
Abstract
Social behavior emerges early in development, a time marked by the onset of neurodevelopmental disorders featuring social deficits, including autism spectrum disorder (ASD). Although social deficits are at the core of the clinical diagnosis of ASD, very little is known about their neural correlates at the time of clinical onset. The nucleus accumbens (NAc), a brain region extensively implicated in social behavior, undergoes synaptic, cellular and molecular alterations in early life, and is particularly affected in ASD mouse models. To explore a link between the maturation of the NAc and neurodevelopmental deficits in social behavior, we compared spontaneous synaptic transmission in NAc shell medium spiny neurons (MSNs) between the highly social C57BL/6J and the idiopathic ASD mouse model BTBR T+Itpr3tf/J at postnatal day (P) 4, P6, P8, P12, P15, P21 and P30. BTBR NAc MSNs display increased spontaneous excitatory transmission during the first postnatal week, and increased inhibition across the first, second and fourth postnatal weeks, suggesting accelerated maturation of excitatory and inhibitory synaptic inputs compared to C57BL/6J mice. BTBR mice also show increased optically evoked medial prefrontal cortex-NAc paired pulse ratios at P15 and P30. These early changes in synaptic transmission are consistent with a potential critical period, which could maximize the efficacy of rescue interventions. To test this, we treated BTBR mice in either early life (P4-P8) or adulthood (P60-P64) with the mTORC1 antagonist rapamycin, a well-established intervention for ASD-like behavior. Rapamycin treatment rescued social interaction deficits in BTBR mice when injected in infancy, but did not affect social interaction in adulthood.
Similar content being viewed by others

Introduction
Social behaviors, which include social communication, interaction, parental and reproductive behaviors [1, 2], enable intraspecies interactions necessary to find food, mate and avoid predation [3]. Consistent with their key role in survival, early deficits in the development of social interaction and communication have devastating consequences [4, 5], and are a hallmark of several neurodevelopmental disorders [6], including Fragile X syndrome [7], schizophrenia [8], and autism spectrum disorder (ASD) [9]. ASD encompasses a set of heterogeneous neurodevelopmental disorders affecting an estimated 1% of the world population [9], and features impairments in social cognition, communication and social perception [10]. Even though deficits in social interaction are at the core of clinical diagnoses for neurodevelopmental disorders such as ASD [11], very little is known about the neural basis of such deficits, particularly at the time of clinical onset and diagnosis [12].
Evidence points to conserved brain regions underlying social processing across species [13]. In particular, the rodent nucleus accumbens (NAc) and its projections [14, 15] have been extensively implicated in multiple facets of social behavior, including social interaction and motivation [16,17,18], social cognition [19], social approach [20, 21], social memory [22,23,24], social-spatial representations [25] and susceptibility to social stress [26,27,28,29,30]. Consistent with its important role in social behavior, imaging studies of ASD patients show abnormal activity in NAc [31, 32], as well as changes in functional connectivity within frontostriatal [33,34,35] and striatal [36] circuits. Accordingly, ASD mouse models display altered NAc synaptic plasticity and transmission [37,38,39], as well as changes in gene expression of inhibitory and excitatory markers [40] in adult animals. Importantly, manipulations targeted to the NAc can mimic [41] or rescue behavior deficits in a range of ASD mouse models [37, 42,43,44], further implicating the NAc in ASD pathophysiology.
Recent evidence has highlighted the value and long-term benefits of early ASD interventions [45, 46], but the precise time window of optimal efficacy for these interventions has not been clearly defined. In the developing sensory cortex, plasticity in response to external stimuli is curtailed within critical periods [47], suggesting that similar mechanisms could guide the efficacy of rescue interventions in other brain regions. As with sensory cortex development, the rodent NAc also undergoes early postnatal changes in synaptic transmission [48,49,50,51,52] and plasticity [53], in the levels of dopamine receptors [54,55,56,57,58], dopamine/serotonin transporters [59, 60], stimulus-evoked dopamine transients [61], complement C3 [58], and microglia [62], which stabilize around the start of adolescence. To determine whether the timing of these developmental changes defines a window of increased efficacy for interventions, they must be examined in animal models featuring early social deficits, paralleling the clinical presentation of ASD.
Several studies have identified rapamycin, an inhibitor of the serine/threonine kinase mammalian target of rapamycin (mTOR), as an effective pharmacological intervention in ASD animal models [63,64,65,66,67,68]. mTOR is a critical regulator of cell growth, proliferation, and metabolism [69, 70], as well as synaptic plasticity and memory consolidation [71,72,73]. mTOR is part of two structurally and functionally distinct multiprotein complexes called mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), of which only mTORC1 shows sensitivity to acute inhibition by rapamycin [70]. Importantly, impaired mTORC1 signaling has been implicated in ASD and related neurodevelopmental disorders [69, 74,75,76], consistent with the success of rapamycin treatment in rescuing social behavior deficits in ASD mouse models [77,78,79]. Rapamycin treatment was shown to reverse cellular [78, 80], synaptic [63, 81,82,83], and behavioral [63, 78, 80, 81, 83,84,85,86,87] deficits in genetic, idiopathic and environmental ASD rodent models. However, rapamycin has not been successful in rescuing behavioral abnormalities in the Fmr1 knockout mice, worsening social interaction deficits and impairing sleep duration [88], or in rescuing the repetitive behavioral deficits in Tsc1 mutant mice [78, 80, 89]. Additionally, some studies found that acute and chronic rapamycin treatment resulted in impaired performance in the Morris water maze [90, 91], object place recognition [92], increased anxiety [90, 93] and deficits in social interaction [94]. Here, we compared the maturation of spontaneous excitatory and inhibitory synaptic transmission of NAc shell medium spiny neurons (MSNs) of C57BL/6J mice and a mouse model of idiopathic ASD, the BTBR T+Itpr3tf/J (BTBR) [95, 96] mouse strain. BTBR mice display face validity to all three core ASD behavioral symptoms: deficits in social communication [97,98,99], deficits in social interaction [96, 100,101,102] and repetitive behaviors [96, 101,102,103]. Importantly, when compared to the highly social C57BL/6J strain, BTBR mice display changes in ultrasonic vocalizations, a well-established metric of social communication [104, 105], starting from infancy, with a remarkably reduced repertoire of calls as early as postnatal day (P)8 [97], matching the early social deficits seen in ASD patients [6]. As other metrics of social behavior (e.g. social interaction) are not technically accessible during infancy, changes in ultrasonic vocalizations may be one of the first indicators of social deficits in early development. We found that both strains show age-dependent increases in NAc shell MSN spontaneous excitatory and inhibitory transmission, particularly at the start of the second postnatal week. Compared to C57BL/6J, BTBR mice show changes in spontaneous transmission from as early as P4, with increased excitation during the first postnatal week and increased inhibition in the first, second and fourth weeks. Additionally, infralimbic cortex (IL) optically evoked transmission onto BTBR NAc MSNs is decreased at P15 and P30 as revealed by paired pulse ratios. Based on these data, we next tested the efficacy of targeting treatment with the mTORC1 inhibitor rapamycin to the age of onset of maturational changes in NAc synaptic transmission. We found that rapamycin treatment was successful at rescuing BTBR deficits in social interaction when applied in infancy, but had no effect in adulthood, suggesting that early changes in synaptic transmission may act as a marker for a critical period maximizing the efficacy of therapeutic interventions.
Materials and methods
Animals
C57BL/6J (Jackson Laboratory) and BTBR T + Itpr3tf/J (Jackson Laboratory; referred to as BTBR for simplicity) mouse strains were bred at the University of Toronto Scarborough and kept on a 12 h light/dark cycle (lights on at 07:00 h) with access to food and water ad libitum. Date of birth was assigned P0. Approximately equal numbers of females and males were used for social behavior. Due to difficulties in balancing animals across sex, age and litter domains only males were used for slice electrophysiology experiments. We acknowledge that as a limitation of this study. Offspring were weaned with same-sex siblings on P21 (2–4 mice per cage). All animal procedures were approved by the Animal Care Committee at the University of Toronto.
Stereotaxic surgery
C57BL/6J and BTBR male mice underwent surgery at P8 or P23. Mice were given an intraperitoneal (IP) injection of ketamine (100 mg/kg) and xylazine (5 mg/kg) and placed in a stereotaxic frame (Stoelting). AAV1.CaMKIIa.hChR2(H134R)-eYFP.WPRE.hGH virus (Addgene) was infused bilaterally in the infralimbic cortex (IL; coordinates relative to bregma: P8: AP + 1.2 mm, ML ± 0.25 mm, and DV − 1.7 mm; P23: AP + 1.8 mm, ML ± 0.25 mm, and DV − 2.6 mm). Viral particles (0.2 µl/hemisphere) were delivered at a rate of 100 nl/minute by way of a motorized microsyringe. After viral infusion, the needle was left in place for five additional minutes to allow for diffusion of the virus, and then was slowly removed from the injection site. Mice were then returned to their home cages for seven days to allow for recovery and viral expression. Whole-cell patch-clamp recording experiments were performed at P15 or P30.
Slice electrophysiology
Electrophysiological recordings were obtained from male C57BL/6J and BTBR mice at different postnatal ages (P4, P6, P8, P12, P15, P21 and P30). Mice were deeply anesthetized using isoflurane and the brain was quickly removed and placed in ice-cold sucrose-based cutting solution containing (in mM): 180 sucrose, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 1 CaCl2, 2 MgCl2, 2 Na+ pyruvate and 0.4 L-Ascorbic acid with 95% O2 and 5% CO2. Coronal brain slices containing the NAc (250 μm- thick) were prepared with a vibratome (Leica VT1000S). Slices were then incubated for 30 min at 30 °C in a recovery solution composed of 50% sucrose-based cutting solution and 50% artificial cerebrospinal fluid (ACSF). The ACSF was composed of (in mM): 120 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 11 glucose, 2 CaCl2, 1 MgCl2, 2 Na+ pyruvate and 0.4 L-Ascorbic acid with 95% O2 and 5% CO2. Slices were then placed in regular ASCF for another 30 min at room temperature before electrophysiology recordings. After recovery, slices were placed in the recording chamber and perfused with 2ml/min oxygenated ACSF at room temperature for recordings.
Whole-cell patch-clamp recordings were obtained from NAc shell MSNs using borosilicate glass pipettes (3–5 ΩM; WPI) filled with intracellular solution containing (in mM): 120 Cs-methanesulfonate, 10 HEPES, 0.5 EGTA, 8 NaCl, 4 Mg-ATP, 1 QX-314, 10 Na-phosphocreatine, and 0.4 Na-GTP, pH 7.3, at 290 mOsmol. Spontaneous excitatory (sEPSCs) and inhibitory (sIPSCs) postsynaptic currents were recorded from the same cell at holding potentials of -60 mV and 0 mV, respectively, and five minutes of stable recordings were analyzed for frequency and amplitude (miniAnalysis; Synaptosoft).
