
Abstract
Background
Neurodegenerative disorders are a group of age-associated diseases characterized by progressive degeneration of the structure and function of the CNS. Two key pathological features of these disorders are blood-brain barrier (BBB) breakdown and protein aggregation.
Main body
The BBB is composed of various cell types and a non-cellular component---the basal lamina (BL). Although how different cells affect the BBB is well studied, the roles of the BL in BBB maintenance and function remain largely unknown. In addition, located in the perivascular space, the BL is also speculated to regulate protein clearance via the meningeal lymphatic/glymphatic system. Recent studies from our laboratory and others have shown that the BL actively regulates BBB integrity and meningeal lymphatic/glymphatic function in both physiological and pathological conditions, suggesting that it may play an important role in the pathogenesis and/or progression of neurodegenerative disorders. In this review, we focus on changes of the BL and its major components during aging and in neurodegenerative disorders, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS). First, we introduce the vascular and lymphatic systems in the CNS. Next, we discuss the BL and its major components under homeostatic conditions, and summarize their changes during aging and in AD, PD, and ALS in both rodents and humans. The functional significance of these alterations and potential therapeutic targets are also reviewed. Finally, key challenges in the field and future directions are discussed.
Conclusions
Understanding BL changes and the functional significance of these changes in neurodegenerative disorders will fill the gap of knowledge in the field. Our goal is to provide a clear and concise review of the complex relationship between the BL and neurodegenerative disorders to stimulate new hypotheses and further research in this field.
Similar content being viewed by others

Background
Neurodegenerative disorders, a group of age-associated diseases, are characterized by progressive neuronal dysfunction and cognitive decline. Three major types of neurological disorders are Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS). Neurodegenerative disorders in total affect more than 7 million people in the US [1]. This number is expected to increase dramatically over the next few decades with our aging population. Unfortunately, there are no effective treatments or disease-modifying therapeutics for these devastating diseases.
One key pathology of neurodegenerative disorders is blood-brain barrier (BBB) breakdown, which has been observed in almost all neurodegenerative disorders in both rodents and humans [2,3,4]. BBB breakdown in AD has been confirmed in multiple independent postmortem human studies [5]. Recent neuroimaging studies have detected BBB breakdown in individuals with mild cognitive impairment (MCI) and early AD, before cognitive decline and/or other brain pathologies [6,7,8,9,10,11,12,13,14,15]. MRI studies also show increased cerebral microbleeds (reflecting loss of cerebrovascular integrity) in 25% of individuals with MCI and 45–78% of individuals with early AD before dementia [16,17,18,19,20,21]. These findings indicate that BBB disruption is not only a consequence but also a cause in AD [4, 7, 9, 22,23,24].
Vascular cognitive impairment (VCI), cognitive deficits associated with cerebrovascular disease, is another common type of dementia [25]. Like in AD, BBB disruption correlates well with VCI. For instance, serum-derived proteins are detected in brain tissue from VCI patients [26, 27]. Increased albumin and laminin levels are detected in the CSF from VCI patients [27,28,29]. MRI studies also demonstrate enhanced BBB leakage in VCI patients [30,31,32,33]. These results suggest an essential role of BBB breakdown in the pathogenesis of VCI.
BBB disruption has also been reported in animal models of PD [34,35,36]. A postmortem study revealed increased BBB permeability in the post commissural putamen of PD patients [37]. A positron emission tomography (PET) study showed dysfunction of the BBB transporter system in PD patients [38], and a dynamic contrast enhanced MRI study demonstrated enhanced BBB leakage in PD patients [39]. These results highlight a critical role of BBB integrity in PD.
Similarly, blood-CNS barrier (BCNSB) disruption has also been reported in ALS. For example, blood-spinal cord barrier (BSCB) breakdown was detected in rodent models of ALS prior to motor neuron degeneration and neuroinflammation and worsens with disease progression [40,41,42,43,44,45], although one human study reported that BSCB leakage was independent of motor neuron pathology in ALS [46]. Postmortem studies revealed structural and functional impairment of the BSCB in gray and white matter microvessels of medulla and spinal cord tissue from ALS patients [45, 47, 48]. Recent neuroimaging studies have demonstrated early BSCB dysfunction in ALS patients [49,50,51]. These findings suggest that barrier disruption contributes to ALS pathogenesis.
It should be noted that, although BBB breakdown is detected in other neurodegenerative diseases such as frontotemporal dementia (FTD) and dementia with Lewy bodies (DLB) [52, 53], little data are available on the role of BBB breakdown in the pathogenesis of these disorders [54, 55].
Together, these findings highlight the important role of BBB in neurodegenerative disorders. Most BBB research focuses on its cellular components, including brain microvascular endothelial cells (BMECs), pericytes, and astrocytes [4, 56]. For example, pericytes have been shown to play a key role in BBB maintenance [57,58,59] and loss of pericytes contributes to BBB disruption and pathogenesis of various neurodegenerative diseases [50, 60,61,62]. Its non-cellular component---the basal lamina (BL), on the other hand, is largely understudied. Recent studies from our laboratory and others show that the BL actively regulates BBB integrity and abnormal BL is observed in various neurological disorders [63,64,65].
Another common pathology of neurodegenerative disorders is protein aggregation. For example, accumulation of β-amyloid (Aβ)/tau, alpha-synuclein, and TDP-43/SOD-1 is a hallmark of AD, PD, and ALS, respectively [66,67,68,69,70]. Although how exactly these aggregates form is not fully understood, reduced clearance is an important mechanism. Recent evidence suggests that receptor-mediated transport across the BBB and the meningeal lymphatic/glymphatic system are two major mechanisms of solute/waste clearance in the brain, and that both mechanisms are affected in aging and neurodegenerative diseases [71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87]. Since the BL is a key constituent of the BBB and meningeal lymphatic/glymphatic system, we hypothesize that BL defects contribute to the development of neurodegenerative disorders via compromising BBB integrity and/or affecting protein aggregate clearance.
