
Abstract
Background
Infection with Salmonella enterica is a major public health concern in developed countries, and multidrug-resistant strains have become increasingly prevalent. S. enterica serovar Typhimurium DT104 (DT104) strains are prevalent in livestock in Japan and include numerous strains of multidrug-resistant S. enterica. Epidemiological analysis of these strains is critical for both agriculture and public health; however, diagnostic tests for these strains have yielded inconsistent results.
Results
We developed a rapid, simple, and inexpensive polymerase chain reaction test to detect multi-drug resistant DT104 strains. We designed primers specific to the prophage ST104 sequence encoded by DT104 strains and assessed the specificity of these primers by assaying a panel of 50 S. enterica isolates. Amplification products of the expected size were generated from the genomes of each of the DT104 strains; however, the ST104 primers failed to amplify products from non-DT104 strains of S. enterica serovar Typhimurium or other S. enterica serovars. Furthermore, a probe generated using the ST104 primers detected a restriction fragment encoding the ST104 region of DT104 by Southern hybridization.
Conclusions
The ST104 primers exhibit specificity to DT104 strains and are suitable for epidemiological applications.
Similar content being viewed by others

Background
Infection with Salmonella enterica is a major public health concern in developed countries. The multidrug-resistant S. enterica serovar Typhimurium (S. enterica Typhimurium) definitive phage type 104 (DT104) strain was first isolated in the late 1980s and has since spread across Japan and through numerous western countries [1–5]. DT104 strains exhibit a core pattern of resistance to ampicillin, chloramphenicol, streptomycin, sulfonamides, and tetracycline; the genes that confer resistance to these antimicrobials are encoded by a chromosomal locus containing Class 1 integrons [6]. Malthe et al. [7] recently reported that DT104 strains are also resistant to ciprofloxacin, making them particularly difficult organisms to control.
DT104 strains have a wide host range and are pathogenic in humans and livestock [8], with cattle, in particular, being considered a major reservoir. Although food-borne illnesses associated with S. enterica Typhimurium decreased in Japan during the 1990s [9], human nontyphoidal salmonellosis caused by DT104 strains remains a serious public health problem [10]. In a previous study, Sameshima et al. [11] demonstrated that DT104 strains have existed in Japanese livestock since 1990 and found that more than half (36 of 68) of the S. enterica Typhimurium isolates resistant to five or more antibiotics were DT104 strains. Furthermore, Esaki et al. [12] reported several pulsotypes of DT104 in multiple animal species in Japan. These findings suggest that, rather than evolving from a single clonal strain, multiple DT104-related strains have been introduced in Japan through various routes, including domestic animals, wild birds, and/or food.
Japanese DT104 isolates are genetically similar to the predominant strains found abroad, and the previous studies have demonstrated that all DT104 isolates contain the same prophage (designated ST104) [10–14]. However, Hermans et al. [15] identified strains that contain additional prophages (ST104B and/or ST64B) that are similar to ST104 but represent distinct DT104 subtypes. While epidemiological investigation of DT104 strains is an important task in agriculture and public health, the identification of S. enterica Typhimurium phage types is time-consuming and requires specially trained personnel [16]. In addition, possession of this phage type is limited to a few centralized laboratories. Several reports have described PCR detection of DT104 [17, 18]; however, these assays are inaccurate, often exhibiting nonspecific and false-positive reactions. In this study, we describe an improved PCR-based method for detecting DT104 strains.
Methods
Polymerase chain reaction (PCR) assays
The ST104forward and ST104reverse primers were designed to amplify a 312-base pair (bp) segment of the erf-like gene within the ST104 sequence (Fig. 1; Table 1) and were used to detect S. enterica Typhimurium DT104 strains. For comparison, we used the DT104F and DT104R primers [17], which amplify a 162-bp cryptic sequence from DT104 strains (Table 1). In addition, primers InvAforward and InvAreverse, which amplify a 512-bp segment of the S. enterica invA gene [15, 19, 20], were used as positive controls for sample preparation and amplification (Table 1).

