
Abstract
Chemical synapses in the brain connect neurons to form neural circuits, providing the structural and functional bases for neural communication. Disrupted synaptic signaling is closely related to a variety of neurological and psychiatric disorders. In the past two decades, proteomics has blossomed as a versatile tool in biological and biomedical research, rendering a wealth of information toward decoding the molecular machinery of life. There is enormous interest in employing proteomic approaches for the study of synapses, and substantial progress has been made. Here, we review the findings of proteomic studies of chemical synapses in the brain, with special attention paid to the key players in synaptic signaling, i.e., the synaptic protein complexes and their post-translational modifications. Looking toward the future, we discuss the technological advances in proteomics such as data-independent acquisition mass spectrometry (DIA-MS), cross-linking in combination with mass spectrometry (CXMS), and proximity proteomics, along with their potential to untangle the mystery of how the brain functions at the molecular level. Last but not least, we introduce the newly developed synaptomic methods. These methods and their successful applications marked the beginnings of the synaptomics era.
Similar content being viewed by others


Introduction
In the nervous system, the term synapse refers to the junction between two neurons or between a neuron and its target effector cells. Chemical synapses, comprising a presynaptic terminal, a postsynaptic terminal, and the synaptic cleft between them, are specialized structures for fast and precise unidirectional signal transduction and are classified primarily according to the type of transmitters involved [1]. As the minimal computational units in the central neural system, synapses interconnect billions of neurons into neural circuits, playing a crucial role in neuronal communication and functioning of the brain. Growing evidence has implicated synaptic abnormalities in various brain disorders, including psychiatric, neurodevelopmental, and neurodegenerative disorders [2, 3].
Proteomics is the characterization of all of the proteins in a biological system, such as an organelle, a cell, a tissue, or even an organism. Classified by research perspectives, there are expression proteomics, structural proteomics, and functional proteomics. Gel-based proteomics studies have been flourishing for a while [4, 5], and beginning from this century, liquid chromatography–mass spectrometry (LC–MS)-based shotgun proteomics studies emerged, with advantages such as robustness, high throughput, low labor requirement, and compatibilities with hydrophobic proteins and labeling strategies [6]. After two decades of rapid development, tens of thousands of proteins can now be studied in a high-throughput manner [7, 8]. Proteomics has already become an indispensable research tool for protein expression profiling, post-translational modifications (PTMs) detection, protein–protein interactions (PPIs) exploration, and biomarker discovery and validation, providing multidimensional insights into dynamic physiological and pathological processes in cells [9].
Proteomics met neuroscience in 1999, when a rat brain protein database containing 210 different proteins was constructed by combining two-dimensional electrophoresis (2-DE) and matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) [10]. The concept “neuroproteomics” was proposed in 2009, with a high expectation that proteomics could explain the complexity of the brain at the molecular level, from aspects including protein expression, function, bioinformatics and clinical application [11]. Over the past 20 years, proteomic technologies have increasingly been applied in neuroscience, and several excellent reviews have covered the achievements and challenges in this area [11,12,13,14,15,16].
Here, we focus on the proteomic studies of chemical synapses for the important role they played in brain development and functioning. We scrutinize the synaptic proteome in a functional context, look into the molecular mechanisms underlying brain disorders, and discuss the current opportunities, challenges, and trends in neuroproteomics. Proteomics approaches are widely used in the search for biomarkers of neurological and psychiatric disorders; however, these studies are beyond the scope of this review, and interested readers are referred to a recent review for an overview of this research field [17].
Mapping the synaptic proteome
Despite being small in size, the synapse is surprisingly complex [18, 19]. It contains thousands of various proteins, whose spatiotemporal expression and dynamics remain a daunting challenge for researchers. In this section, we review the discoveries made over the past 20 years regarding the synaptic proteome, with discussions about sample preparation and the molecular components of the different subsynaptic structures.
Sample preparation
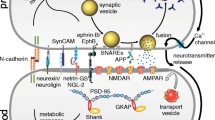
A typical proteomic experiment contains major steps of sample homogenization, protein extraction, protein/peptide separation, and MS detection. In a synaptic proteomic analysis, usually the first step is to prepare synaptic samples from brain tissues or cultured cells. After being extracted from the samples, synaptic proteins can be separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) or 2-DE, and the protein-containing gels are cut into pieces and enzymatically digested; the other path is to digest the collected proteins first, then use chromatographic techniques, usually the combination of strong cation-exchange (SCX) chromatography and capillary reversed-phase high-performance liquid chromatography (RP-HPLC), for peptide fractionation. Usually, the last step is mass spectrometry (MS) detection: properly separated peptides were sent to MS for identification and/or quantification. Data-dependent acquisition (DDA) and data-independent acquisition (DIA) are two modes used for data collection in MS-based proteomic analysis, which we’ll discuss later. Figure 1 illustrated the major steps and methods commonly used in proteomic analysis of synapses.

Major steps and methods in proteomic analysis of chemical synapses
The characterization of subcellular compartments relies primarily on the capability of subcellular fractionation techniques to reduce sample complexity and detect low-abundance proteins [20, 21]. One of two approaches is typically used to prepare synaptic samples: the biochemical fractionation approach utilizes differential and density-gradient centrifugation methods to isolate synaptic vesicles or artificial vesicles derived from synapses based on their unique density, while the protein-based approaches, including immunoisolation [22,23,24,25], affinity purification [26], fluorescence-activated synaptosome sorting [27] and proximity labeling methods [28, 29], isolate synaptic protein complexes or assemblies associated with the targeted proteins (Fig. 1). Both approaches are widely used and have their advantages. Generally speaking, biochemical fractionation enriches synapse-related vesicles with similar biophysical and biochemical properties, while the protein-based approaches have higher selectivity and can discriminate subtypes of synaptic vesicles [27] or synaptic assemblies derived from different types of synapses [22, 23].
Synaptosome
The term synaptosome, created in 1964, refers to the membrane-bound sac containing vesicles that separate from synapses when brain tissue is homogenized under certain conditions [30]. As the synaptosome preserves all the main structural features of the nerve terminals, is relatively homogeneous, and has physical properties resembling those of other subcellular organelles, it has become an important window to examine the molecular machinery of the synapses. In 2003, a study combined MS with several separation techniques, including SDS-PAGE, SCX chromatography, and capillary RP-HPLC, to study the synaptic plasma membrane proteins. The authors identified several relevant synaptic proteins, including various transporters, receptors, ion channels, and enzymes, showing the effectiveness of this multidimensional separation method in profiling the proteome of membrane-associated organelles [31]. The scope of synaptosome proteomics has since expanded from comparing protein components of different synaptic vesicle pools [25] and screening synaptosome subtypes [27] to identifying long-lived synaptic proteins [32] and age- and brain region-specific changes in synaptic protein expression in animal disease models [20], which have greatly enriched our understanding of the molecular basis of synaptic signaling.
Presynaptic components
The presynaptic terminal is the distal termination of an axon where neurotransmitters are released. The function of the presynaptic terminal is precisely regulated and is associated with brain function and diseases such as Alzheimer’s and Parkinson’s [33,34,35]. Integrated proteomics and systems biology approaches have been applied to obtain the molecular map of this neuronal sub-compartment, resulting in a core list of 117 proteins and 92 predicted presynaptic proteins [35]. The presynaptic active zone (PAZ) is a specialized region of the presynaptic plasma membrane, where synaptic vesicles dock and fuse and neurotransmitters are released by exocytosis [36]. Using antibodies against synaptic vesicle protein 2, researchers were able to isolate and analyze the presynaptic compartment containing the PAZ, leading to the identification of 240 proteins, including synaptic vesicle proteins, adhesion molecules, and proteins involved in intracellular signal transduction [24].
The presynaptic proteome becomes dysregulated under certain disease conditions. For example, the expression of several presynaptic markers is reduced in the hippocampal synaptic proteome of mice with a cognitive defect, consistent with the evidence of compromised presynaptic morphology and abnormal synaptic function in this brain region [37]. Proteomics approaches have also been used to reveal the proteins in presynaptic mitochondria that are important during brain development. Specifically, two independent quantitative proteomics approaches, i.e., the sequential window acquisition of all theoretical fragment ion mass spectra (SWATH-MS) and stable isotope labeling by amino acids in cell culture (SILAC), identified 40 mitochondrial proteins that are differentially expressed in mice between postnatal day 7 and day 42, including MitoNEET (Cisd1), which was revealed as a key regulator of mitochondrial function and postnatal brain development [38].
Chemical synapses rely on synaptic vesicles for signal transmission. A review of proteomic studies of synaptic vesicles published in 2007 compared different methods for synaptic vesicle separation and characterization, and highlighted the importance of analyzing subpopulations of synaptic vesicles [39]. A quantitative comparison of glutamatergic and GABAergic synaptic vesicles revealed that only a small portion of their proteins differ from each other (50 of > 450) [23]. Consistent with this result, quantitative comparison of glutamate- and GABA-specific docking complexes revealed only a few proteins that were differentially enriched in these two types of synapses, indicating high conservation of the core molecular machinery of the presynaptic docking sites [22].
Synaptic clefts
The synaptic cleft is the space between pre- and postsynaptic terminals, which is now considered a structurally and functionally integral compartment of the synapse. Although the inability to biochemically isolate the clefts make the characterization of this structure challenging, the emerging proximity labeling proteomics approaches bring opportunities to explore the protein composition and changes in this specialized compartment. By peroxidase-mediated biotin labeling methods, researchers revealed that numerous ion channels, G protein-coupled receptors, adhesion proteins, and transporters reside in the synaptic cleft along with novel cleft candidates, and uncovered differences between excitatory glutamatergic and inhibitory GABAergic synaptic clefts [28, 29]. These studies not only demonstrated the power of proximity labeling approaches for parsing the molecular properties of traditionally unpurifiable structures, but also enriched our understanding of synaptic signaling and inspired biological hypotheses (e.g., the potential role of Mdga2 as a specificity factor at inhibitory synapses [29]).
On the postsynaptic side
The main function of the postsynaptic terminal is to receive signals from the presynaptic terminal and transduce them to intracellular electrical and biochemical signals, thus activating downstream cascades leading to various cellular processes.
The postsynaptic density (PSD) is a complex, dynamic specialized region of the excitatory synapse that is observed as an approximately 30 nm-thick electron-dense membrane-associated structure under an electron microscope [40]. Over the past few decades, effort has been made to obtain a comprehensive picture of this protein-dominant compartment, with more than 2100 proteins identified and quantified [41,42,43,44,45,46,47,48]. The reported PSD proteome comprises various neurotransmitter receptors, cell adhesion molecules, signal enzymes, scaffold proteins, cytoskeleton proteins, and membrane transporters. The PSD composition of excitatory synapses is highly conserved between mice and humans, but the abundances of some key proteins differ, including that of some receptors and adaptor proteins that are reported to be involved in synaptic plasticity [49].
Many of the PSD proteins have been implicated in a wide range of brain disorders [50, 51]. A study in 2011 isolated PSD from human neocortex and identified 1,461 proteins [52]. Bioinformatic analysis revealed that mutations in these proteins were related to 133 neurological and psychiatric diseases and were particularly enriched in neural phenotypes involving cognition and motor functions [52]. Moreover, 143 of 700 proteins identified from PSD fractions of anterior cingulate cortex were differentially expressed in patients with schizophrenia, implicating NMDA-interacting and endocytosis-related proteins in the pathophysiology of schizophrenia [53]. These authors also identified 288 differentially expressed proteins in bipolar disorder, highlighting the important role of synaptic function and energy pathways in psychiatric diseases [54]. The role of PSD components in neurodevelopmental and neurodegenerative disorders has also been investigated [55, 56]. Nevertheless, our current understanding of this highly complex and dynamic structure remains limited and much work is still needed to elucidate its role in various pathological processes. As shown in a meta-analysis of mental disorder-related neuroproteomic studies, identification of key neural pathophysiological pathways was hindered by the poor reproducibility among independent experiments, and there were at least two reasons for this problem: one was technical reasons common to all proteomic experiments and the other was non-technical factors that specific to brain studies, including the high level of heterogeneity in the molecular pathophysiology of mental illnesses and the high biological variability found among human samples [57]. While it’s impossible to change the intrinsic features of biological samples, there are things doable to address the challenges, e.g., to use much larger number of samples (especially when working with human samples), more replicates, highly standardized procedures and detailed reports of parameters throughout the entire experiment from sample collection to data processing and report.
Inhibitory synapses
Normal functioning of the brain depends on the orchestration of excitatory and inhibitory synapses, and an imbalance between them is implicated in many neuropathological processes [58]. Excitatory and inhibitory postsynaptic assemblies differ in protein components, molecular organizations, and biological functions [59]. However, compared with the number of studies on excitatory synapses, there are relatively few studies on inhibitory synapses. An early study once reported that inhibitory synapses lack cell signaling proteins because they didn’t find any in their study of inhibitory synaptic complexes isolated by affinity purification [26]; however, a later study using a more sensitive proximity-labeling approach has revealed that there is a huge and elaborate protein network that reside at inhibitory PSD and mediates postsynaptic inhibition [60]. This network was composed of a wide variety of proteins including neurotransmitter receptors, adhesion molecules, scaffold proteins, transporters and cytoskeletal regulators, and shared a number of signaling proteins such as Cnksr2 and Arhgap39 with excitatory PSD, suggesting a much higher complexity of the inhibitory synapse than previously appreciated [60].
