
Abstract
This narrative review focuses on the role of cholesteryl ester transfer protein (CETP) and peripheral lipoproteins in the vascular contributions to cognitive impairment and dementia (VCID). Humans have a peripheral lipoprotein profile where low-density lipoproteins (LDL) represent the dominant lipoprotein fraction and high-density lipoproteins (HDL) represent a minor lipoprotein fraction. Elevated LDL-cholesterol (LDL-C) levels are well-established to cause cardiovascular disease and several LDL-C-lowering therapies are clinically available to manage this vascular risk factor. The efficacy of LDL-C-lowering therapies to reduce risk of all-cause dementia and AD is now important to address as recent studies demonstrate a role for LDL in Alzheimer’s Disease (AD) as well as in all-cause dementia. The LDL:HDL ratio in humans is set mainly by CETP activity, which exchanges cholesteryl esters for triglycerides across lipoprotein fractions to raise LDL and lower HDL as CETP activity increases. Genetic and pharmacological studies support the hypothesis that CETP inhibition reduces cardiovascular risk by lowering LDL, which, by extension, may also lower VCID. Unlike humans, wild-type mice do not express catalytically active CETP and have HDL as their major lipoprotein fraction. As HDL has potent beneficial effects on endothelial cells, the naturally high HDL levels in mice protect them from vascular disorders, likely including VCID. Genetic restoration of CETP expression in mice to generate a more human-like lipid profile may increase the relevance of murine models for VCID studies. The therapeutic potential of existing and emerging LDL-lowering therapies for VCID will be discussed.
Graphical Abstract
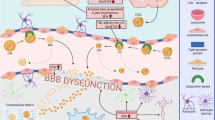
Figure Legend. Cholesteryl Ester Transfer Protein in Alzheimer’s Disease. CETP is mainly produced by the liver, and exchanges cholesteryl esters for triglycerides across lipoprotein fractions to raise circulating LDL and lower HDL as CETP activity increases. Low CETP activity is associated with better cardiovascular health, due to decreased LDL and increased HDL, which may also improve brain health. Although most peripheral lipoproteins cannot enter the brain parenchyma due to the BBB, it is increasingly appreciated that direct access to the vascular endothelium may enable peripheral lipoproteins to have indirect effects on brain health. Thus, lipoproteins may affect the cerebrovasculature from both sides of the BBB. Recent studies show an association between elevated plasma LDL, a well-known cardiovascular risk factor, and a higher risk of AD, and considerable evidence suggests that high HDL levels are associated with reduced CAA and lower neuroinflammation. Considering the potential detrimental role of LDL in AD and the importance of HDL’s beneficial effects on endothelial cells, high CETP activity may lead to compromised BBB integrity, increased CAA deposits and greater neuroinflammation.
Abbreviations: CETP – cholesteryl transfer ester protein; LDL – low-density lipoproteins; HDL – high-density lipoproteins; BBB – blood-brain barrier; CAA – cerebral amyloid angiopathy, SMC – smooth muscle cells, PVM – perivascular macrophages, RBC – red blood cells.

Similar content being viewed by others


Background
Cardiovascular risk factors play an undeniable role in the vascular contributions to cognitive impairment and dementia (VCID) [1], particularly for Alzheimer’s Disease (AD), the most common type of dementia, where cerebral vessels have key roles in clearing amyloid beta (Aβ) peptides from the brain [2, 3]. The issues under discussion for this narrative review include: 1) the association of major peripheral lipoprotein subclasses, focusing on low-density lipoprotein (LDL) and high-density lipoprotein (HDL), on VCID and AD; 2) current challenges in using murine models to investigate mechanisms by which LDL and HDL contribute to VCID; and 3) describing the functions of cholesteryl ester transfer protein (CETP) in regulating the LDL:HDL ratio in humans, including therapeutic strategies targeting CETP. The literature search strategy used focused on the search terms AD, VCID, LDL, HDL, and CETP to identify primary papers and review articles in PubMed and Medline within the last 25 years. As this is a narrative review, formal inclusion and exclusion criteria to guide selection of articles for in-depth review were not used. Nevertheless, both authors reached consensus on the articles reviewed to ensure a balanced representation of primary findings.
Alzheimer’s Disease and the cerebrovasculature
Alzheimer’s Disease (AD), the leading cause of dementia, is definitively diagnosed upon neuropathological evidence of amyloid plaques consisting of fibrillar beta-amyloid (Aβ) peptides that deposit in the brain parenchyma and cerebral vessels, as well as neurofibrillary tangles consisting of aggregated hyperphosphorylated tau protein that deposits within neurons [4]. As neurological health depends on a properly functioning neurovascular unit, it is not surprising that there is a close relationship between vascular health and AD. Despite comprising only 2% of total body mass, the brain consumes approximately 20% of total cardiac output and contains over 400 miles of blood vessels [5]. Age, sex, smoking, blood pressure, physical activity, type 2 diabetes mellitus and dyslipidemia are well-established AD risk factors [6,7,8]. Favorable cardiovascular health and management of modifiable vascular risk factors, especially at midlife, attenuates dementia risk decades later [9,10,11].
Large autopsy studies show that cerebrovascular pathologies are common in AD [12]. Specifically, 60 to 90% of AD brains have evidence of cerebral vessel disease, including arteriole and precapillary deformities [13], reduced vascular density [14, 15], increased vessel tortuosity [15], vessel remnants that lack endothelial cells [16,17,18], and accumulation of Aβ in cerebrovascular arteries, arterioles and capillaries, known as cerebral amyloid angiopathy (CAA) [19]. Studies by the National Alzheimer’s Coordinating Center and the Religious Orders Study and Rush Memory and Aging Project found a greater burden of macro- and micro-infarcts, atherosclerosis, arteriosclerosis and CAA in AD compared to other neurodegenerative diseases [20], and increased AD risk in cases with infarcts and more severe atherosclerosis or arteriosclerosis [21], respectively. CAA is also present in up to 40% of cognitively healthy elderly brains [22]. CAA may reflect impaired clearance of Aβ, which is removed from the brain through multiple pathways including active transport across brain endothelial cells in a process involving LDL receptor related protein (LRP1) [23], p-glycoprotein [24] and LDL receptor [25], and by perivascular drainage in mid- and large-sized arteries along smooth muscle cell basement membranes [26, 27].
Together, this body of literature suggests that strategies that reduce cardiovascular risk and promote cerebrovascular resilience could potentially also reduce AD risk, particularly for patients who have vascular comorbidities. AD has a long prodromal period where neuropathological changes begin to occur 15 to 20 years prior to clinical onset of memory problems, which typically emerge in the 6th and 7th decade of life [28]. This provides a relatively large window of opportunity for interventions that target vascular factors to be evaluated as primary or secondary prevention strategies to delay progression to clinical dementia.
There is tremendous interest in this topic given that several anti-amyloid immunotherapies have been approved by the Food and Drug Administration (FDA) for mild AD [29,30,31,32]. Although these drugs promote clearance of amyloid from the brain largely via perivascular pathways and can slow cognitive decline, they are also associated with safety concerns known as amyloid related imaging abnormalities (ARIA) including edema (ARIA-E) and hemorrhage (ARIA-H) subtypes that reflect damage to cerebral vessels during amyloid clearance [33, 34]. ARIA-E can also arise spontaneously as an autoimmune encephalopathy in patients with CAA who have vascular or perivascular inflammatory infiltrates [35, 36] and CSF anti-Aβ autoantibodies [37]. Increased efforts to screen large cohorts for anti-amyloid clinical trials are beginning to identify rare cases or ARIA-E that appear to be more common in patients with CAA or who carry genetic risk factors for high vascular amyloid burden, such as the presence of apoE4 alleles [38]. It will be important to learn whether improving vascular risk factors, including the LDL:HDL ratio, prior to or concurrent with anti-amyloid immunotherapies, also may improve safety and efficacy.
Lipoproteins in AD
Apolipoprotein E (ApoE): ApoE is the most significant genetic risk factor for sporadic late onset AD, with apoE2 (Cys112, Cys158) protective, apoE3 (Cys112, Arg158) neutral, and apoE4 (Arg112, Arg158) detrimental [39]. The mechanisms by which apoE affects AD risk and age of onset remain incompletely understood. Although apoE4 is most well-known for accelerating amyloid deposition [40, 41], apoE also regulates tau-mediated neurogeneration [42] and has pleiotropic effects on cerebral vessels including regulating BBB integrity [43], amyloid clearance across the BBB [41], vascular transcriptomic and proteomic signatures [44], cerebral blood flow [45], inflammation [46, 47], vascular compliance via modifying endothelial nitric oxide secretion [48, 49], and brain glucose metabolism [50].
In addition to apoE produced within the central nervous system (CNS), apoE is also made by peripheral hepatocytes and macrophages and, as it is an exchangeable lipoprotein, circulates in blood on several peripheral lipoprotein subclasses [51]. Notably, peripheral lipoproteins have largely been ignored in AD as the healthy BBB prevents brain and peripheral apoE pools from mixing [52, 53]. However, it is increasingly appreciated that peripheral lipoproteins, including those containing apoE, can affect brain function via their actions on the cerebrovasculature [54,55,56]. Importantly, the underlying mechanisms by which lipoproteins on either side of the BBB affect cerebrovascular functions, including regulation of cerebrovascular reactivity and amyloid clearance pathways, are largely unknown. It is reasonable to hypothesize that peripheral lipoproteins may be important in regulating endothelial functions as their circulation through the vascular lumen allows direct endothelial contact.