Nucleotide sequence of the ST104 erf-like gene. The ST104 sequence was amplified from Salmonella enterica serovar Typhimurium DT104 by using the ST104 primers
The primers used for PCR analysis of 50 S. enterica isolates are listed in Table 2. The isolates included 30 S. enterica Typhimurium DT104 isolates; 12 non-DT104 S. enterica Typhimurium isolates; two isolates of S. enterica Enteritidis; and one isolate each of S. enterica Brandenburg, S. enterica Dublin, S. enterica Javiana, S. enterica Montevideo, S. enterica Nagoya, and S. enterica Saintpaul. The Salmonella strains were obtained from the National Veterinary Assay Laboratory in Japan (Table 2). The clinical isolates were epidemiologically unrelated. After serological examination, the serovars and phage types were identified. To harvest template DNA, S. enterica strains were grown overnight at 37 °C on brain–heart infusion agar plates and then one loopful of cells of each strain was boiled in 200 µl sterile distilled water for 10 min. After centrifugation, the supernatants were stored at −20 °C. PCR amplification reactions were performed as follows: 1 μl template was added to a 20 μl reaction mixture containing 1× PCR buffer (Takara Bio, Tokyo, Japan), 1 μM of each primer, 200 μM deoxynucleoside triphosphates, 2 mM MgCl2, and 0.5 U of AmpliTaq DNA Polymerase (Takara Bio). Amplification was performed in 0.2 ml tubes in an iCycler PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA) as follows: denaturation at 95 °C for 1 min; 30 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min; and a final extension cycle at 72 °C for 3 min. The reaction mixtures were separated by 2 % agarose gel electrophoresis (Agarose L03; Takara Bio).
Pulsed-field gel electrophoresis
Pulsed-field gel electrophoresis (PFGE) was performed by contour-clamped homogeneous electric-field electrophoresis using a CHEF-DR® II system (Bio-Rad). PFGE was used in combination with Southern blotting to achieve high resolution when detecting ST104 primer-targeted sequences. One of the DT104 strains was grown overnight at 37 °C in lysogeny broth. Cells were harvested by centrifugation for 10 min at 3600×g and resuspended in 0.5 ml of NT buffer [10 mM Tris–HCl (pH 7.5), 1 M NaCl]. An aliquot (0.3 ml) of each suspension was transferred to a microcentrifuge tube, and the cells were pelleted at 12,000×g and washed twice in NT buffer. The cell suspensions were mixed with an equal volume of 1.5 % low-melting point agarose (FMC Bioproducts, Philadelphia, PA, USA) and allowed to solidify in 100 µl plug molds (Bio-Rad). The agarose plugs were incubated overnight at 55 °C in 1 ml lysis buffer [60 mM Tris–HCl (pH 7.5), 50 mM ethylenediaminetetraacetic acid (EDTA), 1 % sodium lauroyl sarcosine, lysozyme (1 mg/ml), RNase (1 µg/ml), proteinase K (1 mg/ml)], washed twice for 30 min with TE buffer [10 mM Tris–HCl (pH 7.5), 0.1 mM EDTA] containing the protease inhibitor phenylmethylsulfonyl fluoride (1 mM), and washed four additional times for 30 min each with 1 ml of TE buffer. A slice of each plug was cut and incubated in 400 µl restriction buffer containing 50 U XbaI (Takara Bio) at 37 °C for 2 h. The restriction enzyme-digested DNA fragments were separated using pulsed-field electrophoresis-certified agarose (Bio-Rad). Electrophoresis was performed for 25 h at 14 °C and 6 V/cm in twofold-diluted TBE buffer with pulse times of 5–80 s. Lambda PFGE DNA markers (New England BioLabs, Ipswich, MA, USA) were used as DNA size markers.
Southern hybridization
A DNA fragment containing the ST104 bacteriophage region was amplified from DT104 with the ST104F and ST104R primers (Table 1). The PCR product was purified using a QIAquick® Gel Extraction Kit (Qiagen, Hilden, Germany) and labeled with digoxigenin (DIG)-11-deoxyuridine triphosphate by random priming using a DIG-High Prime DNA Labeling Kit (Boehringer GmbH, Germany), according to the manufacturer’s instructions. The labeled PCR product was then used as a probe for Southern hybridization analyses. After PFGE of agarose plugs, genomic DNA from S. enterica strains was transferred to positively charged membranes (Boehringer) by capillary action. Prehybridization (>30 min) and hybridization (>16 h) were performed in DIG Easy Hyb Solution (Boehringer) under high-stringency conditions, and hybrid detection was achieved with a DIG Luminescent Detection Kit (Boehringer), according to the manufacturer’s instructions. Hybridization products were detected by exposing Hyperfilm MP (Amersham International, Little Chalfont, UK) to the membranes for 1–10 min at room temperature. Films were developed in a Kodak X-Omat processor (Rochester, NY, USA).
ST104 primer target analysis
BLAST was used for in silico identification of genes with strong homology to the sequence targeted by the ST104 primers (Fig. 1).