For ex vivo optogenetic experiments, IL axon terminals were stimulated using TTL-pulsed microscope objective-coupled LEDs (460 nm, ∼1 mW/mm2; Prizmatix) and light-evoked EPSCs were recorded from MSNs at holding potential of -60 mV. Glutamate release probability from IL terminals to NAc shell MSNs was assessed by measuring paired-pulse ratio by dividing the amplitude of the second light-evoked EPSC by the amplitude of the first EPSC at inter-stimulus intervals of 250 and 100 ms for P15 and P30 respectively. All electrophysiology recordings were obtained in the absence of synaptic blockers.
Electrophysiology data were acquired using a MultiClamp 700B (Molecular Devices), digitized at 10 kHz using Digidata 1550 A and pClamp 10 (Molecular Devices). Recordings were low pass filtered at 4 kHz. Access resistance was regularly assessed during experiments and data were included only if the holding current was stable and access resistance varied < 20% of initial value.
Behavior
The three-chamber social interaction and social memory tests were conducted following established protocols from the Crowley group [106]. Tests took place in a 60 × 20 × 40 cm plexiglass apparatus with three 20 × 20 × 40 cm chambers interconnected by two 7 × 40 cm vertically removable plexiglass doors. Two 8.5 × 8.5 × 40 cm enclosures with three 0.3 cm vertical slits per side were placed at the lateral wall of the right and left chambers and used to hold social target animals as required in the tasks. The chamber was elevated 41 cm off the floor and a camera was mounted 75 cm above the chamber on a metal rack.
On the day prior to behavior testing, C57BL/6J and BTBR mice were socially isolated for an hour in a separate room. On the day of testing, mice were socially isolated for an hour, followed immediately by habituation to the apparatus for 10 min. Following habituation, mice underwent the three-chamber social interaction test, in which an age-, strain- and sex-matched stranger mouse was placed in one of the enclosures in a counterbalanced manner. Experimental mice were placed in the middle chamber at the start of the experiment, and opening of the doors marked the start of the social interaction test. Mice were able to freely explore the three chambers for a total of 10 min.
Social memory tests took place one hour after the social interaction test. For the social memory tests, two mice were placed in the enclosures: the stranger mouse used in the social interaction test (now familiar mouse), and a novel age, strain and sex-matched mouse, that the experimental mouse had never previously encountered (stranger mouse). The experimental mouse was placed in the middle chamber with the doors closed. Following the opening of the doors, the animal was free to explore the three chambers for 10 min.
Social behavior was conducted during the late phase of the light cycle. A subset of animals had undergone social interaction and social memory tests in the dark cycle, but similar to what has been described in the literature [107], we saw no behavioral differences between light and dark cycles for these strains, and therefore consolidated the datasets. Behavior was analyzed using ANY-maze® software, and cross validated through manual scoring by an experimenter blind to experimental conditions.
Drugs
The stock solution of rapamycin (50 mg/kg in 100% ethanol; LC laboratories; Woburn, MA; U.S.A) was stored at -80oC. Stock solution was diluted in 10% of polyethylene glycol 400 (PEG400) and 10% Tween 80 to a final concentration of 1 mg/ml in 2% ethanol and stored at -20oC [66]. Mice received an intraperitoneal injection of rapamycin (0.5 mg/kg) or vehicle (10% PEG400, 10% Tween; 1 mg/kg) [66] for five consecutive days, from either P4 to P8 or P60 to P64.
Perfusions and sectioning
BTBR mice were treated with vehicle or rapamycin from P4-P8 and were perfused at P30 for immunohistochemistry against phosphoS6. At P30, behaviorally naïve mice were injected with Avertin (250 mg/kg, i.p.) and once deeply anesthetized, were transcardially perfused with 0.1 M phosphate buffered saline (PBS), followed by 4% paraformaldehyde (PFA). The brains were then extracted and stored overnight at 4 °C in 4% PFA. The brains were sectioned using a vibratome (VT 1000, Leica) to obtain 50 μm coronal sections, which were stored in a 60% glycerol and 0.01% sodium azide in PBS solution at -20oC.
Immunohistochemistry
Sections containing the NAc shell were treated with 0.3% Triton X-100 in PBS for 15 min, followed by 10% normal goat serum in 0.1% Triton X-100 in PBS for 1 h. Sections were incubated with rabbit monoclonal anti-phospho-S6S240/244 (pS6; 1:1000; Cell Signaling, Beverly, MA, RRID:AB 10,694,233) for 48 h, followed by secondary antibody incubation with Alexa Fluor 594-conjugated goat anti-rabbit IgG (1:500; Jackson Immunoresearch Laboratories) and Hoechst dye (1:1000; Thermo Fisher Scientific) for 90 min. Sections were mounted on glass slides (Thermo Fisher Scientific) with Permafluor aqueous mounting medium (Thermo Fisher Scientific) and coverslipped for storage at 4 °C.
Image acquisition and quantification
Image acquisition was performed using a Nikon Eclipse Ni-U epiluorescence microscope. Immunofluorescence was visualized with an LED illumination system (X-Cite 120 LED Boost, Excelitas Technologies) and captured with a Nikon DS-Qi2 digital camera. All images were acquired at 10x magnification using Plan-Apochromat differential interference contrast (DIC) N1. When brightness and/or contrast adjustments were made in a figure, these changes were made equally to all photomicrographs.
Cell counts were done manually using Fiji/ImageJ software (v. 1.53e) by an experimenter blind to treatment conditions. Three to five sections per subject containing the NAc shell were used for cell counts. For each section, the integrated density in the NAc shell was acquired, corrected for background, and averaged to obtain the mean integrated density per animal.
Statistical analysis
Data are presented as mean ± standard error of the mean. All statistical analyses were performed in Graphpad Prism® version 9. Potential sex differences were first assessed using a two- or three-way, repeated measures ANOVA and given the absence of effects, data were pooled for subsequent analyses. Exploration time in all chambers in the social interaction and social memory tests was analyzed by two-way, repeated-measures ANOVA followed by Sidak’s post-hoc tests. Distance travelled was analyzed using unpaired two-tailed t tests. For electrophysiology experiments and immunohistochemistry, paired t-tests and Wilcoxon signed-rank tests were used for within-group comparisons, unpaired t-tests and one- or two-way repeated measures ANOVA followed by Tukey’s and Sidak’s post-hoc tests respectively were used for normally distributed data and Mann-Whitney Rank Sum test when data did not pass normality. For all analyses, p < 0.05 was considered significant.
Results
Maturation of spontaneous excitatory currents of medium spiny neurons of the nucleus accumbens shell of C57BL/6J and BTBR mice
To examine the maturation of spontaneous excitatory and inhibitory synaptic transmission of the NAc shell in C57BL/6J and BTBR mice, whole cell patch-clamp recordings were obtained from visually identified MSNs during the first postnatal weeks, comprising infant (P4, P6, P8, P12), juvenile (P15, P21) and adolescent (P30) [108] stages similar to our previous work [109] (Fig. 1A). We sampled NAc shell MSN maturation at multiple ages during infancy due to the clinical relevance of that developmental stage for the clinical manifestations of ASD [12], and to match the timing of previously reported ultrasonic vocalization differences between C57BL/6J and BTBR mice [97]. In MSNs from both strains, spontaneous excitatory postsynaptic currents (sEPSCs) were observed as early as P4 and developed gradually during the first postnatal month (Fig. 1). In C57BL/6J mice, sEPSC frequency showed a significant increase at P15 and P21 with a further increase at P30 (Fig. 1B-C, one way ANOVA, F(6, 64) = 25.40 p < 0.0001, Tukey’s multiple comparison tests: P4 vs. P15, p = 0.02; P4 vs. P21, p = 0.0003; P4 vs. P30, p < 0.0001; P6 vs. P15, p = 0.04; P6 vs. P21, p = 0.0004; P6 vs. P30, p < 0.0001; P8 vs. P21, p = 0.0017; P8 vs. P30, p < 0.0001; P12 vs. P30, p < 0.0001; P15 vs. P30, p < 0.0001; P21 vs. P30, p < 0.0001). However, in BTBR mice, a marked increase in sEPSC frequency was observed at P12, with a further increase from P21 (Fig. 1B, D, one way ANOVA, F(6, 66) = 14.86 p < 0.0001, Tukey’s multiple comparison tests: P4 vs. P12, p = 0.007; P4 vs. 21 and P30, p < 0.0001; P6 vs. P21, p = 0.0001; P6 vs. P30, p < 0.0001; P8 vs. P21, p = 0.0007; P8 vs. P30, p < 0.0001; P12 vs. P30, p = 0.0007; P15 vs. P21, p = 0.004; P15 vs. P30, p < 0.0001). Interestingly, sEPSC amplitude displayed a distinct maturation profile. While C57BL/6J mice showed increased sEPSC amplitude between P6-P8 and P15 and P30 (Fig. 1E, one way ANOVA, F(6, 66) = 7.429 p < 0.0001, Tukey’s multiple comparison tests: P6 vs. P15, p = 0.0004; P6 vs. P30, p = 0.0005; P8 vs. P15, p = 0.0003; P8 vs. P30, p = 0.0003), the amplitude of sEPSCs in MSNs from BTBR mice was decreased at P8, P12 and P21 relative to P4 (Fig. 1F, one way ANOVA, F(6, 70) = 3.480 p = 0.0046, Tukey’s multiple comparison tests: P4 vs. P8, p = 0.002; P4 vs. P12, p = 0.03; P4 vs. P21, p = 0.01). Furthermore, sEPSC time constant displayed a marked increase during the second (P12, P15) and third (P21) postnatal weeks in C57BL/6J mice and similarly during the second (P8) and third (P21) postnatal weeks in BTBR mice (Fig. 1G, one way ANOVA, F(6, 67) = 5.602 p < 0.0001, P4 vs. P12, p = 0.006; P4 vs. P15, p = 0.0006; P4 vs. P21, p < 0.0001; Fig. 1H, one way ANOVA, F(6, 70) = 3.121 p = 0.0091; P4 vs. P8, p = 0.04; P4 vs. P21, p = 0.02). Overall, these data show that MSNs from BTBR mice display slightly earlier increases in sEPSC frequency and distinct age-dependent changes in sEPSC amplitude, but broadly equivalent timeline of maturation of sEPSC kinetics.

