
Abstract
Human studies consistently identify bioenergetic maladaptations in brains upon aging and neurodegenerative disorders of aging (NDAs), such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and Amyotrophic lateral sclerosis. Glucose is the major brain fuel and glucose hypometabolism has been observed in brain regions vulnerable to aging and NDAs. Many neurodegenerative susceptible regions are in the topological central hub of the brain connectome, linked by densely interconnected long-range axons. Axons, key components of the connectome, have high metabolic needs to support neurotransmission and other essential activities. Long-range axons are particularly vulnerable to injury, neurotoxin exposure, protein stress, lysosomal dysfunction, etc. Axonopathy is often an early sign of neurodegeneration. Recent studies ascribe axonal maintenance failures to local bioenergetic dysregulation. With this review, we aim to stimulate research in exploring metabolically oriented neuroprotection strategies to enhance or normalize bioenergetics in NDA models. Here we start by summarizing evidence from human patients and animal models to reveal the correlation between glucose hypometabolism and connectomic disintegration upon aging/NDAs. To encourage mechanistic investigations on how axonal bioenergetic dysregulation occurs during aging/NDAs, we first review the current literature on axonal bioenergetics in distinct axonal subdomains: axon initial segments, myelinated axonal segments, and axonal arbors harboring pre-synaptic boutons. In each subdomain, we focus on the organization, activity-dependent regulation of the bioenergetic system, and external glial support. Second, we review the mechanisms regulating axonal nicotinamide adenine dinucleotide (NAD+) homeostasis, an essential molecule for energy metabolism processes, including NAD+ biosynthetic, recycling, and consuming pathways. Third, we highlight the innate metabolic vulnerability of the brain connectome and discuss its perturbation during aging and NDAs. As axonal bioenergetic deficits are developing into NDAs, especially in asymptomatic phase, they are likely exaggerated further by impaired NAD+ homeostasis, the high energetic cost of neural network hyperactivity, and glial pathology. Future research in interrogating the causal relationship between metabolic vulnerability, axonopathy, amyloid/tau pathology, and cognitive decline will provide fundamental knowledge for developing therapeutic interventions.
Similar content being viewed by others

Background
The mammalian brain is energetically demanding: consuming ~ 20% of the body’s total energy production despite accounting for only ~ 2% of body weight [1]. Glucose is the major brain fuel under normal circumstances [2, 3]. Thus, the brain mainly relies on glucose catabolism to generate adenosine triphosphate (ATP) and cannot afford any disruption of glucose and oxygen delivery [4]. Brain glucose uptake and catabolism to generate ATP have been extensively studied and reviewed [3,4,5]. Briefly, glucose metabolism requires glucose uptake into the brain cells, glycolysis in the cytoplasm and oxidative phosphorylation (OXPHO) in the mitochondria (Table 1). Each cycle of glycolysis yields 2 ATPs while OXPHO gives ~ 30 ATPs. In addition to pyruvate, neuronal mitochondria are capable of oxidizing ketone bodies and glutamine as alternative fuels when glucose and pyruvate availability is limited [6,7,8,9].
Recent human studies highlight bioenergetic maladaptation in brains upon aging and neurodegenerative disorders of aging (NDAs), such as Alzheimer’s disease (AD) [17], Parkinson’s disease (PD) [18], and Huntington’s disease (HD) [19]. Aging is considered one of the biggest risk factors for dementia and sporadic late-onset neurodegenerative diseases [20]. Single-cell transcriptomic analysis identified the decreased expression of mitochondrial OXPHO genes as a consistent signature of brain aging, prevalent across neurons and non-neuronal cell types [21, 22]. Meanwhile, proteomic and metabolomic profiling further demonstrate degraded mitochondrial metabolism in aged brains [23,24,25]. Primed by the aging process, neurodegenerative insults such as adverse environmental factors (e.g. unhealthy lifestyle [26], free radical or neurotoxicants exposures [27]), disease-associated genetic mutations [28], or epigenetic modifications [29] result in NDAs. During NDA progression, energy metabolism dysregulation is viciously aggravated along with functional decline [30]. Integrative bioinformatic analysis using brain multi-omics data has illuminated the age-dependent molecular and cellular changes in four major NDAs: AD [17], PD [18], HD [19] and amyotrophic lateral sclerosis (ALS) [31]. These studies consistently identify mitochondrial-related perturbations as a common pre-symptomatic feature.
To gain spatial information on glucose metabolism changes during aging/NDAs, numerous fluorodeoxyglucose-positron emission tomography (FDG-PET) studies have been conducted (selected studies listed in Table 2). A whole-brain metabolic network constructed from a large FDG-PET data set with subsequent diagnosis showed that the metabolic correlation between brain regions is progressively weakened as AD risks increase, and this disruption is more evident in females [32]. Glucose hypometabolism is often detected in AD-susceptible brain regions, which strongly predicts the incidence of mild cognitive impairment (MCI) in later life [33,34,35,36,37]. The majority of AD-susceptible regions identified are located in the default mode network (DMN), the topological central hub of the brain connectome [38, 39], containing densely interconnected long-range cortico-cortical axons [40, 41].
Compelling neuroimaging evidence from NDA patients further supports the prevalence of glucose hypometabolism in highly interconnected brain regions associated with functional impairment (Table 2). Briefly, human FDG-PET and diffusion tensor magnetic resonance imaging studies have revealed: (1) In MCI/AD patients, especially in AD-risk gene carriers, glucose hypometabolism preferentially occurs in DMN regions (such as posterior cingulate cortex, precuneus, anterior hippocampus and parahippocampal gyrus) [42,43,44,45,46,47,48,49], strongly correlated with white matter degeneration in these areas, and cognitive impairment [50,51,52]; (2) PD patients show hypometabolism in premotor and parieto-occipital cortex that correlates with motor dysfunction [53]; (3) HD patients show progressive glucose hypometabolism in the frontal lobe, temporal lobe, and striatum, accompanying white matter volume reduction [54]; (4) ALS patients exhibit complicated and region-diversified alterations in glucose metabolism, such as a decrease in prefrontal and frontal regions and an increase in hippocampus, parahippocampal gyrus and subcortical regions [55, 56].
NDA rodent models on isogenic backgrounds have been developed to gain mechanistic insights on NDA progression while controlling environmental factors. Resembling observations in human patients, dysregulated energy metabolism has been observed in the majority of NDA models starting at presymptomatic ages (Table 3). Using these animal models, significant molecular, cellular, and subcellular details at different stages of disease progressions have been acquired. Axonopathy is often observed simultaneously with bioenergetic impairment upon the onset of neurodegeneration (Table 3). Axons, the longest and the most morphologically complex subcellular compartment of most neurons [57,58,59], are particularly vulnerable to aging [60, 61], environmental insults [62] and neurodegenerative conditions [63,64,65]. Axonal activities require substantial energy supply [2, 66] and recent studies ascribe axonal maintenance failures to the dysregulation of axonal bioenergetics [67, 68]. Neurons cannot store high energy molecules, such as glycogen and fat, and thus they must synthesize ATP on demand [69, 70]. The latest high-resolution imaging studies reveal distinctive distributions and organization of mitochondria, the major ATP synthesizing organelle, in cell bodies, dendrites, and axons (Table 4) [71,72,73,74,75]. Surprisingly, despite the highly energetically demanding activities constantly taking place in axons, axonal mitochondria are not only sparsely distributed but are also smaller in size, have simpler morphology (Table 4), and show weaker metabolic activity compared to dendritic and somatic mitochondria [76]. In addition, axonal mitochondrial motility progressively decreases as neurons mature and age [77,78,79,80]. Notably, spatially confined mitochondrial compartments, consisting of single or multiple mitochondrial filaments with temporal stability of up to 80 min, are exclusively found in dendrites and soma, but not in axons [81]. This remarkable mismatch between the high energy demand and the less complex mitochondrial population in axons raises the following questions: (1) How is bioenergetic machinery organized and regulated across axons to meet local energy needs? (2) How does aging, the major risk factor for neurodegeneration, affect axonal bioenergetics? (3) How does the aged axonal bioenergetic system go further awry as NDAs develop?
In this review, we specifically focus on central nervous system (CNS) axons, which can be divided into three subdomains: axonal initial segments, myelinated segments, and the axonal arbors harboring presynaptic boutons. We will first provide an overview of the subdomain-specific, energy-demanding events and characterize their locally specialized bioenergetic machineries; second, we will summarize the current literature on NAD redox homeostasis in axons, the mainstay of proper energetic metabolism; third, we will describe axonal bioenergetic maladaptation upon aging/NDAs and discuss how network hyperexcitability, NAD redox dysregulation, and pathological glial response could exacerbate axonal bioenergetic failures.
Overview of energy-demanding events and bioenergetic machinery across distinct axonal subdomains
Mathematical modeling based on experimental data suggests energy consuming events vary by regions: in grey matter ~ 43% of energy expenditure is devoted to synaptic activities, while in white matter > 99% of energy is used for maintaining the resting membrane potential and other housekeeping tasks [2, 147, 148]. Among these expenditures, synaptic-related activities consume the highest amount of ATP [2]. As long-range axons in the brain travel through grey and white matter, their dominant energy consuming activities likely vary, because axonal regions in grey and white matter have different compositions of ion channels, organelles, biochemical machineries, and glial partners. Generally, CNS axons can be divided into three domains: (1) the axon initial segment (AIS), located adjacent to the soma and where action potentials originate, (2) the myelinated axon, an axonal shaft wrapped by layers of myelin sheath formed by oligodendrocytes, and (3) axonal collateral/terminal arbors, where axons form a plethora of en passant or terminal presynaptic boutons and are surrounded by astrocytes. Less ultrastructural and energetic data are available for lightly myelinated axons, abundant in the grey matter. For example, serotonergic, dopaminergic, and adrenergic axons which are barely myelinated [149, 150]. Due to a lack of data, we do not review the energetic regulation for these lightly myelinated axons here.
In the glia-neuron lactate shuttle hypothesis, neurons import lactate from astrocytes or oligodendrocytes through monocarboxylic acid transporters (MCTs), rather than directly taking up glucose for neuronal glycolysis. The lactate is then converted into pyruvate as the substrate for mitochondrial tricarboxylic acid (TCA) cycle and OXPHO [151, 152]. However, recent studies show that neuronal glycolysis is required for the ATP synthesis necessary to support neurotransmission and fast axonal transport [153,154,155]. In the neocortex, oligodendrocyte networks predominantly deliver glucose through MCTs to support compound action potentials in callosal myelinated axons [156]. Together, the above evidence suggests the existence of diverse glial metabolic support mechanismsin different axonal subdomains.
Given that glucose metabolism is the dominant bioenergetic pathway for axons under normal dietary conditions, here we summarize the local energy demanding events that are primarily fueled by glucose metabolism and the spatial organization of the glucose metabolism machinery within each axonal subdomain (Fig. 1A). We hope the knowledge summarized here will provide a framework to delineate the sources of metabolic vulnerability of long-range axons.

The spatial organization of glucose metabolism machineries across axon. A. Upper panel: Heat map description of the degree of energy consumption by each axonal activity within three axonal subdomains. Bottom panel: the intensity of each feature/parameter of glucose metabolism machineries within three axonal subdomains. Heat map color from white to dark represents the mild to strong degree/intensity. B.-D. The spatial organization of glucose transporters (GLUTs), monocarboxylic acid transporters (MCTs), glycolytic enzymes, mitochondria, and glial partners in axon initial segment (B), myelinated axonal shaft (C), and axonal arbors (D). D. The activity driven adaption of glucose metabolism machineries in presynaptic compartments. ① Activity dependent recruitment and anchoring of mitochondria to F actin filament at presynaptic sites through the AMPK-PAK-Syntaphilin axis; ② Neuronal stimulation induced mitochondrial ultrastructural changes with wider cristae and more compact or irregular matrices; ③ Surface mobilization of GLUT4, and presumably increased glucose uptake and glucose metabolism; and ④ Increased glucose metabolism through the Hexosamine synthesis pathway to boost O-GlcNAcylation of mitochondrial adaptor protein Milton, which reduce mitochondria mobility
Axon Initial Segment (AIS)
When the somatic membrane potential is sufficiently depolarized, an action potential (AP) is initiated in the AIS, the base of axon [157]. Mathematical modeling estimates the energy cost of AP propagation is ~ fourfold higher in grey matter than white matter [2] and the cost is highest at the AIS [158]. To form the focal point to initiate AP spikes, voltage gated sodium channels are densely anchored in the AIS to lower the voltage threshold for AP generation [159]. Subsequently, the highly amplified Na+ influx poses a high burden on the energy-consuming Na+/K+ ATPase to restore Na+ and K+ gradients. Studies looking at direct measurements of energy consumption during and after AP generation in the AIS remain to be conducted.
Stimulated emission depletion (STED) super resolution imaging and 3-dimensional (3D) electron microscopy (EM) reveal non-uniform mitochondrial coverage in axons. STED imaging of mouse cortical neurons and human induced pluripotent stem cells (iPSC) derived forebrain neurons shows that the proximal, rather than the distal region of the AIS retains an immobile cluster of mitochondria [160] (Fig. 1B). EM studies visualizing layer 2/3 pyramidal neurons of mouse primary visual cortex find that mitochondrial occupancy decreases as the axon extends further away from the soma [71]. Given that AP initiation likely dominates the energetic burden in AIS, it is tempting to ask if these clustered mitochondria are specifically recruited to provide copious ATP to fuel the efficient and accurate initiation and propagation of AP. Sensor imaging in the soma of hippocampal dentate granule neurons showed that increased Na+/K+ pump activity is the predominant activator of neuronal glycolysis upon neuronal stimulation [161]. Paradoxically, glycolytic enzymes are not enriched in the AIS, according to an AIS proteome profiling study [162], despite the enrichment of Na+/K+ pumps and its close proximity to the soma. Future studies are needed to determine the contribution of glycolysis to AIS energetics.
The AIS also plays a critical role in establishing and maintaining axon-dendrite polarity, in which the AIS actively guides selective and polarized protein trafficking to axons, and prevents protein diffusion from the somatodendritic area through the physical barrier formed by AIS-anchored membrane proteins and various cytoskeletal proteins [157]. Tau, a normally axonal microtubule-associated-protein, loses its polarity and is missorted in tauopathy conditions. Tjiang and Zempel recently demonstrated that locally restricted disruption of the membrane potential of mitochondrial clusters located in AIS results in Tau mis-sorting into soma, potentially due to defective microtubule dynamics [160]. It is unclear whether the polarized tau sorting demands substantial energy supplied by AIS-enriched mitochondria. It is also possible that clustered mitochondria provide Ca2+ buffering and/or the interactions with endoplasmic reticulum/endosomes to maintain the axon-dendrite polarity [163].
Supported by single axon EM reconstruction, proximal axons within ~ 100 µm of the soma lack myelination and oligodendroglial support [164]. Inhibitory axo-axonic synapses are selectively enriched onto the AIS region in excitatory pyramidal neurons [165, 166], while ~ 1% of AIS in cortex are associated with microglia [167]. It remains to be determined if glia-axon metabolic coupling exists in the AIS. Interestingly, ~ 75–85% of the AIS surface of rat and cat cerebellar Purkinje neurons are covered by glial processes [168]. The origin of these glial processes and their roles in axonal metabolism remain to be studied. It is also unclear whether extensive AIS glial coverage also occurs for other types of neurons.
Myelinated axonal segments
In mature CNS white matter, most axons are wrapped with a myelin sheath originating from oligodendrocytes. Myelination greatly improves the energetic efficiency of AP propagation, costing less than 1% of white matter’s energy budget [2, 169]. To achieve saltatory AP propagation, therefore improving the energetic efficiency of impulse propagation, CNS oligodendrocytes or peripheral nervous system (PNS) Schwann cells insulate most of the axonal surface with myelin sheath, rendering the nodes of Ranvier the only exposed areas where ion channels are densely embedded to allow AP regeneration. Due to their distinct structural and molecular organizations, the myelinated axonal segments can be further subdivided into nodes, paranodes, juxtaparanodes and internodes (Fig. 1C) [170]. Intriguingly, mitochondrial distribution and the external glial composition surrounding the nodes are drastically different between PNS and CNS axons. In PNS, upon Ca2+ elevation and Na+/K+ ATPase activation induced by electrical impulses, mitochondria are preferentially recruited to and accumulate in the nodes, likely providing on-site ATP synthesis to support rapid AP regenerations [171]. In CNS, mitochondria rarely distribute to the nodes but are present in the juxtaparanodes and internodes (Fig. 1C) [172,173,174]. However, a study in ex vivo cerebellar Purkinje cells shows that mitochondria reduce their motility in the nodes and paranodes in response to the potentiated electrical activity [174]. PNS nodes are solely contacted by the microvilli emanating from Schwann cells [170], while CNS nodes are mostly covered by astrocytic processes and sometimes contacted by oligodendrocyte precursor cells (OPCs) [175].
Within myelinated axonal segments, canonical synapses do not exist, and resting potential maintenance by plasma membrane Na+/K+ ATPase and various non-signaling (so-called ‘housekeeping’) tasks cost ~ 99% of the axon’s total energy budget [2]. Housekeeping activities comprise axonal transport, cytoskeleton rearrangement, protein/lipid synthesis, and mitochondrial proton leakage across the inner membrane [148, 176, 177]. Guedes-Dias P et al. [178] estimated that axonal transport costs less than 1% of the total energy budget for individual neurons. This estimate was based on the assumption that a single kinesin molecule is paired with a cargo and drives the transport process throughout. We hypothesize the energy cost for axonal transport to be much higher when one takes into account the complex molecular machineries involved in cargo-specific, dynamic, and bidirectional transport as well as the variety and large quantity of cargos. Axonal transport occurs in both retrograde and anterograde directions and includes the fast transport of synaptic vesicle precursors, RNA granules, endo-lysosome, autophagosome and mitochondria as well as the slow transport of cytoskeleton components and clathrin packets [179]. Evidence shows that even though kinesin and dynein drive transport in opposite directions, they attach to the same vesicular cargo through modulation by a variety of cargo-specific adaptors and coordinators [180]. The precise mechanism of kinesin and dynein’s cooperation was first explained by “the tug-of-war model”. In this model, the outcome of mechanical competition between kinesin and dynein determines the bidirectional transport behavior. This was, however, later challenged by “the paradox of co-dependence” phenomenon that inhibiting only one type of motors diminishes the motility in both directions [180]. Nevertheless, the exact energetic cost of axonal transport is hard to estimate without a detailed mechanistic understanding.
Emerging EM datasets allow us to compare mitochondrial morphology, size, and subcellular occupancy within the myelinated axonal compartment and unmyelinated en passant boutons (Table 5). In both subdomains, the mitochondria are similar in their morphological simplicity. However, myelinated regions are often occupied by fewer mitochondria with more heterogeneous sizes, including mitochondria of a larger volume than those seen in presynaptic compartments. It is unclear how this mitochondrial heterogeneity in myelinated axons develops and whether it has functional relevance.
The sparse and heterogeneous distribution of mitochondria in myelinated axons raises the question on the energy source that fuels fast axonal transport, which is in constant demand to achieve fast and continuous movement of a large number of vesicular cargos in axons. Studies have shown that glycolytic enzymes are physically associated with fast-moving vesicles through their interaction with Huntingtin and provide onboard ATP synthesis to propel vesicular transport [154, 155] (Fig. 1D). Fast axonal transport in vitro is mainly supported by glycolysis, and OXPHO inhibition exerts a minimal impact on this form of transport [155]. The presence of the glycolytic complex on transporting cargos likely enables the efficient and continuous supply of ATP to overcome the lesser ATP efficiency of glycolysis compared to OXPHO. Future in vivo studies will be critical to determine whether glycolysis is the dominant energy source for fast axonal transport.
Using ex vivo acute brain slices, Meyer et al. showed that oligodendrocytes directly deliver glucose, instead of lactate, potentially through MCT1/2 and GLUT1/3, to axons in the corpus callosum (Fig. 1C), which indispensably supports the compound AP conduction [156]. Interestingly, GLUT1 incorporation into the oligodendroglial myelin compartment is upregulated upon optic nerve spiking activity [15]. It is currently unclear whether the enhancement of glucose uptake by neural activity, either directly through GLUTs on axons [14] or indirectly through oligodendrocytes [156], is a general mechanism to potentiate local glycolysis.