Both the quality and quantity of peripheral apoE may matter in AD pathogenesis. In humans, patients with prodromal and manifest AD have lower plasma apoE levels compared to cognitively healthy controls, and the APOE4 genotype is associated with lower plasma apoE levels regardless of the diagnosis [57]. Moreover, low plasma apoE levels are associated with Aβ pathology, higher cerebrospinal fluid (CSF) total- and phosphorylated-tau and worse cognition [57]. Mice engineered to have inducible liver-specific human apoE3 and apoE4 expression with no detectable apoE in the brain further support the protective effects of peripheral apoE3 and the toxic effects of peripheral apoE4 on brain function [58]. Transcriptomic analysis reveals that peripheral expression of apoE4 leads to endothelial dysfunction and impaired immune function within the glio-vascular unit that disrupts vascular function and BBB integrity [58]. In APP/PS1 mice, peripheral expression of apoE3 reduces brain amyloid pathology while apoE4 exacerbates it [58]. Thus, lipoproteins may affect the cerebrovasculature from either side of the BBB; brain lipoproteins made largely from apoE acting from the abluminal side and peripheral lipoproteins acting from the luminal side. This concept highlights the cerebrovasculature as an important functional bridge between the brain and the rest of the body.
Low-density lipoprotein (LDL): LDL particles exist in subclasses including large, buoyant LDL-I (density 1.019–1.023 g/mL), LDL-II of intermediate size and density (density 1.023–1.034 g/mL), small dense LDL-III (density 1.034–1.044 g/mL), and a fourth subfraction of very small dense LDL-IV (density 1.044–1.063 g/mL), which is present in individuals with elevated triglyceride (TG) levels [59,60,61,62]. In humans, it is well established that elevated levels of LDL cholesterol (LDL-C) are causally related to risk of cardiovascular disease including atherosclerosis, coronary artery disease, stroke, hypertension and type II diabetes [63] and that lowering levels of LDL and other apoB-containing lipoproteins reduces cardiovascular events [64]. In large peripheral vessels, LDL can cross the endothelium and enter the arterial intima [65] via electrostatic interactions with arterial proteoglycans [66], however, whether this process occurs in leptomeningeal or other cerebral vessels is not known. Importantly, oxidized LDL damages the endothelial barrier integrity [67, 68], which may be particularly relevant to the cerebral microvasculature. With respect to the impact of biological sex on AD risk, estrogens reduce LDL transcytosis by downregulating scavenger receptor B1 (SR-B1) in endothelial cells [69] and, if this process also occurs in cerebral vessels, may account for some of the increased risk for AD in post-menopausal women.
A recent meta-meta analysis of plasma lipids in AD supports a significant association between high LDL-C levels with increased AD risk [70]. A UK Biobank study of more than 1.8 million people over two decades revealed a modest association between dementia risk and LDL-C measured in mid-life (< 65 years) [71]. A meta-analysis and systematic review that included this and two additional cohort studies investigating the association between midlife LDL and HDL levels and dementia concluded that for each 1 mmol/L increase in LDL-C, there is an 8% increase in incidence of all-cause dementia [72]. The Religious Orders Study/Memory and Ageing Project further demonstrated that LDL-C is associated with AD neuropathology including Aβ plaques and neurofibrillary tangles and CAA independent of apoE. Therefore, LDL-C is a modifiable risk factor for AD.
High density lipoproteins (HDL): HDL is a peripheral lipoprotein class that regulates reverse cholesterol transport, a process that removes excess cholesterol from the body [73]. The human HDL “particle-ome” contains ~ 200 proteins, ~ 300 lipids, and RNA cargo [74, 75]. HDL composition differs among individuals and disease states [76,77,78,79] and becomes less cardioprotective in women after menopause [80]. Although clinical measures of HDL’s cholesterol content, HDL-C, remains a gold standard to quantify HDL levels, HDL functions are determined by their composition [81], with different proteins driving distinct functions of HDL subpopulations [82]. Circulating HDL has multiple vasoprotective functions including stimulating reverse cholesterol transport, reducing endothelial activation, and mitigating endothelial inflammation [76, 83]. While HDL has been extensively studied in cardiovascular disease [84, 85], its potential roles in AD are less clear [76, 83]. High levels of HDL-cholesterol (HDL-C) or apoA-I (HDL’s major protein) correlate with reduced AD risk [86], improved memory [87, 88] and low brain amyloid [89]. The prospective Honolulu-Aging study found that the highest quartile of plasma apoA-I at baseline correlated with the lowest risk of dementia 16 years later [90]. Similarly, high baseline HDL-C in the Baltimore Longitudinal Study of Aging protected against cognitive impairment and brain volume reductions 20 years later [91]. However, cross-sectional studies including the Framingham Heart Study [92] and others [93], and some prospective studies with short follow-up [94,95,96,97], found no relationship between HDL-C and cognitive impairment. It is likely that baseline age and follow-up length may explain these inconsistencies [90, 95], as studies with > 10 years of follow-up found significant associations between HDL-C levels and AD risk [90, 91], whereas those with < 10 years of follow-up did not [95, 97]. Furthermore, baseline HDL-C levels in middle age were significantly associated with AD risk [87, 90, 98], whereas baseline measures in subjects > 70 years were not [96, 97]. Thus, HDL, like LDL, may exert its greatest influence on AD risk at mid-life. New findings report a significant interaction between low serum HDL-C and cognitive impairment in APOE4 carriers [99], which contrasts with another study that found an association between genetically determined increased HDL-C and higher risk of AD using a Mendelian randomization framework [100]. Contradictory conclusions of HDL’s role in AD pathogenesis may be prompted by the composition and functionality of HDL particles and not simply by HDL-C levels in plasma, as well as when in life HDL is measured.
The importance of understanding the functional properties of plasma HDL subpopulations has gained considerable attention in recent years. ApoE is present in a minor subfraction (6–9%) of total circulating HDL particles and was previously believed to mainly recycle these HDL particles in the liver [101, 102]. However, the fraction of HDL that contains apoE (HDL-apoE) is now believed to promote critical metabolic steps in reverse cholesterol transport that are associated with reduced coronary heart disease risk [82], and plasma HDL-apoE was recently confirmed as a potential biomarker for coronary heart disease [103]. Recently, high HDL-apoE levels were found to be associated with improved cognitive function as measured by the Modified Mini-Mental State Examination in the elderly (> 75 years of age) [104], and high levels of plasma HDL-apoE lacking apoC-III were reported to be associated with better cognitive function and lower dementia risk in a prospective case-cohort of 1351 participants in the Ginkgo Evaluation of Memory Study [105]. HDL-apoE can also reduce vascular stiffening by altering expression of the extracellular matrix proteins collagen-I and fibronectin in vascular smooth muscle cells [106].
Preclinical studies in mice show that deletion of apoA-I in amyloidogenic models drastically reduces HDL levels and exacerbates CAA and cerebrovascular inflammation [33, 38]. Conversely, apoA-I overexpression from its native promoter reduces CAA and neuroinflammation [20], and delivery of recombinant HDL or apoA-I Milano (an atheroprotective apoA-I genetic variant) into the systemic circulation of mice acutely decreases soluble brain Aβ levels and leads to long-lasting lowering of CAA and neuroinflammation, respectively [13, 14], suggesting the potential for HDL to play a role in removing pre-existing vascular amyloid deposits in vivo.
In a scaffold-directed model of perfusable cerebral blood vessels, HDL delivered from the “blood side” facilitates Aβ transport and attenuates Aβ accumulation in the synthetic vascular tissue, particularly for more pathogenic Aβ42 peptide [107], and the HDL-apoE fraction appears particularly potent for this function [108]. Multiple mechanisms appear to underlie HDL’s ability to attenuate Aβ vascular deposition and Aβ-induced endothelial inflammation, namely reducing Aβ binding to collagen-I by forming an HDL-Aβ complex, reducing collagen-I protein levels produced by smooth-muscle cells (SMC), and blocking Aβ uptake into SMC perhaps by reducing LRP1 levels [108]. In this model system, HDL appeared ineffective in reducing preformed vascular Aβ deposits in a 24 h time period [109], suggesting that either more time may be required to attenuate pre-existing CAA or that components in addition to HDL may be required in this model system, including pericytes that have a clear role in CAA pathology [110].
Sex differences in LDL and HDL
It is well-established that two thirds of those with AD are women and that menopause is associated with a shift from a cardioprotective lipid profile to an atherogenic lipid profile characterized by higher LDL-C and lower and more dysfunctional HDL-C in post-menopausal women [111,112,113,114]. A recent study of 5,366 statin users from approximately 50,000 participants in the real-world PharmLines initiative in the Netherlands showed that women had a significantly higher mean percent increase in HDL-C levels upon statin therapy compared to men, with no sex differences observed for LDL-C reduction [115]. However, another well-powered primary care study found that women were less adherent to statin treatment compared to men [116]. Sex but not menopausal differences were also observed in a small real-world study of proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, another class of LDL-C-lowering therapies [117]. Much remains to be learned about how targeting vascular risk factors for primary and secondary prevention of dementia may require sex and menopausal considerations.
Cholesteryl ester transfer protein (CETP)
CETP is a boomerang-shaped glycoprotein produced primarily by the liver [118, 119] that promotes bidirectional transfer of hydrophobic lipids, cholesteryl esters and triglycerides across plasma lipoprotein subclasses [120]. Because most of the cholesteryl esters in plasma are found in HDL and most triglycerides originate from very low density lipoprotein (VLDL) and chylomicrons, the net effect of CETP activity is a net mass transfer of cholesteryl esters from HDL to pro-atherogenic LDL and VLDL particles, and a net mass transfer of triglycerides from triglyceride-rich VLDLs and chylomicrons into HDL and LDL [121]. CETP is primarily expressed in and secreted from in the liver and is catalytically active in humans, non-human primates, rabbits and hamsters, but is absent in many other species [122]. In humans, brain CETP expression is very low in most regions, as the Human Protein Atlas human brain dataset lists less than 2 normalized transcript per million (nTPM), the GTEx human brain RNA-Seq dataset shows < 1 nTPM in all brain regions except for the pituitary, and the FANTOM5 human brain CAGE dataset shows ~ 18 scaled tags per million in retina but less than 2 in brain regions [123, 124]. This lack of clear expression in the human brain suggests that CETP may exert its CNS effects indirectly, likely via the cerebrovasculature.