Synaptic proteomics in a functional context
While mapping of the proteome is an important step toward a complete understanding of synapse, investigation of the organization and regulatory mechanisms of synaptic proteins in a functional context is needed to comprehend synaptic signaling. With the inherent advantages in studying PPIs and PTMs, MS-based proteomic approaches have already become an indispensable tool in parsing the protein complexes and their dynamics in synaptic signaling.
Synaptic protein complexes
Multiprotein complexes play essential roles in the organization and functioning of the synapse. They provide unique intracellular microenvironments for proteins to interact and for reactions to occur precisely and efficiently. This is most obvious in the PSD, as it contains a collection of scaffold proteins which become the hubs of the postsynaptic signaling network [61]. Coupled with subsequent MS identification, affinity pulldown and co-immunoprecipitation (co-IP) are regularly used methods for protein complex characterization. The Grant lab was one of the pioneers in utilizing proteomic tools to study synaptic complexes. They explored the organization and functional implications of the NMDA receptor complex [62, 63] and PSD95 complex [64], and published a series of enlightening reviews in the early days of the proteomic era that predicted the wide use of proteomic approaches in neuroscience research [65,66,67,68]. Later, complexes of other key synaptic proteins were resolved one after another, including PSD95 [69, 70], the SHANKs [71], SynGAP [72], PSD-93 [73], FMRP [74,75,76], and metabotropic glutamate receptors [77,78,79,80]; besides, the interactions among them mediated by some specific domains also attracted much attention [81, 82]. The interactome of GTPase-activating proteins and guanine exchange factors in PSD indicated their close association with the core scaffolding machinery of glutamate receptors, reflecting their active role in regulating GTP signaling from receptors to downstream targets [83]. Another study identified the interactomes of Kalirin-7 and Trio, demonstrating the importance of the interaction between Kalirin-7 and neuroligin-1 for normal synaptic function [84]. At the same time, other groups have expanded the focus from single protein complexes to dynamic PPI networks. In one of our previous work, we conducted a large-scale postsynaptic interactome study and identified 2876 proteins across 41 in vivo interactomes in the mouse cortex, thus presenting a spatiotemporal profile of the PSD signaling network and a glimpse of the assembly process of the PSD core network during development [61].
Although synaptic proteins have been implicated in many brain disorders and injuries [2, 51], their pathophysiology remains largely unknown. In recent years, a number of genetic research and genome-wide association studies have identified groups of synaptic proteins as risk factors in psychiatric disorders such as schizophrenia and autism spectrum disorders (ASD) [85,86,87,88], suggesting the central role of synaptic dysfunction in the etiologies of these diseases. Translation of these genetic findings into a physiological understanding of the underlying molecular mechanisms is a challenge faced by researchers, and proteomics has appeared as a powerful tool to fill the gap. For example, a comparison of glutamate synapse protein interactomes from different autism mouse models revealed specific and overlapping signal networks that molecularly distinguish the disease [89]. More recently, it was reported that PSD-93 interacts with SynGAP and mediates its ubiquitination and degradation, a process that may aggravate ischemic brain injury [73].
PTM analysis
Versatile and dynamic PTMs are key features of the synaptic proteome, and accumulating evidence have indicated that the dysregulation of PTM homeostasis is associated with several pathological processes in the central nervous system, including neurodegenerative and neurodevelopmental disorders [90, 91]. MS-based proteomics is currently the most widely used approach in PTM analysis, shedding light on the functioning of the synaptic signaling machinery [92].
Phosphorylation endows the synaptic signaling network with high information-processing capacity and functional diversity, which are closely related to synaptic functions, including synaptic plasticity [93, 94]. It is also the most widely studied PTM, with mature phosphoproteomic procedures established over the years [95, 96]. Since the first mouse synaptic phosphoproteome map drawn in 2005 [97], much work, including ours, has been done to dissect the phosphorylation sites and changes under different physiological conditions, rendering valuable clues toward the mechanisms underlying synaptic functions [93, 98,99,100,101,102]. One of our previous work compared the phosphorylation status of the PSD proteins from mouse hippocampus before and after the induction of long-term potentiation, and found that the phosphoproteins regulated by long-term potentiation represented “PSD risk factors” for a number of psychiatric disorders [102]. Another study focusing on the presynaptic side used KCl to stimulate cultured hippocampal neurons and identified six temporal patterns of coregulated phosphoproteins, in which PAZ scaffold proteins showed a high level of activity-dependent phosphorylation-based regulation [98]. Phosphoproteomic approaches have been used to investigate the synaptic changes related to sleep by three groups independently, with interesting discoveries showing widespread alterations in the phosphorylation status of synaptic proteins during sleep and the profound molecular disturbances caused by sleep deprivation [99,100,101].
The phosphorylation status of single synaptic proteins and their subsequent functional influences have also been under investigation. The synaptic protein CASK, a member of the membrane-associated guanylate kinase family, can be phosphorylated by the kinase-like protein CDKL5 to promote synaptogenesis [103]. A study of ubiquitin protein ligase UBE3A found that protein kinase A phosphorylates its T485 residue to regulate the development of dendritic spines and forestall the development of ASD [104]. The protein TAOK2, whose genetic locus was associated with ASD and schizophrenia in a copy number variation study [105], mediates PSD-95 stability and dendritic spine maturation through phosphorylation of Septin7 [106].
How the phosphoproteome is regulated with efficiency and precision is a fascinating question. A recent review summarized the phosphoproteomic studies of the PSD over the years, with a focus on the components of the phosphorylation regulatory machinery, i.e., the kinases, phosphatases, and several protein domain modules, and described the general rules of the phosphoproteome-associated PSD signaling organization [107].
Besides phosphorylation, other forms of PTMs in synapses have started to receive attention in recent years. A research published in 2019 reported that DIP2A, encoded by a gene associated with susceptibility for ASD, affects synaptic transmission and morphology by regulating the acetylation of cortactin [108]. Ubiquitination plays multifaceted roles in a number of neuronal processes by controlling the quality and abundance of various proteins and has been implicated in neurodevelopmental disorders such as ASD and Angelman syndrome [109, 110]. A quantitative proteomic study showed that the ubiquitin protein ligase UBE3A mediates the ubiquitination and degradation of phosphotyrosyl phosphatase activator and affects spine morphology through a signaling pathway involving protein phosphatase 2 [111]. Table 1 summarizes proteomic studies of some key PSD proteins, providing classified information of their subcellular locations, associated diseases, molecular functions, PPI and PTM studies, and related references.
Trends in neuroproteomics
Advances in proteomic technologies have created more opportunities for deeper exploration of the brain. With the iterative upgrade of mass spectrometers and the development of novel experimental methods, including DIA, chemical cross-linking in combination with mass spectrometry (CXMS) and proximity proteomics, it will be possible to achieve in-depth analysis of protein PTMs and signaling network dynamics. At the same time, to answer the fascinating biological questions, such as the spatiotemporal landscapes of synaptic proteome and the molecular bases of various brain diseases, the great potential of proteomic tools needs to be further explored (Fig. 2). It is the interplay of advancing proteomic technologies and the emerging scientific questions that form the current trends and driving forces of neuroproteomics, leading to profound insights into neurobiology.

Trends in neuroproteomics
Advances in proteomic technologies
DIA and in-depth PTM analysis
Although the importance of PTMs in cell signaling has long been recognized, our understanding of most PTMs is still in its infancy. PTMs typically feature low abundance, multiple locations, and dynamic changes, which make in-depth analyses extremely difficult. The concept of DIA was proposed in 2004 as a new MS method, in which the spectrum data were acquired based on the sequential isolation and fragmentation of preset precursor windows until a desired mass range was covered, independent of the m/z value or signal strength of the peaks [171]. In the past decade, DIA developed rapidly and has been successfully implemented in many domains of biological research [172], largely due to the development of various DIA schemes (e.g., all-ion fragmentation [173], SWATH [174, 175], multiplexed DIA [176], hyper-reaction monitoring [177], SONAR [178], and BoxCar [179]) and robust data processing tools (e.g., OpenSWATH [180], DIA-Umpire [181, 182], Spectronaut Pulsar [183], and DeepNovo-DIA [184]). Compared with the output from traditional DDA mode, DIA selects and fragments all peptides with a signal stronger than noise, rendering unbiased quantitative data with higher speed, coverage, precision, and reproducibility [185], and making large-scale in-depth PTM analyses a reality. Through targeted optimization of experimental DIA methods and algorithms, a recent study quantified more than 20,000 phosphopeptides in a 15 min liquid chromatography–MS analysis [186]. Besides phosphorylation, DIA has also been utilized to study the changes to histone PTMs induced by the lead compound acridone derivative 8a, presenting quantitative methylation and acetylation data for epigenetic analyses [187]. There is reason to believe that with the continuous improvement of experimental methods and software toolkits, sophisticated DIA workflows will greatly accelerate PTM research in the near future, becoming a powerful tool for the in-depth study of synaptic signaling mechanisms.
CXMS and the investigation of direct PPIs
Most proteins accomplish their tasks through interactions with other proteins. Only with a detailed understanding of a protein’s interactors can we begin to probe its function and the consequences of its mutation. Popular methods for PPI identification include the yeast two-hybrid system [188], Förster resonance energy transfer (FRET) method [189, 190], and MS-based methods such as affinity chromatography [191] and co-IP [192]. However, these methods have limitations. In the yeast two-hybrid system, the yeast cell may not provide a suitable cellular environment for in vivo PPIs that occur in mammalian cells; FRET measured by fluorescence lifetime microscopy is expensive and low throughput, which makes it most suitable for hypothesis-driven experiments; with affinity chromatography and co-IP, it is difficult to tell between direct and indirect PPIs.
CXMS uses chemical agents to link two amino acids that are within a certain spatial distance (usually 7–40 Å, depending on different cross-linkers) and can react with the cross-linker via valence bonds [193]. The cross-linked products are analyzed by MS-based proteomics to identify interactors and direct interface regions [194, 195]. The advantages of CXMS include relatively low cost, suitability for detecting transient and loose interactions, ease of excluding contaminants, and the ability to differentiate between direct and indirect interactions. The last decade has seen an increase in studies employing CXMS for protein complex analysis [196]. The first application of CXMS for the study of synaptic protein complexes was published recently, in which researchers identified nearly 12,000 unique cross-links within and between 2362 proteins [197]. Most cross-links identified here were loop links, i.e., the cross-linker reacted with two amino acids within the same protein; nevertheless, it marked the first direct PPI map of mouse brain synapses and was seen as an illuminating proof of the concept of applying CXMS in complex biological samples.
Proximity proteomics and the exploration of protein neighbors
Another emerging method for studying protein interactions is proximity proteomics. For this method, certain enzymes are attached to the target protein to generate small molecules, which can covalently label proteins. The short half-life of the small molecules ensures that only the close neighbors of the target protein are labeled for subsequent enrichment and analysis [198,199,200]. It has a larger spatial range (from ten to hundreds of nanometers) than chemical cross-linking, enabling its use for exploring weak and transient protein interactions as they occur in vivo [201, 202]. Proximity proteomics has found its arena in the study of synapses, where receptors, scaffolding proteins, kinases, cytoskeleton proteins, and various regulatory molecules are closely arranged to form a precisely controlled, highly orchestrated signaling network, and it has demonstrated its usefulness in the analysis of excitatory synaptic clefts, a subcellular compartment that cannot be isolated by traditional biochemical methods [28]. Based on the enzyme used to generate the small tags, proximity labeling methods can essentially be classified into two categories: peroxidase-based and biotin ligase-based proximity labeling [203]. A landmark study of the application of proximity proteomics in neuroscience was published in 2016, in which unique features of excitatory and inhibitory synaptic clefts was revealed by the peroxidase-based APEX proteomics [29]. In the same year, researchers from Duke University and their collaborators employed a biotin ligase-based proximity labeling approach, the proximity-dependent biotin identification (BioID) approach, to identify inhibitory postsynaptic proteins, and uncovered a functionally diverse protein assemblage that regulates postsynaptic inhibition [60]. Three years later, the same group published their study of nascent synapses by in vivo version of BioID (iBioID) and revealed a novel mechanism underlying excitatory synaptogenesis [204]. The group also shared their protocol for the iBioID proximity proteomics method [205], providing an easily adapted technique for the study of substructures that are difficult to purify. Although most proximity labeling methods are still in the early stages of application and have some limitations such as stimulations to cells or poor labeling efficiency [203], its unique advantages (e.g., not limited by the availability of antibodies, the ability to specifically label target proteins in a certain cell type when combined with conditional gene expression techniques) are increasingly being noticed by researchers. It complements classical IP methods, showing potential for a comprehensive map of synaptic PPI network.
