, Lin JL. Nuclear beta-catenin expression is closely related to ulcerative growth of colorectal carcinoma. Br J Cancer. 2002;86:1124–9.
Konsavage Jr WM, Kyler SL, Rennoll SA, Jin G, Yochum GS. Wnt/beta-catenin signaling regulates Yes-associated protein (YAP) gene expression in colorectal carcinoma cells. J Biol Chem. 2012;287:11730–9.
Cai J, Maitra A, Anders RA, Taketo MM, Pan D. Beta-catenin destruction complex-independent regulation of Hippo-YAP signaling by APC in intestinal tumorigenesis. Genes Dev. 2015;29:1493–506.
Ayadi M, Bouygues A, Ouaret D, Ferrand N, Chouaib S, Thiery JP, Muchardt C, Sabbah M, Larsen AK. Chronic chemotherapeutic stress promotes evolution of stemness and WNT/beta-catenin signaling in colorectal cancer cells: implications for clinical use of WNT-signaling inhibitors. Oncotarget. 2015;6:18518–33.
Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–76.
Monga SP, Mars WM, Pediaditakis P, Bell A, Mule K, Bowen WC, Wang X, Zarnegar R, Michalopoulos GK. Hepatocyte growth factor induces Wnt-independent nuclear translocation of beta-catenin after Met-beta-catenin dissociation in hepatocytes. Cancer Res. 2002;62:2064–71.
Clements WM, Wang J, Sarnaik A, Kim OJ, MacDonald J, Fenoglio-Preiser C, Groden J, Lowy AM. Beta-catenin mutation is a frequent cause of Wnt pathway activation in gastric cancer. Cancer Res. 2002;62:3503–6.
Laurent-Puig P, Legoix P, Bluteau O, Belghiti J, Franco D, Binot F, Monges G, Thomas G, Bioulac-Sage P, Zucman-Rossi J. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology. 2001;120:1763–73.
Wang W, Zhong W, Yuan J, Yan C, Hu S, Tong Y, Mao Y, Hu T, Zhang B, Song G: Involvement of WNT/beta-catenin signaling in the mesenchymal stem cells promote metastatic growth and chemoresistance of cholangiocarcinoma. Oncotarget 2015.
Boulter L, Guest RV, Kendall TJ, Wilson DH, Wojtacha D, Robson AJ, Ridgway RA, Samuel K, Van Rooijen N, Barry ST, et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J Clin Invest. 2015;125:1269–85.
Crago AM, Chmielecki J, Rosenberg M, O'Connor R, Byrne C, Wilder FG, Thorn K, Agius P, Kuk D, Socci ND, et al. Near universal detection of alterations in CTNNB1 and Wnt pathway regulators in desmoid-type fibromatosis by whole-exome sequencing and genomic analysis. Genes Chromosomes Cancer. 2015;54:606–15.
Jang GB, Kim JY, Cho SD, Park KS, Jung JY, Lee HY, Hong IS, Nam JS. Blockade of Wnt/beta-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci Rep. 2015;5:12465.
Ojalvo LS, Whittaker CA, Condeelis JS, Pollard JW. Gene expression analysis of macrophages that facilitate tumor invasion supports a role for Wnt-signaling in mediating their activity in primary mammary tumors. J Immunol. 2010;184:702–12.
Shen T, Zhang K, Siegal GP, Wei S: Prognostic value of E-cadherin and beta-catenin in triple-negative breast cancer. Am J Clin Pathol 2016.
Merino VF, Cho S, Liang X, Park S, Jin K, Chen Q, Pan D, Zahnow CA, Rein AR, Sukumar S. Inhibitors of STAT3, beta-catenin, and IGF-1R sensitize mouse PIK3CA mutant breast cancer to PI3K inhibitors. Mol Oncol. 2017;11(5):552–66.
Gaujoux S, Grabar S, Fassnacht M, Ragazzon B, Launay P, Libe R, Chokri I, Audebourg A, Royer B, Sbiera S, et al. Beta-catenin activation is associated with specific clinical and pathologic characteristics and a poor outcome in adrenocortical carcinoma. Clin Cancer Res. 2011;17:328–36.