In addition to MCT-mediated metabolite exchange and activity-induced GLUT1 incorporation, Fruhbeis et al. shed light on the role of oligodendrocyte-secreted exosomes in sustaining axonal transport upon nutritional deprivation [182] (Fig. 1C). Exosomes can be secreted from diverse types of glia cells. It remains to be studied whether the glia-derived metabolic enzymes, metabolites and ATP can be delivered through exosomes to mediate the glia-axon metabolic coupling. Besides exosomes, a recent study showed that mitovesicles, a novel type of extracellular vesicles originating from mitochondria, are metabolically competent for autonomous ATP synthesis [183]. Proteome analysis suggests that neurons and astrocytes can secrete mitovesicles [183]. There is supporting evidence for the transfer of mitochondria from astrocytes to neurons to increase neuronal ATP level and viability upon cerebral ischemia in an astrocytic CD38-dependent manner [184]. Interestingly, damaged axonal mitochondria can also be released and then taken up by astrocytes for degradation. This has been observed in healthy adult optic axons and is implicated to occur similarly in superficial layers of cerebral cortex [185]. Future studies investigating the physiological roles of mitovesicles are likely to provide more insight into glia-axon metabolic coupling and whether such transcellular mitochondrial exchange takes place in axons as a non-cell autonomous mechanism for metabolic support and quality control.
Axonal arbors harboring pre-synaptic boutons
When axons enter their target zones, they often arborize extensively and form en passant or terminal presynaptic boutons full of synaptic vesicles (SVs) (Fig. 1D). These axonal arbors are mostly located in the grey matter and are rarely myelinated [186, 187]. The development of the genetically-encoded synaptic targeting ATP optical reporter Syn-ATP has enabled quantitative measurements of ATP alterations near SVs during physiological events to evaluate the local energetic costs at rest or during activation [188]. Pulido and Ryan [189] used the Syn-ATP reporter to show that the resting potential maintained by Na+/K+-ATPase only utilizes a minor portion of ATP in boutons [189], in contrast to what was previously believed [190]. Instead, they found that ~ 44% ATP is consumed by SV-resident vacuolar-type ATPase to restore the H+ gradient, which is constantly dissipated in resting state, despite the absence of SV exocytosis and recycling [189]. Upon electrical stimulation, ATP consumption in presynaptic boutons increases tremendously. During this active state, many calcium-driven processes and SV cycling are the major consumers of ATP, while Na+/K+ ATPase still consumes little [188]. Future studies should assess if these in vitro findings also occur in normal mature and aged brains and how NDA conditions impact them. In addition to SV cycling, it is plausible that local protein synthesis, occurring in ~ 40% of presynaptic terminals, also consumes a non-negligible amount of ATP [191] since adding one amino acid to the polypeptide chain requires ~ 2ATP and 2 guanosine triphosphate (GTP) [192], while proper protein folding by molecular chaperones also consumes ATP [193]. Furthermore, the robust anatomical plasticity of axons, reflected by the dynamic gain and loss of axonal arbors and presynaptic boutons, requires substantial protein synthesis as well as cytoskeleton remodeling [194]. Rearrangement of cytoskeletal components such as actin and microtubule occurs within minutes of electrical stimulation [195, 196] or long-term potentiation (LTP) [197]. Both actin and tubulin polymerization require ATP and GTP hydrolysis, respectively [148]. However, the degree of energetic burden posed by cytoskeleton remodeling remains to be determined.
Mitochondria are preferentially captured and stabilized at presynaptic boutons to serve as robust energetic factories [77, 198,199,200,201]. Still, less than 50% of boutons contain mitochondria [202,203,204,205], except lemniscal thalamocortical synapses where 92% of them contain mitochondria [206]. Facilitated by the creatine kinase/phosphocreatine system [207], ATP produced from mitochondria can rapidly diffuse over a certain range, therefore constantly fulfilling local bioenergetic needs in resident boutons, but also transiently compensating for the needs of nearby boutons lacking mitochondria [200, 208]. The dynamics and mobile behaviors of axonal mitochondria also impact energy metabolism. The molecular mechanisms governing these mitochondrial dynamics in neuronal subcellular compartments have been extensively reviewed [209,210,211,212] and thus we only highlight a few studies here.
In the Drosophila motor nerve terminal, the mitochondrial volume and densities within each bouton is positively correlated with the estimated energetic demand of each bouton [213], a correlation presumably caused by the activity-driven mechanism that couples energy consumption and synaptic mitochondrial recruitment. In mammalian neurons, several mechanisms governing synaptic mitochondrial positioning have been discovered. Chen et al. [214] showed that Ca2+ release triggered by action potentials binds to Miro, which then releases the C-terminal tail of KIF5 to bind syntaphilin (SNPH). Such KIF5–SNPH coupling then inhibits adenosine triphosphatase activity of kinesin-1, the molecular motor for mitochondria. In other words, SNPH immobilizes mitochondria at Ca2+ entry sites triggered by action potentials. SNPH also binds to dynein light chain LC8, which then enhances the docking between SNPH and microtubules to further reduce mitochondrial mobility [215]. Li et al. further showed that AMP-activated protein kinase–p21-activated kinase (AMPK-PAK) axis upregulates ATP production at presynapses in response to intense neuronal activity [216]. Upon prolonged electrical stimulation, the substantial energy usage in boutons activates AMPK-PAK axis, which then phosphorylates myosin VI to promote its interaction with SNPH (Fig. 1D). Subsequently, SNPH and myosin VI recruit and anchor mitochondria onto the presynaptic actin filament to support onsite ATP synthesis and Ca2+ buffering [216]. It has also been shown that GLUT4 is mobilized to the surface of axonal boutons in response to electrical stimuli and presumably increases local intracellular glucose concentration [14]. Elevated intracellular glucose promotes the O-linked-N-acetylglucosaminylation (O-GlcNAcylation) of the mitochondrial adaptor protein, Milton, which allows its interaction with four and a half LIM domains protein 2 (FHL2) to anchor mitochondria onto presynaptic actin filaments, therefore arresting mitochondria in the boutons [217, 218]. These arrested mitochondria can further adapt to a non-orthodox organellar configuration, featuring wider cristae and more compact or irregular matrices (Fig. 1D), according to an observation in mouse hippocampi upon LTP induction [219]. These presynaptic anchored mitochondria are considered to play important roles for higher order brain function, given the fact that the quantity of mitochondria per bouton in rhesus monkey’s prefrontal cortex positively correlates with working memory performance [220].
The findings of a positive correlation between the amount of oxidized/aged protein in mitochondrial matrix and the distance away from the soma suggest that mitochondrial protein quality control at distal axons is compromised [221, 222]. From a bioenergetic perspective, the mitochondrial membrane potential also declines in a distance-dependent manner from the soma both in vitro and in vivo [222]. Building on these observations, we speculate that mitochondria in the collateral arbor and terminal tips, far away from the soma, manifest compromised protein turnover and OXPHO capacity compared to peri-somatic mitochondria. EM data from neocortex and hippocampus showed that mitochondria are rarely present in dendritic spine heads or adjacent to the post-synaptic densities [71, 181, 205]. Thus, most mitochondria purified from synaptosomes are likely to arise from presynaptic compartments. Studies found that such presynaptic mitochondria are more susceptible to Ca2+ overload and damage induced by traumatic brain injury compared to non-synaptic mitochondria [223, 224]. Proteomic studies further reveal a differential proteome composition between synaptic and non-synaptic mitochondria [225,226,227]. However, the proteomics data should be interpreted with caution due to inconsistencies. For example, Stauch et al. showed most electron transport chain complex subunits are reduced in synaptic mitochondria including NADH:ubiquinone oxidoreductase core subunit S8 (NDUFS8) of complex I, ATP synthase F1 subunit alpha (ATP5A1) and ATP synthase F1 subunit beta (ATP5B) of complex V [225]. In contrast, these proteins were shown to be increased in synaptic mitochondria according to the findings from Volgyi et al. [226]. It is unclear whether the inconsistency was caused by mouse strain, age differences, or other variables in their studies.
Glycolysis, in parallel to mitochondrial OXPHO, contributes to ATP synthesis in presynaptic boutons during both resting and firing states [188, 189, 228]. In resting presynaptic boutons, the ATP level can be maintained for up to ~ 30 min by glycolysis alone when mitochondrial OXPHO is inhibited in vitro [188]. In autaptic hippocampal neurons, basal synaptic transmission can be fueled solely by glycolysis, while evoked transmission requires mitochondrial OXPHO [228]. In the calyx of Held, glycolysis is preferentially required to shape faithful AP waveforms and sustain synaptic transmission [229]. To cope with hypoxic stress, glycolytic enzymes are recruited to form a “metabolon” in synapses to sustain the synaptic function of serotonergic neurosecretory-motor neurons and preserve the locomotion behavior in C. elegans [230]. Although it is not known whether mammalian brains retain this “glycolytic metabolon” forming capability upon hypoxia, proteomics profiling shows the enrichment of glycolytic enzymes in striatal axon terminals of midbrain dopaminergic neurons in mouse brains [231]. The proteomic characterization of mammalian brain synaptic vesicles also identifies many glycolytic enzymes [232, 233] that physically associate with synaptic vesicles to fuel their transport along axons and their neurotransmitter uptake [154, 155, 234, 235]. Recent studies with cultured hippocampal neurons showed that presynaptic mitochondria possess the metabolic flexibility and capability to sustain synaptic transmission in the absence of glycolysis [236] when only oxidative fuel (lactate and pyruvate) was provided.
Glucose, the first substrate for the glycolytic pathway, is transported into neurons through the neuron-specific glucose transporters, GLUT3 and GLUT4 [11,12,13,14]. The majority of GLUT3 is embedded into the axonal plasma membrane and constantly uptakes glucose [14]. In contrast, only ~ 7.5% of GLUT4 is present on the axonal membrane at resting state, while the rest localizes to endosomal vesicles. In response to electrical stimuli, ∼20% of GLUT4, but not GLUT3, gets mobilized to the bouton surface (Fig. 1D) [14]. This additional GLUT4 likely increases glucose uptake and subsequently boosts glycolysis to support extra energy costs from synaptic activity. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) is the master activator of glycolysis. However, its abundance in adult brain neurons remains low due to its constant degradation via the ubiquitin proteasomal pathway [237]. Intriguingly, juvenile hippocampus expresses higher levels of PFKFB3, which gets further upregulated during learning and is required for long-term memory formation exclusively in the juvenile period [238]. Along with the upregulation of GLUT3 and many other glycolytic enzymes [238], the increase in PFKFB3 likely potentiates glycolytic activity in juvenile hippocampus. However, evidence suggests that ubiquitous upregulation of PFKFB3 can be detrimental to mitochondrial function and neuronal health as it reduces pentose phosphate pathway flux and antioxidant capability [239, 240]. This raises the possibility that increased PFKFB3 in juvenile brain is exclusively restrained to synapses to boost local glycolysis without overtly interfering with the pentose phosphate pathway.
Presynaptic compartments are part of the “tripartite synapse”, in which the physical contact and functional integration of glial processes are an instrumental component [241]. 3D EM reconstruction of synapse-astrocyte contacts shows that more than 50% of presynaptic compartments are closely associated with astrocytes [242]. The metabolic coupling between astrocytes and neurons has been extensively studied and reviewed, mostly focusing on the lactate shuttle hypothesis [243,244,245], which has been questioned by stoichiometric and experimental evidence [246,247,248]. Some evidence suggest that lactate secreted from astrocytes modulates neuronal excitability as a signaling molecule by activating hydroxycarboxylic acid receptor 1 (HCAR1), a putative lactate receptor, in excitatory synapses [249]. It remains to be determined what other metabolites are shuttled from astrocytes to the presynaptic compartments in the “tripartite synapse” setting, whether MCTs or glia-derived exosomes are involved in this exchange, which metabolic pathways are mobilized, and how this metabolic coupling is fine-tuned by synaptic activity. The perspectives from Barros et al. suggest a putative coupling mechanism between astrocyte metabolism and synaptic activity through astrocytic Na+/K+ ATPases and Na+/HCO3− cotransporters [250]. Astrocytes can sense the postsynaptic workload represented by the K+ efflux from postsynaptic compartment into the extracellular space upon the evoked excitatory postsynaptic potentials, then adapt their energy metabolism accordingly. Intriguingly, the recent discovery of the roles of endocannabinoids, a well-known retrograde messenger for synaptic transmission [251], in regulating mitochondrial metabolism in both neurons and astrocytes through mitochondrial type-1 cannabinoid receptor (mtCB1) [252, 253], suggests the potential involvement of the endocannabinoid system in homeostatic regulation of astrocyte-neuron metabolic coupling proportional to neural activity. Future studies will be required to confirm these activity-dependent astrocyte-neuron metabolic coupling hypotheses.
NAD redox homeostasis underlying axonal bioenergetics
Nicotinamide adenine dinucleotide (NAD) is a critical cofactor mediating many redox reactions in glucose metabolism. The oxidized form of NAD (NAD+) acts as an electron acceptor while its reduced form (NADH) acts as an electron donor [254]. Pivotally, the NAD redox potential (NAD+/NADH) determines the thermodynamic driving force for the NAD+ or NADH consuming steps in both glycolysis and OXPHO [255]. Mechanisms involved in maintaining the NAD redox potential in mammalian cells include NAD biosynthesis, recycling, degradation, and subcellular compartmentalization and have been extensively reviewed [254, 256,257,258]. Here we focus on summarizing our current knowledge of NAD redox homeostasis in long-range axons.
NAD biosynthesis
The salvage pathway is the major NAD biosynthetic pathway in the mammalian brain. All salvage pathway enzymes are present in the major brain cell types, while the enzymes involved in the kynurenine and Preiss-Handler pathways are minimally expressed in the brain (Fig. 2A-B; [259, 260]). In neurons, nicotinamide phosphoribosyl-transferase (NAMPT), the rate-limiting enzyme in the salvage pathway, is required for survival, mitochondrial homeostasis, and SV cycling [261,262,263,264]. NAMPT is localized to the cytoplasm and mitochondrial matrix in cortical neurons [265], suggesting the possibility that NAMPT in presynaptic mitochondria engages in local NAD biosynthesis to readily support activity-driven glucose metabolism. Notably, NAMPT can be exchanged transcellularly between cells through extracellular vesicles [266, 267]. Thus, axons can potentially receive additional NAMPT through extracellular vesicles secreted from their glial partners or from peripheral organs to boost local NAD+ production.

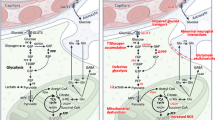
The molecular underpinnings of NAD redox potential maintenance in axons. A. A schematic view of NAD biosynthetic pathways. B. Heat map visualization of the abundance of mRNA and proteins of NAD biosynthetic enzymes. Transcript levels are adapted from [259]; protein levels are adapted from [260]. Abbreviations: Inh. Neu, inhibitory neurons; Exc. Neu, excitatory neurons; Mic. Mac., microglia and macrophages; Oligo, oligodendrocytes; OPC, oligodendrocyte precursor cells; Fibro.-like, fibroblast-like cells; End, endothelial cells. C. The subcellular localization of NMNAT1-3 in the soma and proximal axon. D. The subcellular distribution of NMNAT2, NMNAT3, and proteins involved in NAD+/NADH homeostasis in presynaptic boutons. (D1) NMNAT2, LDH-A, and glycolytic enzymes closely attach to synaptic vesicles; NMNAT2 inhibits SARM1 activation and maintains local NAD redox potential together with LDH-A to support “onboard” glycolysis; (D2) NMNAT3 inside mitochondrial matrix maintains local NAD redox potential together with OXPHO units, SLC25A51 (NAD+ transporter), and Malate-Aspartate shuttle. E. A hypothetical model of mitochondrial independent NAD+ recycling through the glutamine carboxylation pathway. MDH1, cytosolic malate dehydrogenase 1; ACLY, ATP citrate lyase; GLS, glutaminase; Gln, glutamine; Glu, glutamate; GOT1, glutamic-oxaloacetic transaminase 1; α-KG, alpha ketoglutarate; IDH1, isocitrate dehydrogenase (NADP+) 1; OAA, oxaloacetate
At the last step of the salvage pathway, nicotinamide mononucleotide adenylyl transferases (NMNAT) 1–3 catalyze NAD+ synthesis using nicotinamide mononucleotide (NMN) as their substrate. NMNAT1 is mainly localized in the nuclei (Fig. 2C) and is relatively uniformly expressed across different cell types [268, 269]. NMNAT2 is the most abundant NMNAT in the brain and is mainly expressed in neurons [269, 270]. Through palmitoylation, NMNAT2 associates with Golgi membranes and Golgi-derived vesicles including synaptic vesicle precursors in axons [271,272,273]. NMNAT2 is regarded as an axonal maintenance factor because its loss impairs axonal transport and results in axon degeneration in the absence of external insults [274,275,276,277]. Though NMNAT over-expressions can offer axonal protections [269, 278], studies also showed that excess amount of NMNAT can cause adverse consequence on presynaptic function in Drosophila [279] and visual circuit plasticity in mice [280].
Interestingly, OPCs, the unique glia cells forming synapse-like contacts with axons, abundantly express NMNAT2 mRNA [281, 282]. It is unknown whether OPC-NMNAT2 is involved in OPC-axon metabolic coupling. Knowing that OPCs secrete exosomes [283], it will be interesting to investigate whether membrane-bound NMNAT2 can reach axons through OPC-derived exosomes and facilitate axonal NAD biosynthesis.
The literature suggests that NMNAT3 is a mitochondrial-targeted isoform [270, 284]. Despite extremely low NMNAT3 mRNA levels in mouse brain, NMNAT3 protein has been detected in cultured cortical neurons [265], adult mouse brain [260], and human postmortem brain [285]. Future study is required to further confirm NMNAT3’s existence as well as to investigate its physiological function in neurons.
NAD+ recycling from NADH in cytoplasm
In addition to de novo NAD biosynthesis, the NAD redox potential can be replenished via NAD+ recycling from NADH derived from glycolysis and the TCA cycle (Fig. 2D). Based on pharmacological and genetic studies, mitochondrial OXPHO seems to be the major NAD+ recycling pathway in neurons. NAD+ is regenerated from NADH in the mitochondrial matrix through mitochondrial respiratory complex I (NADH-ubiquinone oxidoreductase) and then exchanged to the cytoplasm NAD+ pool through the malate-aspartate shuttle [286,287,288]. However, the low density and immobility of axonal mitochondria in many axonal regions (see above) pose significant challenges for mitochondrial regeneration of NAD+ to restore the axonal NAD redox potential. Knowing that glycolysis supplies ATP in the absence of mitochondria in some axonal segments, mitochondria-independent NAD+ recycling mechanisms are likely to be present in those axonal regions to restore the NAD redox potential. Future studies will be required to test this hypothesis.
One type of mitochondria-independent NAD+ recycling is mediated by lactate dehydrogenase isoform A (LDH-A) that is associated to the fast-moving axonal transport vesicles (Fig. 2D). LDH-A promotes the conversion of pyruvate to lactate. During this enzymatic reaction, it regenerates NAD+ by oxidizing glycolysis derived NADH and therefore facilitates the vesicular glycolysis that is required for fast axonal transport [289, 290]. However, if lactate gets exported from glia to axons following the proposed lactate shuttle hypothesis, lactate dehydrogenase isoform B (LDH-B) will be required to convert lactate into pyruvate for subsequent mitochondrial oxidation. Because LDH-B favors the conversion in reverse direction to LDH-A, that is it consumes NAD+ and produces NADH and pyruvate [291, 292], LDH-B's activity may counteract LDH-A, thus reducing NAD redox potential and restraining glycolysis.
Gaude et al. employed a mammalian cell model of tunable mitochondrial dysfunction to shut down mitochondrial-dependent NAD+ recycling and uncovered that the cytosolic reductive carboxylation of glutamine as an alternative route for NAD+ recycling [293]. The glutamine reductive carboxylation takes over the mitochondrial TCA cycle to provide oxaloacetate as the substrate for malate dehydrogenase 1 (MDH1), which subsequently regenerates NAD+ from NADH and physically couples with GAPDH to facilitate glycolysis (Fig. 2E) [293]. It has also been shown that cultured cortical neurons possess metabolic flexibility to oxidize glutamine when mitochondrial pyruvate uptake is inhibited [9]. The enzymes involved in glutamine carboxylation, such as glutaminase (GLS), glutamic-oxaloacetic transaminase 1 (GOT1), glutamic-oxaloacetic transaminase 2 (GOT2), isocitrate dehydrogenase (NADP+) 1 (IDH1), ATP citrate lyase (ACLY) and MDH1, are highly expressed in neurons (Fig. 2E, [259, 260] and Allen brain single cell transcriptomics explorer). Of note, ACLY is shown to be enriched in vesicles for axonal transports [294]. Whether and in what context glutamine carboxylation contributes to axonal NAD redox homeostasis merits further investigation.