Main body
CNS blood vessels
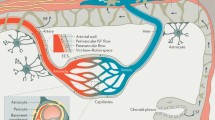
In the CNS, blood vessels exist in both the meninges (dura matter and leptomeninges) and brain/spinal cord parenchyma. Meningeal blood vessels exhibit unique features depending on the location. Specifically, dural blood vessels are fenestrated and lack tight junctions (Fig. 1a), enabling the entry of large molecules and immune cells from the blood into the dura matter [88,89,90]. Echoed with this finding, a large repertoire of immune cells, including T cells, B cells, dendritic cells, macrophages, and mast cells, are found in the dura matter in steady-state conditions [89, 91,92,93,94,95,96,97,98,99]. Leptomeningeal blood vessels are non-fenestrated and have tight junctions (Fig. 1a). Although lacking astrocytic ensheathment, these leptomeningeal vessels are impermeable to large molecules [88, 89, 100]. Similarly, immune cells also exist in the leptomeninges, but to a lesser extent compared to dura matter [89, 91,92,93,94,95,96,97,98,99]. In addition, leptomeningeal vessels are covered by stomata-containing adventitia lining cells (fibroblast-like cells) (Fig. 1a), which may be involved in cerebral spinal fluid (CSF)/interstitial fluid (ISF) exchange [101]. It remains unclear how exactly meningeal blood vessels change during aging and in neurodegenerative disorders. This is an interesting field for future research.

Diagram illustration of the vasculature and BL layers in the CNS. a Scheme of the CNS blood vessels and meningeal lymphatic/glymphatic system. The inset highlights the cross-section view of a meningeal artery. b Scheme of various BL layers and cellular components at the arterial/arteriole, capillary, and post-capillary venule levels in the CNS vasculature. BL: basal lamina; eBL: endothelial basal lamina; EC: endothelial cell; pBL: parenchymal basal lamina; PVM: perivascular macrophage; SMC: smooth muscle cell
Large arteries and veins in the leptomeninges penetrate brain/spinal cord parenchyma and branch into arterioles and venules, which are connected via capillaries (Fig. 1a). The composition of vascular wall changes along the arterial-venous axis, although endothelial cells and astrocytic endfeet are found in all sections of the vasculature. Specifically, smooth muscle cells, perivascular macrophages, and multiple (endothelial, smooth muscle cell [SMC], and parenchymal) BL layers are found in arteries/arterioles (Fig. 1b). Pericytes and tightly packed endothelial and parenchymal BLs exist in capillaries (Fig. 1b). Similar structure is observed in post-capillary venules, with endothelial and parenchymal BLs separated by a perivascular space (Fig. 1b). Accumulating evidence suggests that the structure and function of parenchymal blood vessels are altered during aging and in neurodegenerative disorders. For example, pericyte loss and abnormal BL are observed in AD brains in both rodents and humans [9, 59, 60, 102,103,104,105,106].
CNS lymphatic system
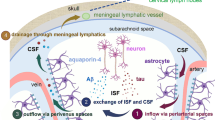
In the brain, lymphatic vessels are located in dural meninges along sinuses (Fig. 1a) [88, 107,108,109,110]. The presence of “leaky” blood vessels and lymphatic vessels enables immune cells to migrate to and from the dura matter, making the meninges an immunologically active barrier tissue [89]. It has been suggested that meningeal lymphatics function to sample and clear CSF/ISF into the cervical lymph nodes [107,108,109,110,111,112,113,114,115].
Unlike the meninges, CNS parenchyma lacks lymphatic vessels. How is waste removed from the CNS? Using mice with intra-cisterna magna injection of fluorescent tracers, Iliff and colleagues proposed a para-vascular route, known as the glymphatic system [71, 116]. The glymphatic system posits that CSF in the subarachnoid space circulates into brain parenchyma along para-arterial spaces via connective flow. It crosses the glia limitans and enters the brain extracellular spaces in an aquaporin-4 (AQP4)-dependent manner. The CSF then mixes with ISF and moves toward the para-venous spaces. Metabolic waste eventually exits the CNS via deep veins, meningeal/cervical lymphatic vessels, and perineural sheaths of cranial/spinal nerves. Consistent with this hypothesis, reduced CSF influx was observed in four different AQP4-/- mouse lines and Snta1-/- mice, which lack perivascular AQP4 [71, 117]. In addition, the clearance of SOD1 oligomer was significantly delayed in AQP4-/- mice compared to the controls [118].
It should be noted, however, that there is also evidence that does not support the proposed glymphatic mechanism or convective pressure-driven CSF flow from para-arterial to para-venous spaces through parenchymal extracellular spaces. For example, in contrast to previous reports [71, 117], one study showed that loss of AQP4 failed to affect solute transport from the subarachnoid space to brain parenchyma in rodents [119]. In addition, recent animal and modeling studies have concluded that fluid flow in brain extracellular spaces is predominantly diffusive rather than convective in nature [119,120,121,122,123,124,125]. Considering all these findings, an updated glymphatic system with convective flow along perivascular spaces of large vessels and diffusion in brain extracellular spaces has been proposed (Fig. 1a) [73, 101, 117, 126, 127]. Given that the BL is a major constituent of the perivascular space [102, 128,129,130] and many BL components regulate AQP4 expression [131,132,133,134], it is speculated that the BL may regulate protein clearance via meningeal lymphatic/glymphatic system in neurodegenerative disorders.