Appealing biological questions
Depicting the spatial molecular landscape of the brain
As the most complex organ in mammals, the brain is anatomically divided into different regions, each with distinct cell types and arrangements, and associated with various physiological functions and pathological processes [206, 207]. A brain region- and cell type-specific proteome map is of unique importance for understanding brain functioning and the molecular basis of related diseases. Large-scale proteome analysis of ten brain regions and cell types in mice revealed that, in line with the functional specificity, protein expression patterns showed cell type- and region-related diversity [208]. Furthermore, subregions within an area can be distinguished, such as the different protein expression profiles for CA2 and CA1 subregions within the hippocampus [209]. In 2008, a quantitative proteomic study comparing PSD preparations from the mouse cortex, midbrain, cerebellum, and hippocampus revealed that the hippocampus had the highest kinase and phosphatase contents, with relatively high overall phosphorylation levels, indicative of a large amount of synaptic signal transduction in this area [210]. Researchers have also found significant molecular heterogeneity between PSD fractions purified from rat forebrain and cerebellum [211]. A more recent study showed high regional diversity of the mouse postsynaptic proteome, with up to 74% of proteins exhibiting differential expression and a unique compositional signature in each region [212]. The functions of specific brain regions in pathological processes have also been under investigation: a team from Switzerland obtained PSD proteomes from striatal and hippocampal tissues of adult Shank3 mutant mice and revealed novel brain region-specific alterations associated with ASD risk genes [160]. Recently, the same group performed proteomic analysis of the pre- and postsynaptic compartments in different brain regions of male and female adult mice and reported new sets of the region- and sex-specific synaptic proteins [213]. Another collaborative study used Shank3B−/− mutant mice to show that during the second and third postnatal weeks, the striatal synaptic proteome is extensively remodeled, implicating the abnormal maturation of striatal circuits in the behavioral deficits exhibited by these mice [214]. Regional profiling of PSD proteins has also been conducted using human neocortex samples and, by integrated analysis of genetic and proteomic data, provided relevant information to identify brain regions involved in behavioral pathology [215].
The brain comprises a complex network of various cell types and fiber bundles, and efforts have been made to reveal the protein components of specific synaptic types, such as the parallel fiber-Purkinje cell synapse [216]. Cell type-specific proteomics is an emerging frontier leading to discoveries of functional and molecular diversities, including recent analyses on brain mitochondria [217] and the psychiatric disorder-related protein DISC1 interactome [218]. Furthermore, proteins can have more than one subcellular location, executing distinct functions with different partners. Many PSD proteins form distinct multiprotein complexes inside and outside the PSD [61], indicating the need for spatial profiling on a more precise scale. The relationships and regulatory mechanisms of these complexes may be the key to understanding synapse-to-nucleus signaling.
Tracing the temporal changes during development
Brain development is a complex and protracted process involving the formation, strengthening, and elimination of synapses [219]. The importance of developmental analyses has long been recognized, and the first large-scale proteomic analysis of synaptic development was published in 2007, which utilized stable isotope labeling in mammals to quantitatively compare synaptic proteins in mouse cerebellum at four postnatal stages (post-natal day 1 (p1), p10, p20, and p45) of development [220]. These researchers used the same approach in a subsequent study of synaptosomal and mitochondrial fractions from three rat brain regions at four postnatal time points to identify novel regulators of neurodevelopment [221]. Other quantitative proteomic approaches, including the multiplexed iTRAQ method [222] and label-free quantitation [61], have also been utilized in the study of the synaptic proteome dynamics during rodent brain development. Findings from the spatiotemporal profiling of the PSD proteome indicated that postnatal day 7 is a key time point in synaptic development when a complete three-layer scaffold structure of PSD first forms [61]. On the timescale of lifespan, tools have been developed to identify age-dependent gene regulatory events and the results showed the postnatal developmental changes in the synaptic proteome and their relevance to the age of disease onset for specific neurological disorders, including schizophrenia [223]. More recently, the molecular and morphological features of five billion excitatory synapses were depicted across the mouse brain from birth to old age, representing the most comprehensive analysis of synapses during development and aging so far [224].
Unveiling the mysteries of brain disorders
Brain disorders, including developmental, psychiatric, and neurodegenerative disorders, affect individuals of different ages and represent a major challenge to human health. Proper brain function depends on orderly and controlled neuronal excitation and inhibition. As the basic connecting and computing unit in the central nervous system, synapses are crucial to all forms of brain activity and synaptic proteins had been implicated in many brain disorders [225]. Thus, the development of effective therapy for these disorders depends on an in-depth understanding of the molecular and functional organization of synapses.
Proteomics has long played an indispensable role in the discovery and verification of biomarkers for brain disorders [17], and studies are increasingly oriented towards exploring the pathological mechanisms underlying various brain disorders. In a recently published work, researchers found that the astrocytic cystine/glutamate antiporter system xc(-) regulated corticostriatal neurotransmission, and influenced social preference and repetitive behavior in mice, providing important clues toward the neural mechanisms related to ASD and obsessive–compulsive disorder [226]. Synaptic proteome abnormalities related to several psychiatric disorders have been revealed in the last decade, and a number of signaling pathways are involved [54, 160, 227, 228]. For example, GABAA receptor blockade in the hippocampus improved synaptic plasticity in an Alzheimer’s disease mouse model, leading to the identification of several proteins that contribute to learning and memory functions in this disease [229]. We have studied the spatial distribution and maturation of PSD interactomes, as well as the phosphorylation network regulated by long-term potentiation, and found that these networks converge at a number of highly connected nodes, which also represent PSD risk factors for many psychiatric disorders [61, 102]. Thus, pathological processes underlying these disorders likely involve the loss of PSD protein interactions and the subsequent dysregulation of synaptic signaling.
Future perspectives
Recent years have seen solid progress in the research field of synapses boosted by the orchestration of genetic, genomic, and proteomic studies. In 2019, a large-scale collaborative study established the Synaptic Gene Ontology (SynGO) database, an evidence-based, expert-curated knowledge base for “discovering the synapse” [230]. It features detailed and traceable annotations of synaptic proteins, offering a universal reference and a powerful yet convenient online platform to facilitate the study of synapses.
In the latest study from one of the leading labs in the field of proteomics, researchers combined technologies such as DIA-proteomics, DIA-phosphoproteomics, proximity labeling, and cross-linking MS to reveal how a mutation in the tumor-driving gene Dyrk2 affects downstream cellular processes [231], demonstrating the great potential of multilayered proteomics for decoding the molecular machinery of life. At the same time, with continued improvements in mass spectrometers and sample processing methods, single-cell proteomics has stepped onto the stage, making more specific and accurate descriptions of molecular dynamics feasible [232]. Specific to the study of synapses, with the combination of genetic labeling and imaging methods, spatial diversity of synaptic proteins could now be seen with single-synapse resolution across all regions of the mouse brain [233, 234]. These new methods have recently been applied on a brain-wide scale to examine the protein composition of individual synapses throughout the mouse lifespan, and spatiotemporal changes of synapse composition with potential relevance to intellectual ability and behavioral disorders have been discovered [224]. These brilliant works represent the beginnings of synaptomics and will be extended with more proteins being examined in various physiological and pathological conditions, delivering new insights into our understanding of the synapses and the brain.
Availability of data and materials
Not applicable.
Abbreviations
- 2-DE:
-
Two-dimensional electrophoresis
- ASD:
-
Autism spectrum disorder
- BioID:
-
Biotin identification
- CXMS:
-
Chemical cross-linking in combination with mass spectrometry
- co-IP:
-
Co-immunoprecipitation
- DDA:
-
Data dependent acquisition
- DIA:
-
Data independent acquisition
- FRET:
-
Förster resonance energy transfer
- LC–MS:
-
Liquid chromatography mass spectrometry
- MALDI-MS:
-
Matrix-assisted laser desorption ionization mass spectrometry
- MS:
-
Mass spectrometry
- PAZ:
-
Presynaptic active zone
- PPI:
-
Protein–protein interaction
- PSD:
-
Postsynaptic density
- PTM:
-
Posttranslational modification
- RP-HPLC:
-
Reversed-phase high performance liquid chromatography
- SCX:
-
Strong cation-exchange
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SILAC:
-
Stable isotope labeling by amino acids in cell culture
- SWATH-MS:
-
Sequential window acquisition of all theoretical fragment ion mass spectra
References
O’Rourke NA, Weiler NC, Micheva KD, Smith SJ. Deep molecular diversity of mammalian synapses: why it matters and how to measure it. Nat Rev Neurosci. 2012;13(6):365–79. https://doi.org/10.1038/nrn3170.
Taoufik E, Kouroupi G, Zygogianni O, Matsas R. Synaptic dysfunction in neurodegenerative and neurodevelopmental diseases: an overview of induced pluripotent stem-cell-based disease models. Open Biol. 2018;8(9):180138. https://doi.org/10.1098/rsob.180138.
Volk L, Chiu SL, Sharma K, Huganir RL. Glutamate synapses in human cognitive disorders. Annu Rev Neurosci. 2015;38:127–49. https://doi.org/10.1146/annurev-neuro-071714-033821.
Chevalier F. Highlights on the capacities of “Gel-based” proteomics. Proteome Sci. 2010;8:23. https://doi.org/10.1186/1477-5956-8-23.
Baggerman G, Vierstraete E, De Loof A, Schoofs L. Gel-based versus gel-free proteomics: a review. Comb Chem High Throughput Screen. 2005;8(8):669–77. https://doi.org/10.2174/138620705774962490.
Chandramouli K, Qian PY. Proteomics: challenges, techniques and possibilities to overcome biological sample complexity. Hum Genomics Proteomics. 2009;2009:1–22. https://doi.org/10.4061/2009/239204.
Ankney JA, Muneer A, Chen X. Relative and absolute quantitation in mass spectrometry-based proteomics. Annu Rev Anal Chem (Palo Alto Calif). 2018;11(1):49–77. https://doi.org/10.1146/annurev-anchem-061516-045357.
Yates JR, Ruse CI, Nakorchevsky A. Proteomics by mass spectrometry: approaches, advances, and applications. Annu Rev Biomed Eng. 2009;11:49–79. https://doi.org/10.1146/annurev-bioeng-061008-124934.
Larance M, Lamond AI. Multidimensional proteomics for cell biology. Nat Rev Mol Cell Biol. 2015;16(5):269–80. https://doi.org/10.1038/nrm3970.
Fountoulakis M, Schuller E, Hardmeier R, Berndt P, Lubec G. Rat brain proteins: two-dimensional protein database and variations in the expression level. Electrophoresis. 1999;20(18):3572–9. https://doi.org/10.1002/(SICI)1522-2683(19991201)20:18%3c3572::AID-ELPS3572%3e3.0.CO;2-T.
Bayes A, Grant SG. Neuroproteomics: understanding the molecular organization and complexity of the brain. Nat Rev Neurosci. 2009;10(9):635–46. https://doi.org/10.1038/nrn2701.
Cookson MR. Proteomics: techniques and applications in neuroscience. J Neurochem. 2019;151(4):394–6. https://doi.org/10.1111/jnc.14867.
Hosp F, Mann M. A primer on concepts and applications of proteomics in neuroscience. Neuron. 2017;96(3):558–71. https://doi.org/10.1016/j.neuron.2017.09.025.
Liao L, McClatchy DB, Yates JR. Shotgun proteomics in neuroscience. Neuron. 2009;63(1):12–26. https://doi.org/10.1016/j.neuron.2009.06.011.
Drabik A, Bierczynska-Krzysik A, Bodzon-Kulakowska A, Suder P, Kotlinska J, Silberring J. Proteomics in neurosciences. Mass Spectrom Rev. 2007;26(3):432–50. https://doi.org/10.1002/mas.20131.
Husi H, Grant SG. Proteomics of the nervous system. Trends Neurosci. 2001;24(5):259–66. https://doi.org/10.1016/s0166-2236(00)01792-6.
Murtaza N, Uy J, Singh KK. Emerging proteomic approaches to identify the underlying pathophysiology of neurodevelopmental and neurodegenerative disorders. Mol Autism. 2020;11(1):27. https://doi.org/10.1186/s13229-020-00334-5.
Bayes A, Collins MO, Reig-Viader R, Gou G, Goulding D, Izquierdo A, Choudhary JS, Emes RD, Grant SG. Evolution of complexity in the zebrafish synapse proteome. Nat Commun. 2017;8:14613. https://doi.org/10.1038/ncomms14613.
Lassek M, Weingarten J, Volknandt W. The synaptic proteome. Cell Tissue Res. 2015;359(1):255–65. https://doi.org/10.1007/s00441-014-1943-4.
Sapp E, Seeley C, Iuliano M, Weisman E, Vodicka P, DiFiglia M, Kegel-Gleason KB. Protein changes in synaptosomes of Huntington’s disease knock-in mice are dependent on age and brain region. Neurobiol Dis. 2020;141:104950. https://doi.org/10.1016/j.nbd.2020.104950.
Stasyk T, Huber LA. Zooming in fractionation strategies in proteomics. Proteomics. 2004;4(12):3704–16. https://doi.org/10.1002/pmic.200401048.
Boyken J, Gronborg M, Riedel D, Urlaub H, Jahn R, Chua JJ. Molecular profiling of synaptic vesicle docking sites reveals novel proteins but few differences between glutamatergic and GABAergic synapses. Neuron. 2013;78(2):285–97. https://doi.org/10.1016/j.neuron.2013.02.027.
Gronborg M, Pavlos NJ, Brunk I, Chua JJ, Munster-Wandowski A, Riedel D, Ahnert-Hilger G, Urlaub H, Jahn R. Quantitative comparison of glutamatergic and GABAergic synaptic vesicles unveils selectivity for few proteins including MAL2, a novel synaptic vesicle protein. J Neurosci. 2010;30(1):2–12. https://doi.org/10.1523/JNEUROSCI.4074-09.2010.
Morciano M, Beckhaus T, Karas M, Zimmermann H, Volknandt W. The proteome of the presynaptic active zone: from docked synaptic vesicles to adhesion molecules and maxi-channels. J Neurochem. 2009;108(3):662–75. https://doi.org/10.1111/j.1471-4159.2008.05824.x.