Discrete NAD+ pool in mitochondria
NAD redox potential in mitochondrial matrix is typically ~ 10–100 fold lower than the potential in the cytoplasm due to the active NADH import through malate-aspartate shuttle [295]. However, the absolute NAD+ pool within mitochondria account for ~ 50–70% of total cellular NAD+ and exhibits relatively long half-life in contrast to the labile cytosolic NAD+, based on NAD sensor studies in mammalian cell lines [296,297,298,299]. The mitochondrial NAD+ transporter, SLC25A51, may be responsible for maintaining such discrete NAD+ pool by actively importing NAD+ from the cytoplasm [300,301,302]. In addition to NAD+ import, NAD synthesis through the Salvage pathway enzymes, NAMPT and NMNAT3, residing inside mitochondria in certain cell types [303, 304], can further increase mitochondrial NAD+. In neurons, the quantity and stability of mitochondrial NAD+ pool across various axonal subdomains have not been determined. These measurements are particularly important for presynaptic boutons where mitochondria support the intense synaptic activities while enduring the potentially incompetent protein turnover and Ca2+ buffering [222, 223]. Single cell transcriptomic and proteomic data suggest the expressions of SLC25A51 and NMNAT3 in neurons (Allen brain single cell transcriptomics explorer and [260]). Future studies are required to elucidate their roles in mitochondrial NAD+ pool maintenance, TCA cycle and OXPHO as well as their requirement for axonal functions.
NAD consuming pathways
Besides glucose metabolism, the fluctuations of NAD+ and its metabolites can elicit a profound impact through NAD+ consuming enzymes, such as Sirtuins (SIRTs) and Poly (adenosine diphosphate-ribose) polymerases (PARPs) [305,306,307]. SIRTs are well known in governing mitochondrial biology and metabolic homeostasis [305, 308, 309]. Noteworthy, Chamberlain et al. discovered a novel form of glia-axon metabolic coupling through SIRT2 [310]. They found that oligodendrocytes provide SIRT2 to myelinated axons through exosomes. Upon reaching axons, SIRT2 deacetylates mitochondrial adenine nucleotide translocase 1/2 and enhances mitochondrial ATP production. PARPs play diverse roles in mRNA processing, ribosomal biogenesis, and protein ubiquitination [311]. It is unclear whether PARPs are present in axons.
Sterile alpha and TIR motif containing protein 1 (SARM1) catalyzes NAD(P) hydrolysis and is a central regulator of an axon-self destruction cascade [312,313,314,315]. Distinct from SIRTs and PARPs, SARM1 is a multifaceted metabolic sensor detecting changes in NAD+, NADP+ and NMN levels. Under normal physiological conditions, when NAD+ and NADP+ levels are actively maintained by NMNAT2 and NAD+ kinase (NADK) [268, 316], SARM1 stays inactive because of substrate inhibitions [313, 317]. The abundance of NMNAT2, the main axonal NAD+ provider, is tightly regulated by proteosome degradation and mitogen-activated protein kinase (MAPK) signaling [318, 319]. Upon axonal injury or mitochondrial insults, NMNAT2 level is reduced and subsequently SARM1 gets activated by the increased ratio of NMN (NMNAT2 substrate) to NAD+ [312, 320,321,322,323]. Activated SARM1 degrades NAD+ and leads to a rapid NAD+ decay, resulting in severe energetic failure in axons. It is yet to be determined whether NADP+ synthesis by NADK is required to suppress SARM1 activity and if SARM1 also mediated NADP+ degradation to exacerbate oxidative stress [324]. Regardless, constraining SARM1 activation seems to be the prerequisite for healthy bioenergetics in axons.
Axonal bioenergetic maladaptation in aging and NDAs
The contribution of axonopathy and synaptopathy to functional decline during aging and the pre-symptomatic stage of NDAs has been extensively studied [60, 325,326,327]. Meta-analysis of 417 studies focusing on synaptopathy in postmortem AD brains reveals a consistent loss of synapses in hippocampus and frontal cortex, in which the presynaptic markers are more affected than the postsynaptic markers [328]. The previous sections of this review have summarized the subdomain-specific organization and regulation of the axonal bioenergetic system that crucially fuels axonal functions. In this section, we will discuss how axonal bioenergetics become altered during aging and NDAs. We first provide a phenotypic overview of axonal bioenergetic maladaptation upon aging and its progressive dysfunction since early-stage NDAs. Second, we will review the following signature aspects in aging/NDAs brains: cortical network hyperexcitability, NAD+ redox homeostasis disruption, and pathological glial responses. We also speculate how these different dysfunctions may further accelerate the failure of axonal bioenergetics.
Phenotypic overview
Increasing literature provides phenotypic evidence at subcellular resolution for aging-induced mitochondrial maladaptation in axons. In myelinated axons of the mouse optic nerve, aging results in reduced axon number, enlarged axon diameter, thickened myelin sheath, and increased nodal/paranodal length [329]. Within these aged optic nerve axons, mitochondrial density, and mitochondria-smooth endoplasmic reticulum contacting area are decreased, while the volume and length of individual mitochondrion are significantly increased [329]. Similar aging induced mitochondrial morphology and distribution changes were also observed in myelinated axons projecting to hippocampal CA1 and dentate gyrus regions [73]. In C. elegans, in vivo analysis of mitochondria in distal neurites throughout adult life reveals a progressive decline in mitochondrial trafficking starting from early adulthood, with mitochondrial size, density, and resistance to oxidative stress undergoing three distinct stages of increase, maintenance and decrease [330]. Reduced mitochondrial transport is also observed in Drosophila wing neuron axons [78, 79], and mouse retina nerve fiber layer myelinated axons [80] during aging.
In presynaptic boutons, aging elicits region-divergent adjustments of mitochondrial contents. Ultrastructural analysis of the hippocampal CA1 area prepared from male rats found significantly reduced numbers of mitochondria per presynaptic bouton, accompanied by an increased individual mitochondrion area in aged rats compared to adolescent and adult rats [331]. These synaptic mitochondria in aged synapses also exhibit functional impairment, including decreased Ca2+ buffering capacity, reduced ATP production, and increased oxidative stress [332,333,334]. Conversely, in the central nucleus of amygdala of aged rats, a brain region less affected by aging and neurodegenerative conditions, the number of mitochondria per bouton is drastically increased, while individual mitochondrion area is decreased compared to adult rats [335]. Similarly, in dorsolateral prefrontal cortex (dlPFC) of female Rhesus monkeys, studies found more mitochondria-containing boutons in aged peri/postmenopausal monkeys than in young and aged premenopausal moneys, with no difference in bouton densities or sizes [220]. Surgical menopause, causing abrupt estrogen loss, results in an increase of boutons containing donut shaped mitochondria and small active zones, and worsened working memory [220]. This finding suggests sexually dimorphic modulation of mitochondria and energy metabolism, a topic being widely reviewed [336,337,338]. In synaptosomal mitochondria samples purified from the whole brain, aging elicits mitochondrial DNA (mtDNA) deletion and activation of nuclear respiratory factor 1 (NRF1) and peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC1A) mediated mitochondrial fitness pathways, accompanied by the adaptive increase of proteins governing OXPHO activity, antioxidant capacity, mitochondrial fusion and mitophagy [339]. These adaptive changes likely reduce the damage caused by mtDNA deletion, preserving mitochondrial respiration in aged brains [339]. Collectively, diverse mitochondrial alterations in myelinated and presynaptic compartments have been associated with aging.
Mitochondrial malfunction significantly worsens in both axonal and presynaptic compartments as NDAs progress. For example, EM studies found a substantial reduction in the density of mitochondria-resident presynapses in AD postmortem auditory association cortex of the temporal lobe, a DMN hub, compared to controls [340]. Interestingly, mitochondrial abnormalities were not detected in the dorsolateral prefrontal cortex, which is not involved in the DMN. A follow-up study from the same group found significantly less OXPHO machinery, but more Sirtuin pathway components, in synaptoneurosomes prepared from the auditory association cortex of AD brains, compared to controls, while no changes were found in the primary visual cortex [341]. Additionally, proteomic profiling of synaptoneurosomes prepared from parietal association cortex from MCI and dementia patients shows a synaptic metabolic shift towards glycolysis, while synaptic metabolism was rewired towards enhanced OXPHO in cognitively normal and resilient individuals [342]. A shift towards glycolysis can be interpreted as attempted compensation for mitochondrial dysfunction. Overall, the above evidence suggests a strong correlation between synaptic mitochondrial dysfunction and presynaptic degeneration, particularly in AD vulnerable brain regions.
Lessons from transgenic AD mouse models mimicking human amyloidogenesis suggest synaptic, but not non-synaptic mitochondria, are defective at pre-symptomatic stages, with defects including decreased mitochondrial respiration, compromised Ca2+ buffering, increased oxidative stress, increased permeability, elevated fission, and activation of Parkin-mediated mitophagy [103, 343,344,345]. A more detailed summary of synaptosomal mitochondrial deficits in current AD mouse models can be found in [345]. Resembling the presynaptic degeneration in AD brains, a preferential loss of excitatory synapses lacking presynaptic mitochondria was observed in the medial prefrontal cortex of pre-symptomatic 5xFAD mice [346]. Additionally, the mitochondrial quantity per presynaptic bouton was significantly reduced in the remaining mitochondria-containing synapses, perhaps due to mitophagy [346].
In postmortem brains of patients with PD and dementia with Lewy bodies (DLB), a similar presynaptic degeneration phenomenon occurs in substantia nigra (SN) dopaminergic neurons, in which the population of presynapses devoid of mitochondria is significantly reduced, despite the increase of mitochondrial proteins within the surviving dopaminergic axons [347]. In addition, the translation of mitochondrial proteins in synapses is dysregulated, revealed by proteomic characterization of synaptosomes from SN pars compacta (SNpc) of human PD brains [348]. Synaptosomal proteomic analysis with SNpc prepared from a PD mouse model overexpressing mutant human α-synuclein further sheds light on the importance of synaptic metabolic rewiring that precedes mitochondrial damage and synaptic pathology [114, 349]. In the mid-stage of this PD model, striatal synaptosomal mitochondria start to exhibit bioenergetic deficits, while no change in the abundance of essential OXPHO components and mitochondrial ultrastructure was detected [114]. Evidence suggests the functional decline could arise from the compromised proteostasis of antioxidant machineries. Overexpressed α-synuclein accumulates in presynaptic sites, interacts with the mitochondrial chaperone heat shock protein 10 (HSP10), and prevents its import into mitochondria, resulting in compromised quality control of essential antioxidant machineries within the mitochondrial matrix which eventually leads to synaptic mitochondrial dysfunction [350].
During the pre-symptomatic stage of HD mouse brains, the mitochondrial protein import deficit also occurs exclusively in the synaptosome, preceding any observable OXPHO impairment [351]. Simultaneously, striatal astrocytes in HD mouse brains acquire a metabolic adaptation towards fatty acid oxidation to compensate for diminished glucose uptake [352]. This leads to subsequent exposure of substantial astrocyte-derived reactive oxygen species which exacerbates the synaptic mitochondrial homeostasis disruption discussed above [352]. Likewise, in an ALS mouse model, defective mitochondrial respiration occurs in presynaptic boutons in the spinal cord at a pre-symptomatic stage without changes in OXPHO machinery abundance and mitochondrial quantity [353]. This study observed an increase in upstream glucose catabolism activity, including glycolysis and TCA cycle, as well as the lipid peroxidation in ALS mouse presynapses [353, 354]. In ALS mouse gliosomes, peri-synaptic astrocytic processes, lipid peroxidation and lactate fermentation were elevated, while mitochondrial respiration were preserved [353, 354].
Consistent across these major NDAs, metabolic alteration and mitochondrial dysfunction arise in the presynaptic compartment, representing the initial metabolic defects in the pre-symptomatic stage. They then interact with a reprogrammed glial environment in a negative cycle and eventually lead to synaptic connectivity deterioration and cognitive decline. Many of the current NDA animal models were generated based on the hypothesis that toxic misfolded protein aggregates are at the root of disease progression. For instance, the amyloid cascade hypothesis has been popular for decades in explaining AD, however the negative clinical outcomes to amyloid-directed therapies call for consideration of alternative hypothesis and models [355]. Here we want to endorse the alternative hypothesis that metabolic failure originating from the presynaptic compartment is one of major causes of neurodegeneration that culminates in cognitive function decline. We call for basic research with novel mouse disease models, covering a broad panel of late-onset NDAs as well as human brain organoid and co-culture system to test this hypothesis. In clinical aspects, we call for the development of multimodal biomarkers to infer the early-onset metabolic alterations in human brains, followed by longitudinal surveillance to elucidate its correlation with disease progression.
Network hyperexcitability
Partly due to reduced GABAergic tone [356,357,358], cortical network hyperexcitability increases during normal aging with excessive APs and increased synaptic transmission, which requires an extravagant energy demand, potentially overwhelming axonal bioenergetic capacity and triggering maladaptations. As axon length increases, the mitochondrial membrane potential declines [222], decreasing energy availability in the distal portion of long-range axons and providing less bioenergetic support for hyperexcitability. In addition, mitochondrial protein turnover is likely to be reduced as axon length increases [221, 222], posing additional difficulties on repairing and replenishing exhausted mitochondria in the synaptic boutons of distal axons.
Developing into the asymptomatic phase of NDAs, network hyperexcitability is further augmented before reaching the ultimate degeneration stage. In the prodromal phase of AD, the resting state activity is aberrantly increased in the medial prefrontal cortex, posterior cingulate cortex, precuneus, and hippocampus—all are connection hubs of DMN manifesting glucose hypometabolism [359, 360]. In apolipoprotein E4 (APOE-ɛ4) allele carriers, the strongest genetic factor for late-onset AD, lateral parietal and precuneus regions of DMN in the right-hemisphere show hyperconnectivity and hyperactivity events developing from a young age of 25.2 ± 6.8 years old [361]. Computational modeling suggests microscale hyperexcitability can increase the power of the lower frequency oscillatory network [362, 363], which is frequently observed in AD patients [363] and may underlie cognitive dysfunction [364]. Pre-symptomatic neuronal hyperexcitability in disease-vulnerable brain regions has also been documented in PD [365, 366], HD [367], and ALS [368].
Even though the casual relationship between hyperexcitability and glucose hypometabolism remains elusive, we speculate the excessively elevated synaptic activity directly poses heavy burden onto axonal bioenergetic system and rapidly consumes NAD+. Furthermore, excitotoxicity triggered by excessive glutamate release and Ca2+ influx can activate a necroptosis pathway that is reported to non-canonically deplete axonal NMNAT2 and activate SARM1, which destabilizes NAD homeostasis, exacerbates glucose hypometabolism, and eventually drives axonal degeneration and network disintegration [369, 370].
NAD redox homeostasis disruption
NAD+ redox potentials decline in aged brains [371, 372]. The mRNA levels of several genes involved in NAD+ biosynthesis, NAD+ recycling and Sirtuin signaling are broadly altered in glutamatergic, GABAergic, dopaminergic, and cholinergic neurons during aging (Fig. 3A; [22]). For example, NMNAT2 mRNA level is decreased in both aged GABAergic and cholinergic neurons, while NMNAT1 mRNA level is reduced in aged glutamatergic and GABAergic neurons (Fig. 3A). Reduced NMNAT1/2 levels are likely to activate SARM1 to further reduce NAD+ levels and result in glucose hypometabolism in these neurons, particularly in GABAergic neurons. SIRT3 signaling has been shown to protect neuronal mitochondria against metabolic and excitatory stress [373]. However, SIRT3 mRNA levels are downregulated in both aged glutamatergic and GABAergic neurons (Fig. 3A). Thus, it is noteworthy that GABAergic neurons are particularly vulnerable to SIRT3 loss [374]. Reduced SIRT3 together with the NAD+ deficiency caused by NMNAT1/2 downregulation is likely to impair GABAergic glucose metabolism and subsequently increase hyperexcitability to further augment mitochondrial metabolic stress in axons.

Aging induced bioenergetic maladaptation in mouse brain. A. Transcriptional fold change of enzymes involved in NAD+ redox potential maintenance in glutamatergic (GLUT), GABAergic (GABA), dopaminergic (DOPA) and cholinergic (CHOL) neurons upon aging, adapted from [22]. Sirt7 in aged cholinergic neurons is increased 5.97-fold, labeled in dark red. B. Transcriptional fold change of glycolytic enzymes and monocarboxylic acid transporters (MCTs) in myelin-forming oligodendrocytes (MF-OLG) and mature oligodendrocytes (MT-OLG). (MT-OLG-1 and MT-OLG-2 are two independent repeats in [22]). HK2 in aged myelin-forming oligodendrocytes is increased 9.72-fold, labeled in purple-red. C. A hypothetical model of the sequential events caused by aging that leads to metabolic failure in long-range axonal projections and glucose hypometabolism in central hubs of the brain connectome
Meta-analyses of human transcriptomic data found defective NAD metabolism in the majority of NDAs [375, 376]. In AD, NMNAT2 expression is significantly decreased in the frontal lobe [377, 378], while LDH-A mRNA level is reduced by almost twofold in both the frontal and temporal lobes [377]. Computational analysis with perturb-Met further predicts the dysregulation of NAD redox homeostasis in cortical layer 2–6 excitatory neurons of AD brains [379]. Similarly, dysregulated NAD homeostasis was also found in an AD mouse model, 3xTg transgenic mice, in a sex-biased manner [380, 381]. Noteworthy, assessed by two-photon lifetime imaging in dissociated neurons from mouse hippocampus of different ages, mitochondrial NADH level is significantly lower in 3xTg neurons, and declines with age [382]. However, due to the concern that cultures of dissociated adult brain neurons may not be disease relevant, more in vivo evidence is required to support this finding. Interestingly, a metabolomic study separating grey and white matter from postmortem brains found that nicotinamide metabolism-relevant metabolites (including NAD+,NADH, NMN, and nicotinamide) are significantly reduced in white matter compared to grey matter (supplementary table of [383]). This finding implies the myelinated compartment (that is, axons and their supporting cells) might be more susceptible to NAD decline. Using preclinical NDA models, numerous studies provide strong evidence supporting the therapeutic benefits of supplementing NAD+ and its precursors [384, 385]. The maintenance of NAD redox potentials is likely to enable optimal axonal bioenergetics and thus protect brain circuits.
To counteract the NAD redox disruption in NDAs, genetic approaches have been applied to enhance NAD salvage/biosynthesis pathways by overexpressing NMNATs, or to suppress NAD degradation by ablating SARM1. It has been shown that overexpressing NMNAT2 results in an increase in NAD redox potential in an AD transgenic cell line [386]. In the Tg2576 AD mouse model, NMNAT2 overexpression activates the AMPK signaling cascade, upregulates α-secretase expression, and attenuates β-secretase dependent amyloidogenesis [386]. The neuroprotective effect of different NMNAT isoforms on various neurodegenerative or neuropathy models has been summarized [387]. The neuroprotection against disease progression by NMNAT overexpression differs across disease models. There is substantial supportive evidence for neuroprotection offered by SARM1 deletion in animal models for traumatic brain injury and retinal degeneration [388,389,390,391,392]. However, there are also several reports showing minimal/no therapeutic benefits of SARM1 loss in PD [393] and ALS models [394, 395]. While the common downstream players of diverse NDAs converge at energy metabolism pathways, the upstream mechanisms are rather disease specific and complex. We hypothesize that targeting multiple upstream pathways that intersect with energy metabolism, together with stabilizing NAD homeostasis, should improve and prolong neuroprotection against NDAs. Recognizing the distinct excitability, energetic demands, mitochondrial regulatory units, proteome composition, and Ca2+ homeostasis in various neuronal subtypes [396, 397], we encourage the revisit of the axon protective effect offered by NMNAT2 and SARM1 manipulation in the most vulnerable neuronal subtypes in individual NDA models to dissect out the subtype specific mechanisms.
Deleterious glial dysfunction
Besides hyperexcitability and NAD+ redox perturbation, aging also elicits region-dependent alterations in glial biology [398,399,400,401]. For instance, oligodendrocyte differentiation is diminished in aged human brains, particularly prominent in the hippocampus and substantia nigra [399]. Additionally, oligodendrocyte numbers and myelin density are decreased in aged frontal cortex, concomitant with the appearance of myelin spheroids and debris [224, 399]. The reduced transcription of most glycolysis and MCTs genes in the oligodendrocytes of aged mouse brain (Fig. 3B) are likely to reduce their glucose metabolism capacity and decrease their metabolic support of myelinated axons.