Results
ST104 primers are specific for DT104 strains
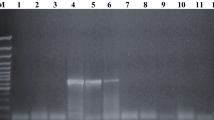
To assess the specificity of the ST104 primers, we performed PCR analysis with genomic DNA from a panel of 50 S. enterica isolates (Table 2). The previously published DT104 primers [17] were used for comparison. While the ST104 primers amplified the expected 312-bp product from each of the 30 DT104 strains tested, PCR yielded no products from the 12 non-DT104 S. enterica Typhimurium strains (Table 2). Likewise, the ST104 primers failed to amplify a product from eight S. enterica serovars (Table 2). Conversely, the DT104 primers amplified the expected 162-bp product from 29 of the DT104 strains (97 %). Although this 162-bp product was not detected in PCR reactions with non-DT104 S. enterica Typhimurium strains, nonspecific amplification was detected for one DT104 strain and in all of the non-DT104 strain reactions. Furthermore, the DT104 primers yielded false-positive products from the S. enterica Montevideo and S. enterica Saintpaul genomes (25 % of the non-Typhimurium serovars tested) and nonspecific products from the DNA of S. enterica Javiana, S. enterica Nagoya, S. enterica Brandenburg, and S. enterica Enteritidis (50 %; Table 2; Fig. 2). The InvA primers, which were used as positive controls for sample preparation and amplification, yielded the expected product from all 50 S. enterica strains (Table 2).

Southern blot hybridization analysis of Salmonella enterica strains. The ST104 sequence was amplified from S. enterica serovar Typhimurium DT104 with the ST104 primers and used as a probe for Southern blot analysis of S. enterica strains. A representative image of the fingerprinting pattern of DT104 after restriction digestion with XbaI is depicted in the left lane. The result of a subsequent Southern blot hybridization experiment, using the digoxigenin (DIG)-11-deoxyuridine triphosphate-labeled ST104 PCR fragment, is depicted in the right lane
Detection of the ST104 region in DNA fragments from DT104 strains by Southern hybridization
To detect the ST104 sequence in DT104 isolates, we performed PFGE followed by Southern hybridization. The Southern blot probe was generated by amplifying the ST104 region from DT104 DNA with the ST104 primers. The ST104 probe hybridized to a region within a genomic DNA restriction fragment of approximately 485 kilobase pairs (kbp) in the DT104 strain (Fig. 2).
In silico analysis of target sequences for the ST104 primers
A 312-bp sequence targeted by the ST104 primers was detected from two non-DT104 Salmonella Typhimurium strains, one Salmonella Pullorum strain, and a bacteriophage [GenBank: CP003836, CP006575, AP011957, and JF900176]. In addition, strong sequence homology above 99 % (310/312) was detected in the S. enterica subsp. arizonae strain [GenBank: CP000880].
Discussion
We aimed to develop a rapid and specific screen for S. enterica serovar Typhimurium DT104. To this end, we developed primers to detect the temperate phage ST104, which is encoded by all DT104 isolates. Because ST104 exhibits a high level of sequence identity to Enterobacteria phage P22, which also exists in S. enterica Typhimurium, the ST104 primers were carefully designed to avoid unintended detection of this phage. Indeed, our results demonstrate that these primers exhibited a high level of sensitivity for DT104 strains, and did not result in false positives or negatives. Moreover, our primers were more sensitive and specific than the previously published DT104 primers [17], which also elicited several false-positive and nonspecific reactions. These results indicated that PCR analysis using the ST104 primers is a reliable and valuable diagnostic tool for the rapid identification of DT104 strains. In addition, we utilized the ST104 primers to generate a probe for Southern blot analysis of DT104 strains. This probe specifically hybridized to a restriction fragment (approximately 485 kbp) present in DT104 strains. While BLAST sequence analysis of the ST104 primer targets showed homology with a few non-DT104 Salmonella strains, the ST104 primers did not result in false-positive or -negative results with Salmonella strains in this study.
Conclusions
PCR analysis with ST104 primers is an inexpensive, rapid, and simple technique for detecting DT104 strains in epidemiological applications. While gene sequences similar to ST104 may exist in non-DT104 strains of S. enterica Typhimurium, our PCR method may facilitate future studies endeavoring to screen for specific phage types.
References
Threlfall EJ, Frost JA, Ward LR, Rowe B. Epidemic in cattle and humans of Salmonella typhimurium DT104 with chromosomally integrated multiple drug resistance. Vet Rec. 1994;134:577.
Ng LK, Khakhria R, Woodward D, Johnson W. National laboratory surveillance of enteric pathogens. Can J Infect Dis. 1997;8:133–6.
Baggesen DL, Aarestrup FM. Characterization of recently emerged multiple antibiotic-resistant Salmonella enterica serovar typhimurium DT104 and other multiresistant phage types from Danish pig herds. Vet Rec. 1998;143:95–7.