Maturation of excitatory synaptic excitation onto medium spiny neurons of the nucleus accumbens shell of C57BL/6J and BTBR mice. A. Schematic representation of the whole-cell patch-clamp recording protocol from an MSN of the NAc shell. B. Representative sEPSC traces obtained from MSNs at different postnatal ages from C57BL/6J mice (black) or BTBR mice (blue). C-D. Maturation of sEPSC frequency. C57BL/6J (C) and BTBR (D) mice show a progressive increase in sEPSC frequency across the first postnatal month. E-F. Maturation of sEPSC amplitude. C57BL/6J mice (E) show an age-dependent increase in sEPSC amplitude, whereas sEPSC amplitude decreases between P4 and P8-12 and P21 in BTBR mice (F). G-H. Maturation of sEPSC tau. Both C57BL6/J (G) and BTBR mice (H) show age-dependent increases in sEPSC tau. I-J. Direct comparison of age-dependent changes in NAc MSN sEPSC frequency and amplitude between C57BL/6J (black circles) and BTBR (blue circles) strains. Compared to C57BL/6J mice, BTBR mice showed reduced sEPSC frequency at P30 (I) and increased sEPSC amplitude at P4 and P6 (J). * p < 0.05. C57BL/6J: P4 (9 cells/4 mice), P6 (9 cells/3 mice), P8 (15 cells/4 mice), P12 (9 cells/ 3 mice), P15 (11 cells/ 4 mice), P21 (9 cells/ 3 mice), P30 (9 cells/ 3 mice); BTBR: P4 (10 cells/3 mice), P6 (10 cells/3 mice), P8 (18 cells/5 mice), P12 (10 cells/ 3 mice), P15 (8 cells/ 3 mice), P21 (8 cells/ 3 mice), P30 (9 cells/ 3 mice)
To better understand strain differences in NAc shell MSN spontaneous transmission in early life, we directly compared MSNs sEPSC parameters between C57BL/6J and BTBR mice across ages. We found that BTBR mice exhibited reduced sEPSC frequency at P30 (Fig. 1I; two way ANOVA, significant effects of age F(6, 130) = 39.16, p < 0.0001, strain F(1, 130) = 5.65, p = 0.02, and age x strain interaction F(6, 130) = 4.9, p = 0.0002; Sidak’s multiple comparisons P30, p < 0.0001) but increased sEPSC amplitude at P4 and P6 relative to C57BL/6J mice (Fig. 1J; two way ANOVA, significant effects of age F(1, 136) = 6.27, p < 0.0001, and age x strain interaction F(6, 136) = 4.36, p = 0.0005; Sidak’s multiple comparisons P4, p = 0.01; P6, p = 0.04).
We then examined the maturation of inhibitory inputs onto MSNs in both strains. Spontaneous inhibitory postsynaptic current (sIPSC) frequency increased significantly during the second postnatal week for both strains and plateaued from P15 (Fig. 2A-B, C57BL/6J: one way ANOVA, F(6, 61) = 10.07 p < 0.0001, Tukey’s multiple comparison tests: P4 vs. P15, p < 0.0001; P4 vs. P21, p = < 0.0001; P4 vs. P30, p = 0.004; P6 vs. P15, p = 0.0003; P6 vs. P21, p = 0.0003; P6 vs. P30, p = 0.01; P8 vs. P15, p = 0.0004; P8 vs. P21, p = 0.0005; P8 vs. P30, p = 0.03; Fig. 2A,C, BTBR, one way ANOVA, F(6, 61) = 5.73 p < 0.0001, Tukey’s multiple comparison tests: P4 vs. P15, p = 0.0031, P4 vs. P30, p = 0.0053; P6 vs. P15, p = 0.007; P6 vs. P30, p = 0.011; P12 vs. P15, p = 0.017, P12 vs. P30, p = 0.025). In contrast, while sIPSC amplitude increased significantly and plateaued from P15 in C57BL/6J mice (Fig. 2D, C57BL/6J: one way ANOVA, F(6, 61) = 7.931 p < 0.0001; Tukey’s multiple comparison tests: P4 vs. P15, p = 0.0031; P6 vs. P15, p < 0.0001; P6 vs. P21, p = 0.0002; P8 vs. P15, p = 0.0009; P8 vs. P21, p = 0.033; P12 vs. P15, p = 0.041), BTBR mice only showed a slight increase at P21 (Fig. 2E, BTBR: one way ANOVA F(6, 62) = 4.04, p = 0.0018; Tukey’s multiple comparison tests: P8 vs. P21, p = 0.023; P15 vs. P21, p = 0.044). Interestingly, sIPSC time constant displayed a distinct maturation profile between MSNs from C57BL/6J and BTBR mice. While in C57BL/6J mice sIPSC time constant was stable across all ages tested (Fig. 2F; one way ANOVA, F(6, 60) = 2.186 p = 0.0567), sIPSC tau showed a significant increase between P4 and P30 in MSNs from BTBR mice (Fig. 2G; one way ANOVA, F(6, 65) = 3.481 p = 0.0048, Tukey’s multiple comparison tests: P4 vs. P30, p = 0.01). Overall, these data show broadly synchronous developmental changes in sIPSC frequency between strains, with BTBR mice showing slightly delayed increases in sIPSC amplitude and distinct maturation of sIPSC decay time.

Maturation of inhibitory synaptic excitation onto medium spiny neurons of the nucleus accumbens shell of C57BL/6J and BTBR mice. A. Representative traces of sIPSCs obtained from MSNs in the NAc shell of C57BL/6J (black) and BTBR mice (blue). B-C. Maturation of sIPSC frequency. Both C57BL/6J (B) and BTBR mice (C) showed an increase in sIPSC frequency across the first postnatal month. D-E. Maturation of sIPSC amplitude. C57BL/6J mice (D) showed a slightly earlier (P15) increase in sEPSC amplitude compared to BTBR mice (E; P21). F-G. Maturation of sIPSC tau. C57BL/6J mice (F) showed stable sIPSC tau from P4, whereas BTBR mice (G) showed an increase in tau between P4 and P30. H-I. Direct comparison of age-dependent changes in NAc MSN sIPSC frequency and amplitude between C57BL/6J (black circles) and BTBR (blue circles) strains. Compared to C57BL/6J mice, BTBR mice showed increased sIPSC frequency at P8 (H) and increased sIPSC amplitude at P6, P12 and P30 (I). *p < 0.05. C57BL/6J: P4 (9 cells/4 mice), P6 (8 cells/3 mice), P8 (14 cells/4 mice), P12 (10 cells/ 3 mice), P15 (11 cells/ 4 mice), P21 (8 cells/ 3 mice), P30 (8 cells/ 3 mice); BTBR: P4 (10 cells/3 mice), P6 (9 cells/3 mice), P8 (16 cells/5 mice), P12 (8 cells/ 3 mice), P15 (10 cells/ 3 mice), P21 (6 cells/ 3 mice), P30 (9 cells/ 3 mice)
Comparison of spontaneous inhibitory synaptic transmission between strains revealed that BTBR mice showed higher sIPSC frequency at P8 and sIPSCs amplitude at P6, P12 and P30 compared to C57BL/6J mice (Fig. 2H-I; sIPSC frequency: two way ANOVA, significant effects of age F(6,122) = 12.90, p < 0.0001, and strain F(6, 122) = 14.2, p = 0.0003; Sidak’s multiple comparisons P8, p = 0.02; sIPSC amplitude: two way ANOVA, significant effects of age F(6, 123) = 7.08, p < 0.0001, strain F(6, 123) = 4.55, p < 0.0001, and age x strain interaction F(6, 123) = 4.55, p = 0.0003; Sidak’s multiple comparisons P6, p = 0.004, P12, p = 0.01, P30, p = 0.009). Consistent with this, comparison of within-cell differences between the frequency of spontaneous excitation and inhibition showed a shift toward synaptic inhibition at P30 in BTBR mice relative to age-matched C57BL/6J mice (Fig. 3; two way ANOVA, significant effects of age F(6, 120) = 5.51, p < 0.0001, strain F(6, 120) = 23.47, p < 0.0001, and age x strain interaction F(6, 120) = 3.74, p = 0.0019; Sidak’s multiple comparisons P30, p < 0.0001). In conclusion, these data show that MSNs from BTBR mice display higher excitatory inputs during the first postnatal week, which is reduced by P30. Inhibitory synaptic inputs onto MSNs from BTBR mice were stronger during the first, second and fourth postnatal weeks than onto MSNs from C57BL/6J mice, suggesting differences in the maturation of excitatory and inhibitory transmission in MSNs of BTBR mice. At P30, C57BL/6J and BTBR mice show opposite profiles of excitation-inhibition balance, with predominant excitation over inhibition for C57BL/6J and inhibition over excitation for BTBR mice. The temporal profile of changes in NAc synaptic transmission in BTBR mice coincides with the onset of social deficits in this strain [97], suggesting these electrophysiological differences might contribute to the early changes in behavior.

Comparison of the developmental profile of spontaneous excitatory and inhibitory frequency between C57BL/6J and BTBR mice. Within-cell comparison of the difference in the frequency of spontaneous excitation and inhibition (E-I frequency) showed a shift toward increased inhibition in P30 BTBR mice relative to C57BL/6J mice. *p < 0.05. sEPSC: C57BL/6J: P4 (9 cells/4 mice), P6 (9 cells/3 mice), P8 (15 cells/4 mice), P12 (9 cells/ 3 mice), P15 (11 cells/ 4 mice), P21 (9 cells/ 3 mice), P30 (9 cells/ 3 mice); BTBR: P4 (10 cells/3 mice), P6 (10 cells/3 mice), P8 (18 cells/5 mice), P12 (10 cells/ 3 mice), P15 (8 cells/ 3 mice), P21 (8 cells/ 3 mice), P30 (9 cells/ 3 mice)
Evidence suggests specialization in the contribution of NAc input and output connections to social processing [20, 110,111,112], with optogenetic manipulation of medial prefrontal cortex (PFC)-NAc projections in particular modulating different aspects of social behavior [23, 27, 30, 113]. To test whether PFC-NAc transmission might be altered in BTBR mice, we injected AAV-ChR2 virus into the infralimbic cortex (IL) subdivision of the PFC of C57BL/6J and BTBR mice one week prior to conducting patch clamp recordings from NAc shell MSNs at P15 and P30 (Fig. 4A). IL-expressing ChR2 terminals onto MSNs were stimulated using a brief light-pulse (5ms) through the objective, and presynaptic transmission at IL-NAc synapses was examined by measuring paired-pulse ratios (PPR). Light-evoked EPSCs from MSNs of BTBR mice showed higher PPR at both P15 and P30 compared to C57BL/6J mice, suggesting a possible reduction in glutamate release probability at IL-NAc synapses (Fig. 4B-C; unpaired t tests, P15: t = 2.13, p = 0.04; P30: t = 5.91, p < 0.0001).

