
Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by the progressive loss of cortical, brain stem and spinal motor neurons that leads to muscle weakness and death. A previous study implicated CACNA1H encoding for Cav3.2 calcium channels as a susceptibility gene in ALS. In the present study, two heterozygous CACNA1H variants were identified by whole genome sequencing in a small cohort of ALS patients. These variants were functionally characterized using patch clamp electrophysiology, biochemistry assays, and molecular modeling. A previously unreported c.454GTAC > G variant produced an inframe deletion of a highly conserved isoleucine residue in Cav3.2 (p.ΔI153) and caused a complete loss-of-function of the channel, with an additional dominant-negative effect on the wild-type channel when expressed in trans. In contrast, the c.3629C > T variant caused a missense substitution of a proline with a leucine (p.P1210L) and produced a comparatively mild alteration of Cav3.2 channel activity. The newly identified ΔI153 variant is the first to be reported to cause a complete loss of Cav3.2 channel function. These findings add to the notion that loss-of-function of Cav3.2 channels associated with rare CACNA1H variants may be risk factors in the complex etiology of ALS.
Similar content being viewed by others

Introduction
Amyotrophic lateral sclerosis (ALS), also known as motor neuron disease or Lou Gehrig’s disease, is a heterogeneous neuromuscular disease characterized by the degeneration of cortical, brain stem and spinal motor neurons that leads to muscle weakness and paralysis. Disease onset averages between 40 and 70 years of age [1], and the annual incidence worldwide is estimated to be between one to three per 100,000 people [2]. ALS is best regarded as a complex genetic disorder with a Mendelian pattern of inheritance in approximately 5–10% of patients (familial ALS, fALS), but most patients have no discernable family history of the disease which is then referred to being “sporadic” or “isolated” in nature (sALS) [3]. However, the observation that established fALS genes are also implicated in sALS makes the distinction between fALS and sALS more abstruse [4]. For instance, mutations in the most common ALS genes (SOD1, FUS, TARDBP, C9orf72, VCP, and PFN1) account for up to 70% of fALS patients and about 10% of sALS [5]. In addition, several genes and loci in apparent sALS patients have been proposed to be associated with an increased risk of ALS, or to modify the onset or progression of the disease, which highlights the importance of genetic risk factors [6]. Among these genes, the most prominent are ATXN2 [7], UNC13A [8], ANG [9], and SMN1 [10]. Recently, whole exome sequence analysis of case-unaffected-parents trios identified two compound heterozygous recessive missense mutations in the gene CACNA1H [11, 12].
In the present study, we report two additional CACNA1H variants (c.3629C > T, p.P1210L and c.454GTAC > G, p.ΔI153) identified using whole genome sequencing of a cohort of 34 sALS patients, with sequencing undertaken at the Genome Institute, Washington University, St Louise USA. The method of whole genome analysis was the same as that reported in a separate study [11]. Whole genome analyses reveal no pathogenetic single nucleotide or structural differences between monozygotic twins discordant for amyotrophic lateral sclerosis [13]. No unaffected parent DNA was subjected to whole genome sequencing, so it was not possible to determine if the variants were recessive or de novo in nature [11]. Functional analysis of these two variants revealed a complete loss of Cav3.2 channel function associated with the ΔI153 variant and a dominant-negative effect of this variant on the wild-type channel when expressed in trans.
Results
Whole genome sequencing identifies heterozygous CACNA1H mutations in ALS patients
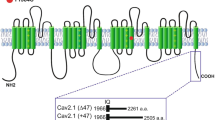
In a previous study, using case-unaffected parents trio exome analyses, we reported an ALS patient with two heterozygous CACNA1H missense mutations causing a partial loss-of-function of Cav3.2 channel, suggesting that rare CACNA1H variants may represent a risk factor for ALS [11, 12]. In the present study, using whole genome sequencing of a small cohort of ALS patients, we identified two additional heterozygous variants in CACNA1H. The first variant (c.3629C > T, p.P1210L) was identified in a man with ALS onset aged 55 years who died aged 62 years. He had no family history of ALS, though his father had Alzheimer’s disease and his mother bipolar disorder. The P1210L variant is located in a non-conserved region of the intracellular linker connecting transmembrane domains II and III (II-III loop) of Cav3.2 (Fig. 1a and b). This variant has previously been reported in 188 out of 240,876 individuals in the gnomAD database (https://gnomad.broadinstitute.org/), including 144 of 193,008 alleles only from individuals who were not ascertained for having a neurological condition in a neurological case/control study. Furthermore, in silico analysis predicted the amino acid change to be neutral (Fig. 1c), suggesting that this variant is likely to not have a major pathological role. The second variant (c.454GTAC > G, p.ΔI153) was identified in a man with ALS onset aged 53 years who died aged 54 years. Although he had no family history of ALS, his mother developed insulin-dependent diabetes mellitus and narcolepsy, and his father presented with early onset dementia, a condition known to precede motor impairment in some people with ALS [14]. This mutation produces an inframe deletion of the isoleucine 153 located in the second transmembrane helix of Cav3.2, a region highly conserved across Cav3.2 channel orthologs (Fig. 1a and b). The ΔI153 variant has only been reported in 1 out of 198,036 individuals in the gnomAD database and this deletion was predicted to be deleterious on the channel (Fig. 1c). Hexanucleotide repeat number in C9orf72, the most common genetic cause of ALS, was normal in both patients.