Recent studies have shown that meningeal immunity (immune cells in the dura matter) is affected during aging and under neurodegenerative conditions [91,92,93,94,95]. The exact role of each immune population, however, remains largely unknown and needs future research. In addition, the CSF/ISF drainage function of the meningeal lymphatic/glymphatic system is substantially impaired in aging. It has been reported that aged mice exhibit decreased para-vascular recirculation of CSF, diminished CSF/ISF exchange, and loss of perivascular AQP4 polarization [76, 107]. Echoed with this report, reduced meningeal lymphatic function is associated with compromised macromolecule drainage and cognitive decline in old mice [107, 114, 135]. Interestingly, VEGF-C, a lymphatic endothelial cell mitogen, is able to increase meningeal lymphatic drainage and improve cognitive function in old mice [135]. Based on these results, it is hypothesized that impaired meningeal lymphatic drainage attenuates the clearance of abnormal protein aggregates and thus aggravates disease pathology/outcome. Consistent with the hypothesis, disruption of meningeal lymphatic vessels in young 5xFAD mice promotes Aβ deposition in the meninges and exacerbates parenchymal Aβ burden [135]. Similarly, significantly reduced meningeal lymphatic flow and delayed deep cervical lymph node perfusion were found in PD patients [136]. Ablation of meningeal lymphatic drainage resulted in meningeal inflammation, enhanced PD pathology, and exacerbated motor/memory deficits in mice injected with α-synuclein preformed fibrils [136]. Echoed with this finding, impaired meningeal drainage caused α-synuclein accumulation, glial activation, increased inflammatory cytokines, and neuronal loss in α-synucleinA53T PD mice [137]. In addition, a 35% decrease in the clearance of intra-cisterna magna-injected ovalbumin was observed in SOD1G93A mouse model of ALS [118]. Together, these results suggest an essential role of the CNS lymphatic system in the pathogenesis of neurodegenerative disorders.
Basal lamina
The BL is a complex amorphous structure with a thickness of 50-100nm. It lays beneath epithelial and endothelial cells, and is a major constituent of the BBB and CNS lymphatic system [128, 138,139,140,141]. In the CNS, multiple BL layers, including endothelial BL, SMC BL, and parenchymal BL, are found between BMECs and astrocytic endfeet [142,143,144] (Fig. 1b). Biochemically, the BL contains four major extracellular matrix (ECM) proteins: collagen IV, laminin, nidogen, and heparan sulfate proteoglycans (HSPGs). Minor BL constituents include fibulins, osteonectin, and netrin-4 [145]. These components are predominantly synthesized by BMECs, pericytes, and astrocytes in the brain. It should be noted that the BL is also found in peripheral blood vessels. Although peripheral BL contains all four major ECM proteins, it lacks astrocyte-derived ECM proteins compared to CNS BL. In this section, we discuss the structure (Fig. 2) and function (Table 1) of major components of the BL in physiological conditions.

Structural illustration of major BL components collagen and laminin. a Collagen IV monomer, trimer, and its suprastructure. 7S, 7S domain; NC1, NC1 domain. b Laminin monomer and its suprastructure. α, β, and γ: laminin-α, -β, and -γ chains, respectively
Collagen IV
Collagen IV is the most abundant component in the BL and is mainly produced by endothelial cells, astrocytes, and pericytes [179,180,181,182,183]. Structurally, collagen IV is a trimeric protein containing three α-chains (Fig. 2a). Currently, six different collagen IV α-chains have been identified (COL4A1-6) [184,185,186]. By dimerization at its NC1 domain and tetramerization at its 7S domain, collagen IV forms a sheet-like suprastructure [187, 188] (Fig. 2a).
Loss-of-function studies reveal critical roles of collagen IV in embryonic development and vascular integrity. Genetic knockout of COL4A1/2 results in embryonic lethality at E10.5 - E11.5 due to BL structural deficiencies, although BL formation at earlier stages is unaffected [64]. Similarly, loss of COL4A1 exon 41 in both alleles causes embryonic lethality and intracerebral hemorrhage (ICH) [146], while its deficiency in one allele leads to perinatal lethality with ICH (~50%) and porencephaly in some survivors [149, 150]. Furthermore, the function of COL4A1 has also been investigated in a cell-specific manner. Deletion of COL4A1 in BMECs or pericytes results in ICH, increased retinal vascular branching, porencephaly, and macroangiopathy [151]. Ablation of COL4A1 in astrocytes, however, causes very mild ICH without defects in retinal vascular branching [151]. Consistent with these findings, missense mutations in COL4A1 and COL4A2 lead to vascular defects and/or brain damage to different degrees [146,147,148]. Collectively, these studies suggest that collagen IV is nonessential for initial BL assembly but plays a key role in BL maintenance and vascular integrity [56].
Laminin
Laminin is a trimeric protein composed of α, β, and γ chains [63, 106, 128] (Fig. 2b). These subunits bind to one another via their C-terminal domains, forming a T- or cross-shaped molecule composed of one long arm and two or three short arms (Fig. 2b). Like collagen IV, laminin is able to form a sheet-like suprastructure by intermolecular self-polymerization via their N-terminal domains [189, 190] (Fig. 2b). Currently, 5 α, 4 β, and 3 γ chains have been identified [128]. Various combinations of these subunit variants create different laminin isoforms. However, it should be noted that not all combinations have been confirmed. Among the 60 theoretical isoforms, only 16 have been identified and 4 proposed based on in vitro and in vivo studies so far [128]. Since distinct laminin isoforms may have different functions, it is important to study laminin’s function in an isoform-specific manner.
In the brain, laminin is predominantly synthesized by BMECs, pericytes, and astrocytes [56]. Interestingly, these cells synthesize and deposit different laminin isoforms to the BL. Specifically, BMECs mainly generate laminin-α4β1γ1 (-411) and laminin-511 [142, 191], and astrocytes predominantly produce laminin-211 [142, 192]. Immunocytochemical analysis from our laboratory shows that brain pericytes primarily generate α4/α5- and γ1-containing laminins [134, 181], although laminin-α2 is also detected in pericytes by single-cell RNAseq analysis [193]. Therefore, laminin-411 and -511 (derived from BMECs) are predominantly found in the endothelial BL, while laminin-211 (derived from astrocytes) is mainly found in the parenchymal BL.