CETP polymorphisms
According to the genome Aggregation Database (gnomAD), hundreds of single nucleotide variants (SNVs) in the CETP gene have been identified, including 125 synonymous SNVs, 290 missense SNVs and 33 predicted loss-of-function variants [125]. Because of CETP’s ability to modulate lipid transport, the natural genetic variations at the CETP locus on CETP activity, protein and lipid levels, and impact on disease have been widely studied. Data from the UK Biobank and the CARDIoGRAM plus C4D consortium show that the CETP genetic score comprising of CETP variants associated with higher HDL (rs3764261, rs1800775, rs708272, rs9939224, rs2303790) have biologically equivalent effects to LDL-C-lowering therapies on reducing the risk of major coronary events when measured per unit change in apoB [126]. In another study aimed to identify regulatory elements affecting CETP mRNA expression, allelic mRNA expression in 56 human livers was shown to be strongly associated with three upstream promoter/enhancer SNPs (rs173539, rs247616, rs3764261) [127]. A closer examination of molecular mechanisms revealed the minor allele of rs247615 and associated high linkage SNPs lead to reduced expression across tissues, potentially because rs247616 alters the putative binding sites of highly expressed transcription factors: Y-box binding protein 1 (YBX1) and CCAAT/enhancer binding protein alpha (CEBPA) [128].
The same group also investigated the common exon 9-lacking alternative splicing isoform (\(\Delta\) 9), which prevents CETP secretion in a dominant negative manner. The \(\Delta\) 9 splice variant was found within 10–48% of total CETP mRNA in 94 livers analysed and was strongly associated with rs5883 and rs9930761 in complete linkage disequilibrium. In males, both rs247616 and rs5883T/rs9930761C were independently associated with elevated HDL-C levels with similar effect sizes. Notably, males with rs5883T/rs9930761C also had significantly increased risk for myocardial infarction (MI), stroke and all-cause mortality. Neither polymorphism had a significant effect in females. This suggests that low CETP activity is associated with poor cardiovascular disease (CVD) outcomes in males and that there are sex-dependent CETP splicing effects independent of HDL levels [127].
A recent study investigating the effect of five single-nucleotide polymorphisms (SNPs; rs1532624, rs5882, rs708272, rs7499892, and rs9989419) and haplotypes on the cardiovascular risk (CVR) in 368 Hungarian/Roma samples showed a significant association between the T allele of rs7499892 and increased CVR estimated by the Framingham Risk Score. Furthermore, three out of 10 haplotypes (H5, H7, and H8) investigated also showed a significant correlation with increased CVR. Interestingly, the H5 effect was mediated via TG and HDL-C levels, while the impact of H7 and H8 was independent of TG and HDL-C, suggesting that more mechanisms need to be explored [129]. Rs708272, which showed no significant association with CVR in the study described above, also failed to show an association with MI risk in 2286 patients from Western Siberia [130] which is inconsistent with an earlier meta-analysis concluding that rs708272 may contribute to MI susceptibility among Caucasians but not Asians [131]. Interestingly, a meta-analysis by Thompson et al. suggests the opposite, a weakly inverse association with coronary risk for rs708272 and two more CETP-inhibiting SNPs (rs5882 and rs1800775) [132].
The discrepancies between the effects of rs708272 and many other CETP polymorphisms (Table 1) can be explained by several challenges. Firstly, the overwhelming majority of genetic studies investigating CETP variants are candidate gene analyses which have been criticized for low statistical power, incomplete coverage of relevant genetic variation within candidate genes and potentially confounding influences such as environmental and ethnic backgrounds [133]. Secondly, CETP’s complex biology may serve as a limitation to genetic association interpretations. Boekholdt and Thompson point out that although CETP activity is often measured to determine the effect of a given polymorphism, protein levels in tissues may contribute to disease outcomes as well. CETP mass measurements could also potentially serve as a limitation if antibodies have different affinities to protein variants [134]. Lastly, most CVDs represent a complex interplay between lifestyle and environmental risk factors with many contributing genes with multiple polymorphisms, suggesting that a given CETP polymorphism may have a strong effect in one population but not necessarily in a different population.
CETP polymorphisms in neurodegeneration and cognitive decline
Several genetic studies support an association between certain CETP polymorphisms with resilience to memory decline [135,136,137]. The Cache County study found that the V allele of the rs5882 (I405V) CETP polymorphism was associated with reduced cognitive decline determined by the Modified Mini-Mental State Examination (3MS) in 4486 subjects who were followed longitudinally for 12 years [136]. A protective effect of the V allele of the rs5882 polymorphism in preserving cognitive function measured with the Ruff Figural Fluency Test was also reported in a population-based cohort sample of 4135 individuals in a study in the Netherlands [138]. An Alzheimer Disease Neuroimaging Initiative (ADNI) study investigating CETP polymorphisms rs5882 (I405V) and (rs1800775) C-629A in 188 controls and 318 AD or mild cognitive impairment (MCI) patients reported that, in APOE4 carriers, the CETP V and A alleles, both of which decrease CETP activity and increase HDL, were associated with greater cortical thickness at baseline and less atrophy over 12 months in the medial temporal lobe. By contrast, for non-APOE4 carriers, the I allele, which increases CETP and decreases HDL, was associated with greater baseline thickness and lower dementia risk, suggesting that APOE genotype may modify the impact of CETP polymorphisms on neurodegeneration and cognitive decline [139]. Diffusion tensor imaging of 403 young adults revealed that the G allele dosage of the rs5882 (A > G) polymorphism was associated with higher fractional anisotropy and lower radial and mean diffusivity, suggesting optimal white matter structural integrity. However, a follow-up analysis of 78 older individuals from the ADNI cohort found an opposite direction of the rs5882 and white matter integrity association, suggesting age-dependent effects on brain [140]. In the Einstein Aging Study, valine homozygosity at the I405V locus was reported to be associated with less incident dementia and slower memory decline determined by the Mini-Mental State Examination in a prospective cohort sample of 608 community-dwelling participants [141]. Another paper from this study showed that APOE4 significantly interacted with CETP I405V, where the V allele buffered the effects of APOE4 memory decline in a sample of 909 community-dwelling adults [142].
However, other genetic studies suggest a more complex scenario. A 2014 meta-analysis of 9 case control studies including 2172 AD patients and 8017 healthy controls suggested a modest detrimental effect of the rs5882 (I405V) polymorphism in increasing AD risk specifically in Caucasians [137]. The Rush Memory and Aging Project and the Religious Order Study reported that the CETP rs5882 polymorphism was associated with increased AD risk in over 1300 participants of European ancestry [143]. A study of 544 AD cases and 5405 controls from the Rotterdam study similarly suggested increased AD risk in VV homozygotes specifically in APOE4 noncarriers [144]. It is possible that the discrepancies among the genetic studies may be explained by interactions among CETP and APOE genetic variation, aging, cardiovascular risk factors, and the presence of multiple neuropathological subtypes of AD. Indeed, a 2020 study using Mendelian randomization of the genetic determinants of blood lipids and cerebral small vessel disease showed that a genetic predisposition to higher HDL-C levels, including CETP variants, was associated with reduced risk of small vessel stroke and white matter hyperintensities that remained after adjustment for LDL-C and triglycerides [145].
Lack of functional CETP expression in mice
Due to a deletion that leads to a nonsense mutation in exon 11 of the rodent CETP gene, both mice and rats have no active CETP protein and thus are functionally equivalent to CETP knockouts. Lack of CETP activity is why mice and rats have naturally high HDL and low LDL levels that leads to an inherent resilience to atherosclerosis and, importantly, potential resistance to VCID. Introducing the CETP gene into mice increases LDL-C levels and decreases HDL-C levels, and is pro-atherogenic in mice fed an atherogenic diet [146], in apoE knock-out mice, [147] in LDL receptor knock-out mice, [147] in APOE*3-Leiden mice [148] and in hypertensive rats [149]. A recent study of commercially available CETP transgenic (Tg) mice (Jax strain 003904) demonstrated transcriptional changes in the liver including reduced Hmgcr, Ldlr, and Lrp1 levels and increased ABCA7 and Trem2, albeit the Trem2 mRNA levels were not associated with increased Trem2 protein levels as measured by Western blot [150]. This group also reported that CETP was expressed in the cortex of CETP transgenic mice and increased Il-1β levels. Upon induction of CETP activity through a high-cholesterol diet, CETP Tg mice displayed a 22% increase in hippocampal cholesterol levels that was reported to be due not by increased cholesteryl synthesis in astrocytes but rather to decreased cholesterol excretion through the BBB. Decreased cholesterol efflux was revealed by a significant reduction in 24S-hydroxycholesterol, a cholesterol metabolite that can freely diffuse over the BBB, in the brains of CETP transgenic mice [150]. Transcriptional profiling of astrocytes of these mice also revealed high upregulation of complement factor C1Q subunit genes which was associated with increased C1Q protein levels throughout the hippocampus. As C1Q has been associated with neuronal cholesterol efflux [151], increased C1Q protein expression may be a compensatory mechanism to excrete excess cholesterol. C1Q is also a major effector of the peripheral immune response and mediates synapse elimination in the developing brain. Importantly, C1Q is increased in the CNS during ischemia–reperfusion injury, in AD, and during normal aging [152,153,154,155], where it is hypothesized to contribute to disrupted hippocampal circuitry [155] and to astrocyte-mediated synapse loss [156]. More work is needed to validate these findings, as a recent systematic review and meta-analysis of 86 studies investigating the complement cascade in AD did not find evidence for consistently altered CSF C1Q levels in AD patients [157], potentially reflecting mixed vascular and parenchymal pathology in the studies reviewed.