Location of ALS-associated Cav3.2 variants. a Schematic representation of the membrane topology of Cav3.2 depicting the position of the ΔI153 (red circle) and P1210L variants (blue circle). b Amino acid sequence alignment of Cav3.2 regions containing the two mutations across several species showing the conservation of the I153 residue. Alignments were performed using UniProt (Homo sapiens O95180; Rattus norvegicus Q9EQ60; Mus musculus O88427; Pan troglodytes H2QA94; Macaca mulatta A0A1D5R8A8; Felis catus M3WP54; Canis lupus F1PQE5; Ficedula albicollis U3KGY9; Xenopus Tropicalis F6U0H3; Alligator sinensis A0A3Q0GL31). c In silico prediction of the potential impact of the P1210L and ΔI153 mutations on the functioning of Cav3.2 channel
The ΔI153 mutation causes a complete loss of Cav3.2 function
In the first series of experiments we assessed the functional expression of Cav3.2 P1210L and ΔI153 channel variants expressed in tsA-201 cells by whole-cell patch clamp electrophysiology. Cells expressing the P1210L channel variant displayed a characteristic low-threshold voltage-activated T-type current (Fig. 2a and b) that only differed from cells expressing the wild-type (WT) channel by a 32% reduction (p = 0.0125, Mann-Whitney test) of the maximal conductance (Gmax) (from 571.3 ± 58.4 pS/pF, n = 42 to 387.7 ± 33.9 pS/pF, n = 41) (Fig. 2c). The main electrophysiological properties, including voltage-dependence of activation and inactivation (Fig. 2d), and recovery from inactivation (Fig. 2e), remained unaffected. In cells expressing the ΔI153 channel variant, we did not record any T-type conductance (Fig. 2a-c). It is noteworthy that experimental conditions known to favor the expression of misfolded proteins, such as treatment of cells with the proteasome inhibitor MG132 or decrease of cell incubation temperature to 30 °C, were used but failed to restore a T-type conductance. Additionally, co-expression of the ΔI153 channel variant with Stac1 or with a calnexin-derived peptide that has previously been reported to potentiate the expression of Cav3.2 in the plasma membrane [15, 16] also failed to restore T-type currents (data not shown). The lack of functional expression of the ΔI153 channel variant could have been inherent in our experimental conditions using recombinant channels, so we aimed to further assess the phenotypic effect of the ΔI153 mutation on native Cav3.2 channels in a neuronal environment. Therefore, we used a CRISPR/Cas9 approach to introduce the ΔI153 mutation in native Cav3.2 channels in cultured dorsal root ganglion (DRG) neurons. DRG neurons were used as a model system since these neurons are known to display a T-type conductance that is almost exclusively carried by Cav3.2 channel subtype [17]. Consistent with our observation with recombinant Cav3.2 channels, T-type currents recorded from Cav3.2 ΔI153 DRG neurons 3 days after gene editing were reduced by 73% (Mann-Whitney p < 0.0001) compared to wild type neurons (from 15.4 ± 2.5 pA/pF, n = 12 to 4.1 ± 0.8 pA/pF, n = 12) (Fig. 2f and g).

Electrophysiological characterization of Cav3.2 P1210L and ΔI153 variants. a Representative T-type current traces recorded in response to 150 ms depolarizing steps to values ranging between − 90 mV and + 30 mV from a holding potential of − 100 mV for wild-type (WT, black traces), P1210L (blue traces), and ΔI153 (red traces) channel variants expressed in tsA-201 cells. b Corresponding mean current-voltage relationship (I/V) for WT (black circles), P1210L (blue circles), and ΔI153 (red circles) channels. c Corresponding mean maximal macroscopic conductance (Gmax) obtained from the fit of the I/V curves with the modified Boltzmann eq. (1). d Mean normalized voltage-dependence of activation and inactivation for WT (black circles) and P1210L channels (blue circles). e Mean normalized recovery from inactivation kinetics. f Representative T-type current traces recorded from WT (black trace) and ΔI153 DRG neurons (red trace) 3 days after editing of Cacna1h by CRISPR/Cas9 in response to 80 ms depolarizing steps to − 25 mV from a holding potential of − 90 mV. g Corresponding mean peak T-type current density at − 25 mV in WT and ΔI153 mutant DRG neurons
Collectively, these data revealed a mild loss of channel function associated with the P1210L variant, and the deleterious effect of the ΔI153 mutation leading to a complete loss of Cav3.2 activity.