Oligodendrocytes also undergo pathological changes as NDAs develop. Such glia alterations likely worsen glia-axon metabolic coupling and aggravates aging-induced maladaptation of axonal bioenergetics. The loss of myelination, oligodendrocytes, and OPCs has been frequently observed in the pre-symptomatic stage of AD and ALS animal models [402,403,404,405]. Consistently, in human postmortem AD brains, the number of mature myelinated oligodendrocytes is decreased, while remyelinating oligodendrocytes are increased [406]. Expanding on the fact that oligodendrocytes are inherently heterogeneous in the CNS [407], Sadick et al. further delineated the subtype-specific transcriptomic profile of oligodendrocytes in human AD brains. The biggest subcluster exhibiting downregulated expression were synaptic cell adhesion molecules, implying the loss of contact between oligodendrocytes and axons [408]. Interestingly, other subclusters show upregulation of putative neuroprotective pathways such as cholesterol metabolism [408], which accelerates Aβ production and in turn kills oligodendrocytes [409]. PD mouse model generated through alpha-synuclein injection into the dorsal striatum also showed the pathological inclusion of alpha-synuclein in oligodendrocytes secondary to neuronal pathology [410]. In iPSC-derived oligodendrocytes from PD patients, the transcriptional programs for maturation and myelination are significantly dampened and shifted towards immune reactive status, likely ascribed to the alpha-synuclein burden within oligodendrocytes [411]. Consistently, the population of myelinating oligodendrocyte is reduced in human PD postmortem midbrains, accompanied by the transcriptomic upregulation of unfolded protein response and downregulation of neuro-supportive pathways [412]. Future studies should be conducted to assess the metabolic decoupling between axons and oligodendrocytes in NDA brains and to determine whether such decoupling poses significant burdens to axonal bioenergetics. Furthermore, the remyelinating and immune reactive process in oligodendrocytes seem to be energy demanding [413, 414], potentially competing for the glucose metabolism substrates and therefore worsening the axonal bioenergetics.
Astrocytes have been shown to be altered in aging brain in a region-specific manner. Neurotoxic astrocyte subtypes found in aged mouse hippocampus and striatum exhibit downregulated genes in mitochondrial function, antioxidant defense, and cholesterol synthesis [400, 401]. The morphological complexity of astrocytes significantly decreases in AD-susceptible brain regions during aging, such as the entorhinal cortex and hippocampal CA1 region [402, 403]. Reduced astrocyte end-feet surface area is likely to diminish astrocytic coverage on synapses, and subsequently retard neurotransmitter uptake, further exacerbating neuronal hyperexcitability, while also impairing the astrocyte-neuron metabolic coupling that is essential for presynaptic bioenergetics. Similarly, as NDAs develop, astrocytes undergo substantial reprogramming in both morphological and metabolic aspects [415,416,417,418,419,420]. However, due to the evolutionarily divergent gene expression profiles, human astrocytes appear distinct from rodent astrocytes in Ca2+ signaling and metabolic activities [421, 422]. Such species-specific features raise the concern on whether astrocytic metabolic changes observed in AD mouse model are relevant to AD patients. For example, glycolytic and the mitochondrial TCA cycle activity are often reported to be decreased in astrocytes derived from AD mouse models [416]. However, the large-scale proteomic analysis of human AD postmortem brains suggests enhanced sugar metabolism in astrocytes and microglia [423]. Transcriptionally, astrocytes from human AD prefrontal cortex also exhibit remarkably differences from mouse 5xFAD cortex, featured by the downregulation of genes coordinating free-fatty-acid transport, lipid droplet storage and ROS detoxification [424]. In addition, the genes responsible for synaptogenesis and astrocyte morphogenesis are downregulated in certain subsets of astrocytes from AD brains, suggesting the trend towards tripartite synapse disintegration [408]. In late-stage PD patients, the interaction between astrocytes and excitatory neurons is estimated to be decreased by ~ 25% in the prefrontal cortex [425]. Given that astrocytic end-feet in tripartite synapses provide indispensable metabolic support, the astrocytic dysfunction mentioned above is anticipated to worsen the presynaptic bioenergetics that is already problematic since the pre-symptomatic stage. Overall, the characteristics and mechanism of glial metabolic alterations in human NDA brains remains largely unknown, as well as its relationship with axonal bioenergetic maladaptation. Therefore, we call for the development of multimodal biomarkers specific for glial metabolism to dissect out the region divergent changes and delineate the longitudinal profile during diseases progression, hoping to shape the direction for mechanistic study. Additionally, developing better experimental models recapitulating axon-glia interaction in human brains (e.g., human brain organoids) are important future directions.
In conclusion, we hypothesize (Fig. 3C) that network hyperexcitability occurs in disease-specific brain regions in NDAs, together with the NAD redox disruption, which triggers the axon-intrinsic metabolic failure that will be inherently determined by its bioenergetic supply system. As the disease progresses, glial alterations take place concomitantly in disease sensitive regions, leading to the destruction of the extrinsic metabolic support system that in turn accelerates the deterioration of axonal bioenergetics, synaptic connectivity, and cognitive function.
Concluding remarks
Axons, key components of the brain connectome, are the most morphologically complex subcellular compartment in neurons and are prone to deterioration upon various insults. Aging, the most prevalent risk factor for many neurodegenerative diseases, is associated with decreased glucose metabolism, preferentially in distal axons and their terminals. This altered metabolism is accompanied by the mitochondrial maladaptation across myelinated axonal shaft and presynaptic boutons. These closely correlated phenotypes have driven the discovery of the molecular underpinnings that mediate age-associated bioenergetic incompetence and extrinsic glial metabolic support breakdown. Manipulating these molecular targets to enhance or normalize bioenergetics in specific cell types in NDA models will help to further interrogate the causal relationship between metabolic vulnerability, axonopathy, disease specific pathology and cognitive decline. Other than innate bioenergetic incompetency, additional efforts are needed to understand the influence of NDA relevant genetic, epigenetic, and environmental factors on neuronal bioenergetics.
Availability of data and materials
Not applicable.
Abbreviations
- 3D:
-
3-Dimensional
- ACLY:
-
ATP citrate lyase
- AD:
-
Alzheimer’s disease
- AIS:
-
Axon initial segment
- ALS:
-
Amyotrophic lateral sclerosis
- AMPK:
-
AMP-activated protein kinase
- AP:
-
Action potential
- APOE:
-
Apolipoprotein E
- APP:
-
Amyloid precursor protein
- ATP:
-
Adenosine triphosphate
- ATP5A1:
-
ATP synthase F1 subunit alpha
- ATP5B:
-
ATP synthase F1 subunit beta
- BACE1:
-
Beta-secretase 1
- C9ORF72:
-
Chromosome 9 open reading frame 72
- CNS:
-
Central nervous system
- COX IV:
-
Cytochrome c oxidase subunit 4
- cx-DHED:
-
Carboxy-dehydroevodiamine∙HCl
- DLB:
-
Dementia with Lewy bodies
- DMN:
-
Default mode network
- Drp1:
-
Dynamin-related protein 1
- DTI:
-
Diffusion tensor imaging
- DVR:
-
Delayed verbal recall task
- EM:
-
Electron microscopy
- FADH2 :
-
Oxidized flavin adenine dinucleotide
- FDG-PET:
-
Fluorodeoxyglucose-positron emission tomography
- FTLD:
-
Frontotemporal lobar degeneration.
- GLP-1:
-
Glucagon-like peptide 1
- GLS:
-
Glutaminase
- GLUT:
-
Glucose transport
- GOT:
-
Glutamic-oxaloacetic transaminase
- GTP:
-
Guanosine triphosphate
- HCAR1:
-
Hydroxycarboxylic acid receptor 1
- HD:
-
Huntington’s disease
- HSP10:
-
Heat shock protein 10
- IDH1:
-
Isocitrate dehydrogenase (NADP +) 1
- iPSC:
-
Induced pluripotent stem cells
- LDH-A:
-
Lactate dehydrogenase isoform A
- LDH-B:
-
Lactate dehydrogenase isoform B
- LRRK2:
-
Leucine-rich repeat kinase 2
- LTP:
-
Long-term potentiation
- MAPK:
-
Mitogen-activated protein kinase
- MCI:
-
Mild-cognitive impairment
- MCT:
-
Monocarboxylic acid transporters
- MDH1:
-
Malate dehydrogenase 1
- MMSE:
-
Mini Mental State Examination
- mPTP:
-
Mitochondrial permeability transition pore
- MRI:
-
Magnetic resonance imaging
- mtCB1 :
-
Mitochondrial type-1 cannabinoid receptor
- mtDNA:
-
Mitochondrial DNA
- NAD:
-
Nicotinamide adenine dinucleotide
- NADH:
-
Oxidized nicotinamide adenine dinucleotide
- NADK:
-
NAD+ kinase
- NAMPT:
-
Nicotinamide phosphoribosyl-transferase
- NDA:
-
Neurodegenerative disorders of aging
- NDUFS8:
-
NADH:ubiquinone oxidoreductase core subunit S8
- NMJ:
-
Neuromuscular junction
- NMN:
-
Nicotinamide mononucleotide
- NMNAT:
-
Nicotinamide mononucleotide adenylyl transferase
- NRF1:
-
Nuclear respiratory factor 1
- O-GlcNAcylation:
-
O-linked-N-acetylglucosaminylation
- OPC:
-
Oligodendrocyte precursor cell
- OSCP:
-
ATP synthase peripheral stalk subunit OSCP (ATP5PO)
- OXPHO:
-
Oxidative phosphorylation
- PAK:
-
P21-activated kinase
- PARP:
-
Poly (adenosine diphosphate-ribose) polymerase
- PCC:
-
Posterior cingulate cortex
- PD:
-
Parkinson’s disease
- PDH-E1α:
-
Pyruvate dehydrogenase E1 subunit alpha 1
- PFKFB3:
-
6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3
- PGC1A:
-
Peroxisome proliferator-activated receptor gamma coactivator 1 alpha
- PiB:
-
Amyloid-beta biomarker
- PPARγ:
-
Peroxisome proliferator-activated receptor gamma
- PSEN1/2:
-
Presenilin1/2
- SARM1:
-
Sterile alpha and TIR motif containing protein 1
- SIRT:
-
Sirtuin
- SN:
-
Substantia nigra
- SNpc:
-
SN pars compacta
- STED:
-
Stimulated emission depletion
- SV:
-
Synaptic vesicle
- TCA:
-
Tricarboxylic acid
- α-Syn:
-
α-Synuclein
References
Erbsloh F, Bernsmeier A, Hillesheim H. The glucose consumption of the brain & its dependence on the liver. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr. 1958;196:611–26.
Harris JJ, Attwell D. The energetics of CNS white matter. J Neurosci. 2012;32:356–71.
Yellen G. Fueling thought: management of glycolysis and oxidative phosphorylation in neuronal metabolism. J Cell Biol. 2018;217:2235–46.
Dienel GA. Brain glucose metabolism: integration of energetics with function. Physiol Rev. 2019;99:949–1045.
Mergenthaler P, Lindauer U, Dienel GA, Meisel A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 2013;36:587–97.
Zielke HR, Zielke CL, Baab PJ. Direct measurement of oxidative metabolism in the living brain by microdialysis: a review. J Neurochem. 2009;109(Suppl 1):24–9.
Silva B, Mantha OL, Schor J, Pascual A, Placais PY, Pavlowsky A, Preat T. Glia fuel neurons with locally synthesized ketone bodies to sustain memory under starvation. Nat Metab. 2022;4:213–24.
Katsu-Jimenez Y, Gimenez-Cassina A. Fibroblast growth Factor-21 promotes ketone body utilization in neurons through activation of AMP-dependent kinase. Mol Cell Neurosci. 2019;101: 103415.
Divakaruni AS, Wallace M, Buren C, Martyniuk K, Andreyev AY, Li E, Fields JA, Cordes T, Reynolds IJ, Bloodgood BL, et al. Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death. J Cell Biol. 2017;216:1091–105.
Morgello S, Uson RR, Schwartz EJ, Haber RS. The human blood-brain barrier glucose transporter (GLUT1) is a glucose transporter of gray matter astrocytes. Glia. 1995;14:43–54.
Leino RL, Gerhart DZ, van Bueren AM, McCall AL, Drewes LR. Ultrastructural localization of GLUT 1 and GLUT 3 glucose transporters in rat brain. J Neurosci Res. 1997;49:617–26.
Ferreira JM, Burnett AL, Rameau GA. Activity-dependent regulation of surface glucose transporter-3. J Neurosci. 2011;31:1991–9.
McClory H, Williams D, Sapp E, Gatune LW, Wang P, DiFiglia M, Li X. Glucose transporter 3 is a rab11-dependent trafficking cargo and its transport to the cell surface is reduced in neurons of CAG140 Huntington’s disease mice. Acta Neuropathol Commun. 2014;2:179.
Ashrafi G, Wu Z, Farrell RJ, Ryan TA. GLUT4 mobilization supports energetic demands of active synapses. Neuron. 2017;93(606–615): e603.
Saab AS, Tzvetavona ID, Trevisiol A, Baltan S, Dibaj P, Kusch K, Mobius W, Goetze B, Jahn HM, Huang W, et al. Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron. 2016;91:119–32.
Wang L, Pavlou S, Du X, Bhuckory M, Xu H, Chen M. Glucose transporter 1 critically controls microglial activation through facilitating glycolysis. Mol Neurodegener. 2019;14:2.
Shokhirev MN, Johnson AA. An integrative machine-learning meta-analysis of high-throughput omics data identifies age-specific hallmarks of Alzheimer’s disease. Ageing Res Rev. 2022;81: 101721.
Glaab E, Schneider R. Comparative pathway and network analysis of brain transcriptome changes during adult aging and in Parkinson’s disease. Neurobiol Dis. 2015;74:1–13.
Christodoulou CC, Zachariou M, Tomazou M, Karatzas E, Demetriou CA, Zamba-Papanicolaou E, Spyrou GM. Investigating the transition of pre-symptomatic to symptomatic huntington’s disease status based on omics data. Int J Mol Sci. 2020;21:7414.
Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, Bohr VA. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15:565–81.
Davie K, Janssens J, Koldere D, De Waegeneer M, Pech U, Kreft L, Aibar S, Makhzami S, Christiaens V, Bravo Gonzalez-Blas C, et al. A single-cell transcriptome atlas of the aging drosophila brain. Cell. 2018;174(982–998): e920.
Ximerakis M, Lipnick SL, Innes BT, Simmons SK, Adiconis X, Dionne D, Mayweather BA, Nguyen L, Niziolek Z, Ozek C, et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat Neurosci. 2019;22:1696–708.
Ivanisevic J, Stauch KL, Petrascheck M, Benton HP, Epstein AA, Fang M, Gorantla S, Tran M, Hoang L, Kurczy ME, et al. Metabolic drift in the aging brain. Aging (Albany NY). 2016;8:1000–20.
Currais A, Huang L, Goldberg J, Petrascheck M, Ates G, Pinto-Duarte A, Shokhirev MN, Schubert D, Maher P. Elevating acetyl-CoA levels reduces aspects of brain aging. Elife. 2019;8:e47866.
Ding J, Ji J, Rabow Z, Shen T, Folz J, Brydges CR, Fan S, Lu X, Mehta S, Showalter MR, et al. A metabolome atlas of the aging mouse brain. Nat Commun. 2021;12:6021.
Popa-Wagner A, Dumitrascu DI, Capitanescu B, Petcu EB, Surugiu R, Fang WH, Dumbrava DA. Dietary habits, lifestyle factors and neurodegenerative diseases. Neural Regen Res. 2020;15:394–400.
Cannon JR, Greenamyre JT. The role of environmental exposures in neurodegeneration and neurodegenerative diseases. Toxicol Sci. 2011;124:225–50.
Bertram L, Tanzi RE. The genetic epidemiology of neurodegenerative disease. J Clin Invest. 2005;115:1449–57.
Berson A, Nativio R, Berger SL, Bonini NM. Epigenetic regulation in neurodegenerative diseases. Trends Neurosci. 2018;41:587–98.
Cunnane SC, Trushina E, Morland C, Prigione A, Casadesus G, Andrews ZB, Beal MF, Bergersen LH, Brinton RD, de la Monte S, et al. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2020;19:609–33.
Pandya VA, Patani R. Decoding the relationship between ageing and amyotrophic lateral sclerosis: a cellular perspective. Brain. 2020;143:1057–72.
Ye F, Funk Q, Rockers E, Shulman JM, Masdeu JC, Pascual B. Alzheimer’s disease neuroimaging I: in alzheimer-prone brain regions, metabolism and risk-gene expression are strongly correlated. Brain Commun. 2022;4:fcac216.
Kalpouzos G, Chetelat G, Baron JC, Landeau B, Mevel K, Godeau C, Barre L, Constans JM, Viader F, Eustache F, Desgranges B. Voxel-based mapping of brain gray matter volume and glucose metabolism profiles in normal aging. Neurobiol Aging. 2009;30:112–24.
Oh H, Madison C, Baker S, Rabinovici G, Jagust W. Dynamic relationships between age, amyloid-beta deposition, and glucose metabolism link to the regional vulnerability to Alzheimer’s disease. Brain. 2016;139:2275–89.
Krell-Roesch J, Syrjanen JA, Vassilaki M, Lowe VJ, Vemuri P, Mielke MM, Machulda MM, Stokin GB, Christianson TJ, Kremers WK, et al. Brain regional glucose metabolism, neuropsychiatric symptoms, and the risk of incident mild cognitive impairment: the mayo clinic study of aging. Am J Geriatr Psychiatry. 2021;29:179–91.
Baran TM, Lin FV. Alzheimer’s disease neuroimaging I: amyloid and FDG PET of successful cognitive aging: global and cingulate-specific differences. J Alzheimers Dis. 2018;66:307–18.
Kelley CM, Ginsberg SD, Liang WS, Counts SE, Mufson EJ. Posterior cingulate cortex reveals an expression profile of resilience in cognitively intact elders. Brain Commun. 2022;4:fcac162.
Leech R, Sharp DJ. The role of the posterior cingulate cortex in cognition and disease. Brain. 2014;137:12–32.
Crossley NA, Mechelli A, Scott J, Carletti F, Fox PT, McGuire P, Bullmore ET. The hubs of the human connectome are generally implicated in the anatomy of brain disorders. Brain. 2014;137:2382–95.
Hagmann P, Cammoun L, Gigandet X, Meuli R, Honey CJ, Wedeen VJ, Sporns O. Mapping the structural core of human cerebral cortex. PLoS Biol. 2008;6: e159.
Lafourcade M, van der Goes MH, Vardalaki D, Brown NJ, Voigts J, Yun DH, Kim ME, Ku T, Harnett MT. Differential dendritic integration of long-range inputs in association cortex via subcellular changes in synaptic AMPA-to-NMDA receptor ratio. Neuron. 2022;110(1532–1546): e1534.
Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann Neurol. 1997;42:85–94.
Yakushev I, Schreckenberger M, Muller MJ, Schermuly I, Cumming P, Stoeter P, Gerhard A, Fellgiebel A. Functional implications of hippocampal degeneration in early Alzheimer’s disease: a combined DTI and PET study. Eur J Nucl Med Mol Imaging. 2011;38:2219–27.
Roy M, Rheault F, Croteau E, Castellano CA, Fortier M, St-Pierre V, Houde JC, Turcotte EE, Bocti C, Fulop T, et al. Fascicle- and glucose-specific deterioration in white matter energy supply in Alzheimer’s disease. J Alzheimers Dis. 2020;76:863–81.
Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804.
Drzezga A, Becker JA, Van Dijk KR, Sreenivasan A, Talukdar T, Sullivan C, Schultz AP, Sepulcre J, Putcha D, Greve D, et al. Neuronal dysfunction and disconnection of cortical hubs in non-demented subjects with elevated amyloid burden. Brain. 2011;134:1635–46.
Small GW, Mazziotta JC, Collins MT, Baxter LR, Phelps ME, Mandelkern MA, Kaplan A, La Rue A, Adamson CF, Chang L, et al. Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA. 1995;273:942–7.
Small GW, Ercoli LM, Silverman DH, Huang SC, Komo S, Bookheimer SY, Lavretsky H, Miller K, Siddarth P, Rasgon NL, et al. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2000;97:6037–42.
Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, Thibodeau SN, Osborne D. Preclinical evidence of Alzheimer’s disease in persons homozygous for the ε4 allele for apolipoprotein E. J N Engl J Med. 1996;334:752–8.
Douaud G, Jbabdi S, Behrens TE, Menke RA, Gass A, Monsch AU, Rao A, Whitcher B, Kindlmann G, Matthews PM, Smith S. DTI measures in crossing-fibre areas: increased diffusion anisotropy reveals early white matter alteration in MCI and mild Alzheimer’s disease. Neuroimage. 2011;55:880–90.
Araque Caballero MA, Suarez-Calvet M, Duering M, Franzmeier N, Benzinger T, Fagan AM, Bateman RJ, Jack CR, Levin J, Dichgans M, et al. White matter diffusion alterations precede symptom onset in autosomal dominant Alzheimer’s disease. Brain. 2018;141:3065–80.
Wen Q, Mustafi SM, Li J, Risacher SL, Tallman E, Brown SA, West JD, Harezlak J, Farlow MR, Unverzagt FW, et al. White matter alterations in early-stage Alzheimer’s disease: a tract-specific study. Alzheimers Dement (Amst). 2019;11:576–87.
Matthews DC, Lerman H, Lukic A, Andrews RD, Mirelman A, Wernick MN, Giladi N, Strother SC, Evans KC, Cedarbaum JM, Even-Sapir E. FDG PET Parkinson’s disease-related pattern as a biomarker for clinical trials in early stage disease. Neuroimage Clin. 2018;20:572–9.