Many loss-of-function studies have been performed to investigate laminin’s function in BBB integrity. Such studies heavily rely on conditional knockout mice, since most global knockouts (e.g. laminin-α5-/-, -β1-/-, and -γ1-/-) are embryonic lethal [154,155,156, 160,161,162], hindering further investigation into their roles in BBB integrity.
Global knockout of laminin-α4 leads to disrupted vascular integrity and hemorrhage at perinatal stage, but not in adulthood [153]. The lack of phenotype in adulthood is believed to be compensated by laminin-511, which is expressed in blood vessels at postnatal stage [153, 194]. To investigate the functional significance of laminin-α5, we and others generated endothelial (Tie2-Cre) laminin-α5 conditional knockout mice. Interestingly, these mutants fail to show a phenotype under homeostatic conditions [157,158,159], suggesting that laminin-411 may be able to compensate for the loss of endothelial laminin-511. To overcome this mutual compensation between laminin-411 and -511 and investigate the role of endothelial laminin, we further generated mice lacking both laminin-α4 and -α5 in endothelial cells. The resulting mutants display moderate BBB breakdown, characterized by enhanced leakage of intravenously injected tracers into brain parenchyma. These findings strongly suggest that endothelial laminins maintain BBB integrity.
To investigate the function of astrocytic laminin (laminin-211) in BBB integrity, we generated conditional knockout mice with laminin-γ1 deficiency in neural progenitor cells (Nestin-Cre) and in neurons (CamK2a-Cre), respectively [132]. The former but not the latter show severe BBB disruption and spontaneous ICH [132, 163], highlighting an important role of glial cell-derived laminin in BBB regulation. Using adenovirus expressing Cre under GFAP promoter, we further validated that loss of astrocytic laminin leads to BBB breakdown [132]. Consistent with our result, mice with global knockout of laminin-α2 demonstrate BBB disruption [132, 152]. Together, these findings suggest an indispensable role of astrocytic laminin in BBB maintenance.
Due to the lack of pericyte-specific markers [62], the function of pericytic laminin in BBB integrity was investigated indirectly using two conditional knockout mouse lines. By using the PDGFRβ-Cre line, we generated mice with laminin deficiency in mural cells, which include both pericytes and SMCs [134]. By using the SM22α-Cre line, we generated mice with laminin deficiency in SMCs only [134, 164]. Although the SMC-specific laminin knockout mice fail to show gross abnormalities [134], the mural cell-specific laminin knockout mice display genetic background-dependent phenotypes. In C57/Bl6-FVB mixed background, the mutants exhibit BBB breakdown and hydrocephalus with incomplete penetrance [134]. In C57Bl6 dominant background, these mutants are grossly normal at young age, but develop mild BBB compromise at old age [133]. Together, these findings suggest that pericyte- rather than SMC-derived laminin also contributes to BBB maintenance, but to a lesser extent compared to astrocytic or endothelial laminin.
Nidogen
Nidogen, also called entactin, is a glycoprotein containing three globular and multiple rod-like domains [195]. Unlike collagen IV and laminin, nidogen is unable to self-polymerize or form a sheet-like suprastructure. Instead, it interacts with both collagen IV and laminin as a crosslinker. It has been suggested that nidogen may play a role in stabilizing collagen IV and laminin networks.
Two nidogen isoforms (nidogen-1/2) have been identified in mammals [165]. Global knockout of either isoform results in a generally normal phenotype [165,166,167,168, 170], indicating possibly mutual compensation. Interestingly, there is a redistribution and upregulation of nidogen-2 in nidogen-1 knockout mice, although nidogen-1 expression is unaffected in nidogen-2 knockout mice [169, 170]. Consistent with the mutual compensation hypothesis, loss of both isoforms simultaneously leads to early perinatal death and severe BL defects [65, 171, 172]. These results suggest that these two isoforms share a compensatory mechanism, and nidogen function in BBB integrity remains unknown due to early perinatal lethality. Nidogen-1/2 double conditional knockout mice may overcome embryonic lethality and enable investigation of its role in BBB maintenance. Future research should focus on generating this mutant line.
HSPG
HSPGs are glycoproteins containing covalently attached heparan sulfate (HS) chains. They are present on cell surface of all tissues and in the BL. Their functions vary depending on the types of HSPGs. Two major HSPGs found in the BL are agrin and perlecan.
Agrin
Agrin is a multidomain HSPG [196,197,198], which can interact with laminin [199]. There are two isoforms of agrin (z+ and z-), but only the z- isoform is present in the BL [196, 200, 201]. It has been shown that agrin accumulates in the BL during BBB development in a correlation study [202], suggesting a possible role of agrin in early BBB development. Global knockout of agrin results in embryonic lethality [173], preventing investigation of its function at later stages. Interestingly, mice with agrin deletion in endothelial cells (Tie2-Cre) display intact BBB structure, although AQP4 expression is reduced [131], suggesting that agrin is dispensable for the structural but not biochemical maintenance of the BBB. The exact function of agrin in BBB integrity needs further investigation.
Perlecan
Perlecan, also called HSPG2, is a large HSPG with five domains and three glycosaminoglycan chains [128, 203]. Like nidogen, perlecan is unable to form a sheet-like suprastructure [203], but can interact with other BL components and/or heparin-binding growth factors [203,204,205,206]. Perlecan global knockout embryos exhibit severe developmental defects in several organs, including the brain, and usually die around E10 - E12 [174,175,176]. Interestingly, BL formation is not affected in these mutants, although BL deterioration is observed in areas with high mechanical stress [175]. This data indicates that perlecan is not required for BL assembly but plays an important role in BL maintenance. To overcome embryonic lethality, several perlecan mutant mouse lines have been generated. For example, mice carrying a C-to-Y mutation at residue 1532 with the neomycin cassette (C1532Yneo) exhibit reduced perlecan secretion and a skeletal phenotype similar to Schwartz-Jampel syndrome, whereas those harboring only the mutation without the neomycin cassette (C1532Y) display comparable perlecan level and no obvious abnormalities [177]. It should be noted that whether the C1532Yneo mice have BBB disruption remains unclear. Recently, a perlecan-knockout rescued line was generated by expressing perlecan gene under the Col2α1 promoter and enhancer in the perlecan-/- background [178]. These mutants are viable and show no defects in BBB integrity [178], indicating a dispensable role of perlecan in BBB maintenance under homeostatic conditions. After ischemic stroke, however, these mutants demonstrate exacerbated BBB damage and attenuated pericyte accumulation [178]. Mechanistic studies reveal that perlecan promotes PDGFRβ signaling and pericyte migration [178]. These results suggest that perlecan contributes to BBB repair in ischemic stroke possibly by regulating pericyte recruitment.