Morciano M, Burre J, Corvey C, Karas M, Zimmermann H, Volknandt W. Immunoisolation of two synaptic vesicle pools from synaptosomes: a proteomics analysis. J Neurochem. 2005;95(6):1732–45. https://doi.org/10.1111/j.1471-4159.2005.03506.x.
Heller EA, Zhang W, Selimi F, Earnheart JC, Slimak MA, Santos-Torres J, Ibanez-Tallon I, Aoki C, Chait BT, Heintz N. The biochemical anatomy of cortical inhibitory synapses. PLoS ONE. 2012;7(6):e39572. https://doi.org/10.1371/journal.pone.0039572.
Biesemann C, Gronborg M, Luquet E, Wichert SP, Bernard V, Bungers SR, Cooper B, Varoqueaux F, Li L, Byrne JA, Urlaub H, Jahn O, Brose N, Herzog E. Proteomic screening of glutamatergic mouse brain synaptosomes isolated by fluorescence activated sorting. EMBO J. 2014;33(2):157–70. https://doi.org/10.1002/embj.201386120.
Cijsouw T, Ramsey AM, Lam TT, Carbone BE, Blanpied TA, Biederer T. Mapping the proteome of the synaptic cleft through proximity labeling reveals new cleft proteins. Proteomes. 2018;6(4):48. https://doi.org/10.3390/proteomes6040048.
Loh KH, Stawski PS, Draycott AS, Udeshi ND, Lehrman EK, Wilton DK, Svinkina T, Deerinck TJ, Ellisman MH, Stevens B, Carr SA, Ting AY. Proteomic analysis of unbounded cellular compartments: synaptic clefts. Cell. 2016;166(5):1295–307. https://doi.org/10.1016/j.cell.2016.07.041.
Whittaker VP, Michaelson IA, Kirkland RJ. The separation of synaptic vesicles from nerve-ending particles ('synaptosomes’). Biochem J. 1964;90(2):293–303. https://doi.org/10.1042/bj0900293.
Stevens SM Jr, Zharikova AD, Prokai L. Proteomic analysis of the synaptic plasma membrane fraction isolated from rat forebrain. Brain Res Mol Brain Res. 2003;117(2):116–28. https://doi.org/10.1016/s0169-328x(03)00282-1.
Heo S, Diering GH, Na CH, Nirujogi RS, Bachman JL, Pandey A, Huganir RL. Identification of long-lived synaptic proteins by proteomic analysis of synaptosome protein turnover. Proc Natl Acad Sci USA. 2018;115(16):E3827–36. https://doi.org/10.1073/pnas.1720956115.
Barthet G, Mulle C. Presynaptic failure in Alzheimer’s disease. Prog Neurobiol. 2020;194:101801. https://doi.org/10.1016/j.pneurobio.2020.101801.
Lee W, Koh S, Hwang S, Kim SH. Presynaptic dysfunction by familial factors in Parkinson disease. Int Neurourol J. 2018;22(Suppl 3):S115-121. https://doi.org/10.5213/inj.1836216.108.
Abul-Husn NS, Bushlin I, Moron JA, Jenkins SL, Dolios G, Wang R, Iyengar R, Ma’ayan A, Devi LA. Systems approach to explore components and interactions in the presynapse. Proteomics. 2009;9(12):3303–15. https://doi.org/10.1002/pmic.200800767.
Volknandt W, Karas M. Proteomic analysis of the presynaptic active zone. Exp Brain Res. 2012;217(3–4):449–61. https://doi.org/10.1007/s00221-012-3031-x.
Lenselink AM, Rotaru DC, Li KW, van Nierop P, Rao-Ruiz P, Loos M, van der Schors R, Gouwenberg Y, Wortel J, Mansvelder HD, Smit AB, Spijker S. Strain differences in presynaptic function: PROTEOMICS, ULTRASTRUCTURE, AND PHYSIOLOGY OF HIPPOCAMPAL SYNAPSES IN DBA/2J AND C57Bl/6J MICE. J Biol Chem. 2015;290(25):15635–45. https://doi.org/10.1074/jbc.M114.628776.
Stauch KL, Villeneuve LM, Totusek S, Lamberty B, Ciborowski P, Fox HS. Quantitative proteomics of presynaptic mitochondria reveal an overexpression and biological relevance of neuronal MitoNEET in postnatal brain development. Dev Neurobiol. 2019;79(4):370–86. https://doi.org/10.1002/dneu.22684.
Burre J, Volknandt W. The synaptic vesicle proteome. J Neurochem. 2007;101(6):1448–62. https://doi.org/10.1111/j.1471-4159.2007.04453.x.
Palay SL. Synapses in the central nervous system. J Biophys Biochem Cytol. 1956;2(4 Suppl):193–202. https://doi.org/10.1083/jcb.2.4.193.
Wilson RS, Rauniyar N, Sakaue F, Lam TT, Williams KR, Nairn AC. Development of targeted mass spectrometry-based approaches for quantitation of proteins enriched in the postsynaptic density (PSD). Proteomes. 2019;7(2):12. https://doi.org/10.3390/proteomes7020012.
Distler U, Schmeisser MJ, Pelosi A, Reim D, Kuharev J, Weiczner R, Baumgart J, Boeckers TM, Nitsch R, Vogt J, Tenzer S. In-depth protein profiling of the postsynaptic density from mouse hippocampus using data-independent acquisition proteomics. Proteomics. 2014;14(21–22):2607–13. https://doi.org/10.1002/pmic.201300520.
Collins MO, Husi H, Yu L, Brandon JM, Anderson CN, Blackstock WP, Choudhary JS, Grant SG. Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J Neurochem. 2006;97(Suppl 1):16–23. https://doi.org/10.1111/j.1471-4159.2005.03507.x.
Peng J, Kim MJ, Cheng D, Duong DM, Gygi SP, Sheng M. Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J Biol Chem. 2004;279(20):21003–11. https://doi.org/10.1074/jbc.M400103200.
Satoh K, Takeuchi M, Oda Y, Deguchi-Tawarada M, Sakamoto Y, Matsubara K, Nagasu T, Takai Y. Identification of activity-regulated proteins in the postsynaptic density fraction. Genes Cells. 2002;7(2):187–97. https://doi.org/10.1046/j.1356-9597.2001.00505.x.
Walikonis RS, Jensen ON, Mann M, Provance DW Jr, Mercer JA, Kennedy MB. Identification of proteins in the postsynaptic density fraction by mass spectrometry. J Neurosci. 2000;20(11):4069–80.
Walsh MJ, Kuruc N. The postsynaptic density: constituent and associated proteins characterized by electrophoresis, immunoblotting, and peptide sequencing. J Neurochem. 1992;59(2):667–78. https://doi.org/10.1111/j.1471-4159.1992.tb09421.x.
Cho KO, Hunt CA, Kennedy MB. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron. 1992;9(5):929–42. https://doi.org/10.1016/0896-6273(92)90245-9.
Bayes A, Collins MO, Croning MD, van de Lagemaat LN, Choudhary JS, Grant SG. Comparative study of human and mouse postsynaptic proteomes finds high compositional conservation and abundance differences for key synaptic proteins. PLoS ONE. 2012;7(10):e46683. https://doi.org/10.1371/journal.pone.0046683.
Roeper J. Closing gaps in brain disease-from overlapping genetic architecture to common motifs of synapse dysfunction. Curr Opin Neurobiol. 2018;48:45–51. https://doi.org/10.1016/j.conb.2017.09.007.
Grant SG. Synaptopathies: diseases of the synaptome. Curr Opin Neurobiol. 2012;22(3):522–9. https://doi.org/10.1016/j.conb.2012.02.002.
Bayes A, van de Lagemaat LN, Collins MO, Croning MD, Whittle IR, Choudhary JS, Grant SG. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat Neurosci. 2011;14(1):19–21. https://doi.org/10.1038/nn.2719.
Focking M, Lopez LM, English JA, Dicker P, Wolff A, Brindley E, Wynne K, Cagney G, Cotter DR. Proteomic and genomic evidence implicates the postsynaptic density in schizophrenia. Mol Psychiatry. 2015;20(4):424–32. https://doi.org/10.1038/mp.2014.63.
Focking M, Dicker P, Lopez LM, Hryniewiecka M, Wynne K, English JA, Cagney G, Cotter DR. Proteomic analysis of the postsynaptic density implicates synaptic function and energy pathways in bipolar disorder. Transl Psychiatry. 2016;6(11):e959. https://doi.org/10.1038/tp.2016.224.
Zolochevska O, Bjorklund N, Woltjer R, Wiktorowicz JE, Taglialatela G. Postsynaptic proteome of non-demented individuals with alzheimer’s disease neuropathology. J Alzheimers Dis. 2018;65(2):659–82. https://doi.org/10.3233/JAD-180179.
Kaizuka T, Takumi T. Postsynaptic density proteins and their involvement in neurodevelopmental disorders. J Biochem. 2018;163(6):447–55. https://doi.org/10.1093/jb/mvy022.
Reig-Viader R, Sindreu C, Bayes A. Synaptic proteomics as a means to identify the molecular basis of mental illness: are we getting there? Prog Neuropsychopharmacol Biol Psychiatry. 2018;84:353–61. https://doi.org/10.1016/j.pnpbp.2017.09.011.
Gao R, Penzes P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr Mol Med. 2015;15(2):146–67. https://doi.org/10.2174/1566524015666150303003028.
Sheng M, Kim E. The postsynaptic organization of synapses. Cold Spring Harb Perspect Biol. 2011;3(12):a005678. https://doi.org/10.1101/cshperspect.a005678.
Uezu A, Kanak DJ, Bradshaw TW, Soderblom EJ, Catavero CM, Burette AC, Weinberg RJ, Soderling SH. Identification of an elaborate complex mediating postsynaptic inhibition. Science. 2016;353(6304):1123–9. https://doi.org/10.1126/science.aag0821.
Li J, Zhang W, Yang H, Howrigan DP, Wilkinson B, Souaiaia T, Evgrafov OV, Genovese G, Clementel VA, Tudor JC, Abel T, Knowles JA, Neale BM, Wang K, Sun F, Coba MP. Spatiotemporal profile of postsynaptic interactomes integrates components of complex brain disorders. Nat Neurosci. 2017;20(8):1150–61. https://doi.org/10.1038/nn.4594.
Delint-Ramirez I, Fernandez E, Bayes A, Kicsi E, Komiyama NH, Grant SG. In vivo composition of NMDA receptor signaling complexes differs between membrane subdomains and is modulated by PSD-95 and PSD-93. J Neurosci. 2010;30(24):8162–70. https://doi.org/10.1523/JNEUROSCI.1792-10.2010.
Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci. 2000;3(7):661–9. https://doi.org/10.1038/76615.
Fernandez E, Collins MO, Uren RT, Kopanitsa MV, Komiyama NH, Croning MD, Zografos L, Armstrong JD, Choudhary JS, Grant SG. Targeted tandem affinity purification of PSD-95 recovers core postsynaptic complexes and schizophrenia susceptibility proteins. Mol Syst Biol. 2009;5:269. https://doi.org/10.1038/msb.2009.27.
Grant SG, Marshall MC, Page KL, Cumiskey MA, Armstrong JD. Synapse proteomics of multiprotein complexes: en route from genes to nervous system diseases. Hum Mol Genet. 2005;14:R225–34. https://doi.org/10.1093/hmg/ddi330.
Grant SG. Synapse signalling complexes and networks: machines underlying cognition. BioEssays. 2003;25(12):1229–35. https://doi.org/10.1002/bies.10381.
Grant SG, Blackstock WP. Proteomics in neuroscience: from protein to network. J Neurosci. 2001;21(21):8315–8.
Grant SG, Husi H. Proteomics of multiprotein complexes: answering fundamental questions in neuroscience. Trends Biotechnol. 2001;19(10):S49-54. https://doi.org/10.1016/S0167-7799(01)01799-1.
Fernandez E, Collins MO, Frank RAW, Zhu F, Kopanitsa MV, Nithianantharajah J, Lempriere SA, Fricker D, Elsegood KA, McLaughlin CL, Croning MDR, McLean C, Armstrong JD, Hill WD, Deary IJ, Cencelli G, Bagni C, Fromer M, Purcell SM, Pocklington AJ, Choudhary JS, Komiyama NH, Grant SGN. Arc requires PSD95 for assembly into postsynaptic complexes involved with neural dysfunction and intelligence. Cell Rep. 2017;21(3):679–91. https://doi.org/10.1016/j.celrep.2017.09.045.
Dosemeci A, Makusky AJ, Jankowska-Stephens E, Yang X, Slotta DJ, Markey SP. Composition of the synaptic PSD-95 complex. Mol Cell Proteomics. 2007;6(10):1749–60. https://doi.org/10.1074/mcp.M700040-MCP200.
Lee Y, Ryu JR, Kang H, Kim Y, Kim S, Zhang Y, Jin C, Cho HM, Kim WK, Sun W, Han K. Characterization of the zinc-induced Shank3 interactome of mouse synaptosome. Biochem Biophys Res Commun. 2017;494(3–4):581–6. https://doi.org/10.1016/j.bbrc.2017.10.143.
Krapivinsky G, Medina I, Krapivinsky L, Gapon S, Clapham DE. SynGAP-MUPP1-CaMKII synaptic complexes regulate p38 MAP kinase activity and NMDA receptor-dependent synaptic AMPA receptor potentiation. Neuron. 2004;43(4):563–74. https://doi.org/10.1016/j.neuron.2004.08.003.