CETP as a therapeutic target
Several CETP inhibitors have been developed and evaluated in clinical trials for cardiovascular outcomes. Torcetrapib inhibited the development of atherosclerosis in rabbits and, in early-phase studies in humans, increased HDL-C by 60 to 100% and lowered LDL-C by up to 20% [158]. However, torcetrapib was associated with increased risk of death and cardiac events due to off-target effects that increased aldosterone, cortisol and endothelin-I, changed serum electrolytes, and increased blood pressure. Since then, other CETP inhibitors have been required to undergo assessments to exclude off-target toxicity and include dedicated ambulatory blood pressure monitoring studies, with dalcetrapib, evacetrapib, anacetrapib and obicetrapib showing no clinically relevant effects on blood pressure or mineralocorticoid levels [159,160,161]. Although dalcetrapib raised HDL-C levels by approximately 30% in phase 2 studies, lack of significant LDL-C lowering led to termination of clinical trials due to futility [162]. Evacetrapib trials were also halted due to futility, however, this could be due to insufficient duration to detect a meaningful reduction in major coronary events [163]. Anacetrapib was found to have subtle LDL-C lowering effects and significantly reduced major coronary events (i.e., coronary death, myocardial infarction, or coronary revascularization) over 6.4 years of follow up, where continued efficacy results from its accumulation in adipose tissue leading to a long half-life [164, 165]. Torcetrapib, anacetrapib, dalcetrapib and evacetrapib are all lipophilic compounds that block transfer of cholesteryl esters by interfering with the connection between the N- and C-terminal pockets in the CETP protein. Evacetrapib has been reported to enter mouse brain tissue [166], although it is not clear at this time whether CNS entry is required for CETP-inhibitors to potentially affect AD-relevant pathways.
Despite the failed trials listed above, one CETP inhibitor, obicetrapib, is advancing through clinical trials mainly for cardiovascular outcomes (NCT05972278, NCT05142722, NCT05425745, NCT4753606, NCT04770389, NCCT05421078, NCT06005597, NCCT05202509, NCT05266586), with one proof-of-concept open label phase 2a trial in early AD patients to evaluate plasma and CSF lipoprotein changes (NCT05161715). Unlike the other CETP inhibitors, obicetrapib is significantly less lipophilic and shows no clinically significant off-target effects on vital signs, blood pressure, and aldosterone, sodium, potassium or bicarbonate concentrations [161]. Obicetrapib also exerts the most potent effects on LDL-C and HDL-C among the CETP inhibitors tested to date with very favourable changes in lipid profile. Specifically, obicetrapib at a 5 mg dose reduced CETP activity at steady state by 91%, reduced LDL-C by 45.3%, reduced apoB by 33.6%, increased HDL-C after 12 weeks by 157.1%, and increased apoA-I by 57.5% [167]. These attributes make obicetrapib an attractive candidate to evaluate in the context of VCID, as it leads to favourable changes in both LDL-C and HDL-C levels. LDL-C reduction is required for lowered cardiovascular risk, which would be expected to reduce LDL-mediated effects on AD pathological changes, and the elevated HDL-C is of interest with respect to its potential beneficial roles in endothelial physiology and amyloid clearance.
Knowledge gaps and considerations for future directions
Improving mouse models for VCID studies
High levels of vasoprotective HDL and low levels of LDL renders mice highly resilient to vascular disorders, and VCID studies in mice need to be interpreted in this context. One approach to improve the relevance of mouse models for VCID studies, especially studies that aim to understand the contribution of peripheral lipoproteins on ADRD, is to reconstitute functional CETP activity to generate a more human-like peripheral lipoprotein profile in mice. This would be expected to exacerbate CAA and BBB dysfunction. One strain of CETP Tg mice that express a human CETP minigene containing both 5’ and 3’ regulatory elements responsive to a high fat high cholesterol diet is commercially available from Jackson Laboratories (Jax strain 003904) and could enable fundamental research studies to investigate several unanswered questions, such as the effect of CETP on BBB and brain expression profiles, inflammatory tone, cerebrovascular physiology, amyloid deposition, amyloid clearance, and tau pathology. Reconstitution of CETP activity in mice will also be essential to address whether CETP inhibition is effective against CAA, parenchymal amyloid, or both, whether CETP inhibition can remove pre-existing vascular or parenchymal amyloid deposits, whether apoE isoform modifies CETP inhibition efficacy, whether CETP inhibition can reduce apoE-mediated tau neurodegeneration, and whether CETP inhibitors need CNS entry to affect vascular, amyloid, tau, and inflammatory pathways relevant to AD.
Improving in vitro models for VCID studies
Although several advanced in vitro models such as vascularized human brain organoids and human BBB models have been developed and applied in VCID studies [168, 169], a major limitation of these models is the absence of human endothelial cells, either primary or iPSC derived, which recapitulate the transcriptomic profiles of human cerebrovascular endothelial cells, including vascular zonation considerations. For multicellular models, much needs to be learned about how transcriptomic profiles of the input cells may change under lengthy co-culture conditions.
Understanding whether peripheral lipoproteins modify anti-amyloid safety and efficacy
Given LDL’s causal role in reducing cardiovascular disease risk and association with AD neuropathology, it is of interest to determine whether LDL-C-lowering therapies may also enable existing amyloid deposits to be more efficiently and safely extracted from the brain, and if so, what mechanisms are involved. It will be important to consider the class of LDL-lowering agents, as statins, PSCK9 inhibitors, cholesterol absorption inhibitors (ezetimibe), bile acid sequestrants, ATP citrate lyase (ACL) inhibitors (bempedoic acid), small interfering RNA (siRNA) therapies (Inclisiran) and CETP inhibitors use different mechanisms to lower LDL-C and have different effects on the LDL:HDL ratio that could be important for VCID. Secondary analyses of residual plasma specimens from anti-amyloid immunotherapy trials could reveal whether the LDL:HDL ratio or HDL composition may modify ARIA risk. Similarly, secondary analysis of residual plasma specimens from CETP inhibitor trials could reveal whether AD-relevant biomarkers such as Aβ42:Aβ40, p-tau-181, p-tau-217, neurofilament light, and glial fibrillary acidic protein are altered after exposure to CETP inhibition and whether the degree of biomarker change correlates with the extent of LDL lowering and/or HDL elevation.
Conclusions
As identification of factors that modify ARIA risk is a major priority, further investigation of peripheral lipoproteins and CETP activity can be considered as attractive candidates. LDL-C, a known cause of cardiovascular disease, is associated with AD pathology and could contribute to cerebrovascular dysfunction in both amyloid-dependent and amyloid-independent ways. How HDL composition affects its known vasoprotective functions is primarily studies in the context of atherosclerosis, and the pre-clinical and in vitro data on the associations of HDL with CAA and Aβ clearance provides a strong rationale for additional studies. There are, however, important challenges to overcome. Many clinical ADRD studies exclude those with vascular disease, and those that do not will have a high proportion of participants with mixed pathology. Better CAA diagnostic and staging tools are needed. Studies in mice need to be interpreted in the context of inherent vascular resilience due to low LDL and high HDL levels. Incorporation of functional CETP into AD animal model studies will improve their relevance to human disease and enable the mechanisms by which lipoproteins and CETP may affect VCID to be understood. An improved understanding of human cerebrovascular endothelial cell physiology in response to lipoprotein exposure will also improve the relevance of human-based in vitro model systems for VCID research.
Availability of data and materials
Not applicable.
Abbreviations
- Aβ:
-
Amyloid beta
- AD:
-
Alzheimer’s Disease
- ADNI:
-
Alzheimer’s Disease Neuroimaging Initiative
- ApoAI:
-
Apolipoprotein A1
- ApoB:
-
Apolipoprotein B
- ApoE:
-
Apolipoprotein E
- ARIA:
-
Amyloid related imaging abnormalities
- BBB:
-
Blood brain barrier
- CAA:
-
Cerebral amyloid angiopathy
- CETP:
-
Cholesteryl ester transfer protein
- CNS:
-
Central nervous system
- HDL:
-
High density lipoprotein
- HDL-C:
-
High density lipoprotein cholesterol
- LDL:
-
Low density lipoprotein
- LDL-C:
-
Low density lipoprotein cholesterol
- LDLR:
-
Low density lipoprotein receptor
- LRP1:
-
Low density lipoprotein receptor related protein 1
- PCSK9:
-
Proprotein convertase subtilisin/kexin type 9
- SR-B1:
-
Scavenger receptor B1
- VCID:
-
Vascular contributions to cognitive impairment and dementia
References
Iadecola C, Smith EE, Anrather J, Gu C, Mishra A, Misra S, et al. The neurovasculome: key roles in brain health and cognitive impairment: a scientific statement from the American Heart Association/American Stroke Association. Stroke. 2023;54:e251.
Fisher RA, Miners JS, Love S. Pathological changes within the cerebral vasculature in Alzheimer’s disease: new perspectives. Brain Pathol. 2022;32(6):e13061.
Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, et al. Clearance systems in the brain—implications for Alzheimer disease. Nat Rev Neurol. 2015;11(8):457–70.
Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14(4):535–62.
Siegel GJ, Agranoff BW, Albers RW, et al., editors. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. 6th edition. Philadelphia: Lippincott-Raven; 1999.
Gottesman RF, Albert MS, Alonso A, Coker LH, Coresh J, Davis SM, et al. Associations between midlife vascular risk factors and 25-year incident dementia in the Atherosclerosis Risk in Communities (ARIC) Cohort. JAMA Neurol. 2017;74(10):1246–54.
Mosconi L, Walters M, Sterling J, Quinn C, McHugh P, Andrews RE, et al. Lifestyle and vascular risk effects on MRI-based biomarkers of Alzheimer’s disease: a cross-sectional study of middle-aged adults from the broader New York City area. BMJ Open. 2018;8(3): e019362.
Norton S, Matthews FE, Barnes DE, Yaffe K, Brayne C. Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol. 2014;13(8):788–94.