The ΔI153 mutation disrupts Cav3.2 biogenesis
The alteration of T-type currents in ALS-associated Cav3.2 variants could originate from an overall decreased expression of channel proteins, reduced channel density in the plasma membrane, altered gating of the channel, or from a combination of several of these. Therefore, we first assessed the expression levels of P1210L and ΔI153 channel variants in tsA-201 cells by western blot (Fig. 3a). Immunoblot analysis from total cell lysates showed that the P1210L channel variant was present at a similar level as the WT channel (Fig. 3b). In contrast, the expression level of the ΔI153 channel variant was reduced by 78% (Mann-Whitney p = 0.0286), suggesting that this variant may undergo extensive degradation (Fig. 3b). Next, we aimed to assess the expression of Cav3.2 channel variants at the cell surface. Therefore, we analyzed charge movements (Q) that refer to the movement of the channel voltage-sensor in the plasma membrane in response to electrical membrane depolarizations. Total charges (Qmax) were assessed at the reversal potential of the ionic current, where we can consider Qrev to be equal to Qmax, providing an accurate assessment of the total number of channels in the plasma membrane (Fig. 3c). In cells expressing the P1210L variant, we observed a 27% reduction of Qmax (t-test p = 0.0467) compared to cells expressing the WT channel (from 12.0 ± 1.3 fC/pF, n = 19 to 8.7 ± 0.8 fC/pF, n = 18) (Fig. 3d). This reduction of Qmax is similar to the reduction of the maximal T-type conductance we previously observed (32%, Fig. 2c), suggesting that the decrease of the T-type conductance in cells expressing the P1210L channel variant is likely caused by a reduced expression of the channel in the plasma membrane. This notion is further supported by the observation that neither the Gmax/Qmax dependency (Fig. 3e), nor the kinetics of charge movements (Fig. 3f), were modified, indicating that the gating properties of the P1210L channel variant remained unaltered. In contrast, we did not detect any charge movement in cells expressing the ΔI153 channel variant (Fig. 3c and d), suggesting that despite being biochemically expressed, this variant is not present in the plasma membrane.

Expression of Cav3.2 P1210L and ΔI153 variants. a Representative immunoblot of Cav3.2 from tsA-201 cells expressing wild-type (WT), P1210L, and ΔI153 channel variants. b Corresponding mean expression levels of P1210L and ΔI153 variants relative to WT channels. c Representative charge movement traces recorded at the ionic reversal potential from cells expressing wild-type (WT, black trace), P1210L (blue trace), and ΔI153 (red trace) channel variants. The dotted line depicts the time course of the integral for each trace. d Corresponding mean Qmax values calculated for each investigated cell. e Corresponding mean Gmax/Qmax ratios. f Corresponding mean 10–90% rise times calculated from the integral time course shown in panel c
Altogether, these data are consistent with a mildly decreased surface expression of the P1210L variant without additional alterations. Importantly, these data demonstrate the profound deleterious effect of the ΔI153 mutation on the biogenesis and surface trafficking of Cav3.2 channels.
Dominant-negative effect of the ΔI153 channel variant
Given the heterozygosity of the ΔI153 mutation and the defective trafficking of the ΔI153 channel variant, we aimed to test whether this variant could have a dominant-negative effect on WT channels. Therefore, we co-expressed the WT and ΔI153 channels in tsA-201 cells in a 1:1 ratio (equal amount of cDNAs) and compared T-type currents with cells expressing the WT channel in combination with a cation-impermeant but trafficking-competent channel (PM). Recording of T-type currents in cells expressing a combination of WT:ΔI153 channels (Fig. 4a) revealed a 35% reduction (Mann-Whitney p = 0.0080) of the maximal T-type conductance compared to cells expressing a combination of WT:PM channels (from 569 ± 73 pS/pF, n = 38 to 372 ± 27 pS/pF, n = 58) (Fig. 4b and c), indicating that the ΔI153 variant produced a dominant-negative effect on the WT channel when expressed in trans. In contrast, the voltage-dependence of activation and inactivation remained unaltered. Given the comparatively mild phenotype produced by the P1210L mutation, the P1210L variant was not tested in combination with the WT channel. Finally, to test whether this dominant-negative effect could be mediated by an interaction between Cav3.2 subunits, we performed co-immunoprecipitations from tsA-201 cells co-expressing Myc-tagged and GFP-tagged Cav3.2 to discriminate between the two channels. We observed that the GFP-tagged Cav3.2 was immunoprecipitated with the Myc-tagged Cav3.2 using a specific anti-Myc antibody, revealing the ability of Cav3.2 channels to dimerize (Fig. 4d).

Electrophysiological characterization of Cav3.2 WT and ΔI153 expressed in trans. a Representative T-type current traces recorded from tsA-201 cells expressing WT channels in combination with either the ΔI153 variant (WT:ΔI153, red traces) or the cation-impermeant but trafficking-competent Cav3.2 pore mutant (WT:PM; grey traces) in a ratio 1:1. b Corresponding mean current-voltage relationship (I/V) for WT:ΔI153 (black|red circles), and WT:PM (black|grey circles) conditions. c Corresponding mean maximal macroscopic conductance (Gmax). d Co-immunoprecipitation of Cav3.2 from tsA-201 cells co-transfected with a Myc-tagged and GFP-tagged Cav3.2. The left panel shows the result of the co-immunoprecipitation of Myc-Cav3.2 with GFP-Cav3.2 using an anti-Myc antibody. The middle and right panels show the immunoblot of GFP-Cav3.2 and Myc-Cav3.2 using and anti-GFP and anti-Myc antibody, respectively
Collectively, these data revealed the dominant-negative effect of the ΔI153 variant on the WT channel, a phenomenon likely to be mediated by the interaction between Cav3.2 subunits.