Basal lamina and normal aging
BL defects are observed during normal aging. The functional significance of these BL alterations, however, is largely unknown. In this section, we summarize (Table 2) and illustrate (Fig. 3) how the BL and its major components change during normal aging.

BL changes in aging and neurological disorders. Diagram illustrations showing changes of BL, endothelial cells, pericytes, microglia, neurons, and protein aggregates in aging and three neurodegenerative disorders (AD, PD, and ALS). BL: basal lamina; EC: endothelial cell; PC: pericytes; As: astrocytes; rM: resting microglia; pM: primed microglia; aM: activated microglia; hN: healthy neurons; iN: injured neurons; Aβ: β-amyloid; α-Syn: α-synuclein; SC: serum components; AD: Alzheimer’s disease; PD: Parkinson’s disease; ALS: amyotrophic lateral sclerosis
The BL is thickened during aging
Accumulating evidence shows that the BL thickens with age in rodents, although the extent of BL thickening is much less during normal aging compared to neurodegenerative disorders [207,208,209]. Consistent with these findings, mild BL thickening is also observed in aged human brains [210,211,212]. Together with slightly increased BL, mild pericyte loss and microglial priming are also observed in aged brains [59, 62, 213, 214]. These results suggest an important role of the BL in aging and age-associated alteration of BL composition.
Collagen IV alteration during aging is controversial
Controversy exists on how collagen IV changes during normal aging. On one hand, increased collagen IV levels have been found in the CNS in both rodents and humans [209,210,211]. In sharp contrast to these reports, one study found decreased collagen IV in aged mouse brain [207]. In addition, unaltered collagen IV levels have also been reported during normal aging in both mice and humans [66, 212]. This discrepancy may be caused by different experimental methods and conditions. Future research should address how exactly collagen IV changes during normal aging.
Laminin expression is decreased during aging
Most studies show that laminin level decreases during normal aging. For example, reduced laminin expression has been reported in aged mice [66, 207] and humans [211]. One study, however, finds increased laminin expression in mouse retina during aging [209]. This disparity may be explained by different laminin antibodies and/or fixation protocols used in these studies. It has been shown that heavy formaldehyde fixation masks laminin antigen, while mild-moderate fixation reveals it [215].
Nidogen alteration during aging is controversial
During normal aging, both decreased [207] and increased [209] nidogen levels have been reported in mouse brains. In postmortem human brains, however, increased nidogen expression is observed during normal aging [212]. It is important to determine how nidogen changes during normal aging in future research.
Agrin alteration during aging is largely unknown
Few studies investigated how normal aging affects agrin levels. One group found increased agrin in human brains during normal aging [211]. This finding needs to be validated in future studies.
Perlecan expression is elevated during aging
Mounting evidence supports that perlecan expression increases during normal aging [66, 207, 209]. It should be noted, however, that these studies were all performed in mice. It is critical to validate this finding in human brains in the future.
Basal lamina and Alzheimer’s disease
AD, first reported in 1907, is the most common form of dementia. Clinically, AD is characterized by brain shrinkage and progressive decline in memory and other cognitive functions, which usually lead to behavioral changes, impaired mobility, hallucinations, seizures, and ultimately death [216, 217]. Pathologically, AD is marked by senile plaques (SPs), neurofibrillary tangles (NFTs), and cerebral amyloid angiopathy (CAA). SPs are extracellular aggregates composed of Aβ peptides, while NFTs are intracellular accumulation of hyperphosphorylated tau protein [216]. CAA, found in ~80% of AD cases, refers to the deposition of Aβ in cerebral and leptomeningeal blood vessel wall [218]. Depending on if there is a genetic cause, AD is categorized into familial AD (fAD) and sporadic AD (sAD). The former is caused by mutations of certain genes, including amyloid precursor protein (APP) and presenilin-1/2 (PSEN1/2) [216]. The latter occurs in a sporadic nature without known causes. The APOE gene is considered to be the biggest risk factor for sAD, with APOE4 heterozygotes and homozygotes having 3x and 12x higher risks, respectively [219].
The pathogenesis of AD remains largely unknown, although many theories have been proposed. One of the most popular hypotheses is the amyloid cascade hypothesis (ACH), which has been the foundation of numerous studies and potential therapies [220]. It proposes that genetic and environmental factors cause deposition of Aβ and formation of SPs and NFTs, eventually leading to neuronal loss and dementia [221]. This hypothesis is supported by the findings that Aβ is the most important constituent of SPs [222] and abnormal processing of APP is an early event of AD [221, 223]. It, however, has been challenged by the following observations. First, a significant amount of elderly people present Aβ plaques without developing any clinical symptoms of AD [224], suggesting that other factors may also contribute to AD pathogenesis. Second, fAD patients have abnormal Aβ production at young age but do not develop AD symptoms until at old age [225]. Third, the presence of SPs/NFTs is quantitatively inconsistent with cognitive impairment, age, and disease progression [226,227,228]. Lastly, no ACH-based therapies have been successfully developed [229], although this could be due to a number of other reasons.