Berthon A, Sahut-Barnola I, Lambert-Langlais S, de Joussineau C, Damon-Soubeyrand C, Louiset E, Taketo MM, Tissier F, Bertherat J, Lefrancois-Martinez AM, et al. Constitutive beta-catenin activation induces adrenal hyperplasia and promotes adrenal cancer development. Hum Mol Genet. 2010;19:1561–76.
Gallagher SJ, Rambow F, Kumasaka M, Champeval D, Bellacosa A, Delmas V, Larue L. Beta-catenin inhibits melanocyte migration but induces melanoma metastasis. Oncogene. 2013;32:2230–8.
Damsky WE, Curley DP, Santhanakrishnan M, Rosenbaum LE, Platt JT, Gould Rothberg BE, Taketo MM, Dankort D, Rimm DL, McMahon M, Bosenberg M. Beta-catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell. 2011;20:741–54.
Yi GZ, Liu YW, Xiang W, Wang H, Chen ZY, Xie SD, Qi ST. Akt and beta-catenin contribute to TMZ resistance and EMT of MGMT negative malignant glioma cell line. J Neurol Sci. 2016;367:101–6.
Lee Y, Lee JK, Ahn SH, Lee J, Nam DH. WNT signaling in glioblastoma and therapeutic opportunities. Lab Invest. 2016;96:137–50.
Wickstrom M, Dyberg C, Milosevic J, Einvik C, Calero R, Sveinbjornsson B, Sanden E, Darabi A, Siesjo P, Kool M, et al. Wnt/beta-catenin pathway regulates MGMT gene expression in cancer and inhibition of Wnt signalling prevents chemoresistance. Nat Commun. 2015;6:8904.
Kruck S, Eyrich C, Scharpf M, Sievert KD, Fend F, Stenzl A, Bedke J. Impact of an altered Wnt1/beta-catenin expression on clinicopathology and prognosis in clear cell renal cell carcinoma. Int J Mol Sci. 2013;14:10944–57.
Arai E, Sakamoto H, Ichikawa H, Totsuka H, Chiku S, Gotoh M, Mori T, Nakatani T, Ohnami S, Nakagawa T, et al. Multilayer-omics analysis of renal cell carcinoma, including the whole exome, methylome and transcriptome. Int J Cancer. 2014;135:1330–42.
Hoang BH, Kubo T, Healey JH, Sowers R, Mazza B, Yang R, Huvos AG, Meyers PA, Gorlick R. Expression of LDL receptor-related protein 5 (LRP5) as a novel marker for disease progression in high-grade osteosarcoma. Int J Cancer. 2004;109:106–11.
Yeung J, Esposito MT, Gandillet A, Zeisig BB, Griessinger E, Bonnet D, So CW. Beta-catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell. 2010;18:606–18.
Islam F, Gopalan V, Smith RA, Lam AK. Translational potential of cancer stem cells: a review of the detection of cancer stem cells and their roles in cancer recurrence and cancer treatment. Exp Cell Res. 2015;335:135–47.
Heidel FH, Bullinger L, Feng Z, Wang Z, Neff TA, Stein L, Kalaitzidis D, Lane SW, Armstrong SA. Genetic and pharmacologic inhibition of beta-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell. 2012;10:412–24.
Mathur R, Sehgal L, Braun FK, Berkova Z, Romaguerra J, Wang M, Rodriguez MA, Fayad L, Neelapu SS, Samaniego F. Targeting Wnt pathway in mantle cell lymphoma-initiating cells. J Hematol Oncol. 2015;8:63.
Lucijanic M, Livun A, Tomasovic-Loncaric C, Stoos-Veic T, Pejsa V, Jaksic O, Prka Z, Kusec R. Canonical Wnt/beta-catenin signaling pathway is dysregulated in patients with primary and secondary myelofibrosis. Clin Lymphoma Myeloma Leuk. 2016;16:523–6.
Geduk A, Atesoglu EB, Tarkun P, Mehtap O, Hacihanefioglu A, Demirsoy ET, Baydemir C. The role of beta-catenin in Bcr/Abl negative myeloproliferative neoplasms: an immunohistochemical study. Clin Lymphoma Myeloma Leuk. 2015;15:785–9.