Discussion
While several common genes are implicated in familial ALS, the occurrence of rare genetic variants in patients with no family history of the disease has emerged as a potential contributing factor in sporadic ALS [11]. In this study, we report two heterozygous CACNA1H variants identified by whole genome sequencing of a small cohort of ALS patients. Functional analysis revealed mild to severe alterations of Cav3.2 variants that were consistent with a loss-of-function of the channels.
The P1210L missense mutation was located in a variable region of Cav3.2 and was not predicted to be deleterious. Our electrophysiological analysis showed a moderate reduction of the expression of the P1210L channel variant at the cell surface and an associated reduction in the T-type conductance. We cannot entirely rule out the possibility that the phenotypic expression of the P1210L variant could have differed when introduced into a different Cav3.2 splice variant [18], or when functionally assessed under different experimental conditions [19], but our experimental data together with the relatively high occurrence of this variant in the general population strongly suggest that it is indeed unlikely to be pathogenic. In contrast, the ΔI153 variant had never been reported and was predicted to be deleterious. Electrophysiological analysis revealed a complete loss of functional expression of the ΔI153 variant, and recording of charge movements suggested that this variant was absent from the cell surface. Furthermore, our biochemical analysis revealed a dramatic decrease of the expression level of the channel protein, suggesting that this variant may have undergone extensive degradation. Of particular importance was the dominant-negative effect produced by the ΔI153 variant on the WT channel when the two channels were expressed in trans. This effect was likely to be mediated by the ability of Cav3.2 subunits to dimerize, which could have prevented the proper trafficking of the WT channel to the cell surface in the presence of the impaired ΔI153 variant. In this regard, it is worth considering that this dominant-negative effect may also have an effect on other ion channels. Indeed, Cav3.2 channels are known to biochemically interact with several calcium- and voltage-activated potassium and sodium channel subunits [20,21,22,23] whose surface trafficking and activity could be affected by the Cav3.2 ΔI153 variant.
The molecular mechanisms underlying the deleterious effect of the ΔI153 variant can be appreciated by examining the 3-dimensional environment of I153, and the possible impact of its deletion in the homology model of Cav3.2 we have developed, using the 3.3 Å CryoEM structure of Cav3.1 [24]. In this model, I153 is located within the transmembrane S2 alpha helix of domain I (Fig. 5a), where it is surrounded by hydrophobic residues near the membrane-cytosol interface (Fig. 5b). The nearby hydrophobic residues are highly conserved between L- and T-type channels and I153 shows a clear involvement in the helical packing (Fig. 5b). Therefore, deletion of I153 that results in a net loss of hydrophobicity within the transmembrane segment is likely to alter helix packing in domain I which would result in the misfolding of the channel. Additionally, deletion of I153 would also affect downstream residues in the helix due to a change in the helical register, thus further affecting the helical packing in the voltage-sensing domain.

Homology model of human Cav3.2. a Cartoon representation of secondary structural elements of human Cav3.2 (Uniprot O95180) homology model (residues 97–1974) based on Cav3.1 (PDB: 6KZO), showing side (left panel) and bottom (right panel) views of the channel. The four domains of Cav3.2 are colored in red, yellow, blue and green. The S1-S6 helices are indicated in red for domain I. Some of the flexible loops connecting the transmembrane helices are not shown, or could not be modeled, due to poor model accuracy or lack of structural information, respectively. The isoleucine 153 (Ile153) is shown in black. b Stereo diagram of Ile153 and nearby hydrophobic residues showing its involvement in the helical packing
From a clinical point of view, the loss-of-channel function associated with the ΔI153 variant could have several pathological implications. First, Cav3.2 is present in several central neurons, including reticular thalamic neurons [25], where they contribute to NMDA receptor-mediated synaptic transmission [26]. Given that gain-of-function mutations associated with childhood absence epilepsy were shown to enhance synaptic activities [26], the reciprocal theory would suggest that loss-of-channel function could, in contrast, decrease synaptic transmission. Along these lines, neuroimaging studies have revealed decreased thalamic activity in ALS [27,28,29,30,31,32], and a recent MRI study reported alterations of thalamic connectivities that mirrored the progressive motor functional decline in ALS [33]. Second, although the functional expression of Cav3.2 in mammalian motor neurons remains elusive, several studies suggest that T-type channels may have a functional role. For instance, Cav3.1 channels are present in turtle spinal motor neurons where they contribute to cellular excitability [34]. In addition, a low-threshold voltage-activated calcium conductance was reported at nodes of Ranvier in mouse spinal motor neurons, suggesting the presence of T-type channels [35]. Third, a T-type channel ortholog is present in motor neurons of the nematode C. elegans [36] where it contributes to motor-related functions [37, 38]. Finally, a recent study documented the role of T-type channels in the maintenance of neuronal progenitor cells [39]. A loss-of-function of Cav3.2 could compromise the architecture of nerve cells and precipitate neuronal degeneration.
In conclusion, this newly identified ΔI153 variant is the first to be reported to cause a complete loss of Cav3.2 channel function [40]. Although its pathogenic role in the context of ALS remains to be established, these findings add to the notion that rare CACNA1H variants represent a risk factor for ALS. Furthermore, several T-type channels blockers are currently being used for the treatment of epilepsy [41]. The question then arises as to whether long term use of these molecules may present a risk to the development of ALS. This notion should be given particular attention, especially considering that several other T-type channel blockers are currently evaluated in clinical trials for the management of epilepsy and chronic pain symptoms.