Vu TT, Zhao L, Liu L, Schiman C, Lloyd-Jones DM, Daviglus ML, et al. Favorable cardiovascular health at young and middle ages and dementia in older age-The CHA study. J Am Heart Assoc. 2019;8(1): e009730.
Tan ECK, Qiu C, Liang Y, Wang R, Bell JS, Fastbom J, et al. Antihypertensive medication regimen intensity and incident dementia in an older population. J Am Med Dir Assoc. 2018;19(7):577–83.
Solomon A, Kivipelto M, Soininen H. Prevention of Alzheimer’s disease: moving backward through the lifespan. J Alzheimer’s Dis : JAD. 2013;33(Suppl 1):S465–9.
Sweeney MD, Montagne A, Sagare AP, Nation DA, Schneider LS, Chui HC, et al. Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimer’s Dementia. 2019;15(1):158–67.
Hassler O. Vascular changes in senile brains. A Micro-Angiograph Study Acta Neuropathol. 1965;5(1):40–53.
Bell MA, Ball MJ. Morphometric comparison of hippocampal microvasculature in ageing and demented people: diameters and densities. Acta Neuropathol. 1981;53(4):299–318.
Fischer VW, Siddiqi A, Yusufaly Y. Altered angioarchitecture in selected areas of brains with Alzheimer’s disease. Acta Neuropathol. 1990;79(6):672–9.
Kalaria RN, Hedera P. Differential degeneration of the cerebral microvasculature in Alzheimer’s disease. NeuroReport. 1995;6(3):477–80.
Zipser BD, Johanson CE, Gonzalez L, Berzin TM, Tavares R, Hulette CM, et al. Microvascular injury and blood-brain barrier leakage in Alzheimer’s disease. Neurobiol Aging. 2007;28(7):977–86.
Challa VR, Thore CR, Moody DM, Anstrom JA, Brown WR. Increase of white matter string vessels in Alzheimer’s disease. J Alzheimer’s Dis: JAD. 2004;6(4):379–83.
Kuhn J, Sharman T. Cerebral Amyloid Angiopathy. [Updated 2023 Jun 5]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023. https://www.ncbi.nlm.nih.gov/books/NBK556105/.
Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s coordinating centre. Brain : A J Neurol. 2013;136(Pt 9):2697–706.
Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA, Schneider JA. Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol. 2016;15(9):934–43.
Biffi A, Greenberg SM. Cerebral amyloid angiopathy: a systematic review. J Clin Neurol (Seoul, Korea). 2011;7(1):1–9.
Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, et al. Clearance of Alzheimer’s amyloid-β1-40 peptide from brain by LDL receptor–related protein-1 at the blood-brain barrier. J Clin Investig. 2000;106(12):1489–99.
Lam FC, Liu R, Lu P, Shapiro AB, Renoir JM, Sharom FJ, et al. β-Amyloid efflux mediated by p-glycoprotein. J Neurochem. 2001;76(4):1121–8.
Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, et al. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular Aβ clearance. Neuron. 2009;64(5):632–44.
Prior R, Wihl G, Urmoneit B. Apolipoprotein E, Smooth Muscle Cells and the Pathogenesis of Cerebral Amyloid Angiopathy: the Potential Role of Impaired Cerebrovascular Abeta Clearance. Annals of the New York Academy of Sciences. 2000;903(1 Vascular fact):180–6.
Hawkes CA, Jayakody N, Johnston DA, Bechmann I, Carare RO. Failure of perivascular drainage of β-amyloid in cerebral amyloid angiopathy. Brain Pathol. 2014;24(4):396–403.
2020 Alzheimer's disease facts and figures. Alzheimers Dement. 2020;16(3):391–460. https://doi.org/10.1002/alz.12068.
van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 2022;388(1):9–21.
Mintun MA, Lo AC, Duggan Evans C, Wessels AM, Ardayfio PA, Andersen SW, et al. Donanemab in early Alzheimer’s disease. N Engl J Med. 2021;384(18):1691–704.
Sims JR, Zimmer JA, Evans CD, Lu M, Ardayfio P, Sparks J, et al. Donanemab in early symptomatic Alzheimer disease: The TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA. 2023;330(6):512–27.
Budd Haeberlein S, Aisen PS, Barkhof F, Chalkias S, Chen T, Cohen S, et al. Two randomized phase 3 studies of Aducanumab in early Alzheimer’s disease. J Prev Alzheimer’s Dis. 2022;9:197.
Ostrowitzki S, Deptula D, Thurfjell L, Barkhof F, Bohrmann B, Brooks DJ, et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012;69(2):198–207.
Boche D, Zotova E, Weller RO, Love S, Neal JW, Pickering RM, et al. Consequence of Abeta immunization on the vasculature of human Alzheimer’s disease brain. Brain : A J Neurol. 2008;131(Pt 12):3299–310.
Eng JA, Frosch MP, Choi K, Rebeck GW, Greenberg SM. Clinical manifestations of cerebral amyloid angiopathy-related inflammation. Ann Neurol. 2004;55(2):250–6.
Scolding NJ, Joseph F, Kirby PA, Mazanti I, Gray F, Mikol J, et al. Aβ-related angiitis: primary angiitis of the central nervous system associated with cerebral amyloid angiopathy. Brain. 2005;128(3):500–15.
Antolini L, DiFrancesco JC, Zedde M, Basso G, Arighi A, Shima A, et al. Spontaneous ARIA-like events in cerebral amyloid angiopathy-related inflammation. A Multicenter Prospect Longitudinal Cohort Study. 2021;97(18):e1809–22.
Sperling RA, Jack CR Jr, Black SE, Frosch MP, Greenberg SM, Hyman BT, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s association research roundtable workgroup. Alzheimers Dement. 2011;7(4):367–85.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science (New York, NY). 1993;261(5123):921–3.
Castellano JM, Deane R, Gottesdiener AJ, Verghese PB, Stewart FR, West T, et al. Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Aβ clearance in a mouse model of β-amyloidosis. Proc Natl Acad Sci U S A. 2012;109(38):15502–7.
Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, et al. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118(12):4002–13.
Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549(7673):523–7.
Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A, et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature. 2020;581(7806):71–6.
Barisano G, Kisler K, Wilkinson B, Nikolakopoulou AM, Sagare AP, Wang Y, et al. A “multi-omics” analysis of blood–brain barrier and synaptic dysfunction in APOE4 mice. J Exp Med. 2022;219(11):20221137.
Sheline YI, Morris JC, Snyder AZ, Price JL, Yan Z, D’Angelo G, et al. APOE4 allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Aβ42. J Neurosci. 2010;30(50):17035–40.
Lynch JR, Tang W, Wang H, Vitek MP, Bennett ER, Sullivan PM, et al. APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response*. J Biol Chem. 2003;278(49):48529–33.
Vitek MP, Brown CM, Colton CA. APOE genotype-specific differences in the innate immune response. Neurobiol Aging. 2009;30(9):1350–60.
Sacre SM, Stannard AK, Owen JS. Apolipoprotein E (apoE) isoforms differentially induce nitric oxide production in endothelial cells. FEBS Lett. 2003;540(1):181–7.
Ulrich V, Konaniah ES, Herz J, Gerard RD, Jung E, Yuhanna IS, et al. Genetic variants of ApoE and ApoER2 differentially modulate endothelial function. Proc Natl Acad Sci. 2014;111(37):13493–8.
Wu L, Zhang X, Zhao L. Human ApoE isoforms differentially modulate brain glucose and ketone body metabolism: implications for Alzheimer’s disease risk reduction and early intervention. J Neurosci. 2018;38(30):6665–81.
Bouchareychas L, Raffai RL. Apolipoprotein E and atherosclerosis: from lipoprotein metabolism to MicroRNA control of inflammation. J Cardiovasc Dev Dis. 2018;5(2):30.
Linton MF, Gish R, Hubl ST, Bütler E, Esquivel C, Bry WI, et al. Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. J Clin Invest. 1991;88(1):270–81.
Liu M, Kuhel DG, Shen L, Hui DY, Woods SC. Apolipoprotein E does not cross the blood-cerebrospinal fluid barrier, as revealed by an improved technique for sampling CSF from mice. Am J Physiol Regul Integr Comp Physiol. 2012;303(9):R903–8.
Bonaterra-Pastra A, Fernández-De-Retana S, Rivas-Urbina A, Puig N, Benítez S, Pancorbo O, et al. Comparison of plasma lipoprotein composition and function in cerebral amyloid angiopathy and Alzheimer’s disease. Biomedicines. 2021;9(1):72.
Van Valkenburgh J, Meuret C, Martinez AE, Kodancha V, Solomon V, Chen K, et al. Understanding the exchange of systemic HDL particles into the brain and vascular cells has diagnostic and therapeutic implications for neurodegenerative diseases. Front Physiol. 2021;12: 700847.
Wang T, Wang X, Yao Y, Zhao C, Yang C, Han Y, et al. Association of plasma apolipoproteins and levels of inflammation-related factors with different stages of Alzheimer’s disease: a cross-sectional study. BMJ Open. 2022;12(4): e054347.
Giannisis A, Al-Grety A, Carlsson H, Patra K, Twohig D, Sando SB, et al. Plasma apolipoprotein E levels in longitudinally followed patients with mild cognitive impairment and Alzheimer’s disease. Alzheimer’s Res Ther. 2022;14(1):115.
Liu C-C, Zhao J, Fu Y, Inoue Y, Ren Y, Chen Y, et al. Peripheral apoE4 enhances Alzheimer’s pathology and impairs cognition by compromising cerebrovascular function. Nat Neurosci. 2022;25(8):1020–33.
Diffenderfer MR, Schaefer EJ. The composition and metabolism of large and small LDL. Curr Opin Lipidol. 2014;25(3):221–6.
Krauss RM. Lipoprotein subfractions and cardiovascular disease risk. Curr Opin Lipidol. 2010;21(4):305–11.