Ciarmiello A, Cannella M, Lastoria S, Simonelli M, Frati L, Rubinsztein DC, Squitieri F. Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington’s disease. J Nucl Med. 2006;47:215–22.
Diehl-Schmid J, Licata A, Goldhardt O, Forstl H, Yakushew I, Otto M, Anderl-Straub S, Beer A, Ludolph AC, Landwehrmeyer GB, et al. FDG-PET underscores the key role of the thalamus in frontotemporal lobar degeneration caused by C9ORF72 mutations. Transl Psychiatry. 2019;9:54.
Cistaro A, Valentini MC, Chio A, Nobili F, Calvo A, Moglia C, Montuschi A, Morbelli S, Salmaso D, Fania P, et al. Brain hypermetabolism in amyotrophic lateral sclerosis: a FDG PET study in ALS of spinal and bulbar onset. Eur J Nucl Med Mol Imaging. 2012;39:251–9.
Wang M, Liu K, Pan J, Li J, Sun P, Zhang Y, Li L, Guo W, Xin Q, Zhao Z, et al. Brain-wide projection reconstruction of single functionally defined neurons. Nat Commun. 2022;13:1531.
Winnubst J, Bas E, Ferreira TA, Wu Z, Economo MN, Edson P, Arthur BJ, Bruns C, Rokicki K, Schauder D, et al. Reconstruction of 1,000 projection neurons reveals new cell types and organization of long-range connectivity in the mouse brain. Cell. 2019;179(268–281): e213.
Peng H, Xie P, Liu L, Kuang X, Wang Y, Qu L, Gong H, Jiang S, Li A, Ruan Z, et al. Morphological diversity of single neurons in molecularly defined cell types. Nature. 2021;598:174–81.
Salvadores N, Sanhueza M, Manque P, Court FA. Axonal degeneration during aging and its functional role in neurodegenerative disorders. Front Neurosci. 2017;11:451.
Groh J, Knöpper K, Arampatzi P, Yuan X, Lößlein L, Saliba A-E, Kastenmüller W, Martini R. Accumulation of cytotoxic T cells in the aged CNS leads to axon degeneration and contributes to cognitive and motor decline. Nature Aging. 2021;1:357–67.
Valentine WM. Toxic peripheral neuropathies: agents and mechanisms. Toxicol Pathol. 2020;48:152–73.
Coleman MP. The challenges of axon survival: introduction to the special issue on axonal degeneration. Exp Neurol. 2013;246:1–5.
Zhang J, Long B, Li A, Sun Q, Tian J, Luo T, Ding Z, Gong H, Li X. Whole-brain three-dimensional profiling reveals brain region specific axon vulnerability in 5xFAD mouse model. Front Neuroanat. 2020;14: 608177.
Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307:1282–8.
Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron. 2012;75:762–77.
Beirowski B. Emerging evidence for compromised axonal bioenergetics and axoglial metabolic coupling as drivers of neurodegeneration. Neurobiol Dis. 2022;170: 105751.
Pacelli C, Giguere N, Bourque MJ, Levesque M, Slack RS, Trudeau LE. Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr Biol. 2015;25:2349–60.
Vilchez D, Ros S, Cifuentes D, Pujadas L, Valles J, Garcia-Fojeda B, Criado-Garcia O, Fernandez-Sanchez E, Medrano-Fernandez I, Dominguez J, et al. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci. 2007;10:1407–13.
Schonfeld P, Reiser G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J Cereb Blood Flow Metab. 2013;33:1493–9.
Turner NL, Macrina T, Bae JA, Yang R, Wilson AM, Schneider-Mizell C, Lee K, Lu R, Wu J, Bodor AL, et al. Reconstruction of neocortex: Organelles, compartments, cells, circuits, and activity. Cell. 2022;185(1082–1100): e1024.
Delgado T, Petralia RS, Freeman DW, Sedlacek M, Wang YX, Brenowitz SD, Sheu SH, Gu JW, Kapogiannis D, Mattson MP, Yao PJ. Comparing 3D ultrastructure of presynaptic and postsynaptic mitochondria. Biol Open. 2019;8:bio044834.
Faitg J, Lacefield C, Davey T, White K, Laws R, Kosmidis S, Reeve AK, Kandel ER, Vincent AE, Picard M. 3D neuronal mitochondrial morphology in axons, dendrites, and somata of the aging mouse hippocampus. Cell Rep. 2021;36: 109509.
Lewis TL Jr, Kwon SK, Lee A, Shaw R, Polleux F. MFF-dependent mitochondrial fission regulates presynaptic release and axon branching by limiting axonal mitochondria size. Nat Commun. 2018;9:5008.
Santuy A, Turegano-Lopez M, Rodriguez JR, Alonso-Nanclares L, DeFelipe J, Merchan-Perez A. A quantitative study on the distribution of mitochondria in the neuropil of the juvenile rat somatosensory cortex. Cereb Cortex. 2018;28:3673–84.
Overly CC, Rieff HI, Hollenbeck PJ. Organelle motility and metabolism in axons vs dendrites of cultured hippocampal neurons. J Cell Sci. 1996;109(Pt 5):971–80.
Lewis TL Jr, Turi GF, Kwon SK, Losonczy A, Polleux F. Progressive decrease of mitochondrial motility during maturation of cortical axons in vitro and in vivo. Curr Biol. 2016;26:2602–8.
Vagnoni A, Hoffmann PC, Bullock SL. Reducing Lissencephaly-1 levels augments mitochondrial transport and has a protective effect in adult Drosophila neurons. J Cell Sci. 2016;129:178–90.
Vagnoni A, Bullock SL. A cAMP/PKA/Kinesin-1 axis promotes the axonal transport of mitochondria in aging drosophila neurons. Curr Biol. 2018;28(1265–1272): e1264.
Takihara Y, Inatani M, Eto K, Inoue T, Kreymerman A, Miyake S, Ueno S, Nagaya M, Nakanishi A, Iwao K, et al. In vivo imaging of axonal transport of mitochondria in the diseased and aged mammalian CNS. Proc Natl Acad Sci U S A. 2015;112:10515–20.
Rangaraju V, Lauterbach M, Schuman EM. Spatially stable mitochondrial compartments fuel local translation during plasticity. Cell. 2019;176(73–84): e15.
Zhang M, Cheng X, Dang R, Zhang W, Zhang J, Yao Z. Lactate deficit in an alzheimer disease mouse model: the relationship with neuronal damage. J Neuropathol Exp Neurol. 2018;77:1163–76.
He K, Nie L, Zhou Q, Rahman SU, Liu J, Yang X, Li S. Proteomic profiles of the early mitochondrial changes in APP/PS1 and ApoE4 transgenic mice models of Alzheimer’s disease. J Proteome Res. 2019;18:2632–42.
Gonzalez-Dominguez R, Garcia-Barrera T, Vitorica J, Gomez-Ariza JL. Region-specific metabolic alterations in the brain of the APP/PS1 transgenic mice of Alzheimer’s disease. Biochim Biophys Acta. 2014;1842:2395–402.
Wirths O, Weis J, Kayed R, Saido TC, Bayer TA. Age-dependent axonal degeneration in an Alzheimer mouse model. Neurobiol Aging. 2007;28:1689–99.
Zhou Q, Zheng H, Chen J, Li C, Du Y, Xia H, Gao H. Metabolic fate of glucose in the brain of APP/PS1 transgenic mice at 10 months of age: a (13)C NMR metabolomic study. Metab Brain Dis. 2018;33:1661–8.
Wirths O, Weis J, Szczygielski J, Multhaup G, Bayer TA. Axonopathy in an APP/PS1 transgenic mouse model of Alzheimer’s disease. Acta Neuropathol. 2006;111:312–9.
Li Z, Zhang Y, Zheng Y, Liu W, Zhang X, Li W, Zhang D, Cai Q, Wang S, Meng X, Huang G. Intranasal 15d-PGJ2 ameliorates brain glucose hypometabolism via PPARgamma-dependent activation of PGC-1alpha/GLUT4 signalling in APP/PS1 transgenic mice. Neuropharmacology. 2021;196: 108685.
Xu YJ, Mei Y, Shi XQ, Zhang YF, Wang XY, Guan L, Wang Q, Pan HF. Albiflorin ameliorates memory deficits in APP/PS1 transgenic mice via ameliorating mitochondrial dysfunction. Brain Res. 2019;1719:113–23.
Liu W, Zhuo P, Li L, Jin H, Lin B, Zhang Y, Liang S, Wu J, Huang J, Wang Z, et al. Activation of brain glucose metabolism ameliorating cognitive impairment in APP/PS1 transgenic mice by electroacupuncture. Free Radic Biol Med. 2017;112:174–90.
Stojakovic A, Trushin S, Sheu A, Khalili L, Chang SY, Li X, Christensen T, Salisbury JL, Geroux RE, Gateno B, et al. Partial inhibition of mitochondrial complex I ameliorates Alzheimer’s disease pathology and cognition in APP/PS1 female mice. Commun Biol. 2021;4:61.
Piquet J, Toussay X, Hepp R, Lerchundi R, Le Douce J, Faivre E, Guiot E, Bonvento G, Cauli B. Supragranular pyramidal cells exhibit early metabolic alterations in the 3xTg-AD mouse model of Alzheimer’s disease. Front Cell Neurosci. 2018;12:216.
Singulani MP, Pereira CPM, Ferreira AFF, Garcia PC, Ferrari GD, Alberici LC, Britto LR. Impairment of PGC-1alpha-mediated mitochondrial biogenesis precedes mitochondrial dysfunction and Alzheimer’s pathology in the 3xTg mouse model of Alzheimer’s disease. Exp Gerontol. 2020;133: 110882.
Nicholson RM, Kusne Y, Nowak LA, LaFerla FM, Reiman EM, Valla J. Regional cerebral glucose uptake in the 3xTG model of Alzheimer’s disease highlights common regional vulnerability across AD mouse models. Brain Res. 2010;1347:179–85.
Desai MK, Sudol KL, Janelsins MC, Mastrangelo MA, Frazer ME, Bowers WJ. Triple-transgenic Alzheimer’s disease mice exhibit region-specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia. 2009;57:54–65.
Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106:14670–5.
Kim J, Choi IY, Michaelis ML, Lee P. Quantitative in vivo measurement of early axonal transport deficits in a triple transgenic mouse model of Alzheimer’s disease using manganese-enhanced MRI. Neuroimage. 2011;56:1286–92.
Stojakovic A, Chang SY, Nesbitt J, Pichurin NP, Ostroot MA, Aikawa T, Kanekiyo T, Trushina E. Partial inhibition of mitochondrial complex I reduces tau pathology and improves energy homeostasis and synaptic function in 3xTg-AD mice. J Alzheimers Dis. 2021;79:335–53.
Correia SC, Machado NJ, Alves MG, Oliveira PF, Moreira PI. Intermittent hypoxic conditioning rescues cognition and mitochondrial bioenergetic profile in the triple transgenic mouse model of Alzheimer’s disease. Int J Mol Sci. 2021;22:461.
Andersen JV, Skotte NH, Christensen SK, Polli FS, Shabani M, Markussen KH, Haukedal H, Westi EW, Diaz-delCastillo M, Sun RC, et al. Hippocampal disruptions of synaptic and astrocyte metabolism are primary events of early amyloid pathology in the 5xFAD mouse model of Alzheimer’s disease. Cell Death Dis. 2021;12:954.
Gowrishankar S, Yuan P, Wu Y, Schrag M, Paradise S, Grutzendler J, De Camilli P, Ferguson SM. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc Natl Acad Sci U S A. 2015;112:E3699-3708.
Hong I, Kang T, Yoo Y, Park R, Lee J, Lee S, Kim J, Song B, Kim SY, Moon M, et al. Quantitative proteomic analysis of the hippocampus in the 5XFAD mouse model at early stages of Alzheimer’s disease pathology. J Alzheimers Dis. 2013;36:321–34.
Wang L, Guo L, Lu L, Sun H, Shao M, Beck SJ, Li L, Ramachandran J, Du Y, Du H. Synaptosomal mitochondrial dysfunction in 5xFAD mouse model of Alzheimer’s disease. PLoS ONE. 2016;11: e0150441.
Beck SJ, Guo L, Phensy A, Tian J, Wang L, Tandon N, Gauba E, Lu L, Pascual JM, Kroener S, Du H. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat Commun. 2016;7:11483.
Chu TH, Cummins K, Sparling JS, Tsutsui S, Brideau C, Nilsson KPR, Joseph JT, Stys PK. Axonal and myelinic pathology in 5xFAD Alzheimer’s mouse spinal cord. PLoS ONE. 2017;12: e0188218.
Sadleir KR, Kandalepas PC, Buggia-Prevot V, Nicholson DA, Thinakaran G, Vassar R. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Abeta generation in Alzheimer’s disease. Acta Neuropathol. 2016;132:235–56.
Franke TN, Irwin C, Bayer TA, Brenner W, Beindorff N, Bouter C, Bouter Y. In vivo imaging With (18)F-FDG- and (18)F-Florbetaben-PET/MRI detects pathological changes in the brain of the commonly used 5XFAD mouse model of Alzheimer’s disease. Front Med (Lausanne). 2020;7:529.
Bouter C, Irwin C, Franke TN, Beindorff N, Bouter Y. Quantitative brain positron emission tomography in female 5XFAD Alzheimer mice: pathological features and sex-specific alterations. Front Med (Lausanne). 2021;8: 745064.
Andersen JV, Christensen SK, Westi EW, Diaz-delCastillo M, Tanila H, Schousboe A, Aldana BI, Waagepetersen HS. Deficient astrocyte metabolism impairs glutamine synthesis and neurotransmitter homeostasis in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2021;148: 105198.
Gauba E, Sui S, Tian J, Driskill C, Jia K, Yu C, Rughwani T, Wang Q, Kroener S, Guo L, Du H. Modulation of OSCP mitigates mitochondrial and synaptic deficits in a mouse model of Alzheimer’s pathology. Neurobiol Aging. 2021;98:63–77.
Whitmore CA, Haynes JR, Behof WJ, Rosenberg AJ, Tantawy MN, Hachey BC, Wadzinski BE, Spiller BW, Peterson TE, Paffenroth KC, et al. Longitudinal consumption of ergothioneine reduces oxidative stress and amyloid plaques and restores glucose metabolism in the 5XFAD mouse model of Alzheimer’s disease. Pharmaceuticals (Basel). 2022;15:742.
Kim J, Kang S, Chang KA. Effect of cx-DHED on abnormal glucose transporter expression induced by AD pathologies in the 5xFAD mouse model. Int J Mol Sci. 2022;23:10602.
Zheng J, Xie Y, Ren L, Qi L, Wu L, Pan X, Zhou J, Chen Z, Liu L. GLP-1 improves the supportive ability of astrocytes to neurons by promoting aerobic glycolysis in Alzheimer’s disease. Mol Metab. 2021;47: 101180.
Merino-Galan L, Jimenez-Urbieta H, Zamarbide M, Rodriguez-Chinchilla T, Belloso-Iguerategui A, Santamaria E, Fernandez-Irigoyen J, Aiastui A, Doudnikoff E, Bezard E, et al. Striatal synaptic bioenergetic and autophagic decline in premotor experimental parkinsonism. Brain. 2022;145:2092–107.
Devrome M, Casteels C, Van der Perren A, Van Laere K, Baekelandt V, Koole M. Identifying a glucose metabolic brain pattern in an adeno-associated viral vector based rat model for Parkinson’s disease using (18)F-FDG PET imaging. Sci Rep. 2019;9:12368.
Koprich JB, Johnston TH, Reyes MG, Sun X, Brotchie JM. Expression of human A53T alpha-synuclein in the rat substantia nigra using a novel AAV1/2 vector produces a rapidly evolving pathology with protein aggregation, dystrophic neurite architecture and nigrostriatal degeneration with potential to model the pathology of Parkinson’s disease. Mol Neurodegener. 2010;5:43.
Chung CY, Koprich JB, Siddiqi H, Isacson O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J Neurosci. 2009;29:3365–73.
Chen L, Xie Z, Turkson S, Zhuang X. A53T human alpha-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. J Neurosci. 2015;35:890–905.
Graham SF, Rey NL, Yilmaz A, Kumar P, Madaj Z, Maddens M, Bahado-Singh RO, Becker K, Schulz E, Meyerdirk LK, et al. Biochemical profiling of the brain and blood metabolome in a mouse model of prodromal parkinson’s disease reveals distinct metabolic profiles. J Proteome Res. 2018;17:2460–9.
Bido S, Soria FN, Fan RZ, Bezard E, Tieu K. Mitochondrial division inhibitor-1 is neuroprotective in the A53T-alpha-synuclein rat model of Parkinson’s disease. Sci Rep. 2017;7:7495.
Choi ML, Chappard A, Singh BP, Maclachlan C, Rodrigues M, Fedotova EI, Berezhnov AV, De S, Peddie CJ, Athauda D, et al. Pathological structural conversion of alpha-synuclein at the mitochondria induces neuronal toxicity. Nat Neurosci. 2022;25:1134–48.
Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, Kuum M, Zharkovsky A, Kaasik A. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem. 2011;286:10814–24.
Fernandes HJR, Patikas N, Foskolou S, Field SF, Park JE, Byrne ML, Bassett AR, Metzakopian E. Single-cell transcriptomics of parkinson’s disease human in vitro models reveals dopamine neuron-specific stress responses. Cell Rep. 2020;33: 108263.
Kouroupi G, Taoufik E, Vlachos IS, Tsioras K, Antoniou N, Papastefanaki F, Chroni-Tzartou D, Wrasidlo W, Bohl D, Stellas D, et al. Defective synaptic connectivity and axonal neuropathology in a human iPSC-based model of familial Parkinson’s disease. Proc Natl Acad Sci U S A. 2017;114:E3679–88.
Koch JC, Bitow F, Haack J, d’Hedouville Z, Zhang JN, Tonges L, Michel U, Oliveira LM, Jovin TM, Liman J, et al. Alpha-Synuclein affects neurite morphology, autophagy, vesicle transport and axonal degeneration in CNS neurons. Cell Death Dis. 2015;6: e1811.
Liu HF, Ho PW, Leung GC, Lam CS, Pang SY, Li L, Kung MH, Ramsden DB, Ho SL. Combined LRRK2 mutation, aging and chronic low dose oral rotenone as a model of parkinson’s disease. Sci Rep. 2017;7:40887.
Tagliaferro P, Kareva T, Oo TF, Yarygina O, Kholodilov N, Burke RE. An early axonopathy in a hLRRK2(R1441G) transgenic model of parkinson disease. Neurobiol Dis. 2015;82:359–71.
Chen ML, Wu RM. Homozygous mutation of the LRRK2 ROC domain as a novel genetic model of parkinsonism. J Biomed Sci. 2022;29:60.
Liu J, Tao X, Zhu Y, Li C, Ruan K, Diaz-Perez Z, Rai P, Wang H, Zhai RG. NMNAT promotes glioma growth through regulating post-translational modifications of P53 to inhibit apoptosis. Elife. 2021;10:e70046.
Wang X, Yan MH, Fujioka H, Liu J, Wilson-Delfosse A, Chen SG, Perry G, Casadesus G, Zhu X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet. 2012;21:1931–44.
Howlett EH, Jensen N, Belmonte F, Zafar F, Hu X, Kluss J, Schule B, Kaufman BA, Greenamyre JT, Sanders LH. LRRK2 G2019S-induced mitochondrial DNA damage is LRRK2 kinase dependent and inhibition restores mtDNA integrity in parkinson’s disease. Hum Mol Genet. 2017;26:4340–51.
Schwab AJ, Sison SL, Meade MR, Broniowska KA, Corbett JA, Ebert AD. Decreased sirtuin deacetylase activity in LRRK2 G2019S iPSC-derived dopaminergic neurons. Stem Cell Reports. 2017;9:1839–52.
Boecker CA, Goldsmith J, Dou D, Cajka GG, Holzbaur ELF. Increased LRRK2 kinase activity alters neuronal autophagy by disrupting the axonal transport of autophagosomes. Curr Biol. 2021;31(2140–2154): e2146.
Tsang TM, Woodman B, McLoughlin GA, Griffin JL, Tabrizi SJ, Bates GP, Holmes E. Metabolic characterization of the R6/2 transgenic mouse model of Huntington’s disease by high-resolution MAS 1H NMR spectroscopy. J Proteome Res. 2006;5:483–92.
Cepeda-Prado E, Popp S, Khan U, Stefanov D, Rodriguez J, Menalled LB, Dow-Edwards D, Small SA, Moreno H. R6/2 Huntington’s disease mice develop early and progressive abnormal brain metabolism and seizures. J Neurosci. 2012;32:6456–67.