BTBR mice show decreased PFC-NAc glutamate release probability in early life. A. Schematic of experimental design. B. Top: P15, representative traces of light-evoked EPSC during paired-pulse stimulation (250 ms) from MSNs of C57BL/6J mice (black) and BTBR mice (blue). Bottom: summary plot showing increased PPR in MSNs from BTBR mice compared to MSNs from C57BL/6J mice. C. Top: P30, representative traces of light-evoked EPSC during paired-pulse stimulation (100 ms) from MSNs of C57BL/6J mice (black) and BTBR mice (blue). Bottom right: summary graph showing increased PPR in MSNs from BTBR mice compared to those from C57BL/6J mice. * p < 0.05. C57BL/6J: P15 (12 cells/ 6 mice), P30 (12 cells/ 4 mice); BTBR: P15 (9 cells/ 4 mice), P30 (7 cells/ 4 mice)
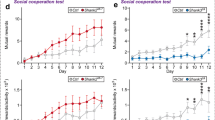
Given the temporal coincidence of alterations in NAc synaptic transmission and behavior in the BTBR strain, targeting intervention strategies to this window may improve treatment outcomes. Informed by our electrophysiological data showing changes in NAc spontaneous transmission as early as P4 (Figs. 1, 2 and 3), we decided to target a well-established rescue strategy for animal models of ASD, the mTORC1 antagonist rapamycin [114], to early development, starting at P4. We first confirmed that BTBR mice displayed social interaction deficits in our lab by testing them on a three-chamber social interaction test, followed by a social memory test. We tested animals during adolescence at P30, a crucial time in social development [115]. In the social interaction test, BTBR mice spent less time with the social target and more time in the empty compartment, compared to C57BL/6J mice (Fig. 5A-B; two-way RM ANOVA, significant effect of chamber F(1.536, 90.61) = 38.79, p < 0.0001 and chamber x strain interaction F(2, 118) = 16.71, p < 0.0001; Sidak’s multiple comparisons test, empty p = 0.0074, middle p = 0.0030, and social p = 0.0006 chambers).

BTBR mice show social interaction deficits but no change in social memory at P30. C57BL/6J and BTBR underwent the three-chamber social interaction test followed by a social memory test at P30. A. Schematic of behavioral testing and heatmaps of time spent in each chamber during testing. B. Three chamber social interaction test. Time spent in the chamber with an empty enclosure (empty), middle chamber and chamber with an age-, strain- and sex-matched stranger mouse in the enclosure (social). C57BL/6J mice spent significantly more time exploring the social chamber (Sidak’s post hoc test, Empty vs. Middle t32 = 10.63 p < 0.0001, Empty vs. Social t32 = 7.365 p < 0.0001, Middle vs. Social t32 = 18.48 p < 0.0001), whereas BTBR mice showed no significant preference for the social chamber (Sidak’s post hoc test, Empty vs. Middle t27 = 4.843 p = 0.0001, Empty vs. Social t27 = 1.76 p = 0.2458, Middle vs. Social t27 = 2.045 p = 0.1446). C57BL/6J, n = 33 (17 females, 16 males); BTBR, n = 28 (17 females, 11 males). C. Schematic of social memory behavioral testing and heatmaps of time spent in each chamber during testing. D. Social memory test. Time spent exploring the chamber with the same mouse from the previous test (familiar), middle chamber, and chamber with a novel, age, strain and sex-matched stranger mouse in the enclosure (stranger). Both C57BL/6J and BTBR mice showed a preference for the stranger mouse (Sidak’s post hoc test, C57BL/6J: Stranger vs. Middle t25 = 9.019 p < 0.0001, Stranger vs. Familiar t25 = 3.366 p = 0.0074, Middle vs. Familiar t25 = 11.14 p < 0.0001; BTBR: Stranger vs. Middle t30 = 5.282 p < 0.0001, Stranger vs. Familiar t30 = 2.533 p = 0.0494, Middle vs. Familiar t30 = 2.237 p = 0.0955. C57BL/6J, n = 26 (13 females, 13 males); BTBR, n = 31 (14 females, 17 males). No sex differences were found for either strain in the three-chamber social interaction test (p > 0.64) or social memory test (p > 0.11). Male individual datapoints are depicted as squares, and female datapoints as circles for transparency. *p < 0.05
While BTBR social interaction deficits consistent with ours have been widely reported [96, 116], social memory deficits in the BTBR strain have a more conflicting literature [77, 117, 118]. We found no differences in social memory, i.e. preference for the novel, unfamiliar mouse, between C57BL/6J and BTBR mice (Fig. 5C-D; two-way RM ANOVA, significant effect of chamber F(2, 110) = 39.63, p < 0.0001 and strain F(1, 55) = 10.23 p = 0.0023; Sidak’s multiple comparisons test showed significant differences between strains only in the middle chamber t = 2.775 p = 0.0268; p > 0.84 for other chambers). Importantly, although our behavioral data showed no sex differences in social interaction (p > 0.64) or social memory (p > 0.11), replicating a widely reported absence of sex differences in social behavior in these strains [96, 101, 102, 106], we presently cannot exclude the possibility of sex differences in the underlying maturation of NAc synaptic transmission.
To test whether the increased synaptic excitation and inhibition onto MSNs from BTBR mice starting at the first postnatal week could signal the start of a sensitive period in the BTBR strain, we targeted rapamycin treatment, a drug known to rescue social deficits in other mouse models of ASD [65, 66], from P4 to P8, and tested animals for social preference at P30 (Fig. 6A). Rapamycin treatment led to a significant increase in time spent in the social chamber (Fig. 6B; two-way RM ANOVA, significant effect of chamber F(2, 48) = 22.42, p < 0.0001, and chamber x treatment interaction F(2, 48) = 9.436, p < 0.0003; Sidak’s multiple comparisons test show significant differences between vehicle and rapamycin treatment in the empty p = 0.0293 and social p = 0.0002 chambers), indicating a rescue of social preference in BTBR mice treated with rapamycin from P4-P8. We saw no difference in distance travelled between vehicle- and rapamycin-treated groups (Fig. 6C; unpaired t test, t19 = 0.2272, p = 0.8227), suggesting that the social interaction effects did not stem from changes in motor activity. These data indicate that early rapamycin treatment was able to rescue BTBR social deficits at adolescence.

Rapamycin treatment in infancy, but not adulthood, rescues BTBR mice social interaction deficits. A-C. BTBR mice were treated with rapamycin or vehicle from P4-P8 and tested in the three-chamber social interaction test at P30. A. Schematic of experimental design and heatmaps of time spent in each chamber during testing. B. Three chamber social interaction test. Time spent in the chamber with an empty enclosure (empty), middle chamber and chamber with an age-, strain- and sex-matched stranger mouse in the enclosure (social). Rapamycin-treated mice showed a preference for the chamber with the social target (Sidak’s post hoc test, Empty vs. Middle t48 = 3.334 p = 0.005, Empty vs. Social t48 = 3.799 p = 0.0012, Middle vs. Social t48 = 7.132 p < 0.0001), which is absent in vehicle-treated BTBR mice (Sidak’s post hoc test, Empty vs. Middle t48 = 3.937 p = 0.0008, Empty vs. Social t48 = 1.982 p = 0.1513, Middle vs. Social t48 = 1.954 p = 0.1601). C. Distance travelled in the social interaction test. There was no difference between vehicle- and rapamycin-treated mice. Vehicle-treated BTBR (VEH), n = 12 (7 females, 5 males); Rapamycin-treated BTBR (RAPA), n = 14 (8 females, 6 males). No sex differences were found for either group (p > 0.516). Male individual datapoints are depicted as squares, and female datapoints as circles for transparency. D-F. BTBR mice were treated with rapamycin or vehicle from P60-P64 and tested in the three-chamber social interaction test at P86. D. Schematic of experimental design and heatmaps of time spent in each chamber during testing. E. Three chamber social interaction test. Time spent in the chamber with an empty enclosure (empty), middle chamber and chamber with an age, strain and sex-matched stranger mouse in the enclosure (social). Neither vehicle or rapamycin treated mice showed a preference for the chamber with the social target over the empty chamber (VEH: Sidak’s post hoc test, Empty vs. Middle t34 = 2.214 p = 0.097, Empty vs. Social t34 = 2.012 p = 0.148, Middle vs. Social t34 = 4.22 p = 0.0005; RAPA: Empty vs. Middle t34 = 2.72 p = 0.030, Empty vs. Social t34 = 0.557 p = 0.93, Middle vs. Social t34 = 2.16 p = 0.11). F. Distance travelled in the social interaction test. There was no difference between vehicle- and rapamycin-treated mice. Vehicle-treated BTBR (VEH), n = 9 (5 females, 4 males); Rapamycin-treated BTBR (RAPA), n = 10 (5 females, 5 males). No sex differences were found for either group (p > 0.1185). Male individual datapoints are depicted as squares, and female datapoints as circles for transparency. G-H. Rapamycin treatment decreases phosphoS6 levels in the NAc of BTBR mice at P30. BTBR mice were treated with rapamycin or vehicle from P4-P8, kept in their homecage and then perfused at P30 for brain processing and immunohistochemistry against phosphoS6. G. Representative images of phosphoS6 staining in the NAc shell. H. Quantification of phosphoS6 intensity shows that rapamycin treatment reduced phosphoS6 levels at P30. Scale bar = 250 μm, n = 4 vehicle, 4 BTBR. *p < 0.05
A study by Burket and colleagues had shown that a four-day rapamycin treatment starting at P28 also rescued social deficits in BTBR mice tested 1 h after the last injection [84]. To test whether the efficacy of the rapamycin rescue was tied to its timing during early life, we injected an additional cohort of BTBR mice with the same rapamycin regimen and interval between treatment and testing, but starting at P60 (Fig. 6D). We found that rapamycin treatment starting at early adulthood did not affect the time spent in the social chamber (Fig. 6E; two-way RM ANOVA, significant effect of chamber F(2, 34) = 11.32, p = 0.0002; Sidak’s multiple comparisons test show no significant differences between vehicle and rapamycin treatment in any of the chambers) or distance travelled (Fig. 6F; unpaired t test, t17 = 0.052, p = 0.9591). As an additional validation, we confirmed that early rapamycin treatment decreased phosphorylation of S6 ribosomal protein, a commonly used measure of mTOR pathway activity [119], in P30 BTBR mice (Fig. 6G-H; unpaired t test, t6 = 3.2, p = 0.01). Overall, these data indicate that P60 rapamycin treatment did not affect social interaction in adulthood, suggesting that early intervention with rapamycin is necessary for improving social interaction in the BTBR strain.