Su N, Wang P, Li Y. Role of Wnt/beta-catenin pathway in inducing autophagy and apoptosis in multiple myeloma cells. Oncol Lett. 2016;12:4623–9.
van Andel H, Kocemba KA, de Haan-Kramer A, Mellink CH, Piwowar M, Broijl A, van Duin M, Sonneveld P, Maurice MM, Kersten MJ, et al.: Loss of CYLD expression unleashes Wnt signaling in multiple myeloma and is associated with aggressive disease. Oncogene 2016.
Ho SY, Keller TH. The use of porcupine inhibitors to target Wnt-driven cancers. Bioorg Med Chem Lett. 2015;25:5472–6.
Miranda M, Galli LM, Enriquez M, Szabo LA, Gao X, Hannoush RN, Burrus LW. Identification of the WNT1 residues required for palmitoylation by Porcupine. FEBS Lett. 2014;588:4815–24.
Boone JD, Arend RC, Johnston BE, Cooper SJ, Gilchrist SA, Oelschlager DK, Grizzle WE, McGwin Jr G, Gangrade A, Straughn Jr JM, Buchsbaum DJ. Targeting the Wnt/beta-catenin pathway in primary ovarian cancer with the porcupine inhibitor WNT974. Lab Invest. 2016;96:249–59.
Rudy SF, Brenner JC, Harris JL, Liu J, Che J, Scott MV, Owen JH, Komarck CM, Graham MP, Bellile EL, et al. In vivo Wnt pathway inhibition of human squamous cell carcinoma growth and metastasis in the chick chorioallantoic model. J Otolaryngol Head Neck Surg. 2016;45:26.
Safholm A, Tuomela J, Rosenkvist J, Dejmek J, Harkonen P, Andersson T. The Wnt-5a-derived hexapeptide Foxy-5 inhibits breast cancer metastasis in vivo by targeting cell motility. Clin Cancer Res. 2008;14:6556–63.
Jorge E. Cortes SF, John Pagel, Chul Won Jung, Sung-Soo Yoon, Youngil Koh, Animesh Dev Pardanani, Robert S Hauptschein, Kyoung-June Lee, Je-Hwan Lee: Phase 1 study of CWP232291 in relapsed/refractory acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). In J Clin Oncol 33, 2015 (suppl; abstr 7044).
Spagnuolo C, Russo GL, Orhan IE, Habtemariam S, Daglia M, Sureda A, Nabavi SF, Devi KP, Loizzo MR, Tundis R, Nabavi SM. Genistein and cancer: current status, challenges, and future directions. Adv Nutr. 2015;6:408–19.
El-Rayes BF, Philip PA, Sarkar FH, Shields AF, Ferris AM, Hess K, Kaseb AO, Javle MM, Varadhachary GR, Wolff RA, Abbruzzese JL. A phase II study of isoflavones, erlotinib, and gemcitabine in advanced pancreatic cancer. Invest New Drugs. 2011;29:694–9.
Lohr JM, Karimi M, Omazic B, Kartalis N, Verbeke CS, Berkenstam A, Frodin JE: A phase I dose escalation trial of AXP107-11, a novel multi-component crystalline form of genistein, in combination with gemcitabine in chemotherapy-naive patients with unresectable pancreatic cancer. Pancreatology. 2016.
Lazarevic B, Hammarstrom C, Yang J, Ramberg H, Diep LM, Karlsen SJ, Kucuk O, Saatcioglu F, Tasken KA, Svindland A. The effects of short-term genistein intervention on prostate biomarker expression in patients with localised prostate cancer before radical prostatectomy. Br J Nutr. 2012;108:2138–47.
Le PN, McDermott JD, Jimeno A. Targeting the Wnt pathway in human cancers: therapeutic targeting with a focus on OMP-54 F28. Pharmacol Ther. 2015;146:1–11.