Gatto RG, Ye AQ, Colon-Perez L, Mareci TH, Lysakowski A, Price SD, Brady ST, Karaman M, Morfini G, Magin RL. Detection of axonal degeneration in a mouse model of Huntington’s disease: comparison between diffusion tensor imaging and anomalous diffusion metrics. MAGMA. 2019;32:461–71.
Perluigi M, Poon HF, Maragos W, Pierce WM, Klein JB, Calabrese V, Cini C, De Marco C, Butterfield DA. Proteomic analysis of protein expression and oxidative modification in r6/2 transgenic mice: a model of Huntington disease. Mol Cell Proteomics. 2005;4:1849–61.
Wade A, Jacobs P, Morton AJ. Atrophy and degeneration in sciatic nerve of presymptomatic mice carrying the Huntington’s disease mutation. Brain Res. 2008;1188:61–8.
Acevedo-Torres K, Berrios L, Rosario N, Dufault V, Skatchkov S, Eaton MJ, Torres-Ramos CA, Ayala-Torres S. Mitochondrial DNA damage is a hallmark of chemically induced and the R6/2 transgenic model of Huntington’s disease. DNA Repair (Amst). 2009;8:126–36.
Hering T, Kojer K, Birth N, Hallitsch J, Taanman JW, Orth M. Mitochondrial cristae remodelling is associated with disrupted OPA1 oligomerisation in the Huntington’s disease R6/2 fragment model. Exp Neurol. 2017;288:167–75.
Johri A, Calingasan NY, Hennessey TM, Sharma A, Yang L, Wille E, Chandra A, Beal MF. Pharmacologic activation of mitochondrial biogenesis exerts widespread beneficial effects in a transgenic mouse model of Huntington’s disease. Hum Mol Genet. 2012;21:1124–37.
Magrane J, Cortez C, Gan WB, Manfredi G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet. 2014;23:1413–24.
Lim MA, Selak MA, Xiang Z, Krainc D, Neve RL, Kraemer BC, Watts JL, Kalb RG. Reduced activity of AMP-activated protein kinase protects against genetic models of motor neuron disease. J Neurosci. 2012;32:1123–41.
Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–40.
Fischer TD, Dash PK, Liu J, Waxham MN. Morphology of mitochondria in spatially restricted axons revealed by cryo-electron tomography. PLoS Biol. 2018;16: e2006169.
Vincent AE, White K, Davey T, Philips J, Ogden RT, Lawless C, Warren C, Hall MG, Ng YS, Falkous G, et al. Quantitative 3D mapping of the human skeletal muscle mitochondrial network. Cell Rep. 2019;26(996–1009): e1004.
Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21:1133–45.
Engl E, Attwell D. Non-signalling energy use in the brain. J Physiol. 2015;593:3417–29.
Call CL, Bergles DE. Cortical neurons exhibit diverse myelination patterns that scale between mouse brain regions and regenerate after demyelination. Nat Commun. 2021;12:4767.
Kapfhammer JP, Schwab ME. Inverse patterns of myelination and GAP-43 expression in the adult CNS: neurite growth inhibitors as regulators of neuronal plasticity? J Comp Neurol. 1994;340:194–206.
Morrison BM, Lee Y, Rothstein JD. Oligodendroglia: metabolic supporters of axons. Trends Cell Biol. 2013;23:644–51.
Philips T, Rothstein JD. Oligodendroglia: metabolic supporters of neurons. J Clin Invest. 2017;127:3271–80.
Diaz-Garcia CM, Yellen G. Neurons rely on glucose rather than astrocytic lactate during stimulation. J Neurosci Res. 2019;97:883–9.
Hinckelmann MV, Virlogeux A, Niehage C, Poujol C, Choquet D, Hoflack B, Zala D, Saudou F. Self-propelling vesicles define glycolysis as the minimal energy machinery for neuronal transport. Nat Commun. 2016;7:13233.
Zala D, Hinckelmann MV, Yu H, da Lyra Cunha MM, Liot G, Cordelieres FP, Marco S, Saudou F. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell. 2013;152:479–91.
Meyer N, Richter N, Fan Z, Siemonsmeier G, Pivneva T, Jordan P, Steinhauser C, Semtner M, Nolte C, Kettenmann H. Oligodendrocytes in the mouse corpus callosum maintain axonal function by delivery of glucose. Cell Rep. 2018;22:2383–94.
Rasband MN. The axon initial segment and the maintenance of neuronal polarity. Nat Rev Neurosci. 2010;11:552–62.
Ju H, Hines ML, Yu Y. Cable energy function of cortical axons. Sci Rep. 2016;6:29686.
Bender KJ, Trussell LO. The physiology of the axon initial segment. Annu Rev Neurosci. 2012;35:249–65.
Tjiang N, Zempel H. A mitochondria cluster at the proximal axon initial segment controls axodendritic TAU trafficking in rodent primary and human iPSC-derived neurons. Cell Mol Life Sci. 2022;79:120.
Meyer DJ, Diaz-Garcia CM, Nathwani N, Rahman M, Yellen G. The Na+/K+ pump dominates control of glycolysis in hippocampal dentate granule cells. Elife. 2022;11:e81645.
Hamdan H, Lim BC, Torii T, Joshi A, Konning M, Smith C, Palmer DJ, Ng P, Leterrier C, Oses-Prieto JA, et al. Mapping axon initial segment structure and function by multiplexed proximity biotinylation. Nat Commun. 2020;11:100.
Rangaraju V, Lewis TL Jr, Hirabayashi Y, Bergami M, Motori E, Cartoni R, Kwon SK, Courchet J. Pleiotropic mitochondria: the influence of mitochondria on neuronal development and disease. J Neurosci. 2019;39:8200–8.
Tomassy GS, Berger DR, Chen HH, Kasthuri N, Hayworth KJ, Vercelli A, Seung HS, Lichtman JW, Arlotta P. Distinct profiles of myelin distribution along single axons of pyramidal neurons in the neocortex. Science. 2014;344:319–24.
Shapson-Coe A, Januszewski M, Berger DR, Pope A, Wu Y, Blakely T, Schalek RL, Li PH, Wang S, Maitin-Shepard J, et al. A connectomic study of a petascale fragment of human cerebral cortex. BioRxiv. 2021.05.29.446289. https://www.biorxiv.org/content/10.1101/2021.05.29.446289v1.
Nathanson AJ, Davies PA, Moss SJ. Inhibitory synapse formation at the axon initial segment. Front Mol Neurosci. 2019;12:266.
Baalman K, Marin MA, Ho TS, Godoy M, Cherian L, Robertson C, Rasband MN. Axon initial segment-associated microglia. J Neurosci. 2015;35:2283–92.
Somogyi P, Hamori J. A quantitative electron microscopic study of the Purkinje cell axon initial segment. Neuroscience. 1976;1:361–5.
Yu Y, Herman P, Rothman DL, Agarwal D, Hyder F. Evaluating the gray and white matter energy budgets of human brain function. J Cereb Blood Flow Metab. 2018;38:1339–53.
Rasband MN, Peles E. The nodes of ranvier: molecular assembly and maintenance. Cold Spring Harb Perspect Biol. 2015;8: a020495.
Zhang CL, Ho PL, Kintner DB, Sun D, Chiu SY. Activity-dependent regulation of mitochondrial motility by calcium and Na/K-ATPase at nodes of Ranvier of myelinated nerves. J Neurosci. 2010;30:3555–66.
Edgar JM, McCulloch MC, Thomson CE, Griffiths IR. Distribution of mitochondria along small-diameter myelinated central nervous system axons. J Neurosci Res. 2008;86:2250–7.
Chiu SY. Matching mitochondria to metabolic needs at nodes of Ranvier. Neuroscientist. 2011;17:343–50.
Ohno N, Kidd GJ, Mahad D, Kiryu-Seo S, Avishai A, Komuro H, Trapp BD. Myelination and axonal electrical activity modulate the distribution and motility of mitochondria at CNS nodes of Ranvier. J Neurosci. 2011;31:7249–58.
Serwanski DR, Jukkola P, Nishiyama A. Heterogeneity of astrocyte and NG2 cell insertion at the node of ranvier. J Comp Neurol. 2017;525:535–52.
Niven JE. Neuronal energy consumption: biophysics, efficiency and evolution. Curr Opin Neurobiol. 2016;41:129–35.
Bordone MP, Salman MM, Titus HE, Amini E, Andersen JV, Chakraborti B, Diuba AV, Dubouskaya TG, Ehrke E, de Espindola Freitas A, et al. The energetic brain - A review from students to students. J Neurochem. 2019;151:139–65.
Guedes-Dias P, Holzbaur ELF. Axonal transport: driving synaptic function. Science. 2019;366:eaww9997.
Maday S, Twelvetrees AE, Moughamian AJ, Holzbaur EL. Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron. 2014;84:292–309.
Hancock WO. Bidirectional cargo transport: moving beyond tug of war. Nat Rev Mol Cell Biol. 2014;15:615–28.
Kasthuri N, Hayworth KJ, Berger DR, Schalek RL, Conchello JA, Knowles-Barley S, Lee D, Vazquez-Reina A, Kaynig V, Jones TR, et al. Saturated reconstruction of a volume of neocortex. Cell. 2015;162:648–61.
Fruhbeis C, Frohlich D, Kramer-Albers EM. Emerging roles of exosomes in neuron-glia communication. Front Physiol. 2012;3:119.
D’Acunzo P, Perez-Gonzalez R, Kim Y, Hargash T, Miller C, Alldred MJ, Erdjument-Bromage H, Penikalapati SC, Pawlik M, Saito M, et al. Mitovesicles are a novel population of extracellular vesicles of mitochondrial origin altered in Down syndrome. Sci Adv. 2021;7:eabe5085.
Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 2016;535:551–5.
Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, Kinebuchi M, Phan S, Zhou Y, Bihlmeyer NA, et al. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A. 2014;111:9633–8.
Stedehouder J, Brizee D, Slotman JA, Pascual-Garcia M, Leyrer ML, Bouwen BL, Dirven CM, Gao Z, Berson DM, Houtsmuller AB, Kushner SA. Local axonal morphology guides the topography of interneuron myelination in mouse and human neocortex. Elife. 2019;8:e48615.
Almeida RG, Williamson JM, Madden ME, Early JJ, Voas MG, Talbot WS, Bianco IH, Lyons DA. Myelination induces axonal hotspots of synaptic vesicle fusion that promote sheath growth. Curr Biol. 2021;31(3743–3754): e3745.
Rangaraju V, Calloway N, Ryan TA. Activity-driven local ATP synthesis is required for synaptic function. Cell. 2014;156:825–35.
Pulido C, Ryan TA. Synaptic vesicle pools are a major hidden resting metabolic burden of nerve terminals. Sci Adv. 2021;7:eabi9027.
Astrup J, Sorensen PM, Sorensen HR. Oxygen and glucose consumption related to Na+-K+ transport in canine brain. Stroke. 1981;12:726–30.
Hafner AS, Donlin-Asp PG, Leitch B, Herzog E, Schuman EM. Local protein synthesis is a ubiquitous feature of neuronal pre- and postsynaptic compartments. Science. 2019;364:eaau3644.
Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010;11:113–27.
Clare DK, Saibil HR. ATP-driven molecular chaperone machines. Biopolymers. 2013;99:846–59.
Chen JL, Nedivi E. Neuronal structural remodeling: is it all about access? Curr Opin Neurobiol. 2010;20:557–62.
Sankaranarayanan S, Atluri PP, Ryan TA. Actin has a molecular scaffolding, not propulsive, role in presynaptic function. Nat Neurosci. 2003;6:127–35.
Colicos MA, Collins BE, Sailor MJ, Goda Y. Remodeling of synaptic actin induced by photoconductive stimulation. Cell. 2001;107:605–16.
Qu X, Kumar A, Blockus H, Waites C, Bartolini F. Activity-dependent nucleation of dynamic microtubules at presynaptic boutons controls neurotransmission. Curr Biol. 2019;29(4231–4240): e4235.
Zhou B, Yu P, Lin MY, Sun T, Chen Y, Sheng ZH. Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. J Cell Biol. 2016;214:103–19.
Moutaux E, Christaller W, Scaramuzzino C, Genoux A, Charlot B, Cazorla M, Saudou F. Neuronal network maturation differently affects secretory vesicles and mitochondria transport in axons. Sci Rep. 2018;8:13429.
Sun T, Qiao H, Pan PY, Chen Y, Sheng ZH. Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Rep. 2013;4:413–9.
Shepherd GM, Harris KM. Three-dimensional structure and composition of CA3–>CA1 axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. J Neurosci. 1998;18:8300–10.
Chavan V, Willis J, Walker SK, Clark HR, Liu X, Fox MA, Srivastava S, Mukherjee K. Central presynaptic terminals are enriched in ATP but the majority lack mitochondria. PLoS ONE. 2015;10: e0125185.
Dufour A, Rollenhagen A, Satzler K, Lubke JHR. Development of synaptic boutons in layer 4 of the barrel field of the rat somatosensory cortex: a quantitative analysis. Cereb Cortex. 2016;26:838–54.
Lees RM, Johnson JD, Ashby MC. Presynaptic boutons that contain mitochondria are more stable. Front Synaptic Neurosci. 2019;11:37.
Zhu Y, Uytiepo M, Bushong E, Haberl M, Beutter E, Scheiwe F, Zhang W, Chang L, Luu D, Chui B, et al. Nanoscale 3D EM reconstructions reveal intrinsic mechanisms of structural diversity of chemical synapses. Cell Rep. 2021;35: 108953.
Rodriguez-Moreno J, Rollenhagen A, Arlandis J, Santuy A, Merchan-Perez A, DeFelipe J, Lubke JHR, Clasca F. Quantitative 3D ultrastructure of thalamocortical synapses from the “lemniscal” ventral posteromedial nucleus in mouse barrel cortex. Cereb Cortex. 2018;28:3159–75.
Andres RH, Ducray AD, Schlattner U, Wallimann T, Widmer HR. Functions and effects of creatine in the central nervous system. Brain Res Bull. 2008;76:329–43.
Pathak D, Shields LY, Mendelsohn BA, Haddad D, Lin W, Gerencser AA, Kim H, Brand MD, Edwards RH, Nakamura K. The role of mitochondrially derived ATP in synaptic vesicle recycling. J Biol Chem. 2015;290:22325–36.
Chamberlain KA, Sheng ZH. Mechanisms for the maintenance and regulation of axonal energy supply. J Neurosci Res. 2019;97:897–913.
Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci. 2012;125:2095–104.
Pekkurnaz G, Wang X. Mitochondrial heterogeneity and homeostasis through the lens of a neuron. Nat Metab. 2022;4:802.
Devine MJ, Kittler JT. Mitochondria at the neuronal presynapse in health and disease. Nat Rev Neurosci. 2018;19:63–80.
Justs KA, Lu Z, Chouhan AK, Borycz JA, Lu Z, Meinertzhagen IA, Macleod GT. Presynaptic mitochondrial volume and packing density scale with presynaptic power demand. J Neurosci. 2022;42:954–67.
Chen Y, Sheng ZH. Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J Cell Biol. 2013;202:351–64.
Chen YM, Gerwin C, Sheng ZH. Dynein light chain LC8 regulates syntaphilin-mediated mitochondrial docking in axons. J Neurosci. 2009;29:9429–38.
Li S, Xiong GJ, Huang N, Sheng ZH. The cross-talk of energy sensing and mitochondrial anchoring sustains synaptic efficacy by maintaining presynaptic metabolism. Nat Metab. 2020;2:1077–95.
Pekkurnaz G, Trinidad JC, Wang X, Kong D, Schwarz TL. Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell. 2014;158:54–68.
Basu H, Pekkurnaz G, Falk J, Wei W, Chin M, Steen J, Schwarz TL. FHL2 anchors mitochondria to actin and adapts mitochondrial dynamics to glucose supply. J Cell Biol. 2021;220:e201912077.
Smith HL, Bourne JN, Cao G, Chirillo MA, Ostroff LE, Watson DJ, Harris KM. Mitochondrial support of persistent presynaptic vesicle mobilization with age-dependent synaptic growth after LTP. Elife. 2016;5:e15275.
Hara Y, Yuk F, Puri R, Janssen WG, Rapp PR, Morrison JH. Presynaptic mitochondrial morphology in monkey prefrontal cortex correlates with working memory and is improved with estrogen treatment. Proc Natl Acad Sci U S A. 2014;111:486–91.
Ferree AW, Trudeau K, Zik E, Benador IY, Twig G, Gottlieb RA, Shirihai OS. MitoTimer probe reveals the impact of autophagy, fusion, and motility on subcellular distribution of young and old mitochondrial protein and on relative mitochondrial protein age. Autophagy. 2013;9:1887–96.
Baranov SV, Baranova OV, Yablonska S, Suofu Y, Vazquez AL, Kozai TDY, Cui XT, Ferrando LM, Larkin TM, Tyurina YY, et al. Mitochondria modulate programmed neuritic retraction. Proc Natl Acad Sci U S A. 2019;116:650–9.
Brown MR, Sullivan PG, Geddes JW. Synaptic mitochondria are more susceptible to Ca2+overload than nonsynaptic mitochondria. J Biol Chem. 2006;281:11658–68.
Hill RA, Li AM, Grutzendler J. Lifelong cortical myelin plasticity and age-related degeneration in the live mammalian brain. Nat Neurosci. 2018;21:683–95.
Stauch KL, Purnell PR, Fox HS. Quantitative proteomics of synaptic and nonsynaptic mitochondria: insights for synaptic mitochondrial vulnerability. J Proteome Res. 2014;13:2620–36.
Volgyi K, Gulyassy P, Haden K, Kis V, Badics K, Kekesi KA, Simor A, Gyorffy B, Toth EA, Lubec G, et al. Synaptic mitochondria: a brain mitochondria cluster with a specific proteome. J Proteomics. 2015;120:142–57.
Graham LC, Eaton SL, Brunton PJ, Atrih A, Smith C, Lamont DJ, Gillingwater TH, Pennetta G, Skehel P, Wishart TM. Proteomic profiling of neuronal mitochondria reveals modulators of synaptic architecture. Mol Neurodegener. 2017;12:77.
Sobieski C, Fitzpatrick MJ, Mennerick SJ. Differential presynaptic ATP supply for basal and high-demand transmission. J Neurosci. 2017;37:1888–99.
Lujan B, Kushmerick C, Banerjee TD, Dagda RK, Renden R. Glycolysis selectively shapes the presynaptic action potential waveform. J Neurophysiol. 2016;116:2523–40.
Jang S, Nelson JC, Bend EG, Rodriguez-Laureano L, Tueros FG, Cartagenova L, Underwood K, Jorgensen EM, Colon-Ramos DA. Glycolytic enzymes localize to synapses under energy stress to support synaptic function. Neuron. 2016;90:278–91.
Hobson BD, Choi SJ, Mosharov EV, Soni RK, Sulzer D, Sims PA. Subcellular proteomics of dopamine neurons in the mouse brain. Elife. 2022;11:e70921.
Burre J, Volknandt W. The synaptic vesicle proteome. J Neurochem. 2007;101:1448–62.
Morciano M, Burre J, Corvey C, Karas M, Zimmermann H, Volknandt W. Immunoisolation of two synaptic vesicle pools from synaptosomes: a proteomics analysis. J Neurochem. 2005;95:1732–45.
Ishida A, Noda Y, Ueda T. Synaptic vesicle-bound pyruvate kinase can support vesicular glutamate uptake. Neurochem Res. 2009;34:807–18.
Ikemoto A, Bole DG, Ueda T. Glycolysis and glutamate accumulation into synaptic vesicles. Role of glyceraldehyde phosphate dehydrogenase and 3-phosphoglycerate kinase. J Biol Chem. 2003;278:5929–40.
Ashrafi G, de Juan-Sanz J, Farrell RJ, Ryan TA. Molecular tuning of the axonal mitochondrial Ca(2+) uniporter ensures metabolic flexibility of neurotransmission. Neuron. 2020;105(678–687): e675.
Herrero-Mendez A, Almeida A, Fernandez E, Maestre C, Moncada S, Bolanos JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol. 2009;11:747–52.
Cruz E, Bessieres B, Magistretti P, Alberini CM. Differential role of neuronal glucose and PFKFB3 in memory formation during development. Glia. 2022;70:2207.
Rodriguez-Rodriguez P, Fernandez E, Almeida A, Bolanos JP. Excitotoxic stimulus stabilizes PFKFB3 causing pentose-phosphate pathway to glycolysis switch and neurodegeneration. Cell Death Differ. 2012;19:1582–9.
Lopez-Fabuel I, Garcia-Macia M, Buondelmonte C, Burmistrova O, Bonora N, Alonso-Batan P, Morant-Ferrando B, Vicente-Gutierrez C, Jimenez-Blasco D, Quintana-Cabrera R, et al. Aberrant upregulation of the glycolytic enzyme PFKFB3 in CLN7 neuronal ceroid lipofuscinosis. Nat Commun. 2022;13:536.