Discussion
In this study, we compared the maturation of NAc shell MSN spontaneous synaptic transmission in C57BL/6J and BTBR strains from infancy to adolescence. In both strains, MSNs displayed excitatory and inhibitory spontaneous currents by P4, suggesting the presence of functional excitatory and inhibitory synapses in the NAc early during development. Excitatory and inhibitory inputs onto MSNs developed gradually up to P12 and underwent a marked maturation by P15, when levels plateaued until P30. These data suggest that synaptic inputs onto NAc MSNs establish early during development with a slow maturation during the first 10–12 postnatal days, followed by a potentiation of synaptic inputs onto MSNs by P15 that persists until P30. This is consistent with other reports showing stable NAc paired pulse ratio (PPR) of AMPA-mediated EPSCs from juvenility [50], and changes in AMPAR- and NMDAR-mediated EPSCs up to the second postnatal week (but decrease of NMDA-EPSC later during development) [52].
Overall, we found age-dependent changes in the maturation of spontaneous excitatory and inhibitory transmission within and between strains, with BTBR mice showing increased excitation during the first postnatal week, which decreased by P30, as well as increased spontaneous inhibition across early development. Additionally, BTBR mice display IL-NAc paired-pulse facilitation at P15 and P30 compared to C57BL/6J mice, suggesting reduced presynaptic glutamate release from IL terminals onto MSNs in BTBR mice. The difference in PPR between C57BL/6J and BTBR mice at P15 and P30 was observed using long (250 ms) and short (100 ms) interstimulus intervals, respectively, which might be influenced by developmental differences in presynaptic short-term plasticity, as reported previously [120,121,122].
In the Shank3B−/− mouse model of ASD, an accelerated maturation of striatal spiny projecting neurons (SPNs) of the dorsomedial striatum (DMS) was observed during early developmental stages (third and fourth postnatal weeks), followed by a decrease in corticostriatal activity during adulthood (P60) [123, 124] - a deficit that was attributed to an overactivation of cortical circuits. Moreover, the authors found that increasing anterior cingulate cortex (ACC) synaptic drive to SPNs of DMS at P8 significantly increased SPN mEPSC frequency, whereas increasing ACC activity from P15 and older reduced SPN mEPSC frequency [124], suggesting a developmental switch in the effect of cortical hyperactivity on SPN synaptic transmission. While our study showed that MSNs from BTBR mice receive weaker synaptic inputs from IL at P15 and P30 relative to MSNs from C57BL/6J mice, it remains unknown whether a similar switch occurs in their response to IL hypofunction.
Given the developmental timeline of NAc shell MSN spontaneous transmission, we hypothesized that early postnatal development might constitute a critical period for the efficacy of rescue manipulations correlating with improved outcomes. To test this, we subjected BTBR mice to rapamycin treatment in infancy (P4-P8) or adulthood (P60-64) and tested its effect on social preference in a three-chamber social interaction test. We found that rapamycin treatment in infancy, but not adulthood, reversed the social interaction deficits in BTBR mice. Our data point to a critical period for the efficacy of rapamycin treatment in BTBR mice from P4-P8 which closes by P60. Work by Burket and colleagues showed that rapamycin rescues BTBR social interaction deficits when administered between P28-P31, with animals tested one hour after the last injection [84]. While this study did not probe the long-term effects of rapamycin treatment on social behavior, it reinforces the value of rapamycin as an effective rescue intervention in BTBR mice. Furthermore, it points to a longer window of efficacy for rapamycin treatment in this strain, likely spanning from infancy to early adolescence. Consistent with this, we found differences in NAc spontaneous transmission between BTBR and C57BL/6J mice up to P30. In the rat, changes in NAc intrinsic excitability and excitatory transmission predominantly occur within the first three postnatal weeks, reaching adult levels by P35 or earlier [50, 51]. A study by Zhang and Warren saw changes in rat NAc excitatory transmission stabilizing by P15, with the amplitude of NMDA responses peaking from P9-12, which they propose is a critical period for this brain region [52]. Additionally, Liu and colleagues reported higher sEPSC frequency and amplitude, as well as lower halfwidth, rise and decay time in the NAc shell of P21-P28 rats compared to adults [125], further pointing to the first postnatal month as a developmental window of heightened changes in NAc synaptic transmission.
Synaptic pruning is a key mechanism during postnatal development, whereby synapses are eliminated by autophagy to ensure the proper wiring of neuronal networks [126]. This mechanism is regulated by mTORC1, and hyperactivation of mTORC1 during early postnatal development leads to a blockade of autophagy, synapse maturation deficits, as well as synaptic and behavioral abnormalities [127, 128]. Therefore, rapamycin-induced decrease of mTORC1 activity during early postnatal development in BTBR mice may enable normative synaptic pruning necessary to prevent synaptic and behavioral abnormalities. Alternatively, rapamycin might modulate excitatory drive onto MSNs, a mechanism known to regulate social behavior deficits in Shank3B−/− mice [123, 124].
Nardou and colleagues had described a critical period for social reward learning in the NAc [129], which peaks at P42, and closes by P98 [129]. This learning window coincides with age-dependent changes in oxytocin-induced long-term synaptic depression in the NAc [16, 129]. Interestingly, NAc oxytocin signaling also modulates social approach [21], suggesting its signaling and developmental time course might also influence social interaction. Another NAc microcircuit regulator, µ-opioid receptor (MOR), modulates social interaction and NAc synaptic transmission [130], as well as social behavior in juveniles such as play behavior [131, 132] and social preference [133]. Furthermore, mice lacking a functional MOR gene (Oprm1−/− mice), an ASD mouse model, display increased oxytocin receptor expression in the NAc, with their social deficits reversed by oxytocin treatment [134]. NAc MOR levels [135, 136] and function [137] also change across development, suggesting that the interplay between NAc MOR and oxytocin might contribute to the shaping of social behavior in early life.
While a wide array of acute or short-term interventions rescue social interaction deficits in adult [77, 116, 138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161] and young [162,163,164,165,166,167] BTBR mice, very few probed their long-term efficacy. Pharmacological rescue interventions yielding long-term rescue of BTBR social deficits predominantly target mice up to the third postnatal week [168,169,170,171,172], with effects lasting until adulthood. To our knowledge, no other study compared the efficacy of a rescue intervention across different ages in BTBR mice. While we did not see an effect of rapamycin treatment in adult BTBR mice, this has been reported in other ASD mouse models [63,64,65]. Unlike our long-term assessment of the effects of rapamycin treatment, rapamycin rescue of adult behavior in those studies occurred within short delays from treatment [63,64,65], with one study seeing reversal of rapamycin effects within one week [65], supporting the possibility that early rapamycin intervention might increase the persistence of the rescue effects. These studies also used a higher rapamycin dose and different treatment regimens [63,64,65] to ours (and different ASD mouse models), raising the possibility that a variation of our treatment design could still rescue BTBR social deficits in adulthood. Although we cannot exclude that possibility, the absence of a rapamycin effect in our adult treatment cohort still points to early life as a period of increased efficacy for rapamycin treatment in rescuing social interaction deficits in BTBR mice.
Deletion of Shank3 in the NAc at a similar time period to ours (< P6) triggered social deficits in adulthood that were not replicated when the manipulation started at P90 [41], corroborating the existence of a critical period for NAc-associated behavior and susceptibility to long-lasting interventions. Similarly, in the Ube3amStop/p+ mouse model, increased MSN excitability and decreased excitatory input are reversed by UBE3A reinstatement at P21 but not at P70 [173]. Interestingly, another study showed that rapamycin administration to Tsc1 mutant mice between P1 and P13 normalized dendritic arborization but not spine maturation, while treatment between P15 and P27 improved spine maturation but not dendritic arborization, suggesting different critical windows for correcting different aspects of cellular abnormalities [174]. These lines of evidence further support the existence of a critical period for striatal circuits during early postnatal development, and the notion that targeting therapeutic interventions to this time window could maximize their effects. Notably, the NAc is not the only region contributing to the regulation of social behavior in the BTBR strain, as seen with adult manipulations of cerebellum [175] and PFC [176], as well as changes in spontaneous transmission in hippocampus [154]. While we saw decreased presynaptic efficacy in the PFC-NAc pathway of BTBR mice, its precise contribution to behavioral deficits in this strain remains unclear.
Our data delineate the maturation of spontaneous synaptic transmission in the NAc shell across early postnatal development, uncovering distinct early maturation signatures between C57BL/6J and BTBR mice, strains that display distinct patterns of NAc-mediated social behavior. These synaptic changes coincide with key developmental changes in social communication, play, affiliative and social approach behavior [177,178,179,180,181], and abridge a period in which environmental conditions and stress bear a disproportionate impact in shaping future social behavior [182,183,184,185,186]. Accordingly, we revealed a restricted time window for the rescue of social interaction deficits in BTBR mice by rapamycin treatment. These findings emphasize the importance of matching experimental conditions to clinically relevant timelines, and highlight the power of leveraging research in early development toward precisely targeted interventions that maximize positive outcomes.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information file.
Abbreviations
- AMPA:
-
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ACC:
-
Anterior cingulate cortex
- ASD:
-
Autism spectrum disorder
- DMS:
-
Dorsomedial striatum
- IL:
-
Infralimbic cortex
- MOR:
-
µ-opioid receptor
- MSN:
-
Medium spiny neuron
- mTOR:
-
Mammalian target of rapamycin
- NAc:
-
Nucleus accumbens
- NMDA:
-
N-methyl-D-aspartate
- PFC:
-
Medial prefrontal cortex
- sEPSC:
-
Spontaneous excitatory postsynaptic current
- sIPSC:
-
Spontaneous inhibitory postsynaptic current
- SPN:
-
Striatal spiny projecting neurons
References
Ko J. Neuroanatomical substrates of Rodent Social Behavior: the medial prefrontal cortex and its projection patterns. Front Neural Circuits. 2017;11:41.
Robinson GE, Fernald RD, Clayton DF. Genes and social behavior. Sci (80-). 2008;322:896–900.
Chen P, Hong W. Neural Circuit Mechanisms of Social Behavior Neuron. 2018;98:16–30.
Rutter M, et al. Quasi-autistic patterns following severe early global privation. English and Romanian Adoptees (ERA) study team. J Child Psychol Psychiatry. 1999;40:537–49.
Volkmar FR, Pauls D, Autism. Lancet. 2003;362:1133–41.
Ozonoff S, Heung K, Byrd R, Hansen R. Hertz-Picciotto, I. The onset of Autism: patterns of Symptom Emergence in the First Years of Life. Autism Res. 2008;1:320–8.
Garber K, Visootsak J, Warren J. Fragile X syndrome. Eur J Hum Genet. 2008;1:666–72.
Penn DL, Corrigan PW, Bentall RP, Racenstein JM, Newman L. Social Cognition in Schizophrenia. Psychol Bull. 1997;121:114–32.
Levy SE, Mandell DS, Schultz RT, Autism. Lancet. 2009;374:1627–38.
Lai M, Lombardo MV. Baron-cohen, S. Autism. Lancet. 2014;383:896–910.