Methods
Plasmids cDNA constructs and site-directed mutagenesis
The Cav3.2 P1210L and ΔI153 channel variants were created by introducing the respective mutations into the human wild-type HA-tagged Cav3.2 in pcDNA3.1 [42] by PCR-based site-directed mutagenesis using Q5® Site-Directed Mutagenesis Kit (New England Biolabs) and the following mutagenic primers: delI153: 5′-TCAAGATGGTGGCCTTGG-3′ (forward) and 5′-CCATCTCCACCGCAAAAAAG-3′ (reverse); P1210L: 5′-GCCGCCCTCCtGCCTACCAAGTGC-3′ (forward) and 5′-CGGCCGCAGGGGCCGTGG-3′ (reverse). The cation-impermeant Cav3.2 channel was generated by replacing the glutamic acid 378 in domain I with a lysine (E378K) by site-directed mutagenesis. Final constructs were verified by sequencing of the coding sequence of the plasmid cDNAs.
Cell culture and heterologous expression
Human embryonic kidney tsA-201 cells were grown in DMEM medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (all media purchased from Invitrogen) and maintained under standard conditions at 37 °C in a humidified atmosphere containing 5% CO2. Heterologous expression of Cav3.2 channels was performed by transfecting cells with 5 μg plasmid cDNAs encoding for Cav3.2 channel variants using the calcium/phosphate method. For experiments aiming at investigating the dominant negative effect of the ΔI153 variant, cells were co-transfected with 2.5 μg plasmid cDNA encoding for WT channels with either 2.5 μg plasmid cDNA encoding for the ΔI153 channel variant or 2.5 μg plasmid cDNA encoding for a non-conducting but trafficking-competent Cav3.2 (PM).
Patch clamp electrophysiology
Patch clamp recordings of T-type currents in tsA-201 cells expressing Cav3.2 channel variants were performed 72 h after transfection in the whole-cell configuration at room temperature (22-24 °C) as previously described [43]. The bath solution contained (in millimolar): 5 BaCl2, 5 KCl, 1 MgCl2, 128 NaCl, 10 TEA-Cl, 10 D-glucose, 10 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.2 with NaOH). Patch pipettes were filled with a solution containing (in millimolar): 110 CsCl, 3 Mg-ATP, 0.5 Na-GTP, 2.5 MgCl2, 5 D-glucose, 10 EGTA, and 10 HEPES (pH 7.4 with CsOH), and had a resistance of 2–4 MΩ. Recordings were performed using an Axopatch 200B amplifier (Axon Instruments) and acquisition and analysis were performed using pClamp 10 and Clampfit 10 software, respectively (Axon Instruments). The linear leak component of the current was corrected online and current traces were digitized at 10 kHz and filtered at 2 kHz. The voltage dependence of activation of Cav3.2 channels was determined by measuring the peak T-type current amplitude in response to 150 ms depolarizing steps to various potentials applied every 10 s from a holding membrane potential of − 100 mV. The current-voltage relationship (I/V) curve was fitted with the following modified Boltzmann eq. (1):
with I(V) being the peak current amplitude at the command potential V, Gmax the maximum conductance, Vrev the reversal potential, V0.5 the half-activation potential, and k the slope factor. The voltage dependence of the whole-cell Ba2+ conductance was calculated using the following modified Boltzmann eq. (2):
with G(V) being the Ba2+ conductance at the command potential V.
The voltage dependence of the steady-state inactivation of Cav3.2 channels was determined by measuring the peak T-type current amplitude in response to a 150 ms depolarizing step to − 20 mV applied after a 5 s-long conditioning prepulse ranging from − 120 mV to − 30 mV. The current amplitude obtained during each test pulse was normalized to the maximal current amplitude and plotted as a function of the prepulse potential. The voltage dependence of the steady-state inactivation was fitted with the following two-state Boltzmann function (3):
with Imax corresponding to the maximal peak current amplitude and V0.5 to the half-inactivation voltage.
The recovery from inactivation was assessed using a double-pulse protocol from a holding potential of − 100 mV. The cell membrane was depolarized for 2 s at 0 mV (inactivating prepulse) to ensure complete inactivation of the channel, and then to − 20 mV for 150 ms (test pulse) after an increasing time period (interpulse) ranging between 0.1 ms and 2 s at − 100 mV. The peak current from the test pulse was plotted as a ratio of the maximum prepulse current versus interpulse interval. The data were fitted with the following single-exponential function (4):
where τ is the time constant for channel recovery from inactivation.