Semyanov A, Verkhratsky A. Astrocytic processes: from tripartite synapses to the active milieu. Trends Neurosci. 2021;44:781–92.
Aten S, Kiyoshi CM, Arzola EP, Patterson JA, Taylor AT, Du Y, Guiher AM, Philip M, Camacho EG, Mediratta D, et al. Ultrastructural view of astrocyte arborization, astrocyte-astrocyte and astrocyte-synapse contacts, intracellular vesicle-like structures, and mitochondrial network. Prog Neurobiol. 2022;213: 102264.
Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–38.
Magistretti PJ, Allaman I. A cellular perspective on brain energy metabolism and functional imaging. Neuron. 2015;86:883–901.
Machler P, Wyss MT, Elsayed M, Stobart J, Gutierrez R, von Faber-Castell A, Kaelin V, Zuend M, San Martin A, Romero-Gomez I,., et al. In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab. 2016;23:94–102.
Dienel GA. Lack of appropriate stoichiometry: Strong evidence against an energetically important astrocyte-neuron lactate shuttle in brain. J Neurosci Res. 2017;95:2103–25.
Diaz-Garcia CM, Mongeon R, Lahmann C, Koveal D, Zucker H, Yellen G. Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab. 2017;26(361–374): e364.
Ivanov AI, Malkov AE, Waseem T, Mukhtarov M, Buldakova S, Gubkina O, Zilberter M, Zilberter Y. Glycolysis and oxidative phosphorylation in neurons and astrocytes during network activity in hippocampal slices. J Cereb Blood Flow Metab. 2014;34:397–407.
Magistretti PJ, Allaman I. Lactate in the brain: from metabolic end-product to signalling molecule. Nat Rev Neurosci. 2018;19:235–49.
Barros LF, Ruminot I, Sotelo-Hitschfeld T, Lerchundi R, Fernandez-Moncada I. Metabolic recruitment in brain tissue. Annu Rev Physiol. 2022;85:115–135. https://doi.org/10.1146/annurev-physiol-021422-091035.
Ohno-Shosaku T, Tanimura A, Hashimotodani Y, Kano M. Endocannabinoids and retrograde modulation of synaptic transmission. Neuroscientist. 2012;18:119–32.
Benard G, Massa F, Puente N, Lourenco J, Bellocchio L, Soria-Gomez E, Matias I, Delamarre A, Metna-Laurent M, Cannich A, et al. Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat Neurosci. 2012;15:558–64.
Jimenez-Blasco D, Busquets-Garcia A, Hebert-Chatelain E, Serrat R, Vicente-Gutierrez C, Ioannidou C, Gomez-Sotres P, Lopez-Fabuel I, Resch-Beusher M, Resel E, et al. Glucose metabolism links astroglial mitochondria to cannabinoid effects. Nature. 2020;583:603–8.
Katsyuba E, Romani M, Hofer D, Auwerx J. NAD(+) homeostasis in health and disease. Nat Metab. 2020;2:9–31.
Jones DP, Sies H. The Redox code. Antioxid Redox Signal. 2015;23:734–46.
Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–13.
Lautrup S, Sinclair DA, Mattson MP, Fang EF. NAD(+) in brain aging and neurodegenerative disorders. Cell Metab. 2019;30:630–55.
Cambronne XA, Kraus WL. Location, location, location: compartmentalization of NAD(+) synthesis and functions in mammalian cells. Trends Biochem Sci. 2020;45:858–73.
Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell. 2018;174(1015–1030): e1016.
Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, Kongi K, Cantuti L, Hanisch UK, Philips MA, et al. Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci. 2015;18:1819–31.
Wang X, Zhang Q, Bao R, Zhang N, Wang Y, Polo-Parada L, Tarim A, Alemifar A, Han X, Wilkins HM, et al. Deletion of nampt in projection neurons of adult mice leads to motor dysfunction, neurodegeneration, and death. Cell Rep. 2017;20:2184–200.
Lundt S, Zhang N, Wang X, Polo-Parada L, Ding S. The effect of NAMPT deletion in projection neurons on the function and structure of neuromuscular junction (NMJ) in mice. Sci Rep. 2020;10:99.
Stein LR, Wozniak DF, Dearborn JT, Kubota S, Apte RS, Izumi Y, Zorumski CF, Imai S. Expression of Nampt in hippocampal and cortical excitatory neurons is critical for cognitive function. J Neurosci. 2014;34:5800–15.
Lin JB, Kubota S, Ban N, Yoshida M, Santeford A, Sene A, Nakamura R, Zapata N, Kubota M, Tsubota K, et al. NAMPT-Mediated NAD(+) biosynthesis is essential for vision in mice. Cell Rep. 2016;17:69–85.
Wang X, Zhang Z, Zhang N, Li H, Zhang L, Baines CP, Ding S. Subcellular NAMPT-mediated NAD(+) salvage pathways and their roles in bioenergetics and neuronal protection after ischemic injury. J Neurochem. 2019;151:732–48.
Yoshida M, Satoh A, Lin JB, Mills KF, Sasaki Y, Rensing N, Wong M, Apte RS, Imai SI. Extracellular vesicle-contained eNAMPT delays aging and extends lifespan in mice. Cell Metab. 2019;30(329–342): e325.
Lu YB, Chen CX, Huang J, Tian YX, Xie X, Yang P, Wu M, Tang C, Zhang WP. Nicotinamide phosphoribosyltransferase secreted from microglia via exosome during ischemic injury. J Neurochem. 2019;150:723–37.
Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–41.
Ali YO, Li-Kroeger D, Bellen HJ, Zhai RG, Lu HC. NMNATs, evolutionarily conserved neuronal maintenance factors. Trends Neurosci. 2013;36:632–40.
Yan T, Feng Y, Zheng J, Ge X, Zhang Y, Wu D, Zhao J, Zhai Q. Nmnat2 delays axon degeneration in superior cervical ganglia dependent on its NAD synthesis activity. Neurochem Int. 2010;56:101–6.
Milde S, Gilley J, Coleman MP. Subcellular localization determines the stability and axon protective capacity of axon survival factor Nmnat2. PLoS Biol. 2013;11: e1001539.
Mayer PR, Huang N, Dewey CM, Dries DR, Zhang H, Yu G. Expression, localization, and biochemical characterization of nicotinamide mononucleotide adenylyltransferase 2. J Biol Chem. 2010;285:40387–96.
Hung CO, Coleman MP. KIF1A mediates axonal transport of BACE1 and identification of independently moving cargoes in living SCG neurons. Traffic. 2016;17:1155–67.
Gilley J, Coleman MP. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8: e1000300.
Gilley J, Adalbert R, Yu G, Coleman MP. Rescue of peripheral and CNS axon defects in mice lacking NMNAT2. J Neurosci. 2013;33:13410–24.
Niou Z-X, Yang S, Sri A, Rodriquez HC, Gilley J, Coleman MP, et al. NMNAT2 in cortical glutamatergic neurons exerts both cell and non-cell autonomous influences to shape cortical development and to maintain neuronal health. bioRxiv 2022.02.05.479195. https://www.biorxiv.org/content/10.1101/2022.02.05.479195v1.full.
Yang S, Niou Z-X, Enriquez A, LaMar J, Huang J-Y, Ling K, et al. NAD homeostasis maintained by NMNAT2 supports vesicular glycolysis and fuels fast axonal transport in distal axons of cortical glutamatergic neurons in mice. Biorxiv. 2022.02.06.479307. https://www.biorxiv.org/content/10.1101/2022.02.06.479307v2.
Coleman MP, Hoke A. Programmed axon degeneration: from mouse to mechanism to medicine. Nat Rev Neurosci. 2020;21:183–96.
Russo A, Goel P, Brace EJ, Buser C, Dickman D, DiAntonio A. The E3 ligase highwire promotes synaptic transmission by targeting the NAD-synthesizing enzyme dNmnat. EMBO Rep. 2019;20:e46975.
van Lier M, Smit-Rigter L, Krimpenfort R, Saiepour MH, Ruimschotel E, Kamphuis W, Heimel JA, Levelt CN. NMNAT proteins that limit wallerian degeneration also regulate critical period plasticity in the visual cortex. eNeuro. 2019;6:ENEURO.0277.
Nishiyama A, Komitova M, Suzuki R, Zhu X. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat Rev Neurosci. 2009;10:9–22.
Zhang X, Liu Y, Hong X, Li X, Meshul CK, Moore C, Yang Y, Han Y, Li WG, Qi X, et al. NG2 glia-derived GABA release tunes inhibitory synapses and contributes to stress-induced anxiety. Nat Commun. 2021;12:5740.
Goncalves MB, Wu Y, Clarke E, Grist J, Hobbs C, Trigo D, Jack J, Corcoran JPT. Regulation of myelination by exosome associated retinoic acid release from NG2-positive cells. J Neurosci. 2019;39:3013–27.
Galindo R, Banks Greenberg M, Araki T, Sasaki Y, Mehta N, Milbrandt J, Holtzman DM. NMNAT3 is protective against the effects of neonatal cerebral hypoxia-ischemia. Ann Clin Transl Neurol. 2017;4:722–38.
Bai B, Wang X, Li Y, Chen PC, Yu K, Dey KK, Yarbro JM, Han X, Lutz BM, Rao S, et al. deep multilayer brain proteomics identifies molecular networks in Alzheimer’s disease progression. Neuron. 2020;105(975–991): e977.
Zhao Y, Jin J, Hu Q, Zhou HM, Yi J, Yu Z, Xu L, Wang X, Yang Y, Loscalzo J. Genetically encoded fluorescent sensors for intracellular NADH detection. Cell Metab. 2011;14:555–66.
Lee CF, Caudal A, Abell L, Nagana Gowda GA, Tian R. Targeting NAD(+) metabolism as interventions for mitochondrial disease. Sci Rep. 2019;9:3073.
Zhang L, Zhang S, Maezawa I, Trushin S, Minhas P, Pinto M, Jin LW, Prasain K, Nguyen TDT, Yamazaki Y, et al. Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s disease. EBioMedicine. 2019;42:532.
Urbanska K, Orzechowski A. Unappreciated role of LDHA and LDHB to control apoptosis and autophagy in tumor cells. Int J Mol Sci. 2019;20:2085.
Mc cluskey M. thesis: “Study of vesicular glycolysis in health and Huntington’s Disease” Université Grenoble Alpes [2020]. [GIN] Grenoble Institut des Neurosciences; 2021.
Kimelberg HK. The role of hypotheses in current research, illustrated by hypotheses on the possible role of astrocytes in energy metabolism and cerebral blood flow: from Newton to now. J Cereb Blood Flow Metab. 2004;24:1235–9.
Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A. 1994;91:10625–9.
Gaude E, Schmidt C, Gammage PA, Dugourd A, Blacker T, Chew SP, Saez-Rodriguez J, O’Neill JS, Szabadkai G, Minczuk M, Frezza C. NADH shuttling couples cytosolic reductive carboxylation of glutamine with glycolysis in cells with mitochondrial dysfunction. Mol Cell. 2018;69(581–593): e587.
Even A, Morelli G, Turchetto S, Shilian M, Bail RL, Laguesse S, Krusy N, Brisker A, Brandis A, Inbar S, et al. ATP-citrate lyase promotes axonal transport across species. Nat Commun. 2021;12:5878.
Stein LR, Imai S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab. 2012;23:420–8.
Pittelli M, Formentini L, Faraco G, Lapucci A, Rapizzi E, Cialdai F, Romano G, Moneti G, Moroni F, Chiarugi A. Inhibition of nicotinamide phosphoribosyltransferase: cellular bioenergetics reveals a mitochondrial insensitive NAD pool. J Biol Chem. 2010;285:34106–14.
Cambronne XA, Stewart ML, Kim D, Jones-Brunette AM, Morgan RK, Farrens DL, Cohen MS, Goodman RH. Biosensor reveals multiple sources for mitochondrial NAD(+). Science. 2016;352:1474–7.
Zhu Y, Liu J, Park J, Rai P, Zhai RG. Subcellular compartmentalization of NAD(+) and its role in cancer: a sereNADe of metabolic melodies. Pharmacol Ther. 2019;200:27–41.
Magali V, Lena H, Jörn D, Ingvill T, Lars S, Marc N, Camila C-W, van den Barbara H, Øyvind S, Roland S, et al. Chronic depletion of subcellular NAD pools reveals their interconnectivity and a buffering function of mitochondria. Nature Portfolio; 2022.
Luongo TS, Eller JM, Lu MJ, Niere M, Raith F, Perry C, Bornstein MR, Oliphint P, Wang L, McReynolds MR, et al. SLC25A51 is a mammalian mitochondrial NAD(+) transporter. Nature. 2020;588:174–9.
Kory N, UitBos J, van der Rijt S, Jankovic N, Gura M, Arp N, Pena IA, Prakash G, Chan SH, Kunchok T, et al. MCART1/SLC25A51 is required for mitochondrial NAD transport. Sci Adv. 2020;6:eabe5310.
Girardi E, Agrimi G, Goldmann U, Fiume G, Lindinger S, Sedlyarov V, Srndic I, Gurtl B, Agerer B, Kartnig F, et al. Epistasis-driven identification of SLC25A51 as a regulator of human mitochondrial NAD import. Nat Commun. 2020;11:6145.
Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–107.
Zhang X, Kurnasov OV, Karthikeyan S, Grishin NV, Osterman AL, Zhang H. Structural characterization of a human cytosolic NMN/NaMN adenylyltransferase and implication in human NAD biosynthesis. J Biol Chem. 2003;278:13503–11.
Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13:225–38.
Anderson KA, Madsen AS, Olsen CA, Hirschey MD. Metabolic control by sirtuins and other enzymes that sense NAD(+), NADH, or their ratio. Biochim Biophys Acta Bioenerg. 2017;1858:991–8.
Cohen MS. Interplay between compartmentalized NAD(+) synthesis and consumption: a focus on the PARP family. Genes Dev. 2020;34:254–62.
Verdin E, Hirschey MD, Finley LW, Haigis MC. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci. 2010;35:669–75.
Ji Z, Liu GH, Qu J. Mitochondrial sirtuins, metabolism, and aging. J Genet Genomics. 2021;49:287.
Chamberlain KA, Huang N, Xie Y, LiCausi F, Li S, Li Y, Sheng ZH. Oligodendrocytes enhance axonal energy metabolism by deacetylation of mitochondrial proteins through transcellular delivery of SIRT2. Neuron. 2021;109(3456–3472): e3458.
Kim DS, Challa S, Jones A, Kraus WL. PARPs and ADP-ribosylation in RNA biology: from RNA expression and processing to protein translation and proteostasis. Genes Dev. 2020;34:302–20.
Figley MD, Gu W, Nanson JD, Shi Y, Sasaki Y, Cunnea K, Malde AK, Jia X, Luo Z, Saikot FK, et al. SARM1 is a metabolic sensor activated by an increased NMN/NAD(+) ratio to trigger axon degeneration. Neuron. 2021;109(1118–1136): e1111.
Angeletti C, Amici A, Gilley J, Loreto A, Trapanotto AG, Antoniou C, Merlini E, Coleman MP, Orsomando G. SARM1 is a multi-functional NAD (P) ase with prominent base exchange activity, all regulated bymultiple physiologically relevant NAD metabolites. iScience. 2022;25:103812.
Krauss R, Bosanac T, Devraj R, Engber T, Hughes RO. Axons matter: the promise of treating neurodegenerative disorders by targeting SARM1-mediated axonal degeneration. Trends Pharmacol Sci. 2020;41:281–93.
Sambashivan S, Freeman MR. SARM1 signaling mechanisms in the injured nervous system. Curr Opin Neurobiol. 2021;69:247–55.
Love NR, Pollak N, Dolle C, Niere M, Chen Y, Oliveri P, Amaya E, Patel S, Ziegler M. NAD kinase controls animal NADP biosynthesis and is modulated via evolutionarily divergent calmodulin-dependent mechanisms. Proc Natl Acad Sci U S A. 2015;112:1386–91.
Jiang Y, Liu T, Lee CH, Chang Q, Yang J, Zhang Z. The NAD(+)-mediated self-inhibition mechanism of pro-neurodegenerative SARM1. Nature. 2020;588:658–63.
Desbois M, Crawley O, Evans PR, Baker ST, Masuho I, Yasuda R, Grill B. PAM forms an atypical SCF ubiquitin ligase complex that ubiquitinates and degrades NMNAT2. J Biol Chem. 2018;293:13897–909.
Walker LJ, Summers DW, Sasaki Y, Brace EJ, Milbrandt J, DiAntonio A. MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2. Elife. 2017;6:22540.
Summers DW, Frey E, Walker LJ, Milbrandt J, DiAntonio A. DLK activation synergizes with mitochondrial dysfunction to downregulate axon survival factors and promote SARM1-dependent axon degeneration. Mol Neurobiol. 2020;57:1146–58.
Loreto A, Hill CS, Hewitt VL, Orsomando G, Angeletti C, Gilley J, Lucci C, Sanchez-Martinez A, Whitworth AJ, Conforti L, et al. Mitochondrial impairment activates the Wallerian pathway through depletion of NMNAT2 leading to SARM1-dependent axon degeneration. Neurobiol Dis. 2020;134: 104678.
Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J. SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science. 2015;348:453–7.
Yang J, Wu Z, Renier N, Simon DJ, Uryu K, Park DS, Greer PA, Tournier C, Davis RJ, Tessier-Lavigne M. Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell. 2015;160:161–76.
Agledal L, Niere M, Ziegler M. The phosphate makes a difference: cellular functions of NADP. Redox Rep. 2010;15:2–10.
Dillman AA, Majounie E, Ding J, Gibbs JR, Hernandez D, Arepalli S, Traynor BJ, Singleton AB, Galter D, Cookson MR. Transcriptomic profiling of the human brain reveals that altered synaptic gene expression is associated with chronological aging. Sci Rep. 2017;7:16890.
Tzioras M, McGeachan RI, Durrant CS, Spires-Jones TL. Synaptic degeneration in Alzheimer disease. Nat Rev Neurol. 2022;19:19.
Wishart TM, Parson SH, Gillingwater TH. Synaptic vulnerability in neurodegenerative disease. J Neuropathol Exp Neurol. 2006;65:733–9.
de Wilde MC, Overk CR, Sijben JW, Masliah E. Meta-analysis of synaptic pathology in Alzheimer’s disease reveals selective molecular vesicular machinery vulnerability. Alzheimers Dement. 2016;12:633–44.
Stahon KE, Bastian C, Griffith S, Kidd GJ, Brunet S, Baltan S. Age-related changes in axonal and mitochondrial ultrastructure and function in white matter. J Neurosci. 2016;36:9990–10001.
Morsci NS, Hall DH, Driscoll M, Sheng ZH. Age-related phasic patterns of mitochondrial maintenance in adult caenorhabditis elegans neurons. J Neurosci. 2016;36:1373–85.
Lomidze N, Zhvania MG, Tizabi Y, Japaridze N, Pochkhidze N, Rzayev F, Lordkipanidze T. Aging affects cognition and hippocampal ultrastructure in male Wistar rats. Dev Neurobiol. 2021;81:833–46.
Lores-Arnaiz S, Bustamante J. Age-related alterations in mitochondrial physiological parameters and nitric oxide production in synaptic and non-synaptic brain cortex mitochondria. Neuroscience. 2011;188:117–24.
Lores-Arnaiz S, Lombardi P, Karadayian AG, Orgambide F, Cicerchia D, Bustamante J. Brain cortex mitochondrial bioenergetics in synaptosomes and non-synaptic mitochondria during aging. Neurochem Res. 2016;41:353–63.
Olesen MA, Torres AK, Jara C, Murphy MP, Tapia-Rojas C. Premature synaptic mitochondrial dysfunction in the hippocampus during aging contributes to memory loss. Redox Biol. 2020;34: 101558.
Lomidze N, Zhvania MG, Tizabi Y, Japaridze N, Pochkhidze N, Rzayev F, Gasimov E. Age-related behavioral and ultrastructural changes in the rat amygdala. Dev Neurobiol. 2020;80:433–42.
Yao J, Brinton RD. Estrogen regulation of mitochondrial bioenergetics: implications for prevention of Alzheimer’s disease. Adv Pharmacol. 2012;64:327–71.
Gaignard P, Liere P, Therond P, Schumacher M, Slama A, Guennoun R. Role of sex hormones on brain mitochondrial function, with special reference to aging and neurodegenerative diseases. Front Aging Neurosci. 2017;9:406.
Torrens-Mas M, Pons DG, Sastre-Serra J, Oliver J, Roca P. Sexual hormones regulate the redox status and mitochondrial function in the brain Pathological implications. Redox Biol. 2020;31: 101505.
Stauch KL, Purnell PR, Fox HS. Aging synaptic mitochondria exhibit dynamic proteomic changes while maintaining bioenergetic function. Aging (Albany NY). 2014;6:320–34.