Zhang Q, Yang H, Gao H, Liu X, Li Q, Rong R, Liu Z, Wei XE, Kong L, Xu Y, Rong L. PSD-93 interacts with SynGAP and promotes SynGAP Ubiquitination and Ischemic Brain injury in mice. Transl Stroke Res. 2020;11(5):1137–47. https://doi.org/10.1007/s12975-020-00795-z.
Zhang J, Wang G, He WW, Losh M, Berry-Kravis E, Funk WE. Expression and characterization of human fragile X mental retardation protein isoforms and interacting proteins in human cells. Proteomics Insights. 2019. https://doi.org/10.1177/1178641818825268.
Pasciuto E, Bagni C. SnapShot: FMRP interacting proteins. Cell. 2014;159(1):218–218. https://doi.org/10.1016/j.cell.2014.08.036.
Schenck A, Bardoni B, Moro A, Bagni C, Mandel JL. A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc Natl Acad Sci USA. 2001;98(15):8844–9. https://doi.org/10.1073/pnas.151231598.
Pandya NJ, Klaassen RV, van der Schors RC, Slotman JA, Houtsmuller A, Smit AB, Li KW. Group 1 metabotropic glutamate receptors 1 and 5 form a protein complex in mouse hippocampus and cortex. Proteomics. 2016;16(20):2698–705. https://doi.org/10.1002/pmic.201500400.
Ramos C, Chardonnet S, Marchand CH, Decottignies P, Ango F, Daniel H, Le Marechal P. Native presynaptic metabotropic glutamate receptor 4 (mGluR4) interacts with exocytosis proteins in rat cerebellum. J Biol Chem. 2012;287(24):20176–86. https://doi.org/10.1074/jbc.M112.347468.
Francesconi A, Kumari R, Zukin RS. Proteomic analysis reveals novel binding partners of metabotropic glutamate receptor 1. J Neurochem. 2009;108(6):1515–25. https://doi.org/10.1111/j.1471-4159.2009.05913.x.
Farr CD, Gafken PR, Norbeck AD, Doneanu CE, Stapels MD, Barofsky DF, Minami M, Saugstad JA. Proteomic analysis of native metabotropic glutamate receptor 5 protein complexes reveals novel molecular constituents. J Neurochem. 2004;91(2):438–50. https://doi.org/10.1111/j.1471-4159.2004.02735.x.
Zeng M, Shang Y, Guo T, He Q, Yung WH, Liu K, Zhang M. A binding site outside the canonical PDZ domain determines the specific interaction between Shank and SAPAP and their function. Proc Natl Acad Sci USA. 2016;113(22):E3081-3090. https://doi.org/10.1073/pnas.1523265113.
Cui H, Hayashi A, Sun HS, Belmares MP, Cobey C, Phan T, Schweizer J, Salter MW, Wang YT, Tasker RA, Garman D, Rabinowitz J, Lu PS, Tymianski M. PDZ protein interactions underlying NMDA receptor-mediated excitotoxicity and neuroprotection by PSD-95 inhibitors. J Neurosci. 2007;27(37):9901–15. https://doi.org/10.1523/JNEUROSCI.1464-07.2007.
Wilkinson B, Li J, Coba MP. Synaptic GAP and GEF complexes cluster proteins essential for GTP signaling. Sci Rep. 2017;7(1):5272. https://doi.org/10.1038/s41598-017-05588-3.
Paskus JD, Tian C, Fingleton E, Shen C, Chen X, Li Y, Myers SA, Badger JD, Bemben MA, Herring BE, Roche KW. Synaptic kalirin-7 and trio interactomes reveal a GEF protein-dependent neuroligin-1 mechanism of action. Cell Rep. 2019;29(10):2944–52. https://doi.org/10.1016/j.celrep.2019.10.115.
Zhao B, Luo T, Li T, Li Y, Zhang J, Shan Y, Wang X, Yang L, Zhou F, Zhu Z, Alzheimer’s Disease Neuroimaging, I., Pediatric Imaging, N., Genetics, Zhu H. Genome-wide association analysis of 19,629 individuals identifies variants influencing regional brain volumes and refines their genetic co-architecture with cognitive and mental health traits. Nat Genet. 2019;51(11):1637–44. https://doi.org/10.1038/s41588-019-0516-6.
Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, Pallesen J, Agerbo E, Andreassen OA, Anney R, Awashti S, Belliveau R, Bettella F, Buxbaum JD, Bybjerg-Grauholm J, Baekvad-Hansen M, Cerrato F, Chambert K, Christensen JH, Churchhouse C, Dellenvall K, Demontis D, De Rubeis S, Devlin B, Djurovic S, Dumont AL, Goldstein JI, Hansen CS, Hauberg ME, Hollegaard MV, Hope S, Howrigan DP, Huang H, Hultman CM, Klei L, Maller J, Martin J, Martin AR, Moran JL, Nyegaard M, Naerland T, Palmer DS, Palotie A, Pedersen CB, Pedersen MG, dPoterba T, Poulsen JB, Pourcain BS, Qvist P, Rehnstrom K, Reichenberg A, Reichert J, Robinson EB, Roeder K, Roussos P, Saemundsen E, Sandin S, Satterstrom FK, Davey Smith G, Stefansson H, Steinberg S, Stevens CR, Sullivan PF, Turley P, Walters GB, Xu X, Autism Spectrum Disorder Working Group of the Psychiatric Genomics, C., Bupgen, Major Depressive Disorder Working Group of the Psychiatric Genomics, C., andMe Research, T, Stefansson K, Geschwind DH, Nordentoft M, Hougaard DM, Werge T, Mors O, Mortensen PB, Neale BM, Daly MJ, Borglum AD. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet. 2019;51(3):431–44. https://doi.org/10.1038/s41588-019-0344-8.
Li Z, Chen J, Yu H, He L, Xu Y, Zhang D, Yi Q, Li C, Li X, Shen J, Song Z, Ji W, Wang M, Zhou J, Chen B, Liu Y, Wang J, Wang P, Yang P, Wang Q, Feng G, Liu B, Sun W, Li B, He G, Li W, Wan C, Xu Q, Li W, Wen Z, Liu K, Huang F, Ji J, Ripke S, Yue W, Sullivan PF, O’Donovan MC, Shi Y. Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat Genet. 2017;49(11):1576–83. https://doi.org/10.1038/ng.3973.
Kanazawa T, Bousman CA, Liu C, Everall IP. Schizophrenia genetics in the genome-wide era: a review of Japanese studies. NPJ Schizophr. 2017;3(1):27. https://doi.org/10.1038/s41537-017-0028-2.
Brown EA, Lautz JD, Davis TR, Gniffke EP, VanSchoiack AAW, Neier SC, Tashbook N, Nicolini C, Fahnestock M, Schrum AG, Smith SEP. Clustering the autisms using glutamate synapse protein interaction networks from cortical and hippocampal tissue of seven mouse models. Mol Autism. 2018;9:48. https://doi.org/10.1186/s13229-018-0229-1.
Prieto M, Folci A, Martin S. Post-translational modifications of the Fragile X Mental Retardation Protein in neuronal function and dysfunction. Mol Psychiatry. 2020;25(8):1688–703. https://doi.org/10.1038/s41380-019-0629-4.
Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: implications for Alzheimer’s disease. Neurochem Int. 2011;58(4):458–71. https://doi.org/10.1016/j.neuint.2010.12.023.
Jeong J, Paskus JD, Roche KW. Posttranslational modifications of neuroligins regulate neuronal and glial signaling. Curr Opin Neurobiol. 2017;45:130–8. https://doi.org/10.1016/j.conb.2017.05.017.
Coba MP, Pocklington AJ, Collins MO, Kopanitsa MV, Uren RT, Swamy S, Croning MD, Choudhary JS, Grant SG. Neurotransmitters drive combinatorial multistate postsynaptic density networks. Sci Signal. 2009;2(68):ra19. https://doi.org/10.1126/scisignal.2000102.
Lee HK. Synaptic plasticity and phosphorylation. Pharmacol Ther. 2006;112(3):810–32. https://doi.org/10.1016/j.pharmthera.2006.06.003.
von Stechow L, Francavilla C, Olsen JV. Recent findings and technological advances in phosphoproteomics for cells and tissues. Expert Rev Proteomics. 2015;12(5):469–87. https://doi.org/10.1586/14789450.2015.1078730.
Batalha IL, Lowe CR, Roque AC. Platforms for enrichment of phosphorylated proteins and peptides in proteomics. Trends Biotechnol. 2012;30(2):100–10. https://doi.org/10.1016/j.tibtech.2011.07.004.
Collins MO, Yu L, Coba MP, Husi H, Campuzano I, Blackstock WP, Choudhary JS, Grant SG. Proteomic analysis of in vivo phosphorylated synaptic proteins. J Biol Chem. 2005;280(7):5972–82. https://doi.org/10.1074/jbc.M411220200.
Engholm-Keller K, Waardenberg AJ, Muller JA, Wark JR, Fernando RN, Arthur JW, Robinson PJ, Dietrich D, Schoch S, Graham ME. The temporal profile of activity-dependent presynaptic phospho-signalling reveals long-lasting patterns of poststimulus regulation. PLoS Biol. 2019;17(3):e3000170. https://doi.org/10.1371/journal.pbio.3000170.
Bruning F, Noya SB, Bange T, Koutsouli S, Rudolph JD, Tyagarajan SK, Cox J, Mann M, Brown SA, Robles MS. Sleep-wake cycles drive daily dynamics of synaptic phosphorylation. Science. 2019;366(6462):eaav3617. https://doi.org/10.1126/science.aav3617.
Wang Z, Ma J, Miyoshi C, Li Y, Sato M, Ogawa Y, Lou T, Ma C, Gao X, Lee C, Fujiyama T, Yang X, Zhou S, Hotta-Hirashima N, Klewe-Nebenius D, Ikkyu A, Kakizaki M, Kanno S, Cao L, Takahashi S, Peng J, Yu Y, Funato H, Yanagisawa M, Liu Q. Quantitative phosphoproteomic analysis of the molecular substrates of sleep need. Nature. 2018;558(7710):435–9. https://doi.org/10.1038/s41586-018-0218-8.
Diering GH, Nirujogi RS, Roth RH, Worley PF, Pandey A, Huganir RL. Homer1a drives homeostatic scaling-down of excitatory synapses during sleep. Science. 2017;355(6324):511–5. https://doi.org/10.1126/science.aai8355.
Li J, Wilkinson B, Clementel VA, Hou J, O’Dell TJ, Coba MP. Long-term potentiation modulates synaptic phosphorylation networks and reshapes the structure of the postsynaptic interactome. Sci Signal. 2016;9(440):rs8. https://doi.org/10.1126/scisignal.aaf6716.
Samuels BA, Hsueh YP, Shu T, Liang H, Tseng HC, Hong CJ, Su SC, Volker J, Neve RL, Yue DT, Tsai LH. Cdk5 promotes synaptogenesis by regulating the subcellular distribution of the MAGUK family member CASK. Neuron. 2007;56(5):823–37. https://doi.org/10.1016/j.neuron.2007.09.035.
Yi JJ, Berrios J, Newbern JM, Snider WD, Philpot BD, Hahn KM, Zylka MJ. An autism-linked mutation disables phosphorylation control of UBE3A. Cell. 2015;162(4):795–807. https://doi.org/10.1016/j.cell.2015.06.045.
Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, Platt OS, Ruderfer DM, Walsh CA, Altshuler D, Chakravarti A, Tanzi RE, Stefansson K, Santangelo SL, Gusella JF, Sklar P, Wu BL, Daly MJ, Autism C. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358(7):667–75. https://doi.org/10.1056/NEJMoa075974.
Yadav S, Oses-Prieto JA, Peters CJ, Zhou J, Pleasure SJ, Burlingame AL, Jan LY, Jan YN. TAOK2 kinase MEDIATES PSD95 stability and dendritic spine maturation through Septin7 phosphorylation. Neuron. 2017;93(2):379–93. https://doi.org/10.1016/j.neuron.2016.12.006.
Coba MP. Regulatory mechanisms in postsynaptic phosphorylation networks. Curr Opin Struct Biol. 2019;54:86–94. https://doi.org/10.1016/j.sbi.2019.01.003.
Ma J, Zhang LQ, He ZX, He XX, Wang YJ, Jian YL, Wang X, Zhang BB, Su C, Lu J, Huang BQ, Zhang Y, Wang GY, Guo WX, Qiu DL, Mei L, Xiong WC, Zheng YW, Zhu XJ. Autism candidate gene DIP2A regulates spine morphogenesis via acetylation of cortactin. PLoS Biol. 2019;17(10):e3000461. https://doi.org/10.1371/journal.pbio.3000461.
Liu F, Walters KJ. Multitasking with ubiquitin through multivalent interactions. Trends Biochem Sci. 2010;35(6):352–60. https://doi.org/10.1016/j.tibs.2010.01.002.
Khatri N, Man HY. The Autism and angelman syndrome protein Ube3A/E6AP: the gene, E3 ligase ubiquitination targets and neurobiological functions. Front Mol Neurosci. 2019;12:109. https://doi.org/10.3389/fnmol.2019.00109.
Wang J, Lou SS, Wang T, Wu RJ, Li G, Zhao M, Lu B, Li YY, Zhang J, Cheng X, Shen Y, Wang X, Zhu ZC, Li MJ, Takumi T, Yang H, Yu X, Liao L, Xiong ZQ. UBE3A-mediated PTPA ubiquitination and degradation regulate PP2A activity and dendritic spine morphology. Proc Natl Acad Sci USA. 2019;116(25):12500–5. https://doi.org/10.1073/pnas.1820131116.