Recently, an alternative “two-hit” hypothesis has been proposed. This hypothesis highlights the importance of vascular factors, in addition to genetic and environmental factors [5, 22]. It proposes that vascular damage causes BBB dysfunction and hypoperfusion, which compromise the clearance of Aβ and other neurotoxins, leading to neurodegeneration and dementia [22, 230]. The vascular and neurodegenerative pathways can act independently or synergistically [5, 22]. This hypothesis is supported by several findings. First, BBB disruption and hypoperfusion occur prior to Aβ accumulation and dementia in both APPswe/E693G mice and AD patients [231]. Next, BMECs and pericytes are activated/degenerated in the early symptomatic stage of AD, which impairs Aβ clearance and blood flow [4, 10, 60, 61], leading to accumulation of metabolic waste and toxins, microglial activation, and eventually neuronal death.
Understanding how exactly BBB integrity and function are regulated in AD will shed light on the pathogenesis of AD and may identify novel targets with therapeutic potential. Given that the BL is actively involved in BBB maintenance [63, 102, 106, 128, 129, 218] and Aβ clearance [71, 77, 88, 102, 130]. it is logical to speculate that BL defects may contribute to AD pathogenesis. Here, we summarize (Table 3) and illustrate (Fig. 3) how the BL and its major constituents change in AD, and discuss their functional significance.
The BL is thickened in AD
Mounting evidence suggests that the BL thickens in AD brains in both transgenic mouse models (regardless of transgenic genes) and postmortem human samples [210, 212, 231,232,233,234,235,236,237,238,239,240,241,242], although one study finds BL thinning in postmortem human brains [243]. Interestingly, BL thickening in AD brains displays a region-specific pattern: it is mainly observed in the cerebral cortex, hippocampus, and thalamus [207, 237]. Furthermore, an immuno-electron microscopy study shows that BL thickening predominantly occurs in parenchymal BL rather than endothelial BL [237]. These findings establish a direct association of BL thickening with AD.
Based on that BL thickening occurs before Aβ deposition in the vessel wall [231], it is hypothesized that BL thickening compromises Aβ clearance. Two major pathways are responsible for Aβ clearance in the CNS: receptor-mediated transport across the BBB and meningeal lymphatic/glymphatic system. It has been shown that 85% of Aβ is cleared via transport across the BBB and 15% via ISF bulk flow under physiological conditions [80,81,82,83,84,85,86,87]. In AD brains, however, the receptor-mediated trans-vascular mechanism is severely compromised. One major receptor that mediates Aβ uptake and transport across the BBB is low-density receptor-related protein 1 (LRP1) [80, 84, 251]. Both animal [81, 252, 253] and human [80, 81, 254] studies demonstrate that LRP1 expression at the BBB is significantly reduced in aging and AD. Additionally, inactivation of endothelial LRP1 activates the cyclophilin A-matrix metalloproteinase 9 pathway in endothelial cells, which reduces collagen IV and tight junction proteins, resulting in BBB impairment and cognitive deficits [255]. Similarly, activation of the same pathway in pericytes causes BBB breakdown in mice deficient in murine ApoE or those expressing human ApoE4, which poorly interacts with LRP1 [104].
In addition, Aβ clearance via the meningeal lymphatic/glymphatic system is also impaired in AD. First, APOE4 transgenic mice show severely disrupted perivascular drainage of Aβ and brain accumulation of Aβ [244]. Next, decreased CSF influx and Aβ clearance rate are observed in both aged AD mice with existing Aβ plaques and young AD mice without visible Aβ plaques [66, 256]. Third, reduced glymphatic flow is found in WT mice with intraventricular or intra-hippocampal injection of Aβ [207, 256], suggesting that Aβ itself can disrupt glymphatic flow. Like in AD mice, AD patients show reduced CSF uptake and attenuated clearance of tau tracer F-THK5117, which are associated with increased Aβ levels in the brain [109, 257]. Together, these results suggest that BL thickening is an early event, which contributes to AD pathogenesis by impairing Aβ clearance.
Tauopathy is another key feature of AD. Accumulating evidence suggests that tau pathology is also correlated with BBB dysfunction [258]. For example, truncated tau has been shown to increase endothelial permeability via activating glial cells in an in vitro BBB model, although it is not directly toxic to endothelial cells [259]. Tau-induced glial activation has been shown to enhance the expression of endothelial adhesion molecules and increase the transmigration of leukocytes across the BBB [260]. In addition, mice expressing human tau protein in astrocytes exhibit BBB disruption in areas with robust astrocytic tau pathology [261]. Progressive IgG leakage and T cell infiltration are also observed in brain tissue from the tetracycline-regulatable rTg4510 tau transgenic mice [262]. Interestingly, this BBB impairment correlates with the appearance of perivascular tau around major hippocampal blood vessels and is reversible when tau expression was suppressed [262]. Furthermore, BBB breakdown has also been reported in tauopathies without Aβ pathology, including progressive supranuclear palsy [262]. Similarly, BBB disruption is detected in regions of dense perivascular p-Tau accumulation in chronic traumatic encephalopathy [263]. These findings suggest that tau alone can compromise BBB integrity. It remains unclear, however, that how BL thickening affects tau pathology in AD. This question needs to be answered in future studies.
What causes BL thickening in AD brains? One possibility is that imbalance of different BL components leads to BL thickening [264]. Echoed with this hypothesis, it has been shown that APOE4 is able to bind BL components, including laminin and HSPGs [265, 266], and affect BL composition [244]. In addition, all BL constituents are able to interact with Aβ and affect its biochemical properties by inhibiting, disassembling, or strengthening Aβ fibrillation. Here, we discuss the changes and functions of four major components of the BL in AD individually.