Constantino JN, Charman T. Diagnosis of autism spectrum disorder: reconciling the syndrome, its diverse origins, and variation in expression. Lancet Neurol. 2016;15:279–91.
Courchesne E, et al. Mapping early Brain Development in Autism. Neuron. 2007;56:399–413.
Insel TR, Fernald RD. How the brain processes social information: searching for the Social Brain. Annu Rev Neurosci. 2004;27:697–722.
Beckstead RM. An Autoradiographic Examination of Corticocortical and Subcortical Projections of the Mediodorsal-Projection (Prefrontal) Cortex in the Rat. J Comp Neurol 43–62 (1979).
Oades RD, Halliday GM. Ventral tegmental (A10) system: neurobiology. 1. Anatomy and connectivity. Brain Res Rev. 1987;12:117–65.
Dölen G, Darvishzadeh A, Huang KW, Malenka RC. Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature. 2013;501:179–84.
Gunaydin LA, et al. Natural neural projection dynamics underlying social behavior. Cell. 2014;157:1535–51.
Yizhar O, et al. Neocortical excitation / inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–8.
Bicks LK, Koike H, Akbarian S, Morishita H. Prefrontal Cortex and Social Cognition in Mouse and Man. Front Psychol. 2015;6:1–15.
Rogers-Carter MM, Djerdjaj A, Gribbons KB, Varela JA, Christianson JP. Insular cortex projections to nucleus accumbens core mediate social approach to stressed juvenile rats. J Neurosci. 2019;39:8717–29.
Williams AV, et al. Social approach and social vigilance are differentially regulated by oxytocin receptors in the nucleus accumbens. Neuropsychopharmacology. 2020;45:1423–30.
Okuyama T, Kitamura T, Roy D, Itohara S, Tonegawa S. Ventral CA1 neurons store social memory. Sci (80-). 2016;353:1536–41.
Park G, et al. Social isolation impairs the prefrontal-nucleus accumbens circuit subserving social recognition in mice. Cell Rep. 2021;35:109104.
Xing B, et al. A subpopulation of prefrontal cortical neurons is required for Social Memory. Biol Psychiatry. 2021;89:521–31.
Murugan M et al. Combined Social and Spatial Coding in a Descending Projection from the Prefrontal Cortex Combined Social and Spatial Coding in a Descending Projection from the Prefrontal Cortex. Cell 1–15 (2017) doi:https://doi.org/10.1016/j.cell.2017.11.002.
Heshmati M, et al. Depression and social defeat stress are associated with inhibitory synaptic changes in the nucleus accumbens. J Neurosci. 2020;40:6228–33.
Bagot RC, et al. Ventral hippocampal afferents to the nucleus accumbens regulate susceptibility to depression. Nat Commun. 2015;6:7062.
Nam H, et al. Reduced nucleus accumbens enkephalins underlie vulnerability to social defeat stress. Neuropsychopharmacology. 2019;44:1876–85.
Chaudhury D, et al. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature. 2013;493:532–6.
Vialou V, et al. Prefrontal cortical circuit for Depression- and anxiety- related behaviors mediated by cholecystokinin: role of FosB. J Neurosci. 2014;34:3878–87.
Dichter GS, et al. Reward circuitry function in autism spectrum disorders. SCAN. 2012;160–72. https://doi.org/10.1093/scan/nsq095.
Nehrkorn B, et al. Reward system dysfunction in autism spectrum disorders. SCAN. 2013;565–72. https://doi.org/10.1093/scan/nss033.
Zeeland AAS, Dapretto M, Ghahremani DG, Poldrack RA, Bookheimer SY. Reward Processing in Autism. Autism Res. 2010;3:53–67.
D’Cruz A-M, Mosconi MW, Ragozzino ME, Cook EH, Sweeney JA. Alterations in the functional neural circuitry supporting fl exible choice behavior in autism spectrum disorders. Transl Psychiatry 1–9 (2016) doi:https://doi.org/10.1038/tp.2016.161.
Langen M et al. Fronto-striatal circuitry and inhibitory control in autism: Findings from diffusion tensor imaging tractography. Cortex 48, (2012).
Martino A, Di, et al. Aberrant Striatal Functional Connectivity in Children with Autism. BPS. 2010;69:847–56.
Rothwell PE, et al. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell. 2014;158:198–212.
Neuhofer D, et al. Functional and structural deficits at accumbens synapses in a mouse model of Fragile X. Front Cell Neurosci. 2015;9:1–15.
Platt RJ, et al. Chd8 mutation leads to autistic-like behaviors and impaired Striatal circuits. Cell Rep. 2017;19:335–50.
Bukatova S, et al. Shank3 Deficiency is Associated with altered Profile of neurotransmission markers in pups and adult mice. Neurochem Res. 2021;46:3342–55.
Tzanoulinou S, et al. Inhibition of Trpv4 rescues circuit and social deficits unmasked by acute inflammatory response in a Shank3 mouse model of Autism. Mol Psychiatry. 2022. https://doi.org/10.1038/s41380-021-01427-0.
Walsh JJ, et al. 5-HT release in nucleus accumbens rescues social deficits in mouse autism model. Nature. 2018;560:589–94.
Folkes OM, et al. An endocannabinoid-regulated basolateral amygdala-nucleus accumbens circuit modulates sociability. J Clin Invest. 2020;130:1728–42.
Choe KY, et al. Oxytocin normalizes altered circuit connectivity for social rescue of the Cntnap2 knockout mouse. Neuron. 2022;110:795–808e6.
Pickles A et al. Parent-mediated social communication therapy for young children with autism (PACT): long-term follow-up of a randomised controlled trial. Lancet 388, 2501–9.
Estes A, et al. Long-term outcomes of early intervention in 6-Year-old children with Autism Spectrum Disorder. J Am Acad Child Adolesc Psychiatry. 2015;54:580–7.
Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–88.
Huppe-Gourgues F, O’Donnell P. Periadolescent changes of D(2) -AMPA interactions in the rat nucleus accumbens. Synapse. 2012;66:1–8.
Huppé-Gourgues F, O’donnell P. D 1-NMDA receptor interactions in the rat nucleus accumbens change during adolescence. Synapse. 2012;66:584–91.
Kasanetz F, Manzoni OJ. Maturation of excitatory synaptic transmission of the rat nucleus accumbens from juvenile to adult. J Neurophysiol. 2009;101:2516–27.
Belleau ML, Warren RA. Postnatal development of electrophysiological properties of nucleus accumbens neurons. J Neurophysiol. 2000;84:2204–16.
Zhang L, Warren RA. Postnatal development of excitatory postsynaptic currents in nucleus accumbens medium spiny neurons. Neuroscience. 2008;154:1440–9.
Schramm NL, Egli RE, Winder DG. LTP in the mouse nucleus accumbens is developmentally regulated. Synapse. 2002;45:213–9.
Stanwood GD, McElligot S, Lu L, McGonigle P. Ontogeny of dopamine D3 receptors in the nucleus accumbens of the rat. Neurosci Lett. 1997;223:13–6.
Tarazi FI, Baldessarini RJ. Comparative postnatal development of dopamine D1, D2, and D4 receptors in rat forebrain. Int J Dev Neurosci. 2000;18:29–37.
Leslie CA, Robertson MW, Cutler AJ, Bennett JP. Postnatal development of D 1 dopamine receptors in the medial prefrontal cortex, striatum and nucleus accumbens of normal and neonatal 6-hydroxydopamine treated rats: a quantitative autoradiographic analysis. Dev Brain Res. 1991;62:109–14.
Teicher MH, Andersen SL, Hostetter JC. Evidence for dopamine receptor pruning between adolescence and adulthood in striatum but not nucleus accumbens. Dev Brain Res. 1995;89:167–72.
Kopec AM, Smith CJ, Ayre NR, Sweat SC, Bilbo S. D. Microglial dopamine receptor elimination defines sex-specific nucleus accumbens development and social behavior in adolescent rats. Nat Commun 9, (2018).
Tarazi FI, Tomasini EC, Baldessarini RJ. Postnatal development of dopamine and serotonin transporters in rat caudate-putamen and nucleus accumbens septi. Neurosci Lett. 1998;254:21–4.
Coulter CL, Happe HK, Murrin LC. Postnatal development of the dopamine transporter: a quantitative autoradiographic study. Dev Brain Res. 1996;92:172–81.
Robinson DL, Zitzman DL, Smith KJ, Spear L. P. fast dopamine release events in the nucleus accumbens of early adolescent rats. Neuroscience. 2011;176:296–307.
Hope KT, Hawes IA, Moca EN, Bonci A, De Biase. L. M. Maturation of the microglial population varies across mesolimbic nuclei. Eur J Neurosci. 2020;52:3689–709.
Ehninger D, et al. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat Med. 2008;14:843–8.
Sato A, et al. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat Commun. 2012;3:1292.
Xing X, et al. Suppression of Akt-mTOR pathway rescued the social behavior in Cntnap2-deficient mice. Sci Rep. 2019;9:3041.
Amegandjin CA, et al. Sensitive period for rescuing parvalbumin interneurons connectivity and social behavior deficits caused by TSC1 loss. Nat Commun. 2021;12:1–18.
Ehninger D, Silva AJ. Rapamycin for treating tuberous sclerosis and autism spectrum disorders. Trends Mol Med. 2011;17:78–87.
Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab. 2014;19:373–9.
Costa-Mattioli M, Monteggia LM. mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat Neurosci. 2013;16:1537–43.
Laplante M, Sabatini DM. MTOR signaling in growth control and disease. Cell. 2012;149:274–93.
Tischmeyer W, et al. Rapamycin-sensitive signalling in long-term consolidation of auditory cortex-dependent memory. Eur J Neurosci. 2003;18:942–50.
Dash PK, Orsi SA, Moore AN. Spatial memory formation and memory-enhancing effect of glucose involves activation of the tuberous sclerosis complex-mammalian target of rapamycin pathway. J Neurosci. 2006;26:8048–56.
Tang SJ, et al. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:467–72.
Hooshmandi M, Wong C, Khoutorsky A. Dysregulation of translational control signaling in autism spectrum disorders. Cell Signal 75, (2020).
Costa V, et al. MTORC1 inhibition corrects neurodevelopmental and synaptic alterations in a human stem cell model of tuberous sclerosis. Cell Rep. 2016;15:86–95.
Huber KM, Klann E, Costa-Mattioli M, Zukin RS. Dysregulation of mammalian target of rapamycin signaling in mouse models of autism. J Neurosci. 2015;35:13836.
Steinmetz AB, Stern SA, Kohtz AS, Descalzi G, Alberini CM. Insulin-like growth factor II targets the mTOR pathway to reverse autism-like phenotypes in mice. J Neurosci. 2018;38:1015–29.