Brechet A, Buchert R, Schwenk J, Boudkkazi S, Zolles G, Siquier-Pernet K, Schaber I, Bildl W, Saadi A, Bole-Feysot C, Nitschke P, Reis A, Sticht H, Al-Sanna’a N, Rolfs A, Kulik A, Schulte U, Colleaux L, Abou Jamra R, Fakler B. AMPA-receptor specific biogenesis complexes control synaptic transmission and intellectual ability. Nat Commun. 2017;8:15910. https://doi.org/10.1038/ncomms15910.
Shanks NF, Cais O, Maruo T, Savas JN, Zaika EI, Azumaya CM, Yates JR 3rd, Greger I, Nakagawa T. Molecular dissection of the interaction between the AMPA receptor and cornichon homolog-3. J Neurosci. 2014;34(36):12104–20. https://doi.org/10.1523/JNEUROSCI.0595-14.2014.
Shanks NF, Savas JN, Maruo T, Cais O, Hirao A, Oe S, Ghosh A, Noda Y, Greger IH, Yates JR 3rd, Nakagawa T. Differences in AMPA and kainate receptor interactomes facilitate identification of AMPA receptor auxiliary subunit GSG1L. Cell Rep. 2012;1(6):590–8. https://doi.org/10.1016/j.celrep.2012.05.004.
Schwenk J, Harmel N, Brechet A, Zolles G, Berkefeld H, Muller CS, Bildl W, Baehrens D, Huber B, Kulik A, Klocker N, Schulte U, Fakler B. High-resolution proteomics unravel architecture and molecular diversity of native AMPA receptor complexes. Neuron. 2012;74(4):621–33. https://doi.org/10.1016/j.neuron.2012.03.034.
Kang MG, Nuriya M, Guo Y, Martindale KD, Lee DZ, Huganir RL. Proteomic analysis of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor complexes. J Biol Chem. 2012;287(34):28632–45. https://doi.org/10.1074/jbc.M111.336644.
Falsafi SK, Ghafari M, Pollak A, Hoger H, Lubec G. Hippocampal AMPA-type receptor complexes containing GluR3 and GluR4 are paralleling training in the Multiple T-Maze. Neurochem Int. 2012;60(4):425–30. https://doi.org/10.1016/j.neuint.2012.01.005.
Santos SD, Manadas B, Duarte CB, Carvalho AL. Proteomic analysis of an interactome for long-form AMPA receptor subunits. J Proteome Res. 2010;9(4):1670–82. https://doi.org/10.1021/pr900766r.
Schwenk J, Harmel N, Zolles G, Bildl W, Kulik A, Heimrich B, Chisaka O, Jonas P, Schulte U, Fakler B, Klocker N. Functional proteomics identify cornichon proteins as auxiliary subunits of AMPA receptors. Science. 2009;323(5919):1313–9. https://doi.org/10.1126/science.1167852.
Klemmer P, Smit AB, Li KW. Proteomics analysis of immuno-precipitated synaptic protein complexes. J Proteomics. 2009;72(1):82–90. https://doi.org/10.1016/j.jprot.2008.10.005.
Ghafari M, Keihan Falsafi S, Hoger H, Bennett KL, Lubec G. Identification of new phosphorylation sites of AMPA receptors in the rat hippocampus—a resource for neuroscience research. Proteomics Clin Appl. 2015;9(9–10):808–16. https://doi.org/10.1002/prca.201400057.
Zhang F, Guo A, Liu C, Comb M, Hu B. Phosphorylation and assembly of glutamate receptors after brain ischemia. Stroke. 2013;44(1):170–6. https://doi.org/10.1161/STROKEAHA.112.667253.
Huang Y, Man HY, Sekine-Aizawa Y, Han Y, Juluri K, Luo H, Cheah J, Lowenstein C, Huganir RL, Snyder SH. S-nitrosylation of N-ethylmaleimide sensitive factor mediates surface expression of AMPA receptors. Neuron. 2005;46(4):533–40. https://doi.org/10.1016/j.neuron.2005.03.028.
Schwenk J, Baehrens D, Haupt A, Bildl W, Boudkkazi S, Roeper J, Fakler B, Schulte U. Regional diversity and developmental dynamics of the AMPA-receptor proteome in the mammalian brain. Neuron. 2014;84(1):41–54. https://doi.org/10.1016/j.neuron.2014.08.044.
Chen N, Pandya NJ, Koopmans F, Castelo-Szekelv V, van der Schors RC, Smit AB, Li KW. Interaction proteomics reveals brain region-specific AMPA receptor complexes. J Proteome Res. 2014;13(12):5695–706. https://doi.org/10.1021/pr500697b.
Campos-Jurado Y, Marti-Prats L, Moron JA, Polache A, Granero L, Hipolito L. Dose-dependent induction of CPP or CPA by intra-pVTA ethanol: role of mu opioid receptors and effects on NMDA receptors. Prog Neuropsychopharmacol Biol Psychiatry. 2020;100:109875. https://doi.org/10.1016/j.pnpbp.2020.109875.
Salek AB, Edler MC, McBride JP, Baucum AJ. Spinophilin regulates phosphorylation and interactions of the GluN2B subunit of the N-methyl-d-aspartate receptor. J Neurochem. 2019;151(2):185–203. https://doi.org/10.1111/jnc.14831.
Lu F, Shao G, Wang Y, Guan S, Burlingame AL, Liu X, Liang X, Knox R, Ferriero DM, Jiang X. Hypoxia-ischemia modifies postsynaptic GluN2B-containing NMDA receptor complexes in the neonatal mouse brain. Exp Neurol. 2018;299:65–74. https://doi.org/10.1016/j.expneurol.2017.10.005.
Wills TA, Baucum AJ 2nd, Holleran KM, Chen Y, Pasek JG, Delpire E, Tabb DL, Colbran RJ, Winder DG. Chronic intermittent alcohol disrupts the GluN2B-associated proteome and specifically regulates group I mGlu receptor-dependent long-term depression. Addict Biol. 2017;22(2):275–90. https://doi.org/10.1111/adb.12319.
Frank RA, Komiyama NH, Ryan TJ, Zhu F, O’Dell TJ, Grant SG. NMDA receptors are selectively partitioned into complexes and supercomplexes during synapse maturation. Nat Commun. 2016;7:11264. https://doi.org/10.1038/ncomms11264.
Ghafari M, Whittle N, Miklosi AG, Kotlowski C, Schmuckermair C, Berger J, Bennett KL, Singewald N, Lubec G. Dietary magnesium restriction reduces amygdala-hypothalamic GluN1 receptor complex levels in mice. Brain Struct Funct. 2015;220(4):2209–21. https://doi.org/10.1007/s00429-014-0779-8.
Bajaj G, Hau AM, Hsu P, Gafken PR, Schimerlik MI, Ishmael JE. Identification of an atypical calcium-dependent calmodulin binding site on the C-terminal domain of GluN2A. Biochem Biophys Res Commun. 2014;444(4):588–94. https://doi.org/10.1016/j.bbrc.2014.01.111.
Gautam V, Trinidad JC, Rimerman RA, Costa BM, Burlingame AL, Monaghan DT. Nedd4 is a specific E3 ubiquitin ligase for the NMDA receptor subunit GluN2D. Neuropharmacology. 2013;74:96–107. https://doi.org/10.1016/j.neuropharm.2013.04.035.
Al-Hallaq RA, Conrads TP, Veenstra TD, Wenthold RJ. NMDA di-heteromeric receptor populations and associated proteins in rat hippocampus. J Neurosci. 2007;27(31):8334–43. https://doi.org/10.1523/JNEUROSCI.2155-07.2007.
Nishioka T, Amano M, Funahashi Y, Tsuboi D, Yamahashi Y, Kaibuchi K. In vivo identification of protein kinase substrates by kinase-oriented substrate screening (KIOSS). Curr Protoc Chem Biol. 2019;11(1):e60. https://doi.org/10.1002/cpch.60.
Ghafari M, Hoger H, Keihan Falsafi S, Russo-Schlaff N, Pollak A, Lubec G. Mass spectrometrical identification of hippocampal NMDA receptor subunits NR1, NR2A-D and five novel phosphorylation sites on NR2A and NR2B. J Proteome Res. 2012;11(3):1891–6. https://doi.org/10.1021/pr201099u.
Krishnan B, Scott MT, Pollandt S, Schroeder B, Kurosky A, Shinnick-Gallagher P. Fear potentiated startle increases phospholipase D (PLD) expression/activity and PLD-linked metabotropic glutamate receptor mediated post-tetanic potentiation in rat amygdala. Neurobiol Learn Mem. 2016;128:65–79. https://doi.org/10.1016/j.nlm.2015.12.009.
Kato AS, Knierman MD, Siuda ER, Isaac JT, Nisenbaum ES, Bredt DS. Glutamate receptor delta2 associates with metabotropic glutamate receptor 1 (mGluR1), protein kinase Cgamma, and canonical transient receptor potential 3 and regulates mGluR1-mediated synaptic transmission in cerebellar Purkinje neurons. J Neurosci. 2012;32(44):15296–308. https://doi.org/10.1523/JNEUROSCI.0705-12.2012.
Cao Y, Posokhova E, Martemyanov KA. TRPM1 forms complexes with nyctalopin in vivo and accumulates in postsynaptic compartment of ON-bipolar neurons in mGluR6-dependent manner. J Neurosci. 2011;31(32):11521–6. https://doi.org/10.1523/JNEUROSCI.1682-11.2011.
Francesconi A, Kumari R, Zukin RS. Regulation of group I metabotropic glutamate receptor trafficking and signaling by the caveolar/lipid raft pathway. J Neurosci. 2009;29(11):3590–602. https://doi.org/10.1523/JNEUROSCI.5824-08.2009.
Moritz A, Scheschonka A, Beckhaus T, Karas M, Betz H. Metabotropic glutamate receptor 4 interacts with microtubule-associated protein 1B. Biochem Biophys Res Commun. 2009;390(1):82–6. https://doi.org/10.1016/j.bbrc.2009.09.070.
Zhang J, Cheng S, Xiong Y, Ma Y, Luo D, Jeromin A, Zhang H, He J. A novel association of mGluR1a with the PDZ scaffold protein CAL modulates receptor activity. FEBS Lett. 2008;582(30):4117–24. https://doi.org/10.1016/j.febslet.2008.10.054.
Paquet M, Asay MJ, Fam SR, Inuzuka H, Castleberry AM, Oller H, Smith Y, Yun CC, Traynelis SF, Hall RA. The PDZ scaffold NHERF-2 interacts with mGluR5 and regulates receptor activity. J Biol Chem. 2006;281(40):29949–61. https://doi.org/10.1074/jbc.M602262200.
Raka F, Di Sebastiano AR, Kulhawy SC, Ribeiro FM, Godin CM, Caetano FA, Angers S, Ferguson SS. Ca(2+)/calmodulin-dependent protein kinase II interacts with group I metabotropic glutamate and facilitates receptor endocytosis and ERK1/2 signaling: role of beta-amyloid. Mol Brain. 2015;8:21. https://doi.org/10.1186/s13041-015-0111-4.
Rademacher N, Kuropka B, Kunde SA, Wahl MC, Freund C, Shoichet SA. Intramolecular domain dynamics regulate synaptic MAGUK protein interactions. Elife. 2019. https://doi.org/10.7554/eLife.41299.
Waaijers S, Munoz J, Berends C, Ramalho JJ, Goerdayal SS, Low TY, Zoumaro-Djayoon AD, Hoffmann M, Koorman T, Tas RP, Harterink M, Seelk S, Kerver J, Hoogenraad CC, Bossinger O, Tursun B, van den Heuvel S, Heck AJ, Boxem M. A tissue-specific protein purification approach in Caenorhabditis elegans identifies novel interaction partners of DLG-1/Discs large. BMC Biol. 2016;14:66. https://doi.org/10.1186/s12915-016-0286-x.
Lauks J, Klemmer P, Farzana F, Karupothula R, Zalm R, Cooke NE, Li KW, Smit AB, Toonen R, Verhage M. Synapse associated protein 102 (SAP102) binds the C-terminal part of the scaffolding protein neurobeachin. PLoS ONE. 2012;7(6):e39420. https://doi.org/10.1371/journal.pone.0039420.
Baucum AJ 2nd, Shonesy BC, Rose KL, Colbran RJ. Quantitative proteomics analysis of CaMKII phosphorylation and the CaMKII interactome in the mouse forebrain. ACS Chem Neurosci. 2015;6(4):615–31. https://doi.org/10.1021/cn500337u.
Yoshimura Y, Shinkawa T, Taoka M, Kobayashi K, Isobe T, Yamauchi T. Identification of protein substrates of Ca(2+)/calmodulin-dependent protein kinase II in the postsynaptic density by protein sequencing and mass spectrometry. Biochem Biophys Res Commun. 2002;290(3):948–54. https://doi.org/10.1006/bbrc.2001.6320.
Zybura AS, Baucum AJ 2nd, Rush AM, Cummins TR, Hudmon A. CaMKII enhances voltage-gated sodium channel Nav16 activity and neuronal excitability. J Biol Chem. 2020;295(33):11845–65. https://doi.org/10.1074/jbc.RA120.014062.