Collagen IV alteration in AD is controversial
Collagen IV shows inconsistent alterations in transgenic AD mouse models. For example, increased collagen IV expression is found in the BL of 3xTG (APPswe/PSEN1M146V/TauP301L) transgenic mice [232, 234], decreased collagen IV level is observed in APPswe (APPK670N, M671L) [245] and APOE4 [244] transgenic mice, while unaltered collagen IV level is reported in PSEN1P117L mice [233]. Unlike mouse studies, the vast majority of postmortem human studies reveal increased collagen IV levels in AD brains [210, 235, 237,238,239, 241, 246, 247], although one study finds unchanged collagen IV in AD brains [212]. Interestingly, the upregulation of collagen IV in AD brains is region-specific. Collagen IV is mainly increased in the frontal and temporal cortex in both subclinical (Braak stage 3-4) and AD (Braak stage 5-6) patients [235, 241], while it is not severely altered in the neocerebellum [267]. The discrepancy on collagen IV changes in AD may be explained by different genetic models and tissues (mouse vs. human). It should be noted that each genetic model has its own limitations and that no animal models can fully replicate AD pathology in patients.
The functional significance of collagen IV changes in AD is unknown. In vitro studies show that collagen IV is able to bind APP with high affinity [268, 269], prevent the formation of β-sheet structured Aβ40 aggregates [270], and induce disassembly of Aβ42 fibrils [271]. These results suggest that enhanced expression of collagen IV is a protective mechanism, and that increasing collagen IV expression may have a therapeutic potential in AD. The exact function of collagen IV in AD, however, needs to be investigated in vivo using both loss-of-function and gain-of-function studies in the future. Collagen IV conditional knockouts or hypomorphs may be useful in loss-of-function studies, while mice overexpressing collagen IV or recombinant collagen IV may be useful in gain-of-function studies.
Laminin alteration in AD is controversial
Similarly, controversial results exist on how laminin changes in AD. One report finds decreased laminin expression in the BL in APOE4 mouse brains [244], while other studies show unaltered [233] or increased [231, 248, 249] laminin expression in AD brains. One possible reason for this discrepancy is that pan-laminin rather than subunit-specific antibodies were used in these studies. Pan-laminin antibodies, such as the polyclonal antibody against Engelbreth Holm-Swarm mouse sarcoma-derived laminin, are unable to distinguish different laminin subunits in the BL, and thus cannot reflect changes of individual laminin isoforms. Given that distinct laminin isoforms exert different functions [63, 106, 128, 129], it is important to determine how each individual laminin isoform changes. It has been reported that laminin-α1 and -γ1 levels are substantially increased in human AD brains, primarily in reactive astrocytes and/or SPs [248, 249]. Future studies should determine how other laminin subunits change in AD using subunit-specific antibodies.
Although the function of laminin in AD remains largely unclear, there is evidence suggesting that laminin may negatively regulate AD pathogenesis by promoting Aβ clearance. First, it has been reported that laminin is able to inhibit the formation of Aβ40 fibrils [272,273,274] and disrupt fibrils that have already formed in vitro [275]. Next, laminin at high concentration induces a random structural transition of Aβ42 fibrils, disassembling the β-sheet structure and inhibiting fibrillation [271]. Consistent with these findings, laminin is able to interact with amyloid precursor protein [276] and co-localizes with Aβ or Aβ-APOE4 complex in AD brains [250, 277]. In addition, it has been proposed that APOE regulates Aβ clearance in an isoform-specific manner, possibly due to distinct affinity for laminin [84, 277]. With the generation of various laminin conditional knockout mouse lines, the function of each laminin isoform in AD pathogenesis can be investigated in vivo in future studies. Currently, there are no known disease-modifying therapies for AD. Laminin may be a promising target, given its ability to inhibit the formation of Aβ40/Aβ42 fibrils and promote Aβ clearance. Further studies, however, are needed to investigate the therapeutic potential of laminin in AD.
Nidogen alteration in AD is largely unknown
How nidogen changes in AD is mostly unknown. There is only one such study, which finds decreased nidogen expression in CAA in postmortem human brains [212]. This knowledge gap should be addressed in future research.
Our understanding of nidogen’s role in AD predominantly comes from in vitro studies. It has been shown that nidogen dose-dependently inhibits Aβ40 fibril formation and induces disassembly of Aβ40 and/or Aβ42 fibrils in vitro [271], suggesting a beneficial role of nidogen in AD. Elucidating nidogen’s function in AD in vivo relies on loss-of-function studies. There are two nidogen isoforms in mammals [165]. Genetic ablation of either isoform results in a normal phenotype [165,166,167,168, 170], suggesting functional compensation between these isoforms. Knockout of both isoforms simultaneously, however, leads to perinatal lethality [128], preventing investigation of nidogen’s role in AD in vivo. Although both single knockout mice fail to show gross abnormalities under homeostatic conditions, they may display exacerbated vascular and/or neurological damage in AD background. In addition, conditional knockout of both nidogen isoforms may overcome perinatal lethality and enable investigation of nidogen’s function in AD pathogenesis. Future research should focus on generating these mutant mice.
HSPG in general is increased in AD
It has been reported that HSPG in general has a 9.3-fold increase in the hippocampus and 6.6-fold increase in the superior frontal gyrus in AD patients [278].
Unlike other major BL constituents, which inhibit Aβ fibrillation, HSPGs have been found to accelerate Aβ fibrillation contributing to AD pathogenesis [279,280,281]. First, HSPGs are able to bind Aβ and affect its fibrillation via their HS chains [282]. It has been shown that the sulfation pattern of HS is associated with its affinity for Aβ [283] and Aβ fibrillation capability [284]. For example, sulfated HS has a higher affinity for Aβ, while desulfated HS loses Aβ-binding capability almost entirely [283]. Removal of heparin’s O-sulfate partially inhibits Aβ aggregation, while deletion of all sulfate groups completely abolishes Aβ fibrillation [284]. Next, HSPGs also inhibit Aβ degradation [285]. In vitro studies have demonstrated that HSPGs inhibit the proteolytic degradation of fibrillar Aβ [286], and Aβ prevents proteolytic breakdown of HSPGs by inhibiting heparanase, the enzyme that degrades HS [287]. These findings suggest that Aβ-HS interaction protects each other from degradation [288]. Consistent with this observation, overexpression of heparanase lowers Aβ burden in APP transgenic mice [289]. Subsequent mechanistic study reveals that heparanase exerts this function by either releasing Aβ from the plaques or inhibiting tau fibril formation and propagation [288]. Together, these results suggest that HSPGs contribute to almost every stage of AD pathogenesis, including Aβ aggregation and clearance, and that decreasing HSPGs or increasing heparanase activity may have a therapeutic potential in AD.