Tsai P, et al. Autistic-like behavior and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature. 2012;488:647–51.
Pagani M et al. mTOR-related synaptic pathology causes autism spectrum disorder-associated functional hyperconnectivity. Nat Commun 12, (2021).
Tsai PT, et al. Sensitive periods for cerebellar-mediated autistic-like behaviors. Cell Rep. 2018;25:357–367e4.
Haji N, et al. Tsc1 haploinsufficiency in Nkx2.1 cells upregulates hippocampal interneuron mTORC1 activity, impairs pyramidal cell synaptic inhibition, and alters contextual fear discrimination and spatial working memory in mice. Mol Autism. 2020;11:29.
von der Brelie C, Waltereit R, Zhang L, Beck H, Kirschstein T. Impaired synaptic plasticity in a rat model of tuberous sclerosis. Eur J Neurosci. 2006;23:686–92.
Iezzi D, et al. Acute rapamycin rescues the hyperexcitable phenotype of accumbal medium spiny neurons in the valproic acid rat model of autism spectrum disorder. Pharmacol Res. 2022;183:106401.
Burket JA, Benson AD, Tang AH, Deutsch S. I. Rapamycin improves sociability in the BTBR T + Itpr3tf/J mouse model of autism spectrum disorders. Brain Res Bull. 2014;100:70–5.
Kotajima-Murakami H, et al. Effects of rapamycin on social interaction deficits and gene expression in mice exposed to valproic acid in utero. Mol Brain. 2019;12:3.
Zhou J, et al. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific pten knock-out mice. J Neurosci. 2009;29:1773–83.
Kwon CH, Zhu X, Zhang J, Baker SJ. mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc Natl Acad Sci U S A. 2003;100:12923–8.
Sare RM, et al. Negative Effects of Chronic Rapamycin Treatment on Behavior in a mouse model of Fragile X Syndrome. Front Mol Neurosci. 2017;10:452.
Gibson JM, et al. A critical period for development of cerebellar-mediated autism-relevant Social Behavior. J Neurosci. 2022;42:2804–23.
Lu Z, et al. Effect of Chronic Administration of Low Dose Rapamycin on Development and Immunity in Young rats. PLoS ONE. 2015;10:e0135256.
Hadamitzky M, et al. Acute systemic rapamycin induces neurobehavioral alterations in rats. Behav Brain Res. 2014;273:16–22.
Zhou M, et al. mTOR inhibition ameliorates cognitive and affective deficits caused by Disc1 knockdown in adult-born dentate granule neurons. Neuron. 2013;77:647–54.
Russo E, et al. Everolimus improves memory and learning while worsening depressive- and anxiety-like behavior in an animal model of depression. J Psychiatr Res. 2016;78:1–10.
Reith RM, et al. Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiol Dis. 2013;51:93–103.
Meyza KZ, Blanchard DC. The BTBR mouse model of idiopathic autism – current view on mechanisms. Neurosci Biobehav Rev. 2017;76:99–110.
Mcfarlane HG, Kusek GK, Yang M. Autism-like behavioral phenotypes in BTBR T 1 tf / J mice. Genes Brain Behav. 2008;7:152–63.
Scattoni ML, Gandhy SU, Ricceri L, Crawley JN. Unusual repertoire of vocalizations in the BTBR T + tf / J mouse model of Autism. PLoS ONE. 2008;3:48–52.
Scattoni ML, Ricceri L, Crawley JN, Elena VR, Superiore I. Unusual repertoire of vocalizations in adult BTBR T + tf / J mice during three types of social encounters. Genes Brain Behav. 2011;10:44–56.
Wohr M, Roullet FI, Crawley JN. Reduced scent marking and ultrasonic vocalizations in the BTBR T + tf / J mouse model of autism. Genes Brain Behav. 2011;10:35–43.
Bolivar VJ, Walters SR, Phoenix JL. Assessing autism-like behavior in mice: Variations in social interactions among inbred strains. 176, 21–6 (2007).
Yang M, Zhodzishsky V, Crawley JN. Social deficits in BTBR T + tf / J mice are unchanged by cross-fostering with C57BL / 6J mothers. Int J Devl Neurosci. 2007;25:515–21.
Yang M, Clarke AM, Crawley JN. Postnatal lesion evidence against a primary role for the corpus callosum in mouse sociability. Eur J Neurosci. 2009;29:1663–77.
Silverman JL, Tolu SS, Barkan CL, Crawley JN. Repetitive self-grooming behavior in the BTBR mouse model of Autism is blocked by the mGluR5 antagonist MPEP. Neuropsychopharmacology. 2009;35:976–89.
Scattoni ML, Crawley J, Ricceri L. Ultrasonic vocalizations: a tool for behavioural phenotyping of mouse models of neurodevelopmental disorders. Neurosci Biobehav Rev. 2009;33:508–15.
Segbroeck M, Van, et al. NeuroResource MUPET — Mouse Ultrasonic Profile ExTraction: a Signal Processing Tool for Rapid and unsupervised analysis of Ultrasonic Vocalizations NeuroResource MUPET — Mouse Ultrasonic Profile ExTraction : a Signal Processing Tool for Rapid and Unsuper. Neuron. 2017;94:465–485e5.
Silverman JL, Yang M, Lord C, Crawley J. N. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci 11, (2010).
Yang M, et al. Social approach behaviors are similar on conventional versus reverse lighting cycles, and in replications across cohorts, in BTBR T + tf/J, C57BL/6J, and vasopressin receptor 1B mutant mice. Front Behav Neurosci. 2007;1:1–9.
Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev. 2000;24:417–63.
Arruda-Carvalho M, Wu W, Cummings KA, Clem RL. Optogenetic examination of prefrontal-amygdala synaptic development. J Neurosci. 2017. https://doi.org/10.1523/JNEUROSCI.3097-16.2017.
Folkes OM, et al. An endocannabinoid-regulated basolateral amygdala-nucleus accumbens circuit modulates sociability. J Clin Invest. 2020;130:1728–42.
Park G et al. Social isolation impairs the prefrontal-nucleus accumbens circuit subserving social recognition in mice. Cell Rep 35, (2021).
Qi J, et al. VTA glutamatergic inputs to nucleus accumbens drive aversion by acting on GABAergic interneurons. Nat Neurosci. 2016;19:725–33.
Murugan M, et al. Combined social and spatial coding in a descending projection from the Prefrontal Cortex. Cell. 2017;171:1663–1677e16.
Rapamycin R, Lamming DW. Inhibition of the mechanistic target of. Cold Spring Harb Perspect Med. 2016;6:1–14.
Sachser N, Zimmermann TD, Hennessy MB, Kaiser S. Sensitive phases in the development of rodent social behavior. Curr Opin Behav Sci. 2020;36:63–70.
Ruskin DN, et al. Ketogenic Diet improves core symptoms of autism in BTBR mice. PLoS ONE. 2013;8:4–9.
Cope EC, et al. Atypical perineuronal nets in the CA2 region interfere with social memory in a mouse model of social dysfunction. Mol Psychiatry. 2022;27:3520–31.
Zilkha N, Kuperman Y, Kimchi T. High-fat diet exacerbates cognitive rigidity and social deficiency in the BTBR mouse model of autism. Neuroscience. 2017;345:142–54.
Biever A, Valjent E, Puighermanal E. Ribosomal protein S6 phosphorylation in the nervous system: from regulation to function. Front Mol Neurosci. 2015;8:75.
Bornschein G, Brachtendorf S, Schmidt H. Developmental increase of neocortical presynaptic efficacy via maturation of vesicle replenishment. Front Synaptic Neurosci. 2020;11:1–10.
Kim J, Alger BE. Paired-pulse facilitation. J Neurosci. 2001;21:9608–18.
Speed HE, Dobrunz LE. Developmental changes in short-term facilitation are opposite at temporoammonic synapses compared to schaffer collateral synapses onto CA1 pyramidal cells. Hippocampus. 2009;19:187–204.
Peixoto RT, Wang W, Croney DM, Kozorovitskiy Y, Sabatini BL. Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B(-/-) mice. Nat Neurosci. 2016;19:716–24.
Peixoto RT, et al. Abnormal Striatal Development underlies the early onset of behavioral deficits in Shank3B(-/-) mice. Cell Rep. 2019;29:2016–2027e4.
Liu XD, et al. Different physiological properties of spontaneous excitatory postsynaptic currents in nucleus accumbens shell neurons between adult and juvenile rats. Neurosci Lett. 2011;496:80–3.
Paolicelli RC, et al. Synaptic pruning by microglia is necessary for normal brain development. Sci (80-). 2011. https://doi.org/10.1126/science.1202529.
Tang G, et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron. 2014;83:1131–43.
Lieberman OJ, et al. mTOR suppresses Macroautophagy during striatal postnatal development and is hyperactive in mouse models of Autism Spectrum Disorders. Front Cell Neurosci. 2020;14:70.
Nardou R, et al. Oxytocin-dependent reopening of a social reward learning critical period with MDMA. Nature. 2019;569:116–20.
Toddes C, Lefevre EM, Brandner DD, Zugschwert L, Rothwell PE. Μ-Opioid receptor (Oprm1) Copy Number Influences Nucleus Accumbens Microcircuitry and reciprocal Social Behaviors. J Neurosci. 2021;41:7965–77.
Trezza V, Damsteegt R, Marijke Achterberg EJ, Vanderschuren LJ. M. J. Nucleus accumbens µ-opioid receptors mediate social reward. J Neurosci. 2011;31:6362–70.
Manduca A, et al. Interacting cannabinoid and opioid receptors in the nucleus accumbens core control adolescent social play. Front Behav Neurosci. 2016;10:1–17.
Smith CJW, Wilkins KB, Li S, Tulimieri MT, Veenema AH. Nucleus accumbens mu opioid receptors regulate context-specific social preferences in the juvenile rat. Psychoneuroendocrinology. 2018;89:59–68.
Gigliucci V, et al. Region specific up-regulation of oxytocin receptors in the opioid Oprm1 -/- mouse model of autism. Front Pediatr. 2014;2:1–12.
Chang L, et al. Early life stress alters opioid receptor mRNA levels within the nucleus accumbens in a sex-dependent manner. Brain Res. 2019;1710:102–8.
Albores-Garcia D, McGlothan JL, Bursac Z, Guilarte TR. Chronic developmental lead exposure increases µ-opiate receptor levels in the adolescent rat brain. Neurotoxicology. 2021;82:119–29.
Talbot JN, Happe HK, Murrin LC. µ opioid receptor coupling to Gi/o proteins increases during postnatal development in rat brain. J Pharmacol Exp Ther. 2005;314:596–602.
Deng W, et al. Effect of metformin in autistic BTBR T + Itpr3tf/J mice administered a high-fat diet. Brain Res Bull. 2022;183:172–83.