Salling MC, Faccidomo SP, Li C, Psilos K, Galunas C, Spanos M, Agoglia AE, Kash TL, Hodge CW. Moderate alcohol drinking and the amygdala proteome: identification and validation of calcium/calmodulin dependent kinase II and ampa receptor activity as novel molecular mechanisms of the positive reinforcing effects of alcohol. Biol Psychiatry. 2016;79(6):430–42. https://doi.org/10.1016/j.biopsych.2014.10.020.
Rich MT, Abbott TB, Chung L, Gulcicek EE, Stone KL, Colangelo CM, Lam TT, Nairn AC, Taylor JR, Torregrossa MM. Phosphoproteomic analysis reveals a novel mechanism of CaMKIIalpha regulation inversely induced by cocaine memory extinction versus reconsolidation. J Neurosci. 2016;36(29):7613–27. https://doi.org/10.1523/JNEUROSCI.1108-16.2016.
Bemben MA, Shipman SL, Hirai T, Herring BE, Li Y, Badger JD 2nd, Nicoll RA, Diamond JS, Roche KW. CaMKII phosphorylation of neuroligin-1 regulates excitatory synapses. Nat Neurosci. 2014;17(1):56–64. https://doi.org/10.1038/nn.3601.
Bostrom SL, Dore J, Griffith LC. CaMKII uses GTP as a phosphate donor for both substrate and autophosphorylation. Biochem Biophys Res Commun. 2009;390(4):1154–9. https://doi.org/10.1016/j.bbrc.2009.10.107.
Liu W, Xia Y, Kuang H, Wang Z, Liu S, Tang C, Yin D. Proteomic profile of carbonylated proteins screen the regulation of calmodulin-dependent protein kinases-AMPK-Beclin1 in aerobic exercise-induced autophagy in middle-aged rat hippocampus. Gerontology. 2019;65(6):620–33. https://doi.org/10.1159/000500742.
Raynaud F, Janossy A, Dahl J, Bertaso F, Perroy J, Varrault A, Vidal M, Worley PF, Boeckers TM, Bockaert J, Marin P, Fagni L, Homburger V. Shank3-Rich2 interaction regulates AMPA receptor recycling and synaptic long-term potentiation. J Neurosci. 2013;33(23):9699–715. https://doi.org/10.1523/JNEUROSCI.2725-12.2013.
Bockers TM, Mameza MG, Kreutz MR, Bockmann J, Weise C, Buck F, Richter D, Gundelfinger ED, Kreienkamp HJ. Synaptic scaffolding proteins in rat brain Ankyrin repeats of the multidomain Shank protein family interact with the cytoskeletal protein alpha-fodrin. J Biol Chem. 2001;276(43):40104–12. https://doi.org/10.1074/jbc.M102454200.
Wang L, Pang K, Han K, Adamski CJ, Wang W, He L, Lai JK, Bondar VV, Duman JG, Richman R, Tolias KF, Barth P, Palzkill T, Liu Z, Holder JL Jr, Zoghbi HY. An autism-linked missense mutation in SHANK3 reveals the modularity of Shank3 function. Mol Psychiatry. 2020;25(10):2534–55. https://doi.org/10.1038/s41380-018-0324-x.
Modi ME, Brooks JM, Guilmette ER, Beyna M, Graf R, Reim D, Schmeisser MJ, Boeckers TM, O’Donnell P, Buhl DL. Hyperactivity and hypermotivation associated with increased striatal mGluR1 signaling in a Shank2 rat model of Autism. Front Mol Neurosci. 2018;11:107. https://doi.org/10.3389/fnmol.2018.00107.
Reim D, Distler U, Halbedl S, Verpelli C, Sala C, Bockmann J, Tenzer S, Boeckers TM, Schmeisser MJ. Proteomic analysis of post-synaptic density fractions from Shank3 mutant mice reveals brain region specific changes relevant to Autism spectrum disorder. Front Mol Neurosci. 2017;10:26. https://doi.org/10.3389/fnmol.2017.00026.
Lee Y, Kang H, Lee B, Zhang Y, Kim Y, Kim S, Kim WK, Han K. Integrative analysis of brain region-specific Shank3 interactomes for understanding the heterogeneity of neuronal pathophysiology related to SHANK3 mutations. Front Mol Neurosci. 2017;10:110. https://doi.org/10.3389/fnmol.2017.00110.
McMahon AC, Barnett MW, O’Leary TS, Stoney PN, Collins MO, Papadia S, Choudhary JS, Komiyama NH, Grant SG, Hardingham GE, Wyllie DJ, Kind PC. SynGAP isoforms exert opposing effects on synaptic strength. Nat Commun. 2012;3:900. https://doi.org/10.1038/ncomms1900.
Walkup WG, Sweredoski MJ, Graham RL, Hess S, Kennedy MB. Phosphorylation of synaptic GTPase-activating protein (synGAP) by polo-like kinase (Plk2) alters the ratio of its GAP activity toward HRas, Rap1 and Rap2 GTPases. Biochem Biophys Res Commun. 2018;503(3):1599–604. https://doi.org/10.1016/j.bbrc.2018.07.087.
Walkup WG, Washburn L, Sweredoski MJ, Carlisle HJ, Graham RL, Hess S, Kennedy MB. Phosphorylation of synaptic GTPase-activating protein (synGAP) by Ca2+/calmodulin-dependent protein kinase II (CaMKII) and cyclin-dependent kinase 5 (CDK5) alters the ratio of its GAP activity toward Ras and Rap GTPases. J Biol Chem. 2015;290(8):4908–27. https://doi.org/10.1074/jbc.M114.614420.
Gou G, Roca-Fernandez A, Kilinc M, Serrano E, Reig-Viader R, Araki Y, Huganir RL, de Quintana-Schmidt C, Rumbaugh G, Bayes A. SynGAP splice variants display heterogeneous spatio-temporal expression and subcellular distribution in the developing mammalian brain. J Neurochem. 2020;154(6):618–34. https://doi.org/10.1111/jnc.14988.
Goulding SP, Szumlinski KK, Contet C, MacCoss MJ, Wu CC. A mass spectrometry-based proteomic analysis of Homer2-interacting proteins in the mouse brain. J Proteomics. 2017;166:127–37. https://doi.org/10.1016/j.jprot.2017.07.008.
Ye R, Quinlan MA, Iwamoto H, Wu HH, Green NH, Jetter CS, McMahon DG, Veestra-VanderWeele J, Levitt P, Blakely RD. Physical interactions and functional relationships of neuroligin 2 and midbrain serotonin transporters. Front Synaptic Neurosci. 2015;7:20. https://doi.org/10.3389/fnsyn.2015.00020.
Kang Y, Ge Y, Cassidy RM, Lam V, Luo L, Moon KM, Lewis R, Molday RS, Wong RO, Foster LJ, Craig AM. A combined transgenic proteomic analysis and regulated trafficking of neuroligin-2. J Biol Chem. 2014;289(42):29350–64. https://doi.org/10.1074/jbc.M114.549279.
Sterky FH, Trotter JH, Lee SJ, Recktenwald CV, Du X, Zhou B, Zhou P, Schwenk J, Fakler B, Sudhof TC. Carbonic anhydrase-related protein CA10 is an evolutionarily conserved pan-neurexin ligand. Proc Natl Acad Sci USA. 2017;114(7):E1253–62. https://doi.org/10.1073/pnas.1621321114.
Savas JN, Ribeiro LF, Wierda KD, Wright R, DeNardo-Wilke LA, Rice HC, Chamma I, Wang YZ, Zemla R, Lavallee-Adam M, Vennekens KM, O’Sullivan ML, Antonios JK, Hall EA, Thoumine O, Attie AD, Yates JR 3rd, Ghosh A, de Wit J. The sorting receptor SorCS1 regulates trafficking of neurexin and AMPA receptors. Neuron. 2015;87(4):764–80. https://doi.org/10.1016/j.neuron.2015.08.007.
Venable JD, Dong MQ, Wohlschlegel J, Dillin A, Yates JR. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat Methods. 2004;1(1):39–45. https://doi.org/10.1038/nmeth705.
Zhang F, Ge W, Ruan G, Cai X, Guo T. Data-independent acquisition mass spectrometry-based proteomics and software tools: a glimpse in 2020. Proteomics. 2020;20(17–18):e1900276. https://doi.org/10.1002/pmic.201900276.
Geiger T, Cox J, Mann M. Proteomics on an Orbitrap benchtop mass spectrometer using all-ion fragmentation. Mol Cell Proteomics. 2010;9(10):2252–61. https://doi.org/10.1074/mcp.M110.001537.
Lambert JP, Ivosev G, Couzens AL, Larsen B, Taipale M, Lin ZY, Zhong Q, Lindquist S, Vidal M, Aebersold R, Pawson T, Bonner R, Tate S, Gingras AC. Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition. Nat Methods. 2013;10(12):1239–45. https://doi.org/10.1038/nmeth.2702.
Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012. https://doi.org/10.1074/mcp.O111.016717.
Egertson JD, Kuehn A, Merrihew GE, Bateman NW, MacLean BX, Ting YS, Canterbury JD, Marsh DM, Kellmann M, Zabrouskov V, Wu CC, MacCoss MJ. Multiplexed MS/MS for improved data-independent acquisition. Nat Methods. 2013;10(8):744–6. https://doi.org/10.1038/nmeth.2528.
Bruderer R, Bernhardt OM, Gandhi T, Miladinovic SM, Cheng LY, Messner S, Ehrenberger T, Zanotelli V, Butscheid Y, Escher C, Vitek O, Rinner O, Reiter L. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol Cell Proteomics. 2015;14(5):1400–10. https://doi.org/10.1074/mcp.M114.044305.
Moseley MA, Hughes CJ, Juvvadi PR, Soderblom EJ, Lennon S, Perkins SR, Thompson JW, Steinbach WJ, Geromanos SJ, Wildgoose J, Langridge JI, Richardson K, Vissers JPC. Scanning quadrupole data-independent acquisition, part A: qualitative and quantitative characterization. J Proteome Res. 2018;17(2):770–9. https://doi.org/10.1021/acs.jproteome.7b00464.
Meier F, Geyer PE, Virreira Winter S, Cox J, Mann M. BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat Methods. 2018;15(6):440–8. https://doi.org/10.1038/s41592-018-0003-5.
Rost HL, Rosenberger G, Navarro P, Gillet L, Miladinovic SM, Schubert OT, Wolski W, Collins BC, Malmstrom J, Malmstrom L, Aebersold R. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat Biotechnol. 2014;32(3):219–23. https://doi.org/10.1038/nbt.2841.
Tsou CC, Tsai CF, Teo GC, Chen YJ, Nesvizhskii AI. Untargeted, spectral library-free analysis of data-independent acquisition proteomics data generated using Orbitrap mass spectrometers. Proteomics. 2016;16(15–16):2257–71. https://doi.org/10.1002/pmic.201500526.
Tsou CC, Avtonomov D, Larsen B, Tucholska M, Choi H, Gingras AC, Nesvizhskii AI. DIA-umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat Methods. 2015;12(3):258–64. https://doi.org/10.1038/nmeth.3255.
Bernhardt O, Selevsek N, Gillet L, Rinner O, Reiter L (2014). Spectronaut: a fast and efficient algorithm for MRM-like processing of data independent acquisition (SWATH-MS) data. In: Paper presented at the 60th American Society for Mass Spectometry Conference 2012.
Tran NH, Qiao R, Xin L, Chen X, Liu C, Zhang X, Shan B, Ghodsi A, Li M. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nat Methods. 2019;16(1):63–6. https://doi.org/10.1038/s41592-018-0260-3.
Doerr A. DIA mass spectrometry. Nat Methods. 2014;12(1):35–35.
Bekker-Jensen DB, Bernhardt OM, Hogrebe A, Martinez-Val A, Verbeke L, Gandhi T, Kelstrup CD, Reiter L, Olsen JV. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat Commun. 2020;11(1):787. https://doi.org/10.1038/s41467-020-14609-1.
Wang Y, Xu C, Zhong B, Zhan D, Liu M, Gao D, Wang Y, Qin J. Comparative proteomic analysis of histone modifications upon acridone derivative 8a-induced CCRF-CEM cells by data independent acquisition. J Proteome Res. 2020;19(2):819–31. https://doi.org/10.1021/acs.jproteome.9b00650.
Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340(6230):245–6. https://doi.org/10.1038/340245a0.
Jares-Erijman EA, Jovin TM. Imaging molecular interactions in living cells by FRET microscopy. Curr Opin Chem Biol. 2006;10(5):409–16. https://doi.org/10.1016/j.cbpa.2006.08.021.
Hoppe A, Christensen K, Swanson JA. Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys J. 2002;83(6):3652–64. https://doi.org/10.1016/S0006-3495(02)75365-4.
Gingras AC, Gstaiger M, Raught B, Aebersold R. Analysis of protein complexes using mass spectrometry. Nat Rev Mol Cell Biol. 2007;8(8):645–54. https://doi.org/10.1038/nrm2208.
ten Have S, Boulon S, Ahmad Y, Lamond AI. Mass spectrometry-based immuno-precipitation proteomics—the user’s guide. Proteomics. 2011;11(6):1153–9. https://doi.org/10.1002/pmic.201000548.
Fan SB, Wu YJ, Yang B, Chi H, Meng JM, Lu S, Zhang K, Wu L, Sun R, Dong M-Q, He SM. A new approach to protein structure and interaction research: chemical cross-linking in combination with mass spectrometry. Prog Biochem Biophys. 2014;41:1109–25. https://doi.org/10.3724/SP.J.1206.2013.00484.