Krauss RM, Burke DJ. Identification of multiple subclasses of plasma low density lipoproteins in normal humans. J Lipid Res. 1982;23(1):97–104.
Griffin BA, Caslake MJ, Yip B, Tait GW, Packard CJ, Shepherd J. Rapid isolation of low density lipoprotein (LDL) subfractions from plasma by density gradient ultracentrifugation. Atherosclerosis. 1990;83(1):59–67.
Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic epidemiologic, and clinical studies. A consensus statement from the European atherosclerosis society consensus panel. Eur Heart J. 2017;38(32):2459–72.
Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk: the task force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur Heart J. 2019;41(1):111–88.
Mundi S, Massaro M, Scoditti E, Carluccio MA, van Hinsbergh VWM, Iruela-Arispe ML, et al. Endothelial permeability, LDL deposition, and cardiovascular risk factors-a review. Cardiovasc Res. 2018;114(1):35–52.
Borén J, Olin K, Lee I, Chait A, Wight TN, Innerarity TL. Identification of the principal proteoglycan-binding site in LDL. A single-point mutation in apo-B100 severely affects proteoglycan interaction without affecting LDL receptor binding. J Clin Invest. 1998;101(12):2658–64.
Vink H, Constantinescu AA, Spaan JA. Oxidized lipoproteins degrade the endothelial surface layer : implications for platelet-endothelial cell adhesion. Circulation. 2000;101(13):1500–2.
Li W, Wang C, Zhang D, Zeng K, Xiao S, Chen F, et al. Azilsartan ameliorates ox-LDL-induced endothelial dysfunction via promoting the expression of KLF2. Aging (Albany NY). 2021;13(9):12996–3005.
Ghaffari S, Naderi Nabi F, Sugiyama MG, Lee WL. Estrogen inhibits LDL (Low-Density Lipoprotein) transcytosis by human coronary artery endothelial cells via GPER (G-Protein-Coupled Estrogen Receptor) and SR-BI (Scavenger Receptor Class B Type 1). Arterioscler Thromb Vasc Biol. 2018;38(10):2283–94.
Sáiz-Vazquez O, Puente-Martínez A, Ubillos-Landa S, Pacheco-Bonrostro J, Santabárbara J. Cholesterol and Alzheimer’s disease risk: a meta-meta-analysis. Brain Sci. 2020;10(6):386.
Iwagami M, Qizilbash N, Gregson J, Douglas I, Johnson M, Pearce N, et al. Blood cholesterol and risk of dementia in more than 1·8 million people over two decades: a retrospective cohort study. Lancet Healthy Longev. 2021;2(8):e498–506.
Wee J, Sukudom S, Bhat S, Marklund M, Peiris NJ, Hoyos CM, et al. The relationship between midlife dyslipidemia and lifetime incidence of dementia: A systematic review and meta-analysis of cohort studies. Alzheimer’s Dementia: Diagn, Assess Dis Monitor. 2023;15(1): e12395.
Mineo C, Shaul PW. Novel biological functions of high-density lipoprotein cholesterol. Circ Res. 2012;111(8):1079–90.
Michell DL, Vickers KC. Lipoprotein carriers of microRNAs. Biochimica et Biophysica Acta (BBA) - Mol Cell Biol Lipids. 2016;1861(12):2069–74.
Davidson WS, Shah AS, Sexmith H, Gordon SM. The HDL proteome watch: compilation of studies leads to new insights on HDL function. Biochim Biophys Acta Mol Cell Biol Lipids. 2022;1867(2): 159072.
Boyce G, Button E, Soo S, Wellington C. The pleiotropic vasoprotective functions of high density lipoproteins (HDL). J Biomed Res. 2017;32(3):164–82.
Shah AS, Tan L, Long JL, Davidson WS. Proteomic diversity of high density lipoproteins: our emerging understanding of its importance in lipid transport and beyond. J Lipid Res. 2013;54(10):2575–85.
Toth PP, Barter PJ, Rosenson RS, Boden WE, Chapman MJ, Cuchel M, et al. High-density lipoproteins: a consensus statement from the national lipid association. J Clin Lipidol. 2013;7(5):484–525.
Chiesa ST, Charakida M. High-density lipoprotein function and dysfunction in health and disease. Cardiovasc Drugs Ther. 2019;33(2):207–19.
El Khoudary SR, Ceponiene I, Samargandy S, Stein JH, Li D, Tattersall MC, et al. HDL (High-Density Lipoprotein) metrics and atherosclerotic risk in women. Arterioscler Thromb Vasc Biol. 2018;38(9):2236–44.
Furtado JD, Yamamoto R, Melchior JT, Andraski AB, Gamez-Guerrero M, Mulcahy P, et al. Distinct proteomic signatures in 16 HDL (High-Density Lipoprotein) subspecies. Arterioscler Thromb Vasc Biol. 2018;38(12):2827–42.
Morton AM, Koch M, Mendivil CO, Furtado JD, Tjønneland A, Overvad K, et al. Apolipoproteins E and CIII interact to regulate HDL metabolism and coronary heart disease risk. JCI Insight. 2018;3(4):e98045.
Button EB, Robert J, Caffrey TM, Fan J, Zhao W, Wellington CL. HDL from an Alzheimer’s disease perspective. Curr Opin Lipidol. 2019;30(3):224–34.
Soppert J, Lehrke M, Marx N, Jankowski J, Noels H. Lipoproteins and lipids in cardiovascular disease: from mechanistic insights to therapeutic targeting. Adv Drug Deliv Rev. 2020;159:4–33.
Kontush A. HDL and Reverse Remnant-Cholesterol Transport (RRT): relevance to cardiovascular disease. Trends Mol Med. 2020;26(12):1086–100.
Zuliani G, Cavalieri M, Galvani M, Volpato S, Cherubini A, Bandinelli S, et al. Relationship between low levels of high-density lipoprotein cholesterol and dementia in the elderly. The InChianti study. J Gerontol A Biol Sci Med Sci. 2010;65(5):559–64.
Shih YH, Tsai KJ, Lee CW, Shiesh SC, Chen WT, Pai MC, et al. Apolipoprotein C-III is an amyloid-β-binding protein and an early marker for Alzheimer’s disease. J Alzheimers Dis. 2014;41(3):855–65.
Merched A, Xia Y, Visvikis S, Serot JM, Siest G. Decreased high-density lipoprotein cholesterol and serum apolipoprotein AI concentrations are highly correlated with the severity of Alzheimer’s disease. Neurobiol Aging. 2000;21(1):27–30.
Reed B, Villeneuve S, Mack W, Decarli C, Chui HC, Jagust W. Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol. 2014;71(2):195.
Saczynski JS, White L, Peila RL, Rodriguez BL, Launer LJ. The relation between apolipoprotein A-I and dementia: the Honolulu-Asia aging study. Am J Epidemiol. 2007;165(9):985–92.
Armstrong NM, An Y, Beason-Held L, Doshi J, Erus G, Ferrucci L, et al. Predictors of neurodegeneration differ between cognitively normal and subsequently impaired older adults. Neurobiol Aging. 2019;75:178–86.
Tan ZS, Seshadri S, Beiser A, Wilson PWF, Kiel DP, Tocco M, et al. Plasma total cholesterol level as a risk factor for Alzheimer disease. Arch Intern Med. 2003;163(9):1053.
Formiga F, Ferrer A, Chivite D, Pinto X, Cuerpo S, Pujol R. Serum high-density lipoprotein cholesterol levels, their relationship with baseline functional and cognitive status, and their utility in predicting mortality in nonagenarians. Geriatr Gerontol Int. 2011;11(3):358–64.
Marcum ZA, Walker R, Bobb JF, Sin MK, Gray SL, Bowen JD, et al. Serum cholesterol and incident Alzheimer’s disease: findings from the adult changes in thought study. J Am Geriatr Soc. 2018;66(12):2344–52.
Li G, Shofer JB, Kukull WA, Peskind ER, Tsuang DW, Breitner JC, et al. Serum cholesterol and risk of Alzheimer disease: a community-based cohort study. Neurology. 2005;65(7):1045–50.
Mielke MM, Xue QL, Zhou J, Chaves PH, Fried LP, Carlson MC. Baseline serum cholesterol is selectively associated with motor speed and not rates of cognitive decline: the women’s health and aging study II. J Gerontol A Biol Sci Med Sci. 2008;63(6):619–24.
Yaffe K, Barrett-Connor E, Lin F, Grady D. Serum lipoprotein levels, statin use, and cognitive function in older women. Arch Neurol. 2002;59(3):378.
Bates KA, Sohrabi HR, Rainey-Smith SR, Weinborn M, Bucks RS, Rodrigues M, et al. Serum high-density lipoprotein is associated with better cognitive function in a cross-sectional study of aging women. Int J Neurosci. 2017;127(3):243–52.
Wei S, Gao L, Jiang Y, Shang S, Chen C, Dang L, et al. The Apolipoprotein E ε4 Allele-dependent relationship between serum lipid levels and cognitive function: a population-based cross-sectional study. Front Aging Neurosci. 2020;12:44.
Luo J, Thomassen JQ, Bellenguez C, Grenier-Boley B, De Rojas I, Castillo A, et al. Genetic associations between modifiable risk factors and Alzheimer disease. JAMA Netw Open. 2023;6(5): e2313734.
Heeren J, Grewal T, Laatsch A, Rottke D, Rinninger F, Enrich C, et al. Recycling of apoprotein E is associated with cholesterol efflux and high density lipoprotein internalization. J Biol Chem. 2003;278(16):14370–8.
Annema W, Dikkers A, de Boer JF, Gautier T, Rensen PC, Rader DJ, et al. ApoE promotes hepatic selective uptake but not RCT due to increased ABCA1-mediated cholesterol efflux to plasma. J Lipid Res. 2012;53(5):929–40.