Lv H, et al. Nanoformulated bumetanide ameliorates Social Deficiency in BTBR mice model of Autism Spectrum Disorder. Front Immunol. 2022;13:1–13.
Wei H, et al. Inhibition of IL-6 trans-signaling in the brain increases sociability in the BTBR mouse model of autism. Biochim Biophys Acta - Mol Basis Dis. 2016;1862:1918–25.
Albekairi NA et al. Pharmacology, Biochemistry and Behavior CXCR2 antagonist SB332235 mitigates deficits in social behavior and dysregulation of Th1 / Th22 and T regulatory cell-related transcription factor signaling in male BTBR T + Itpr3 tf / J mouse model of autism. 217, (2022).
Kazdoba TM, Hagerman RJ, Zolkowska D, Rogawski MA, Crawley J. N. evaluation of the neuroactive steroid ganaxolone on social and repetitive behaviors in the BTBR mouse model of autism. Psychopharmacology. 2016;233:309–23.
Chadman KK. Fluoxetine but not risperidone increases sociability in the BTBR mouse model of autism. Pharmacol Biochem Behav. 2011;97:586–94.
Yoshimura RF, et al. Allosteric modulation of nicotinic and GABAA receptor subtypes differentially modify autism-like behaviors in the BTBR mouse model. Neuropharmacology. 2017;126:38–47.
Jayaprakash P, et al. Curcumin potentiates α7 nicotinic acetylcholine receptors and alleviates autistic-like social deficits and brain oxidative stress status in mice. Int J Mol Sci. 2021;22:1–17.
Chao OY, et al. Altered dopaminergic pathways and therapeutic effects of intranasal dopamine in two distinct mouse models of autism. Mol Brain. 2020;13:1–16.
Wei D, et al. Enhancement of anandamide-mediated Endocannabinoid Signaling Corrects Autism-Related Social Impairment. Cannabis Cannabinoid Res. 2016;1:81–9.
Mahmood HM, et al. The role of nicotinic receptors in the attenuation of autism-related behaviors in a murine BTBR T + tf/J autistic model. Autism Res. 2020;13:1311–34.
Wang L, et al. Modulation of social deficits and repetitive behaviors in a mouse model of autism: the role of the nicotinic cholinergic system. Psychopharmacology. 2015;232:4303–16.
Gould GG, et al. Acetaminophen differentially enhances social behavior and cortical cannabinoid levels in inbred mice. Prog Neuro-Psychopharmacology Biol Psychiatry. 2012;38:260–9.
Karvat G, Kimchi T. Acetylcholine elevation relieves cognitive rigidity and social deficiency in a mouse model of autism. Neuropsychopharmacology. 2014;39:831–40.
Silverman JL, et al. GABA B receptor agonist R-Baclofen reverses social deficits and reduces repetitive behavior in two mouse models of Autism. Neuropsychopharmacology. 2015;40:2228–39.
Kratsman N, Getselter D, Elliott E. Sodium butyrate attenuates social behavior deficits and modifies the transcription of inhibitory/excitatory genes in the frontal cortex of an autism model. Neuropharmacology. 2016;102:136–45.
Han S, Tai C, Jones CJ, Scheuer T, Catterall WA. Enhancement of inhibitory neurotransmission by GABAA receptors having α2,3-subunits ameliorates behavioral deficits in a mouse model of autism. Neuron. 2014;81:1282–9.
Cai Y, et al. Citalopram attenuates social behavior deficits in the BTBR T + Itpr3tf/J mouse model of autism. Brain Res Bull. 2019;150:75–85.
Venkatachalam K et al. The histamine H3R and dopamine D2R/D3R antagonist ST-713 ameliorates autism-like behavioral features in BTBR T + tf/J mice by multiple actions. Biomed Pharmacother 138, (2021).
Eissa N, et al. Simultaneous blockade of histamine h3 receptors and inhibition of acetylcholine esterase alleviate autistic-like behaviors in btbr t + tf/j mouse model of autism. Biomolecules. 2020;10:1–20.
Nadeem A, et al. Nrf2 activator, sulforaphane ameliorates autism-like symptoms through suppression of Th17 related signaling and rectification of oxidant-antioxidant imbalance in periphery and brain of BTBR T + tf/J mice. Behav Brain Res. 2019;364:213–24.
Cai Y, et al. Autism-like behavior in the BTBR mouse model of autism is improved by propofol. Neuropharmacology. 2017;118:175–87.
Silverman JL, Oliver CF, Karras MN, Gastrell PT, Crawley JN. Neuropharmacology AMPAKINE enhancement of social interaction in the BTBR mouse model of autism. 64, 268–82 (2013).
Cristiano C, et al. Palmitoylethanolamide counteracts autistic-like behaviours in BTBR T + tf/J mice: contribution of central and peripheral mechanisms. Brain Behav Immun. 2018;74:166–75.
Wu H, et al. Selenium inhibits ferroptosis and ameliorates autistic-like behaviors of BTBR mice by regulating the Nrf2/GPx4 pathway. Brain Res Bull. 2022;183:38–48.
Perets N, Hertz S, London M, Offen D. Intranasal administration of exosomes derived from mesenchymal stem cells ameliorates autistic-like behaviors of BTBR mice. Mol Autism. 2018;9:1–12.
Hagen E, et al. Autism-like behavior in BTBR mice is improved by Electroconvulsive Therapy. Neurotherapeutics. 2015;12:657–66.
Bales KL, et al. Long-term exposure to intranasal oxytocin in a mouse autism model. Transl Psychiatry. 2014;4:1–10.
Queen NJ et al. Environmental enrichment improves metabolic and behavioral health in the BTBR mouse model of autism. Psychoneuroendocrinology 111, (2020).
PARK HY, et al. Humulus japonicus rescues autistic-like behaviours in the BTBR T + Itpr3tf/J mouse model of autism. Mol Med Rep. 2021;23:1–8.
Cui J, et al. General Anesthesia during Neurodevelopment reduces autistic behavior in adult BTBR mice, a murine model of Autism. Front Cell Neurosci. 2021;15:1–10.
Wang L, Cai Y, Fan X. Metformin administration during early postnatal life rescues autistic-like behaviors in the BTBR T + Itpr3tf/J mouse model of autism. Front Behav Neurosci. 2018;12:1–10.
Zhong H, et al. Neonatal curcumin treatment restores hippocampal neurogenesis and improves autism-related behaviors in a mouse model of autism. Psychopharmacology. 2020;237:3539–52.
Cai Y, et al. The liver X receptor agonist TO901317 ameliorates behavioral deficits in two mouse models of autism. Front Cell Neurosci. 2019;13:1–12.
Nettleton JE et al. Prebiotic, probiotic, and synbiotic consumption alter behavioral variables and intestinal permeability and microbiota in btbr mice. Microorganisms 9, (2021).
Rotaru DC, Wallaard I, de Vries M, van der Bie J, Elgersma Y. UBE3A expression during early postnatal brain development is required for proper dorsomedial striatal maturation. JCI Insight 8, (2023).
Cox RL, de Anda C, Mangoubi F, T., Yoshii A. Multiple critical periods for Rapamycin treatment to correct structural defects in Tsc-1-Suppressed Brain. Front Mol Neurosci. 2018;11:409.
Chao OY, et al. Targeting inhibitory cerebellar circuitry to alleviate behavioral deficits in a mouse model for studying idiopathic autism. Neuropsychopharmacology. 2020;45:1159–70.
Wang X, et al. Overexpression of mGluR7 in the Prefrontal Cortex attenuates autistic behaviors in mice. Front Cell Neurosci. 2021;15:1–11.
Branchi I, Santucci D, Alleva E. Ultrasonic vocalisation emitted by infant rodents: a tool for assessment of neurobehavioural development. Behav Brain Res. 2001;125:49–56.
Nitschke W, Bell RW, Zachman T. Distress vocalizations of young in three inbred strains of mice. Dev Psychobiol. 1972;5:363–70.
Wolff RJ. Solitary and social play in wild Mus musculus (Mammalia). J Zool. 1981;195:405–12.
Panksepp J. The ontogeny of play in rats. Dev Psychobiol. 1981;14:327–32.
Ferri SL, et al. Sociability development in mice with cell-specific deletion of the NMDA receptor NR1 subunit gene. Genes Brain Behav. 2020;19:1–12.
Deitchman R, Lavine A, Burkholder J. Litter size, emotionality, learning, and social interaction in C57BL/6j mice. Prycbological Rep. 1976;39:27–31.
Bicks LK et al. Prefrontal parvalbumin interneurons require juvenile social experience to establish adult social behavior. Nat Commun 11, (2020).
Branchi I, D’Andrea I, Santarelli S, Bonsignore LT, Alleva E. The richness of social stimuli shapes developmental trajectories: are laboratory mouse pups impoverished? Prog Neuro-Psychopharmacology Biol Psychiatry. 2011;35:1452–60.
Walker DM, Cunningham AM, Gregory JK, Nestler EJ. Long-term behavioral effects of post-weaning social isolation in males and females. Front Behav Neurosci. 2019;13:1–20.
Li DC, Hinton EA, Gourley SL. Persistent behavioral and neurobiological consequences of social isolation during adolescence. Semin Cell Dev Biol. 2021;118:73–82.
Acknowledgements
We thank Mehreen Inayat and Ryan Appings for help with mouse breeding and Jennifer Wilkin for help with the design of figure schematics. We acknowledge resources and support from the Centre for the Neurobiology of Stress (CNS) Core Facility at the University of Toronto Scarborough.
Funding
This work was supported by a University of Toronto Scarborough Postdoctoral Fellowship to MM, and grants from the SickKids Foundation and Canadian Institutes of Health Research (CIHR) – Institute of Human Development, Child and Youth Health (NI19-1132R), CIHR (PJT 399790), Human Frontier Science Program Organization (CDA00009/2018 and RGY0072/2019), and Natural Sciences and Engineering Research Council of Canada (RGPIN-2017-06344) to MAC. A Canada Foundation for Innovation grant (#493864) was used to establish the Centre for the Neurobiology of Stress (CNS) Core Facility.
Author information
Authors and Affiliations
Contributions
MM, AK, CJS and MAC designed research. MM, AK, CJS, MD, UM and MAC performed research. MM, CJS, AK, MD and MAC analyzed/interpreted data. AK and MAC wrote the paper. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal procedures were approved by the Animal Care Committee at the University of Toronto.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Matthiesen, M., Khlaifia, A., Steininger, C.F.D. et al. Maturation of nucleus accumbens synaptic transmission signals a critical period for the rescue of social deficits in a mouse model of autism spectrum disorder. Mol Brain 16, 46 (2023). https://doi.org/10.1186/s13041-023-01028-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-023-01028-8