Antonio Jimeno MSG, Rashmi C, Messersmith WA, Mendelson DS, Jakob D, Stagg RJ, Ann K, Lu X, Rainer Karl B, Smith DC. A first-in-human phase 1 study of anticancer stem cell agent OMP-54F28 (FZD8-Fc), decoy receptor for WNT ligands, in patients with advanced solid tumors. In: ASCO Annual Meeting, vol. J Clin Oncol 32:5s, 2014 (suppl; abstr 2505). Chicago. 2014.
Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–20.
Zhang Y, Liu S, Mickanin C, Feng Y, Charlat O, Michaud GA, Schirle M, Shi X, Hild M, Bauer A, et al. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat Cell Biol. 2011;13:623–9.
Waaler J, Machon O, Tumova L, Dinh H, Korinek V, Wilson SR, Paulsen JE, Pedersen NM, Eide TJ, Machonova O, et al. A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012;72:2822–32.
Zhong Y, Katavolos P, Nguyen T, Lau T, Boggs J, Sambrone A, Kan D, Merchant M, Harstad E, Diaz D, et al. Tankyrase inhibition causes reversible intestinal toxicity in mice with a therapeutic index < 1. Toxicol Pathol. 2016;44:267–78.
Arques O, Chicote I, Puig I, Tenbaum SP, Argiles G, Dienstmann R, Fernandez N, Caratu G, Matito J, Silberschmidt D, et al. Tankyrase inhibition blocks Wnt/beta-catenin pathway and reverts resistance to PI3K and AKT inhibitors in the treatment of colorectal cancer. Clin Cancer Res. 2016;22:644–56.
Eguchi M, Nguyen C, Lee SC, Kahn M. ICG-001, a novel small molecule regulator of TCF/beta-catenin transcription. Med Chem. 2005;1:467–72.
Arensman MD, Telesca D, Lay AR, Kershaw KM, Wu N, Donahue TR, Dawson DW. The CREB-binding protein inhibitor ICG-001 suppresses pancreatic cancer growth. Mol Cancer Ther. 2014;13:2303–14.
Lazarova DL, Chiaro C, Wong T, Drago E, Rainey A, O'Malley S, Bordonaro M. CBP activity mediates effects of the histone deacetylase inhibitor butyrate on WNT activity and apoptosis in colon cancer cells. J Cancer. 2013;4:481–90.
Won HS, Lee KM, Oh JE, Nam EM, Lee KE. Inhibition of beta-catenin to overcome endocrine resistance in tamoxifen-resistant breast cancer cell line. PLoS One. 2016;11:e0155983.
Gala MK, Chan AT. Molecular pathways: aspirin and Wnt signaling-a molecularly targeted approach to cancer prevention and treatment. Clin Cancer Res. 2015;21:1543–8.
Algra AM, Rothwell PM. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012;13:518–27.
Bos CL, Kodach LL, van den Brink GR, Diks SH, van Santen MM, Richel DJ, Peppelenbosch MP, Hardwick JC. Effect of aspirin on the Wnt/beta-catenin pathway is mediated via protein phosphatase 2A. Oncogene. 2006;25:6447–56.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
Iglesia MD, Parker JS, Hoadley KA, Serody JS, Perou CM, Vincent BG. Genomic analysis of immune cell infiltrates across 11 tumor types. J Natl Cancer Inst. 2016;108:djw144.
Meng X, Huang Z, Teng F, Xing L, Yu J. Predictive biomarkers in PD-1/PD-L1 checkpoint blockade immunotherapy. Cancer Treat Rev. 2015;41:868–76.
Gajewski TF, Schreiber H, Fu Y-x. Innate and adaptive immune cells in the tumor microenvironment. 2013. p. 14.
Gajewski TF. The next hurdle in cancer immunotherapy—overcoming the non-T-cell-inflamed tumor microenvironment. Semin Oncol. 2015;42:663–71.
Fu C, Liang X, Cui W, Ober-Blobaum JL, Vazzana J, Shrikant PA, Lee KP, Clausen BE, Mellman I, Jiang A. Beta-catenin in dendritic cells exerts opposite functions in cross-priming and maintenance of CD8+ T cells through regulation of IL-10. Proc Natl Acad Sci U S A. 2015;112:2823–8.