Measurement of charge movements
Recording of charge movements was performed 72 h after transfection as previously described [44, 45]. The bath solution contained (in millimolar): CsCl 95; TEACl 40, BaCl2 5; MgCl2 1; HEPES 10; glucose 10; pH 7.4 (adjusted with CsOH). Patch pipettes had a resistance ranging from 1.8 MΩ to 2.2 MΩ when filled with a solution containing (in millimolar): CH3SO3Cs 130; Na-ATP 5; TEACl 10; HEPES 10; EGTA 10; MgCl2 5; pH 7.4 (adjusted with CsOH). Osmolarity of the intracellular solution was approximately 300 mOsmol/L. Osmolarity of the extracellular solution was adjusted by adding sucrose so that the final value was about 2–3 mOsmol/L lower than the osmolarity of the corresponding intracellular solution. Recordings were performed using HEKA EPC10 amplifier (HEKA Electronics). Acquisition and analysis were performed using Patchmaster v90.2 and Fitmaster v2x73.1 and Origin Pro 2015 software, respectively. Only cells with an input resistance less than 5 MΩ were considered. The input resistance and capacity transients were compensated by up to 70% with in-built circuits of the EPC 10 amplifier. Remaining artifacts were subtracted using a -P/8 procedure. ON-gating currents were recorded in response to a series of 5 depolarizing pulses at the reversal potential of the ionic current assessed for each cell, and total gating charge QON was calculated as the integral of area below the averaged current traces.
CRISPR/Cas9 genome editing in DRG neurons
Male rats (6-week-old) were purchased from Charles River and DRG neurons were harvested as described previously [46]. The next day, neurons were transfected with Crispr-Cas9 plasmids (Cas9-sgRNA plasmid and donor plasmid purchased from GeneCopoeia) using Lipofectamine 2000 from Invitrogen (Cat. 11,668–019). The sequence of Crispr RNA was CGTGGAGATGGTGATCAAGA. The donor plasmid contained the homologous arms of the genomic DNA without I153. Whole-cell voltage-clamp recordings of T-type currents were performed 3 days post transfection. The external solution contained (in mM): 40 TEACl, 65 CsCl, 20 BaCl2, 1 MgCl2, 10 HEPES, 10 D-glucose, pH 7.4. The internal solution contained (in mM): 140 CsCl, 2.5 CaCl2, 1 MgCl2, 5 EGTA, 10 HEPES, 2 Na-ATP, 0.3 Na-GTP, pH 7.3. We used GFP fluorescence to specifically identify neurons that were transfected with the CRISPR plasmids. The overall percentage of GFP positive neurons in a dish was relatively low, and hence we cannot use bulk genomic sequencing for verification. However, given the large functional effect on current densities, we are confident that the use of GFP fluorescence is an appropriate means of identifying neurons that were targeted with these plasmids. We specifically targeted medium diameter neurons for our analysis. The mean capacitance of the neurons that we recorded from was 24.79 ± 4.40 pF for control neurons versus 21.89 ± 1.31 pF for CRISPR-edited neurons.
SDS-PAGE and immunoblot analysis
Immunoblot of HA-tagged Cav3.2 channel was performed as previously described [16]. Briefly, total cell lysate from tsA-201 cells expressing HA-Cav3.2 channels was separated on a 5–20% gradient SDS-PAGE gel and transferred onto PVDF membrane (Millipore). Detection of HA-Cav3.2 was performed using a primary rat monoclonal anti-HA antibody (1:1000, Roche) and secondary HRP-conjugated antibody (1:10,000, Jackson ImmunoResearch). Immunoreactive products were detected by enhanced chemiluminescence and analyzed using ImageJ software.
For co-immunoprecipitation, cell lysates containing GFP-tagged and Myc-tagged Cav3.2 were incubated for 3 h with a biotinylated mouse monoclonal anti-Myc antibody (Santa Cruz Biotechnology), and then for 45 min with streptavidin beads (Invitrogen) at 4 °C, and washed with PBS/Tween-20 buffer. Beads were resuspended in Laemmli buffer and immunoprecipitation samples were separated on SDS-PAGE gel.
Generation of human Cav3.2 homology model
The homology model of the human Cav3.2 channel was prepared using the Cav3.1 structure as a template (PDB: 6KZO) in conjunction with Swiss-Model server (https://swissmodel.expasy.org/) [47]. Figures were prepared using Pymol (v2.2 Schrödinger, LLC.).
Statistics
Data values are presented as mean ± SEM for n measurements. Statistical analysis was performed using GraphPad Prism 7. For datasets passing the D’Agostino & Pearson omnibus normality test, statistical significance was determined using either Student’s t-test or a Mann-Whitney test. Datasets were considered significantly different for p ≤ 0.05.
Availability of data and materials
The data used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- DRG:
-
Dorsal root ganglia
- GFP:
-
Green fluorescent protein
- G max :
-
Maximal macroscopic conductance
- MRI:
-
Magnetic resonance imaging
- PM:
-
Pore mutant
- Q max :
-
Maximal charge movements
- Q rev :
-
Charge movements at the reversal calcium potential
- WT:
-
Wild-type
References
Marin B, Fontana A, Arcuti S, Copetti M, Boumédiene F, Couratier P, et al. Age-specific ALS incidence: a dose-response meta-analysis. Eur J Epidemiol. 2018;33(7):621–34.
Marin B, Boumédiene F, Logroscino G, Couratier P, Babron MC, Leutenegger AL, et al. Variation in worldwide incidence of amyotrophic lateral sclerosis: a meta-analysis. Int J Epidemiol. 2017;46(1):57–74.
Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17071.
Talbot K. Familial versus sporadic amyotrophic lateral sclerosis--a false dichotomy. Brain. 2011;134(Pt 12):3429–31.
Nguyen HP, Van Broeckhoven C, van der Zee J. ALS genes in the genomic era and their implications for FTD. Trends Genet. 2018;34(6):404–23.