Glynn MK, Bopp C, Dewitt W, Dabney P, Mokhtar M, Angulo FJ. Emergence of multidrug-resistant Salmonella enterica serotype Typhimurium DT104 infections in the United States. N Engl J Med. 1998;338:1333–8.
Helms M, Ethelberg S, Molbak K. DT104 Study Group. International Salmonella Typhimurium DT104 infections, 1992–2001. Emerg Infect Dis. 2005;11:859–67.
Briggs CE, Fratamico PM. Molecular characterization of an antibiotic resistance gene cluster of Salmonella typhimurium DT104. Antimicrob Agents Chemother. 1999;43:846–9.
Malthe A, Kristiansen M, Sandvang D, Rasmussen TB. In vivo development of quinolone resistance in Salmonella enterica serotype Typhimurium DT104. J Clin Microbiol. 2003;41:4462–4.
Besser TE, Gay CC, Gay JM, Hancock DD, Rice D, Pritchett LC, et al. Salmonellosis associated with S. typhimurium DT104 in the USA. Vet Rec. 1997;140:75.
National Institute of Health and Infectious Disease Control Division, Ministry of Health and Welfare of Japan. Salmonella, Japan, 1994-1996. Infect Agents Surveill Rep. 1997;18: 51–2.
Izumiya H, Terashima J, Matsushita S, Tamura K, Watanabe H. Characterization of multidrug-resistant Salmonella enterica serovar Typhimurium isolated in Japan. J Clin Microbiol. 2001;39:2700–3.
Sameshima T, Akiba M, Izumiya H, Terajima J, Tamura K, Watanabe H, et al. Salmonella typhimurium DT104 from livestock in Japan. Jpn J Infect Dis. 2000;53:15–6.
Esaki H, Morioka A, Kojima A, Ishihara K, Asai T, Tamura Y, et al. Epidemiological characterization of Salmonella Typhimurium DT104 prevalent among food-producing animals in the Japanese veterinary antimicrobial resistance monitoring program (1999–2001). Microbiol Immunol. 2004;48:553–6.
Tamada Y, Nakaoka Y, Nishimori K, Doi A, Kumaki T, Uemura N, et al. Molecular typing and epidemiological study of Salmonella enterica serotype Typhimurium isolates from cattle by fluorescent amplified-fragment length polymorphism fingerprinting and pulsed-field gel electrophoresis. J Clin Microbiol. 2001;39:1057–66.
Tanaka K, Nishimori K, Makino S, Nishimori T, Kanno T, Ishihara R, et al. Molecular characterization of a prophage of Salmonella enterica serotype Typhimurium DT104. J Clin Microbiol. 2004;42:1807–12.
Hermans AP, Beuling AM, van Hoek AH, Aarts HJ, Abee T, Zwietering MH. Distribution of prophages and SGI-1 antibiotic-resistance genes among different Salmonella enterica serovar Typhimurium isolates. Microbiology. 2006;152:2137–47.
Anderson ES, Ward LR, Saxe MJ, de Sa JD. Bacteriophage-typing designations of Salmonella typhimurium. J Hyg. 1977;78:297–300.
Pritchett LC, Konkel ME, Gay JM, Besser TE. Identification of DT104 and U302 phage types among Salmonella enterica serotype Typhimurium isolates by PCR. J Clin Microbiol. 2000;38:3484–8.
Alvarez J, Sota M, Vivanco AB, Perales I, Cisterna R, Rementeria A, et al. Development of a multiplex PCR technique for detection and epidemiological typing of Salmonella in human clinical samples. J Clin Microbiol. 2004;42:1734–8.
Chiu CH, Ou JT. Rapid identification of Salmonella serovars in feces by specific detection of virulence genes, invA and spvC, by an enrichment broth culture-multiplex PCR combination assay. J Clin Microbiol. 1996;34:2619–22.
Boyd EF, Li J, Ochman H, Selander RK. Comparative genetics of the inv-spa invasion gene complex of Salmonella enterica. J Bacteriol. 1997;179:1985–91.
Authors’ contributions
SY carried out this study. IU and YT participated in the study design. KT and IU provided assistance. YT supplied the Salmonella strains. SY drafted the manuscript. All authors read and approved the final manuscript.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yukawa, S., Tamura, Y., Tanaka, K. et al. Rapid detection of Salmonella enterica serovar Typhimurium DT104 strains by the polymerase chain reaction. Acta Vet Scand 57, 59 (2015). https://doi.org/10.1186/s13028-015-0143-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13028-015-0143-x