Yang B, Wu YJ, Zhu M, Fan SB, Lin J, Zhang K, Li S, Chi H, Li YX, Chen HF, Luo SK, Ding YH, Wang LH, Hao Z, Xiu LY, Chen S, Ye K, He SM, Dong MQ. Identification of cross-linked peptides from complex samples. Nat Methods. 2012;9(9):904–6. https://doi.org/10.1038/nmeth.2099.
Tabb DL. Evaluating protein interactions through cross-linking mass spectrometry. Nat Methods. 2012;9(9):879–81. https://doi.org/10.1038/nmeth.2139.
Tran BQ, Goodlett DR, Goo YA. Advances in protein complex analysis by chemical cross-linking coupled with mass spectrometry (CXMS) and bioinformatics. Biochim Biophys Acta. 2016;1864(1):123–9. https://doi.org/10.1016/j.bbapap.2015.05.015.
Gonzalez-Lozano MA, Koopmans F, Sullivan PF, Protze J, Krause G, Verhage M, Li KW, Liu F, Smit AB. Stitching the synapse: cross-linking mass spectrometry into resolving synaptic protein interactions. Sci Adv. 2020;6(8):eaax5783. https://doi.org/10.1126/sciadv.aax5783.
Kotani N, Gu J, Isaji T, Udaka K, Taniguchi N, Honke K. Biochemical visualization of cell surface molecular clustering in living cells. Proc Natl Acad Sci USA. 2008;105(21):7405–9. https://doi.org/10.1073/pnas.0710346105.
Osbourn JK. Proximity-guided (ProxiMol) antibody selection. Methods Mol Biol. 2002;178:201–5. https://doi.org/10.1385/1-59259-240-6:201.
Bendayan M. Tech.Sight. Worth its weight in gold. Science. 2001;291(5507):1363–5. https://doi.org/10.1126/science.291.5507.1363.
Trinkle-Mulcahy L. Recent advances in proximity-based labeling methods for interactome mapping. F1000Res. 2019. https://doi.org/10.12688/f1000research.16903.1.
Rees JS, Li XW, Perrett S, Lilley KS, Jackson AP. Protein neighbors and proximity proteomics. Mol Cell Proteomics. 2015;14(11):2848–56. https://doi.org/10.1074/mcp.R115.052902.
Han S, Li J, Ting AY. Proximity labeling: spatially resolved proteomic mapping for neurobiology. Curr Opin Neurobiol. 2018;50:17–23. https://doi.org/10.1016/j.conb.2017.10.015.
Spence EF, Dube S, Uezu A, Locke M, Soderblom EJ, Soderling SH. In vivo proximity proteomics of nascent synapses reveals a novel regulator of cytoskeleton-mediated synaptic maturation. Nat Commun. 2019;10(1):386. https://doi.org/10.1038/s41467-019-08288-w.
Uezu A, Soderling S. Identifying synaptic proteins by in vivo BioID from mouse brain. Methods Mol Biol. 2019;2008:107–19. https://doi.org/10.1007/978-1-4939-9537-0_9.
Genon S, Reid A, Langner R, Amunts K, Eickhoff SB. How to characterize the function of a brain region. Trends Cogn Sci. 2018;22(4):350–64. https://doi.org/10.1016/j.tics.2018.01.010.
Siskova Z. How structure shapes (dys)function: a perspective to understanding brain region-specific degeneration in prion disease. Prion. 2013;7(4):291–3.
Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, Kongi K, Cantuti L, Hanisch UK, Philips MA, Rossner MJ, Mann M, Simons M. Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci. 2015;18(12):1819–31. https://doi.org/10.1038/nn.4160.
Gerber KJ, Dammer EB, Duong DM, Deng Q, Dudek SM, Seyfried NT, Hepler JR. Specific proteomes of hippocampal regions CA2 and CA1 reveal proteins linked to the unique physiology of area CA2. J Proteome Res. 2019;18(6):2571–84. https://doi.org/10.1021/acs.jproteome.9b00103.
Trinidad JC, Thalhammer A, Specht CG, Lynn AJ, Baker PR, Schoepfer R, Burlingame AL. Quantitative analysis of synaptic phosphorylation and protein expression. Mol Cell Proteomics. 2008;7(4):684–96. https://doi.org/10.1074/mcp.M700170-MCP200.
Cheng D, Hoogenraad CC, Rush J, Ramm E, Schlager MA, Duong DM, Xu P, Wijayawardana SR, Hanfelt J, Nakagawa T, Sheng M, Peng J. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol Cell Proteomics. 2006;5(6):1158–70. https://doi.org/10.1074/mcp.D500009-MCP200.
Roy M, Sorokina O, McLean C, Tapia-Gonzalez S, DeFelipe J, Armstrong JD, Grant SGN. Regional diversity in the postsynaptic proteome of the mouse brain. Proteomes. 2018;6(3):31. https://doi.org/10.3390/proteomes6030031.
Distler U, Schumann S, Kesseler HG, Pielot R, Smalla KH, Sielaff M, Schmeisser MJ, Tenzer S. Proteomic analysis of brain region and sex-specific synaptic protein expression in the adult mouse brain. Cells. 2020;9(2):313. https://doi.org/10.3390/cells9020313.
Peixoto RT, Chantranupong L, Hakim R, Levasseur J, Wang W, Merchant T, Gorman K, Budnik B, Sabatini BL. Abnormal striatal development underlies the early onset of behavioral deficits in Shank3B(-/-) mice. Cell Rep. 2019;29(7):2016–27. https://doi.org/10.1016/j.celrep.2019.10.021.
Roy M, Sorokina O, Skene N, Simonnet C, Mazzo F, Zwart R, Sher E, Smith C, Armstrong JD, Grant SGN. Proteomic analysis of postsynaptic proteins in regions of the human neocortex. Nat Neurosci. 2018;21(1):130–8. https://doi.org/10.1038/s41593-017-0025-9.
Selimi F, Cristea IM, Heller E, Chait BT, Heintz N. Proteomic studies of a single CNS synapse type: the parallel fiber/purkinje cell synapse. PLoS Biol. 2009;7(4):e83. https://doi.org/10.1371/journal.pbio.1000083.
Fecher C, Trovo L, Muller SA, Snaidero N, Wettmarshausen J, Heink S, Ortiz O, Wagner I, Kuhn R, Hartmann J, Karl RM, Konnerth A, Korn T, Wurst W, Merkler D, Lichtenthaler SF, Perocchi F, Misgeld T. Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat Neurosci. 2019;22(10):1731–42. https://doi.org/10.1038/s41593-019-0479-z.
Wilkinson B, Evgrafov OV, Zheng D, Hartel N, Knowles JA, Graham NA, Ichida JK, Coba MP. Endogenous cell type-specific disrupted in schizophrenia 1 interactomes reveal protein networks associated with neurodevelopmental disorders. Biol Psychiatry. 2019;85(4):305–16. https://doi.org/10.1016/j.biopsych.2018.05.009.
Jiang X, Nardelli J. Cellular and molecular introduction to brain development. Neurobiol Dis. 2016;92:3–17. https://doi.org/10.1016/j.nbd.2015.07.007.
McClatchy DB, Liao L, Park SK, Venable JD, Yates JR. Quantification of the synaptosomal proteome of the rat cerebellum during post-natal development. Genome Res. 2007;17(9):1378–88. https://doi.org/10.1101/gr.6375007.
McClatchy DB, Liao L, Lee JH, Park SK, Yates JR 3rd. Dynamics of subcellular proteomes during brain development. J Proteome Res. 2012;11(4):2467–79. https://doi.org/10.1021/pr201176v.
Gonzalez-Lozano MA, Klemmer P, Gebuis T, Hassan C, van Nierop P, van Kesteren RE, Smit AB, Li KW. Dynamics of the mouse brain cortical synaptic proteome during postnatal brain development. Sci Rep. 2016;6:35456. https://doi.org/10.1038/srep35456.
Skene NG, Roy M, Grant SG. A genomic lifespan program that reorganises the young adult brain is targeted in schizophrenia. Elife. 2017. https://doi.org/10.7554/eLife.17915.
Cizeron M, Qiu Z, Koniaris B, Gokhale R, Komiyama NH, Fransen E, Grant SGN. A brainwide atlas of synapses across the mouse life span. Science. 2020;369(6501):270–5. https://doi.org/10.1126/science.aba3163.
Grant SGN. Synapse diversity and synaptome architecture in human genetic disorders. Hum Mol Genet. 2019;28(R2):R219–25. https://doi.org/10.1093/hmg/ddz178.
Bentea E, Villers A, Moore C, Funk AJ, O’Donovan SM, Verbruggen L, Lara O, Janssen P, De Pauw L, Declerck NB, DePasquale EAK, Churchill MJ, Sato H, Hermans E, Arckens L, Meshul CK, Ris L, McCullumsmith RE, Massie A. Corticostriatal dysfunction and social interaction deficits in mice lacking the cystine/glutamate antiporter. Mol Psychiatry. 2020. https://doi.org/10.1038/s41380-020-0751-3.
Al Shweiki MR, Oeckl P, Steinacker P, Barschke P, Dorner-Ciossek C, Hengerer B, Schonfeldt-Lecuona C, Otto M. Proteomic analysis reveals a biosignature of decreased synaptic protein in cerebrospinal fluid of major depressive disorder. Transl Psychiatry. 2020;10(1):144. https://doi.org/10.1038/s41398-020-0825-7.
Pennington K, Beasley CL, Dicker P, Fagan A, English J, Pariante CM, Wait R, Dunn MJ, Cotter DR. Prominent synaptic and metabolic abnormalities revealed by proteomic analysis of the dorsolateral prefrontal cortex in schizophrenia and bipolar disorder. Mol Psychiatry. 2008;13(12):1102–17. https://doi.org/10.1038/sj.mp.4002098.
Kadoyama K, Matsuura K, Takano M, Otani M, Tomiyama T, Mori H, Matsuyama S. Proteomic analysis involved with synaptic plasticity improvement by GABAA receptor blockade in hippocampus of a mouse model of Alzheimer’s disease. Neurosci Res. 2020. https://doi.org/10.1016/j.neures.2020.04.004.
Koopmans F, van Nierop P, Andres-Alonso M, Byrnes A, Cijsouw T, Coba MP, Cornelisse LN, Farrell RJ, Goldschmidt HL, Howrigan DP, Hussain NK, Imig C, de Jong APH, Jung H, Kohansalnodehi M, Kramarz B, Lipstein N, Lovering RC, MacGillavry H, Mariano V, Mi H, Ninov M, Osumi-Sutherland D, Pielot R, Smalla KH, Tang H, Tashman K, Toonen RFG, Verpelli C, Reig-Viader R, Watanabe K, van Weering J, Achsel T, Ashrafi G, Asi N, Brown TC, De Camilli P, Feuermann M, Foulger RE, Gaudet P, Joglekar A, Kanellopoulos A, Malenka R, Nicoll RA, Pulido C, de Juan-Sanz J, Sheng M, Sudhof TC, Tilgner HU, Bagni C, Bayes A, Biederer T, Brose N, Chua JJE, Dieterich DC, Gundelfinger ED, Hoogenraad C, Huganir RL, Jahn R, Kaeser PS, Kim E, Kreutz MR, McPherson PS, Neale BM, O’Connor V, Posthuma D, Ryan TA, Sala C, Feng G, Hyman SE, Thomas PD, Smit AB, Verhage M. SynGO: an evidence-based, expert-curated knowledge base for the synapse. Neuron. 2019;103(2):217–34. https://doi.org/10.1016/j.neuron.2019.05.002.
Mehnert M, Ciuffa R, Frommelt F, Uliana F, van Drogen A, Ruminski K, Gstaiger M, Aebersold R. Multi-layered proteomic analyses decode compositional and functional effects of cancer mutations on kinase complexes. Nat Commun. 2020;11(1):3563. https://doi.org/10.1038/s41467-020-17387-y.
Marx V. A dream of single-cell proteomics. Nat Methods. 2019;16(9):809–12. https://doi.org/10.1038/s41592-019-0540-6.
Zhu F, Cizeron M, Qiu Z, Benavides-Piccione R, Kopanitsa MV, Skene NG, Koniaris B, DeFelipe J, Fransen E, Komiyama NH, Grant SGN. Architecture of the mouse brain synaptome. Neuron. 2018;99(4):781–99. https://doi.org/10.1016/j.neuron.2018.07.007.
Curran OE, Qiu Z, Smith C, Grant SGN. A single-synapse resolution survey of PSD95-positive synapses in twenty human brain regions. Eur J Neurosci. 2020. https://doi.org/10.1111/ejn.14846.
Acknowledgements
Not applicable.
Funding
This work was supported by the Natural Science Foundation of Shandong Province (No. ZR2019LZL001 to D.W.), the People's Livelihood Science and Technology Project of Qingdao (No. 19-6-1-44-nsh to D.W.), and the Major Research Program of the National Natural Science Foundation of China (No.91849209 to P.L.).
Author information
Authors and Affiliations
Contributions
JL conceived the paper, YX, XS and JL wrote the paper, DW, YW and PL revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xu, Y., Song, X., Wang, D. et al. Proteomic insights into synaptic signaling in the brain: the past, present and future. Mol Brain 14, 37 (2021). https://doi.org/10.1186/s13041-021-00750-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-021-00750-5