Gibson SB, Downie JM, Tsetsou S, Feusier JE, Figueroa KP, Bromberg MB, et al. The evolving genetic risk for sporadic ALS. Neurology. 2017;89(3):226–33.
Sproviero W, Shatunov A, Stahl D, Shoai M, van Rheenen W, Jones AR, et al. ATXN2 trinucleotide repeat length correlates with risk of ALS. Neurobiol Aging. 2017;51:178.e1–9.
van Es MA, Veldink JH, Saris CG, Blauw HM, van Vught PW, Birve A, et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 2009;41(10):1083–7.
Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, et al. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet. 2006;38(4):411–3.
Corcia P, Camu W, Halimi JM, Vourc’h P, Antar C, Vedrine S, et al. SMN1 gene, but not SMN2, is a risk factor for sporadic ALS. Neurology. 2006;67(7):1147–50.
Steinberg KM, Yu B, Koboldt DC, Mardis ER, Pamphlett R. Exome sequencing of case-unaffected-parents trios reveals recessive and de novo genetic variants in sporadic ALS. Sci Rep. 2015;5:9124.
Rzhepetskyy Y, Lazniewska J, Blesneac I, Pamphlett R, Weiss N. CACNA1H missense mutations associated with amyotrophic lateral sclerosis alter Cav3.2 T-type calcium channel activity and reticular thalamic neuron firing. Channels (Austin). 2016;10(6):466–77.
Meltz Steinberg K, Nicholas TJ, Koboldt DC, Yu B, Mardis E, Pamphlett R. Whole genome analyses reveal no pathogenetic single nucleotide or structural differences between monozygotic twins discordant for amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(5–6):385–92.
Nitrini R. Frontotemporal dementia and amyotrophic lateral sclerosis: revisiting one of the first case reports with neuropathology examination. Dement Neuropsychol. 2014;8(1):83–6.
Rzhepetskyy Y, Lazniewska J, Proft J, Campiglio M, Flucher BE, Weiss N. A Cav3.2/Stac1 molecular complex controls T-type channel expression at the plasma membrane. Channels (Austin). 2016;10(5):346–54.
Proft J, Rzhepetskyy Y, Lazniewska J, Zhang FX, Cain SM, Snutch TP, et al. The Cacna1h mutation in the GAERS model of absence epilepsy enhances T-type Ca2+ currents by altering calnexin-dependent trafficking of Cav3.2 channels. Sci Rep. 2017;7(1):11513.
Bourinet E, Alloui A, Monteil A, Barrère C, Couette B, Poirot O, et al. Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J. 2005;24(2):315–24.
Powell KL, Cain SM, Ng C, Sirdesai S, David LS, Kyi M, et al. A Cav3.2 T-type calcium channel point mutation has splice-variant-specific effects on function and segregates with seizure expression in a polygenic rat model of absence epilepsy. J Neurosci. 2009;29(2):371–80.
Souza IA, Gandini MA, Wan MM, Zamponi GW. Two heterozygous Cav3.2 channel mutations in a pediatric chronic pain patient: recording condition-dependent biophysical effects. Pflugers Arch. 2016;468(4):635–42.
Anderson D, Mehaffey WH, Iftinca M, Rehak R, Engbers JD, Hameed S, et al. Regulation of neuronal activity by Cav3-Kv4 channel signaling complexes. Nat Neurosci. 2010;13(3):333–7.
Engbers JD, Anderson D, Asmara H, Rehak R, Mehaffey WH, Hameed S, et al. Intermediate conductance calcium-activated potassium channels modulate summation of parallel fiber input in cerebellar Purkinje cells. Proc Natl Acad Sci U S A. 2012;109(7):2601–6.
Rehak R, Bartoletti TM, Engbers JD, Berecki G, Turner RW, Zamponi GW. Low voltage activation of KCa1.1 current by Cav3-KCa1.1 complexes. PLoS One. 2013;8(4):e61844.
Garcia-Caballero A, Gandini MA, Huang S, Chen L, Souza IA, Dang YL, et al. Cav3.2 calcium channel interactions with the epithelial sodium channel ENaC. Mol Brain. 2019;12(1):12.
Zhao Y, Huang G, Wu Q, Wu K, Li R, Lei J, et al. Cryo-EM structures of apo and antagonist-bound human Cav3.1. Nature. 2019;576(7787):492–7.
Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. 1999;19(6):1895–911.
Wang G, Bochorishvili G, Chen Y, Salvati KA, Zhang P, Dubel SJ, et al. CaV3.2 calcium channels control NMDA receptor-mediated transmission: a new mechanism for absence epilepsy. Genes Dev. 2015;29(14):1535–51.
Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis. 2004;15(3):601–9.
Chang JL, Lomen-Hoerth C, Murphy J, Henry RG, Kramer JH, Miller BL, et al. A voxel-based morphometry study of patterns of brain atrophy in ALS and ALS/FTLD. Neurology. 2005;65(1):75–80.
Sharma KR, Saigal G, Maudsley AA, Govind V. 1H MRS of basal ganglia and thalamus in amyotrophic lateral sclerosis. NMR Biomed. 2011;24(10):1270–6.