Pickett EK, Rose J, McCrory C, McKenzie CA, King D, Smith C, Gillingwater TH, Henstridge CM, Spires-Jones TL. Region-specific depletion of synaptic mitochondria in the brains of patients with Alzheimer’s disease. Acta Neuropathol. 2018;136:747–57.
Hesse R, Hurtado ML, Jackson RJ, Eaton SL, Herrmann AG, Colom-Cadena M, Tzioras M, King D, Rose J, Tulloch J, et al. Comparative profiling of the synaptic proteome from Alzheimer’s disease patients with focus on the APOE genotype. Acta Neuropathol Commun. 2019;7:214.
Carlyle BC, Kandigian SE, Kreuzer J, Das S, Trombetta BA, Kuo Y, Bennett DA, Schneider JA, Petyuk VA, Kitchen RR, et al. Synaptic proteins associated with cognitive performance and neuropathology in older humans revealed by multiplexed fractionated proteomics. Neurobiol Aging. 2021;105:99–114.
Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A. 2010;107:18670–5.
Espino de la Fuente Munoz C, Rosas-Lemus M, Moreno-Castilla P, Bermudez-Rattoni F, Uribe-Carvajal S, Arias C. Age-dependent decline in synaptic mitochondrial function is exacerbated in vulnerable brain regions of female 3xTg-AD mice. Int J Mol Sci. 2020;21:8727.
Ahmad F, Liu P. Synaptosome as a tool in Alzheimer’s disease research. Brain Res. 2020;1746: 147009.
Seo NY, Kim GH, Noh JE, Shin JW, Lee CH, Lee KJ. Selective regional loss of cortical synapses lacking presynaptic mitochondria in the 5xFAD mouse model. Front Neuroanat. 2021;15: 690168.
Reeve AK, Grady JP, Cosgrave EM, Bennison E, Chen C, Hepplewhite PD, Morris CM. Mitochondrial dysfunction within the synapses of substantia nigra neurons in Parkinson’s disease. NPJ Parkinsons Dis. 2018;4:9.
Plum S, Eggers B, Helling S, Stepath M, Theiss C, Leite REP, Molina M, Grinberg LT, Riederer P, Gerlach M, et al. Proteomic characterization of synaptosomes from human substantia Nigra indicates altered mitochondrial translation in parkinson’s disease. Cells. 2020;9:2580.
Prieto GA, Cotman CW. Early bioenergetic and autophagy impairments at the Parkinson’s disease synapse. Brain. 2022;145:1877–9.
Szego EM, Dominguez-Meijide A, Gerhardt E, Konig A, Koss DJ, Li W, Pinho R, Fahlbusch C, Johnson M, Santos P, et al. Cytosolic trapping of a mitochondrial heat shock protein is an early pathological event in synucleinopathies. Cell Rep. 2019;28(65–77): e66.
Yano H, Baranov SV, Baranova OV, Kim J, Pan Y, Yablonska S, Carlisle DL, Ferrante RJ, Kim AH, Friedlander RM. Inhibition of mitochondrial protein import by mutant huntingtin. Nat Neurosci. 2014;17:822–31.
Polyzos AA, Lee DY, Datta R, Hauser M, Budworth H, Holt A, Mihalik S, Goldschmidt P, Frankel K, Trego K, et al. Metabolic reprogramming in astrocytes distinguishes region-specific neuronal susceptibility in huntington mice. Cell Metab. 2019;29(1258–1273): e1211.
Ravera S, Bonifacino T, Bartolucci M, Milanese M, Gallia E, Provenzano F, Cortese K, Panfoli I, Bonanno G. Characterization of the mitochondrial aerobic metabolism in the pre- and perisynaptic Districts of the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Mol Neurobiol. 2018;55:9220–33.
Ravera S, Torazza C, Bonifacino T, Provenzano F, Rebosio C, Milanese M, Usai C, Panfoli I, Bonanno G. Altered glucose catabolism in the presynaptic and perisynaptic compartments of SOD1(G93A) mouse spinal cord and motor cortex indicates that mitochondria are the site of bioenergetic imbalance in ALS. J Neurochem. 2019;151:336–50.
Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. 2015;18:794–9.
Rozycka A, Liguz-Lecznar M. The space where aging acts: focus on the GABAergic synapse. Aging Cell. 2017;16:634–43.
Lockwood CT, Duffy CJ. Hyperexcitability in aging is lost in Alzheimer’s: what is all the excitement about? Cereb Cortex. 2020;30:5874–84.
Stargardt A, Swaab DF, Bossers K. Storm before the quiet: neuronal hyperactivity and Abeta in the presymptomatic stages of Alzheimer’s disease. Neurobiol Aging. 2015;36:1–11.
Busche MA, Konnerth A. Neuronal hyperactivity–a key defect in Alzheimer’s disease? BioEssays. 2015;37:624–32.
Targa Dias Anastacio H, Matosin N, Ooi L. Neuronal hyperexcitability in Alzheimer’s disease: what are the drivers behind this aberrant phenotype? Transl Psychiatry. 2022;12:257.
Koelewijn L, Lancaster TM, Linden D, Dima DC, Routley BC, Magazzini L, Barawi K, Brindley L, Adams R, Tansey KE, et al. Oscillatory hyperactivity and hyperconnectivity in young APOE-varepsilon4 carriers and hypoconnectivity in Alzheimer’s disease. Elife. 2019;8:e36011.
van Nifterick AM, Gouw AA, van Kesteren RE, Scheltens P, Stam CJ, de Haan W. A multiscale brain network model links Alzheimer’s disease-mediated neuronal hyperactivity to large-scale oscillatory slowing. Alzheimers Res Ther. 2022;14:101.
Dauwels J, Srinivasan K, Ramasubba Reddy M, Musha T, Vialatte FB, Latchoumane C, Jeong J, Cichocki A. Slowing and loss of complexity in Alzheimer’s EEG: two sides of the same coin? Int J Alzheimers Dis. 2011;2011: 539621.
Liu C, Han T, Xu Z, Liu J, Zhang M, Du J, Zhou Q, Duan Y, Li Y, Wang J, et al. Modulating gamma oscillations promotes brain connectivity to improve cognitive impairment. Cereb Cortex. 2022;32:2644–56.
Gruntz K, Bloechliger M, Becker C, Jick SS, Fuhr P, Meier CR, Ruegg S. Parkinson disease and the risk of epileptic seizures. Ann Neurol. 2018;83:363–74.
Bishop MW, Chakraborty S, Matthews GA, Dougalis A, Wood NW, Festenstein R, Ungless MA. Hyperexcitable substantia nigra dopamine neurons in PINK1- and HtrA2/Omi-deficient mice. J Neurophysiol. 2010;104:3009–20.
Cepeda C, Oikonomou KD, Cummings D, Barry J, Yazon VW, Chen DT, Asai J, Williams CK, Vinters HV. Developmental origins of cortical hyperexcitability in Huntington’s disease: review and new observations. J Neurosci Res. 2019;97:1624–35.
Higashihara M, Pavey N, van den Bos M, Menon P, Kiernan MC, Vucic S. Association of cortical hyperexcitability and cognitive impairment in patients with amyotrophic lateral sclerosis. Neurology. 2021;96:e2090–7.
Hernandez DE, Salvadores NA, Moya-Alvarado G, Catalan RJ, Bronfman FC, Court FA. Axonal degeneration induced by glutamate excitotoxicity is mediated by necroptosis. J Cell Sci. 2018;131:jcs214684.
Ko KW, Milbrandt J, DiAntonio A. SARM1 acts downstream of neuroinflammatory and necroptotic signaling to induce axon degeneration. J Cell Biol. 2020;219:e201912047.
Zhu XH, Lu M, Lee BY, Ugurbil K, Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci U S A. 2015;112:2876–81.
Braidy N, Poljak A, Grant R, Jayasena T, Mansour H, Chan-Ling T, Guillemin GJ, Smythe G, Sachdev P. Mapping NAD(+) metabolism in the brain of ageing Wistar rats: potential targets for influencing brain senescence. Biogerontology. 2014;15:177–98.
Cheng A, Yang Y, Zhou Y, Maharana C, Lu D, Peng W, Liu Y, Wan R, Marosi K, Misiak M, et al. Mitochondrial SIRT3 mediates adaptive responses of neurons to exercise and metabolic and excitatory challenges. Cell Metab. 2016;23:128–42.
Cheng A, Wang J, Ghena N, Zhao Q, Perone I, King TM, Veech RL, Gorospe M, Wan R, Mattson MP. SIRT3 haploinsufficiency aggravates loss of GABAergic interneurons and neuronal network hyperexcitability in an Alzheimer’s disease model. J Neurosci. 2020;40:694–709.
Noori A, Mezlini AM, Hyman BT, Serrano-Pozo A, Das S. Systematic review and meta-analysis of human transcriptomics reveals neuroinflammation, deficient energy metabolism, and proteostasis failure across neurodegeneration. Neurobiol Dis. 2021;149: 105225.
Bennett JP, Keeney PM. RNA-sequencing reveals similarities and differences in gene expression in vulnerable brain tissues of Alzheimer’s and parkinson’s diseases. J Alzheimers Dis Rep. 2018;2:129–37.
Patel H, Dobson RJB, Newhouse SJ. A meta-analysis of Alzheimer’s disease brain transcriptomic data. J Alzheimers Dis. 2019;68:1635–56.
Ali YO, Allen HM, Yu L, Li-Kroeger D, Bakhshizadehmahmoudi D, Hatcher A, McCabe C, Xu J, Bjorklund N, Taglialatela G, et al. NMNAT2:HSP90 complex mediates proteostasis in proteinopathies. PLoS Biol. 2016;14: e1002472.
Wang Y. Identifying neuron subtype-specific metabolic network changes in single cell transcriptomics of Alzheimer’s Disease using perturb-Met. BioRxiv. 2021.01.18.427154. https://www.biorxiv.org/content/10.1101/2021.01.18.427154v1.full.
Ghosh D, LeVault KR, Barnett AJ, Brewer GJ. A reversible early oxidized redox state that precedes macromolecular ROS damage in aging nontransgenic and 3xTg-AD mouse neurons. J Neurosci. 2012;32:5821–32.
van der Velpen V, Rosenberg N, Maillard V, Teav T, Chatton JY, Gallart-Ayala H, Ivanisevic J. Sex-specific alterations in NAD+ metabolism in 3xTg Alzheimer’s disease mouse brain assessed by quantitative targeted LC-MS. J Neurochem. 2021;159:378–88.
Dong Y, Digman MA, Brewer GJ. Age- and AD-related redox state of NADH in subcellular compartments by fluorescence lifetime imaging microscopy. Geroscience. 2019;41:51–67.
Hammond TC, Xing X, Yanckello LM, Stromberg A, Chang YH, Nelson PT, Lin AL. Human gray and white matter metabolomics to differentiate APOE and stage dependent changes in Alzheimer’s disease. J Cell Immunol. 2021;3:397–412.
Hikosaka K, Yaku K, Okabe K, Nakagawa T. Implications of NAD metabolism in pathophysiology and therapeutics for neurodegenerative diseases. Nutr Neurosci. 2021;24:371–83.
Wang X, He HJ, Xiong X, Zhou S, Wang WW, Feng L, Han R, Xie CL. NAD+ in Alzheimer’s disease: molecular mechanisms and systematic therapeutic evidence obtained in vivo. Front Cell Dev Biol. 2021;9: 668491.
Cheng XS, Shi FX, Zhao KP, Lin W, Li XY, Zhang J, Bu YY, Zhu R, Li XH, Duan DX, et al. Nmnat2 attenuates amyloidogenesis and up-regulates ADAM10 in AMPK activity-dependent manner. Aging (Albany NY). 2021;13:23620–36.
Conforti L, Gilley J, Coleman MP. Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat Rev Neurosci. 2014;15:394–409.
Henninger N, Bouley J, Sikoglu EM, An J, Moore CM, King JA, Bowser R, Freeman MR, Brown RH Jr. Attenuated traumatic axonal injury and improved functional outcome after traumatic brain injury in mice lacking Sarm1. Brain. 2016;139:1094–105.
Marion CM, McDaniel DP, Armstrong RC. Sarm1 deletion reduces axon damage, demyelination, and white matter atrophy after experimental traumatic brain injury. Exp Neurol. 2019;321: 113040.
Bradshaw DV Jr, Knutsen AK, Korotcov A, Sullivan GM, Radomski KL, Dardzinski BJ, Zi X, McDaniel DP, Armstrong RC. Genetic inactivation of SARM1 axon degeneration pathway improves outcome trajectory after experimental traumatic brain injury based on pathological, radiological, and functional measures. Acta Neuropathol Commun. 2021;9:89.
Ozaki E, Gibbons L, Neto NG, Kenna P, Carty M, Humphries M, Humphries P, Campbell M, Monaghan M, Bowie A, Doyle SL. SARM1 deficiency promotes rod and cone photoreceptor cell survival in a model of retinal degeneration. Life Sci Alliance. 2020;3:e201900618.
Finnegan LK, Chadderton N, Kenna PF, Palfi A, Carty M, Bowie AG, Millington-Ward S, Farrar GJ. SARM1 ablation is protective and preserves spatial vision in an in vivo mouse model of retinal ganglion cell degeneration. Int J Mol Sci. 2022;23:1606.
Peters OM, Weiss A, Metterville J, Song L, Logan R, Smith GA, Schwarzschild MA, Mueller C, Brown RH, Freeman M. Genetic diversity of axon degenerative mechanisms in models of Parkinson’s disease. Neurobiol Dis. 2021;155: 105368.
Peters OM, Lewis EA, Osterloh JM, Weiss A, Salameh JS, Metterville J, Brown RH, Freeman MR. Loss of Sarm1 does not suppress motor neuron degeneration in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Hum Mol Genet. 2018;27:3761–71.
Collins JM, Atkinson RAK, Matthews LM, Murray IC, Perry SE, King AE. Sarm1 knockout modifies biomarkers of neurodegeneration and spinal cord circuitry but not disease progression in the mSOD1(G93A) mouse model of ALS. Neurobiol Dis. 2022;172: 105821.
Fu H, Hardy J, Duff KE. Selective vulnerability in neurodegenerative diseases. Nat Neurosci. 2018;21:1350–8.
Markus NM, Hasel P, Qiu J, Bell KF, Heron S, Kind PC, Dando O, Simpson TI, Hardingham GE. Expression of mRNA encoding Mcu and other mitochondrial calcium regulatory genes depends on cell type, neuronal subtype, and Ca2+ signaling. PLoS ONE. 2016;11: e0148164.
Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, Freeman TC, Summers KM, McColl BW. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci. 2016;19:504–16.
Soreq L, Consortium UKBE, North American Brain Expression C, Rose J, Soreq E, Hardy J, Trabzuni D, Cookson MR, Smith C, Ryten M, et al. Major shifts in glial regional identity are a transcriptional hallmark of human brain aging. Cell Rep. 2017;18:557–70.
Boisvert MM, Erikson GA, Shokhirev MN, Allen NJ. The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. 2018;22:269–85.
Clarke LE, Liddelow SA, Chakraborty C, Munch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U S A. 2018;115:E1896–905.
Nasrabady SE, Rizvi B, Goldman JE, Brickman AM. White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol Commun. 2018;6:22.
Mot AI, Depp C, Nave KA. An emerging role of dysfunctional axon-oligodendrocyte coupling in neurodegenerative diseases. Dialogues Clin Neurosci. 2018;20:283–92.
Butt AM, De La Rocha IC, Rivera A. Oligodendroglial cells in Alzheimer’s disease. Adv Exp Med Biol. 2019;1175:325–33.
Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, Rothstein JD, Bergles DE. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013;16:571–9.
Lau SF, Cao H, Fu AKY, Ip NY. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2020;117:25800–9.
Marques S, Zeisel A, Codeluppi S, van Bruggen D, Mendanha Falcao A, Xiao L, Li H, Haring M, Hochgerner H, Romanov RA, et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science. 2016;352:1326–9.
Sadick JS, O’Dea MR, Hasel P, Dykstra T, Faustin A, Liddelow SA. Astrocytes and oligodendrocytes undergo subtype-specific transcriptional changes in Alzheimer’s disease. Neuron. 2022;110(1788–1805): e1710.
Xu J, Chen S, Ahmed SH, Chen H, Ku G, Goldberg MP, Hsu CY. Amyloid-beta peptides are cytotoxic to oligodendrocytes. J Neurosci. 2001;21:RC118.
Uemura N, Uemura MT, Lo A, Bassil F, Zhang B, Luk KC, Lee VM, Takahashi R, Trojanowski JQ. Slow progressive accumulation of oligodendroglial Alpha-Synuclein (alpha-Syn) pathology in synthetic alpha-syn fibril-induced mouse models of synucleinopathy. J Neuropathol Exp Neurol. 2019;78:877–90.
Azevedo C, Teku G, Pomeshchik Y, Reyes JF, Chumarina M, Russ K, Savchenko E, Hammarberg A, Lamas NJ, Collin A, et al. Parkinson’s disease and multiple system atrophy patient iPSC-derived oligodendrocytes exhibit alpha-synuclein-induced changes in maturation and immune reactive properties. Proc Natl Acad Sci U S A. 2022;119: e2111405119.
Smajic S, Prada-Medina CA, Landoulsi Z, Ghelfi J, Delcambre S, Dietrich C, Jarazo J, Henck J, Balachandran S, Pachchek S, et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain. 2022;145:964–78.
Jeffries MA, McLane LE, Khandker L, Mather ML, Evangelou AV, Kantak D, Bourne JN, Macklin WB, Wood TL. mTOR signaling regulates metabolic function in oligodendrocyte precursor cells and promotes efficient brain remyelination in the cuprizone model. J Neurosci. 2021;41:8321–37.
Backes H, Walberer M, Ladwig A, Rueger MA, Neumaier B, Endepols H, Hoehn M, Fink GR, Schroeter M, Graf R. Glucose consumption of inflammatory cells masks metabolic deficits in the brain. Neuroimage. 2016;128:54–62.
Chen Z, Yuan Z, Yang S, Zhu Y, Xue M, Zhang J, Leng L. Brain energy metabolism: astrocytes in neurodegenerative diseases. CNS Neurosci Ther. 2022;29:24.
Andersen JV, Schousboe A, Verkhratsky A. Astrocyte energy and neurotransmitter metabolism in Alzheimer’s disease: Integration of the glutamate/GABA-glutamine cycle. Prog Neurobiol. 2022;217: 102331.
Mulica P, Grunewald A, Pereira SL. Astrocyte-neuron metabolic crosstalk in neurodegeneration: a mitochondrial perspective. Front Endocrinol (Lausanne). 2021;12: 668517.
Bantle CM, Hirst WD, Weihofen A, Shlevkov E. Mitochondrial dysfunction in astrocytes: a role in parkinson’s disease? Front Cell Dev Biol. 2020;8: 608026.
Allen SP, Hall B, Woof R, Francis L, Gatto N, Shaw AC, Myszczynska M, Hemingway J, Coldicott I, Willcock A, et al. C9orf72 expansion within astrocytes reduces metabolic flexibility in amyotrophic lateral sclerosis. Brain. 2019;142:3771–90.
Khakh BS, Beaumont V, Cachope R, Munoz-Sanjuan I, Goldman SA, Grantyn R. Unravelling and exploiting astrocyte dysfunction in Huntington’s disease. Trends Neurosci. 2017;40:422–37.
Oberheim Bush NA, Nedergaard M. Do evolutionary changes in astrocytes contribute to the computational power of the hominid brain? Neurochem Res. 2017;42:2577–87.
Li J, Pan L, Pembroke WG, Rexach JE, Godoy MI, Condro MC, Alvarado AG, Harteni M, Chen YW, Stiles L, et al. Conservation and divergence of vulnerability and responses to stressors between human and mouse astrocytes. Nat Commun. 2021;12:3958.
Johnson ECB, Dammer EB, Duong DM, Ping L, Zhou M, Yin L, Higginbotham LA, Guajardo A, White B, Troncoso JC, et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med. 2020;26:769–80.
Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med. 2020;26:131–42.
Zhu B, Park J-M, Coffey S, Hsu I-U, Lam TT, Gopal PP, et al. Single-cell transcriptomic and proteomic analysis of Parkinson’s disease Brains. BioRxiv. 2022.02.14.480397. https://www.biorxiv.org/content/10.1101/2022.02.14.480397v1.
Acknowledgements
We are grateful to Drs. Ken Mackie, Gergo Szanda and Miss. Andrea Enriquez for their helpful discussions and comments.
Funding
SY was supported by Linda and Jack Gill Fellowship from Indiana Univ. Bloomington. HCL was funded by National Institutes of Health (NIH) NINDS R01NS086794.
Author information
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, S., Park, J.H. & Lu, HC. Axonal energy metabolism, and the effects in aging and neurodegenerative diseases. Mol Neurodegeneration 18, 49 (2023). https://doi.org/10.1186/s13024-023-00634-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-023-00634-3