Spranger S, Gajewski TF. A new paradigm for tumor immune escape: beta-catenin-driven immune exclusion. J Immunother Cancer. 2015;3:43.
Augustin I, Dewi DL, Hundshammer J, Rempel E, Brunk F, Boutros M. Immune cell recruitment in teratomas is impaired by increased Wnt secretion. Stem Cell Res. 2016;17:607–15.
LLuke JJ, Bao R, Spranger S, Sweis RF, Gajewski T. Correlation of WNT/β-catenin pathway activation with immune exclusion across most human cancers [abstract]. J Clin Oncol. 2016;34(suppl; abstr 3004).
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10.
Golovina TN, Vonderheide RH. Regulatory T cells: overcoming suppression of T-cell immunity. Cancer J. 2010;16:342–7.
Sojka DK, Huang YH, Fowell DJ. Mechanisms of regulatory T-cell suppression—a diverse arsenal for a moving target. Immunology. 2008;124:13–22.
Hong Y, Manoharan I, Suryawanshi A, Majumdar T, Angus-Hill ML, Koni PA, Manicassamy B, Mellor AL, Munn DH, Manicassamy S. Beta-catenin promotes regulatory T-cell responses in tumors by inducing vitamin A metabolism in dendritic cells. Cancer Res. 2015;75:656–65.
Alves CH, Ober-Blobaum JL, Brouwers-Haspels I, Asmawidjaja PS, Mus AM, Razawy W, Molendijk M, Clausen BE, Lubberts E. Dendritic cell-specific deletion of beta-catenin results in fewer regulatory T-cells without exacerbating autoimmune collagen-induced arthritis. PLoS One. 2015;10:e0142972.
van Loosdregt J, Fleskens V, Tiemessen MM, Mokry M, van Boxtel R, Meerding J, Pals CE, Kurek D, Baert MR, Delemarre EM, et al. Canonical Wnt signaling negatively modulates regulatory T cell function. Immunity. 2013;39:298–310.
Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, Wrzesinski C, Boni A, Cassard L, Garvin LM, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15:808–13.
Gattinoni L, Ji Y, Restifo NP. Wnt/beta-catenin signaling in T-cell immunity and cancer immunotherapy. Clin Cancer Res. 2010;16:4695–701.
Swafford D, Manicassamy S. Wnt signaling in dendritic cells: its role in regulation of immunity and tolerance. Discov Med. 2015;19:303–10.
Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–5.
Ding Y, Shen S, Lino AC, Curotto de Lafaille MA, Lafaille JJ. Beta-catenin stabilization extends regulatory T cell survival and induces energy in nonregulatory T cells. Nat Med. 2008;14:162–9.
Manicassamy S, Reizis B, Ravindran R, Nakaya H, Salazar-Gonzalez RM, Wang YC, Pulendran B. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science. 2010;329:849–53.
Kaler P, Augenlicht L, Klampfer L. Macrophage-derived IL-1beta stimulates Wnt signaling and growth of colon cancer cells: a crosstalk interrupted by vitamin D3. Oncogene. 2009;28:3892–902.
Kaler P, Augenlicht L, Klampfer L. Activating mutations in beta-catenin in colon cancer cells alter their interaction with macrophages; the role of snail. PLoS One. 2012;7:e45462.
Acknowledgements
The authors would like to acknowledge the Woman’s Board of Northwestern Memorial Hospital
Funding
None
Authors’ contributions
SGP, BAC, JMM, and FJG drafted the manuscript. SGP, BAC, JMM, RBS, and JBK performed the search of literature. SGP, RC, CAL, YKC, and JBK collected the related references. SGP, JMM, and CAL prepared the figures. BAC, JBK, RBS, and FJG revised the language. SGP, BAC, YKC, and FJG carried out the design of the review. All authors contributed to this manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable
Ethics approval and consent to participate
Not applicable
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pai, S.G., Carneiro, B.A., Mota, J.M. et al. Wnt/beta-catenin pathway: modulating anticancer immune response. J Hematol Oncol 10, 101 (2017). https://doi.org/10.1186/s13045-017-0471-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-017-0471-6