Qi Y, Liu J, Wang W, Wang M, Zhao F, Sun J, et al. Apolipoprotein E-containing high-density lipoprotein (HDL) modifies the impact of cholesterol-overloaded HDL on incident coronary heart disease risk: a community-based cohort study. J Clin Lipidol. 2018;12(1):89–98.e2.
Koch M, DeKosky ST, Goodman M, Sun J, Furtado JD, Fitzpatrick AL, et al. High density lipoprotein and its apolipoprotein-defined subspecies and risk of dementia. J Lipid Res. 2020;61(3):445–54.
Koch M, DeKosky ST, Goodman M, Sun J, Furtado JD, Fitzpatrick AL, et al. Association of Apolipoprotein E in Lipoprotein subspecies with risk of dementia. JAMA Netw Open. 2020;3(7): e209250.
Kothapalli D, Liu SL, Bae YH, Monslow J, Xu T, Hawthorne EA, et al. Cardiovascular protection by ApoE and ApoE-HDL linked to suppression of ECM gene expression and arterial stiffening. Cell Rep. 2012;2(5):1259–71.
Robert J, Button EB, Yuen B, Gilmour M, Kang K, Bahrabadi A, et al. Clearance of beta-amyloid is facilitated by apolipoprotein E and circulating high-density lipoproteins in bioengineered human vessels. Elife. 2017;6:2e9595.
Robert J, Button EB, Martin EM, McAlary L, Gidden Z, Gilmour M, et al. Cerebrovascular amyloid Angiopathy in bioengineered vessels is reduced by high-density lipoprotein particles enriched in Apolipoprotein E. Mol Neurodegenerat. 2020;15(1):23.
Robert J, Weilinger NL, Cao L-P, Cataldi S, Button EB, Stukas S, et al. An in vitro bioengineered model of the human arterial neurovascular unit to study neurodegenerative diseases. Mol Neurodegenerat. 2020;15(1):70.
Blanchard JW, Bula M, Davila-Velderrain J, Akay LA, Zhu L, Frank A, et al. Reconstruction of the human blood–brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nat Med. 2020;26(6):952–63.
Schaefer EJ, Lamon-Fava S, Cohn SD, Schaefer MM, Ordovas JM, Castelli WP, et al. Effects of age, gender, and menopausal status on plasma low density lipoprotein cholesterol and apolipoprotein B levels in the Framingham offspring study. J Lipid Res. 1994;35(5):779–92.
Jensen J, Nilas L, Christiansen C. Influence of menopause on serum lipids and lipoproteins. Maturitas. 1990;12(4):321–31.
Bonithon-Kopp C, Scarabin PY, Darne B, Malmejac A, Guize L. Menopause-related changes in lipoproteins and some other cardiovascular risk factors. Int J Epidemiol. 1990;19(1):42–8.
Cífková R, Krajčoviechová A. Dyslipidemia and cardiovascular disease in women. Curr Cardiol Rep. 2015;17(7):609.
Hunt NB, Emmens JE, Irawati S, de Vos S, Bos JHJ, Wilffert B, et al. Sex disparities in the effect of statins on lipid parameters: The pharmlines initiative. Medicine (Baltimore). 2022;101(2): e28394.
Olmastroni E, Boccalari MT, Tragni E, Rea F, Merlino L, Corrao G, et al. Sex-differences in factors and outcomes associated with adherence to statin therapy in primary care: need for customisation strategies. Pharmacol Res. 2020;155: 104514.
Paquette M, Faubert S, Saint-Pierre N, Baass A, Bernard S. Sex differences in LDL-C response to PCSK9 inhibitors: a real world experience. J Clin Lipidol. 2023;17(1):142–9.
Drayna D, Jarnagin AS, McLean J, Henzel W, Kohr W, Fielding C, et al. Cloning and sequencing of human cholesteryl ester transfer protein cDNA. Nature. 1987;327(6123):632–4.
Hesler CB, Swenson TL, Tall AR. Purification and characterization of a human plasma cholesteryl ester transfer protein. J Biol Chem. 1987;262(5):2275–82.
Barter PJ, Hopkins GJ, Calvert GD. Transfers and exchanges of esterified cholesterol between plasma lipoproteins. Biochem J. 1982;208(1):1–7.
Cannon CP, Shah S, Dansky HM, Davidson M, Brinton EA, Gotto AM, et al. Safety of Anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363(25):2406–15.
Ha YC, Barter PJ. Differences in plasma cholesteryl ester transfer activity in sixteen vertebrate species. Comp Biochem Physiol B. 1982;71(2):265–9.
Sjöstedt E, Zhong W, Fagerberg L, Karlsson M, Mitsios N, Adori C, et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science. 2020;367(6482):eaay5947.
Human Protein Atlas. Available from: proteinatlas.org.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43.
Ference BA, Kastelein JJP, Ginsberg HN, Chapman MJ, Nicholls SJ, Ray KK, et al. Association of genetic variants related to CETP inhibitors and statins with Lipoprotein levels and cardiovascular risk. JAMA. 2017;318(10):947–56.
Papp AC, Pinsonneault JK, Wang D, Newman LC, Gong Y, Johnson JA, et al. Cholesteryl Ester Transfer Protein (CETP) polymorphisms affect mRNA splicing, HDL levels, and sex-dependent cardiovascular risk. PLoS ONE. 2012;7(3): e31930.
Suhy A, Hartmann K, Papp AC, Wang D, Sadee W. Regulation of cholesteryl ester transfer protein expression by upstream polymorphisms. Pharmacogenet Genomics. 2015;25(8):394–401.
Piko P, Jenei T, Kosa Z, Sandor J, Kovacs N, Seres I, et al. Association of CETP gene Polymorphisms and Haplotypes with cardiovascular risk. Int J Mol Sci. 2023;24(12):10281.
Semaev S, Shakhtshneider E, Shcherbakova L, Orlov P, Ivanoshchuk D, Malyutina S, et al. Association of common variants APOE, CETP, and the 9p21.3 of and the chromosomal region with the risk of myocardial infarction a prospective study. Int J Mol Sci. 2023;24(13):10908.
Wang Q, Zhou SB, Wang LJ, Lei MM, Wang Y, Miao C, et al. Seven functional polymorphisms in the CETP gene and myocardial infarction risk: a meta-analysis and meta-regression. PLoS ONE. 2014;9(2): e88118.
Thompson A, Di Angelantonio E, Sarwar N, Erqou S, Saleheen D, Dullaart RPF, et al. Association of Cholesteryl Ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. JAMA. 2008;299(23):2777–88.
Johnson EC, Border R, Melroy-Greif WE, de Leeuw CA, Ehringer MA, Keller MC. No evidence that Schizophrenia candidate genes are more associated with Schizophrenia than noncandidate genes. Biol Psychiat. 2017;82(10):702–8.
Boekholdt SM, Thompson JF. Natural genetic variation as a tool in understanding the role of CETP in lipid levels and disease. J Lipid Res. 2003;44(6):1080–93.
Sundermann EE, Wang C, Katz M, Zimmerman ME, Derby CA, Hall CB, et al. Cholesteryl ester transfer protein genotype modifies the effect of apolipoprotein ε4 on memory decline in older adults. Neurobiol Aging. 2016;41:200.e7-.e12.
Lythgoe C, Perkes A, Peterson M, Schmutz C, Leary M, Ebbert MT, et al. Population-based analysis of cholesteryl ester transfer protein identifies association between I405V and cognitive decline: the Cache county study. Neurobiol Aging. 2015;36(1):547.e1–3.
Chen JJ, Li YM, Zou WY, Fu JL. Relationships between CETP genetic polymorphisms and Alzheimer’s disease risk: a meta-analysis. DNA Cell Biol. 2014;33(11):807–15.
Izaks GJ, van der Knaap AM, Gansevoort RT, Navis G, Slaets JP, Dullaart RP. Cholesteryl Ester Transfer Protein (CETP) genotype and cognitive function in persons aged 35 years or older. Neurobiol Aging. 2012;33(8):1851.e7–16.
Murphy EA, Roddey JC, McEvoy LK, Holland D, Hagler DJ Jr, Dale AM, et al. CETP polymorphisms associate with brain structure, atrophy rate, and Alzheimer’s disease risk in an APOE-dependent manner. Brain Imaging Behav. 2012;6(1):16–26.
Warstadt NM, Dennis EL, Jahanshad N, Kohannim O, Nir TM, McMahon KL, et al. Serum cholesterol and variant in cholesterol-related gene CETP predict white matter microstructure. Neurobiol Aging. 2014;35(11):2504–13.
Barzilai N, Atzmon G, Derby CA, Bauman JM, Lipton RB. A genotype of exceptional longevity is associated with preservation of cognitive function. Neurology. 2006;67(12):2170–5.
Sanders AE, Wang C, Katz M, Derby CA, Barzilai N, Ozelius L, et al. Association of a functional polymorphism in the cholesteryl ester transfer protein (CETP) gene with memory decline and incidence of dementia. JAMA. 2010;303(2):150–8.
Yu L, Shulman JM, Chibnik L, Leurgans S, Schneider JA, De Jager PL, et al. The CETP I405V polymorphism is associated with an increased risk of Alzheimer’s disease. Aging Cell. 2012;11(2):228–33.
Arias-Vásquez A, Isaacs A, Aulchenko YS, Hofman A, Oostra BA, Breteler M, et al. The cholesteryl ester transfer protein (CETP) gene and the risk of Alzheimer’s disease. Neurogenetics. 2007;8(3):189–93.
Georgakis MK, Malik R, Anderson CD, Parhofer KG, Hopewell JC, Dichgans M. Genetic determinants of blood lipids and cerebral small vessel disease: role of high-density lipoprotein cholesterol. Brain. 2020;143(2):597–610.
Marotti KR, Castle CK, Boyle TP, Lin AH, Murray RW, Melchior GW. Severe atherosclerosis in transgenic mice expressing simian cholesteryl ester transfer protein. Nature. 1993;364(6432):73–5.