Sharma KR, Sheriff S, Maudsley A, Govind V. Diffusion tensor imaging of basal ganglia and thalamus in amyotrophic lateral sclerosis. J Neuroimaging. 2013;23(3):368–74.
Bede P, Elamin M, Byrne S, McLaughlin RL, Kenna K, Vajda A, et al. Basal ganglia involvement in amyotrophic lateral sclerosis. Neurology. 2013;81(24):2107–15.
Menke RA, Körner S, Filippini N, Douaud G, Knight S, Talbot K, et al. Widespread grey matter pathology dominates the longitudinal cerebral MRI and clinical landscape of amyotrophic lateral sclerosis. Brain. 2014;137(Pt 9):2546–55.
Tu S, Menke RAL, Talbot K, Kiernan MC, Turner MR. Regional thalamic MRI as a marker of widespread cortical pathology and progressive frontotemporal involvement in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2018;89(12):1250–8.
Canto-Bustos M, Loeza-Alcocer E, González-Ramírez R, Gandini MA, Delgado-Lezama R, Felix R. Functional expression of T-type Ca2+ channels in spinal motoneurons of the adult turtle. PLoS One. 2014;9:e108187.
Zhang Z, David G. Stimulation-induced Ca (2+) influx at nodes of Ranvier in mouse peripheral motor axons. J Physiol. 2016;594(1):39–57.
Shtonda B, Avery L. CCA-1, EGL-19 and EXP-2 currents shape action potentials in the Caenorhabditis elegans pharynx. J Exp Biol. 2005;208(Pt 11):2177–90.
Steger KA, Shtonda BB, Thacker C, Snutch TP, Avery L. The C. elegans T-type calcium channel CCA-1 boosts neuromuscular transmission. J Exp Biol. 2005;208(Pt 11):2191–203.
Nicoletti M, Loppini A, Chiodo L, Folli V, Ruocco G, Filippi S. Biophysical modeling of C. elegans neurons: Single ion currents and whole-cell dynamics of AWCon and RMD. PLoS One. 2019;14(7):e0218738.
Kim JW, Oh HA, Lee SH, Kim KC, Eun PH, Ko MJ, et al. T-type calcium channels are required to maintain viability of neural progenitor cells. Biomol Ther (Seoul). 2018;26(5):439–45.
Weiss N, Zamponi GW. Genetic T-type calcium channelopathies. J Med Genet. 2020;57(1):1–10.
Weiss N, Zamponi GW. T-type calcium channels: from molecule to therapeutic opportunities. Int J Biochem Cell Biol. 2019;108:34–9.
Dubel SJ, Altier C, Chaumont S, Lory P, Bourinet E, Nargeot J. Plasma membrane expression of T-type calcium channel alpha (1) subunits is modulated by high voltage-activated auxiliary subunits. J Biol Chem. 2004;279(28):29263–9.
Carter MT, McMillan HJ, Tomin A, Weiss N. Compound heterozygous CACNA1H mutations associated with severe congenital amyotrophy. Channels (Austin). 2019;13(1):153–61.
Ondacova K, Karmazinova M, Lazniewska J, Weiss N, Lacinova L. Modulation of Cav3.2 T-type calcium channel permeability by asparagine-linked glycosylation. Channels (Austin). 2016;10(3):175–84.
Jurkovicova-Tarabova B, Cmarko L, Rehak R, Zamponi GW, Lacinova L, Weiss N. Identification of a molecular gating determinant within the carboxy terminal region of Cav3.3 T-type channels. Mol Brain. 2019;12(1):34.
Altier C, Khosravani H, Evans RM, Hameed S, Peloquin JB, Vartian BA, et al. ORL1 receptor-mediated internalization of N-type calcium channels. Nat Neurosci. 2006;9(1):31–40.
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–303.
Acknowledgements
We thank the patients and family members for their contribution to this study.
Funding
N.W. is supported by the Institute of Organic Chemistry and Biochemistry. L.L. is supported by a grant VEGA 2/0143/19. N.W. and L.L. are supported by a bilateral SAS-CAS project (SAV-18-22). G.W.Z. is supported by a grant from the Natural Sciences and Engineering Research Council and holds a Canada Research Chair.
Author information
Authors and Affiliations
Contributions
N.W., L.L., F.V.P., G.W.Z., and R.P. designed and conceptualized the study. R.N.S., R.I., B.J.T., O.H., S.H., I.A.S., A.L., Y.R., and N.W. collected data, performed analysis and interpreted the results. N.W. and R.P. wrote the manuscript. All authors critically revised the manuscript and contributed significantly to this work. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The whole genome sequencing of white blood cell DNA that gave rise to the finding of the genetic variants further characterised in the present study was undertaken by RP in a joint University of Sydney (Australia) and the Genome Institute Washington University (St Louis, USA) project using DNA samples from the Australian Motor Neuron Disease DNA Bank, with approval from the Sydney South West Area Health Service Human Research Ethics Committee. Informed written consent was obtained from each individual for their DNA to be used for research purposes.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Stringer, R.N., Jurkovicova-Tarabova, B., Huang, S. et al. A rare CACNA1H variant associated with amyotrophic lateral sclerosis causes complete loss of Cav3.2 T-type channel activity. Mol Brain 13, 33 (2020). https://doi.org/10.1186/s13041-020-00577-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-020-00577-6