Plump AS, Masucci-Magoulas L, Bruce C, Bisgaier CL, Breslow JL, Tall AR. Increased atherosclerosis in ApoE and LDL receptor gene knock-out mice as a result of human cholesteryl ester transfer protein transgene expression. Arterioscler Thromb Vasc Biol. 1999;19(4):1105–10.
Westerterp M, van der Hoogt CC, de Haan W, Offerman EH, Dallinga-Thie GM, Jukema JW, et al. Cholesteryl ester transfer protein decreases high-density lipoprotein and severely aggravates atherosclerosis in APOE*3-Leiden mice. Arterioscler Thromb Vasc Biol. 2006;26(11):2552–9.
Herrera VL, Makrides SC, Xie HX, Adari H, Krauss RM, Ryan US, et al. Spontaneous combined hyperlipidemia, coronary heart disease and decreased survival in Dahl salt-sensitive hypertensive rats transgenic for human cholesteryl ester transfer protein. Nat Med. 1999;5(12):1383–9.
Oestereich F, Yousefpour N, Yang E, Phénix J, Nezhad ZS, Nitu A, et al. The cholesteryl ester transfer protein (CETP) raises cholesterol levels in the brain. J Lipid Res. 2022;63(9): 100260.
Benoit ME, Tenner AJ. Complement protein C1q-mediated neuroprotection is correlated with regulation of neuronal gene and microRNA expression. J Neurosci. 2011;31(9):3459–69.
Silverman SM, Kim B-J, Howell GR, Miller J, John SWM, Wordinger RJ, et al. C1q propagates microglial activation and neurodegeneration in the visual axis following retinal ischemia/reperfusion injury. Mol Neurodegen. 2016;11(1):24.
Tooyama I, Sato H, Yasuhara O, Kimura H, Konishi Y, Shen Y, et al. Correlation of the expression level of C1q mRNA and the number of C1q-positive plaques in the Alzheimer disease temporal cortex. Analysis of mrna and its protein using adjacent or nearby sections. Dement Geriatr Cogn Disord. 2001;12(4):237–42.
Reichwald J, Danner S, Wiederhold KH, Staufenbiel M. Expression of complement system components during aging and amyloid deposition in APP transgenic mice. J Neuroinflammation. 2009;6:35.
Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, et al. A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci. 2013;33(33):13460–74.
Dejanovic B, Wu T, Tsai M-C, Graykowski D, Gandham VD, Rose CM, et al. Complement C1q-dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer’s disease mouse models. Nature Aging. 2022;2(9):837–50.
Krance SH, Wu C-Y, Zou Y, Mao H, Toufighi S, He X, et al. The complement cascade in Alzheimer’s disease: a systematic review and meta-analysis. Mol Psychiatry. 2021;26(10):5532–41.
Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357(21):2109–22.
Suico JG, Friedrich S, Krueger KA, Zhang W. Evacetrapib at a supratherapeutic steady state concentration does not prolong QT in a thorough QT/QTc study in healthy participants. J Cardiovasc Pharmacol Ther. 2014;19(3):283–9.
Krishna R, Anderson MS, Bergman AJ, Jin B, Fallon M, Cote J, et al. Effect of the cholesteryl ester transfer protein inhibitor, anacetrapib, on lipoproteins in patients with dyslipidaemia and on 24-h ambulatory blood pressure in healthy individuals: two double-blind, randomised placebo-controlled phase I studies. Lancet. 2007;370(9603):1907–14.
Hovingh GK, Kastelein JJ, van Deventer SJ, Round P, Ford J, Saleheen D, et al. Cholesterol ester transfer protein inhibition by TA-8995 in patients with mild dyslipidaemia (TULIP): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet. 2015;386(9992):452–60.
Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367(22):2089–99.
Tall AR, Rader DJ. Trials and tribulations of CETP inhibitors. Circ Res. 2018;122(1):106–12.
Bowman L, Hopewell JC, Chen F, Wallendszus K, Stevens W, Collins R, et al. Effects of Anacetrapib in patients with atherosclerotic vascular disease. N Engl J Med. 2017;377(13):1217–27.
Late-Breaking Science Abstracts and Featured Science Abstracts From the American Heart Association’s Scientific Sessions 2019 and Late-Breaking Abstracts in Resuscitation Science From the Resuscitation Science Symposium 2019. Circulation. 2019;140(25):e965–e1011.
Phénix J, Côté J, Dieme D, Recinto SJ, Oestereich F, Efrem S, et al. CETP inhibitor evacetrapib enters mouse brain tissue. Front Pharmacol. 2023;14:1171937.
Nicholls SJ, Ditmarsch M, Kastelein JJ, Rigby SP, Kling D, Curcio DL, et al. Lipid lowering effects of the CETP inhibitor obicetrapib in combination with high-intensity statins: a randomized phase 2 trial. Nat Med. 2022;28(8):1672–8.
Cerneckis J, Bu G, Shi Y. Pushing the boundaries of brain organoids to study Alzheimer’s disease. Trends Mol Med. 2023;29(8):659–72.
Helman AM, Murphy MP. Vascular cognitive impairment: modeling a critical neurologic disease in vitro and in vivo. Biochimica et Biophysica Acta Mol Basis Dis. 2016;1862(5):975–82.
Smith EN, Chen W, Kähönen M, Kettunen J, Lehtimäki T, Peltonen L, et al. Longitudinal genome-wide association of cardiovascular disease risk factors in the bogalusa heart study. PLoS Genet. 2010;6(9): e1001094.
Iwanicka J, Iwanicki T, Niemiec P, Balcerzyk A, Krauze J, Górczyńska-Kosiorz S, et al. Relationship between CETP gene polymorphisms with coronary artery disease in Polish population. Mol Biol Rep. 2018;45(6):1929–35.
Peloso GM, Lee SJ, Sims R, Lee SJ, Naj AC, Bellenguez C, et al. Genetically elevated high-density lipoprotein cholesterol through the cholesteryl ester transfer protein gene does not associate with risk of Alzheimer’s disease. Alzheimer’s Dementia: Diagn Assesst Dis Monitor. 2018;10(1):595–8.
Webb TR, Erdmann J, Stirrups KE, Stitziel NO, Masca NGD, Jansen H, et al. Systematic evaluation of pleiotropy identifies 6 further loci associated with coronary artery disease. J Am Coll Cardiol. 2017;69(7):823–36.
Johannsen TH, Frikke-Schmidt R, Schou J, Nordestgaard BG, Tybjærg-Hansen A. Genetic inhibition of CETP, ischemic vascular disease and mortality, and possible adverse effects. J Am Coll Cardiol. 2012;60(20):2041–8.
Qureischie H, Heun R, Lütjohann D, Popp J, Jessen F, Ledschbor-Frahnert C, et al. CETP polymorphisms influence cholesterol metabolism but not Alzheimer’s disease risk. Brain Res. 2008;1232:1–6.
Rodríguez E, Mateo I, Infante J, Llorca J, Berciano J, Combarros O. Cholesteryl ester transfer protein (CETP) polymorphism modifies the Alzheimer’s disease risk associated with APOE ε4 allele. J Neurol. 2006;253(2):181–5.
Ridker PM, Paré G, Parker AN, Zee RY, Miletich JP, Chasman DI. Polymorphism in the CETP gene region, HDL cholesterol, and risk of future myocardial infarction: Genomewide analysis among 18 245 initially healthy women from the women’s genome health study. Circ Cardiovasc Genet. 2009;2(1):26–33.
Xiao Z, Wang J, Chen W, Wang P, Zeng H, Chen W. Association studies of several cholesterol-related genes (ABCA1, CETP and LIPC) with serum lipids and risk of Alzheimer’s disease. Lipids Health Dis. 2012;11(1):163.
Li Q, Huang P, He Q-C, Lin Q-Z, Wu J, Yin R-X. Association between the CETP polymorphisms and the risk of Alzheimer’s disease, carotid atherosclerosis, longevity, and the efficacy of statin therapy. Neurobiol Aging. 2014;35(6):1513.e13-.e23.
Yehya A, Irshaid Y, Saleh AA. Cholesteryl ester transfer protein rs1532624 gene polymorphism is associated with reduced response to statin therapy. Curr Mol Pharmacol. 2013;6(3):156–62.
Adams LA, Marsh JA, Ayonrinde OT, Olynyk JK, Ang WQ, Beilin LJ, et al. Cholesteryl ester transfer protein gene polymorphisms increase the risk of fatty liver in females independent of adiposity. J Gastroenterol Hepatol. 2012;27(9):1520–7.
Cyrus C, Vatte C, Al-Nafie A, Chathoth S, Al-Ali R, Al-Shehri A, et al. The impact of common polymorphisms in CETP and ABCA1 genes with the risk of coronary artery disease in Saudi Arabians. Human Genomics. 2016;10(1):8.
Peloso GM, Demissie S, Collins D, Mirel DB, Gabriel SB, Cupples LA, et al. Common genetic variation in multiple metabolic pathways influences susceptibility to low HDL-cholesterol and coronary heart disease. J Lipid Res. 2010;51(12):3524–32.
Acknowledgements
Not applicable.
Funding
This review was funded by a grant to CW from the Cure Alzheimer Fund.
Author information
Authors and Affiliations
Contributions
CW prepared the manuscript. TP contributed to manuscript finalization and prepared the visual abstract.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
CW is a paid consultant for NewAmsterdam Pharma, Kisbee Therapeutics and Genevant Sciences, and serves as an unpaid member of the Scientific Advisory Board for CLEAR and ProMIS Neurosciences.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Poliakova, T., Wellington, C.L. Roles of peripheral lipoproteins and cholesteryl ester transfer protein in the vascular contributions to cognitive impairment and dementia. Mol Neurodegeneration 18, 86 (2023). https://doi.org/10.1186/s13024-023-00671-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-023-00671-y