Agrin alteration in AD is controversial
There are controversial findings on how agrin changes in AD brains. Specifically, while unaltered agrin expression is observed in APOE4 mice [244], both decreased [243] and increased [235, 250] agrin levels have been reported in AD patients. Biochemical analysis shows that all agrin from normal brains is soluble in 1% SDS, whereas a large fraction of agrin from AD brains is insoluble [290]. Since fibrillar Aβ exhibits similar solubility properties [290], it is suggested that agrin may interact with Aβ. This is consistent with the observation that agrin is detected in SPs, NFTs, and CAA in AD brains [250, 279, 280, 291, 292]. Future studies should determine how exactly agrin changes in AD brains in both mice and humans.
In contrast to HSPGs, agrin has been shown to inhibit Aβ deposition in the brain. For example, overexpression of agrin leads to decreased Aβ [131]. Consistent with this finding, deletion of endothelial agrin in APPswe/PSEN1dE9 mice results in increased Aβ40 deposition, although loss of neuron-associated agrin in these mutants fails to affect Aβ [131]. In addition, agrin may be also involved in Aβ clearance via the glymphatic system. It has been shown that ablation of endothelial agrin in APPswe/PSEN1dE9 mice reduces AQP4 expression in astrocytic endfeet [131], and that deletion of AQP4 in AD mice results in Aβ accumulation without affecting the expression of Aβ-degrading proteases [293]. These results are consistent with previous reports that the function of glymphatic system is AQP4-dependent [294,295,296]. Together, these findings suggest that agrin negatively regulates AD pathogenesis by inhibiting Aβ deposition and promoting Aβ clearance, and that upregulating agrin expression may be able to improve AD outcome.
Perlecan alteration in AD is controversial
Perlecan has been shown to increase in human AD brains [235], but unaltered in APOE4 mice [244]. It remains unclear whether perlecan is a key component of SPs, NFTs, and CAA in AD brains. Several studies fail to detect perlecan in SPs, NFTs, and CAA in AD brains [291, 292, 297], while a group reports its presence in these structures [298]. This discrepancy may be caused by different perlecan antibodies and/or experimental approaches. How perlecan changes in mouse and human AD brains should be clarified using various antibodies and standard protocols.
Like agrin, perlecan also shows a neuroprotective role against Aβ toxicity. It has been reported that perlecan domain V inhibits neurotoxic signaling cascade by blocking Aβ-integrin-α2β1 interaction in vitro [299, 300]. In addition, perlecan domain V is able to reverse Aβ toxicity in endothelial cells and restore angiogenesis in vitro [301]. These results suggest a therapeutic potential of perlecan in AD. It should be noted, however, that it remains unclear how perlecan regulates Aβ deposition/clearance and AD pathology in vivo. This gap of knowledge can be addressed by using perlecan hypomorphic (C1532Yneo) mice and/or perlecan-knockout rescued mutants in future studies.
Basal lamina and Parkinson’s disease
PD, first medically described by James Parkinson in 1817, is the second most common neurodegenerative disease after AD [302]. Typical symptoms of PD include resting tremor, gait disorders, and bradykinesia [303]. In addition, various nonmotor symptoms, such as olfactory dysfunction, depression/anxiety, and dementia, are also found in PD patients [303, 304]. Although there is no cure for PD, some symptomatic treatments have been found beneficial. Among all PD cases, approximately 95% occur sporadically without known genetic causes, while 5% can be attributed to gene mutations [305, 306]. These genes include SNCA (α-synuclein), LRRK2 (leucine rich repeat kinase 2), VPS35 (vacuolar protein sorting 35 homolog), and GBA (glucocerebrosidase) [304, 307, 308].
Pathologically, PD is characterized by loss of dopaminergic neurons in the brain, most dramatically in the pars compacta of the substantia nigra and locus coeruleus [309]. A hallmark of PD is the formation of Lewy bodies, which contain aggregated and post-translationally modified α-synuclein. Various studies report lower α-synuclein level in the CSF of PD patients [310,311,312,313], suggesting that defective transport/clearance of α-synuclein may contribute to PD pathogenesis. This is consistent with previous findings that α-synuclein can be transported bidirectionally across the BBB [310], and that α-synuclein is overexpressed in melanized neurons in PD patients [314]. There is evidence showing that BBB integrity is compromised in PD brains [37, 315,316,317]. Given the important functions of BL in BBB maintenance and the meningeal lymphatic/glymphatic system, it is reasonable to speculate that BL defects may contribute to PD pathogenesis. Here, we summarize (Table 4) and illustrate (Fig. 3) how the BL and its major components change in PD, and discuss their functional significance.
The BL is thickened and collapsed in PD
Although there are no reports on BL changes in PD mouse models, BL abnormalities have been observed in PD patients. Specifically, capillary BL thickening [241, 318] and collapsed BL without endothelium [319] are found in postmortem brains of PD patients. The degeneration of endothelial cells exposes the BL to serum proteins, which leads to abnormal accumulation of plasma proteins at the BL, degeneration of pericytes, activation of glial cells, and eventually neuronal death [319]. Future studies should elucidate how BL changes in various PD mouse models.
The functional significance of BL changes in PD remains unknown. On one hand, BL thickening may be a protective mechanism to attenuate PD pathology. For example, it is possible that the BL thickens to enhance α-synuclein clearance via the meningeal lymphatic/glymphatic system. On the other hand, BL thickening may be the consequence of α-synuclein accumulation in the vasculature. In this case, BL thickening has a deleterious role in PD. Future studies should distinguish these possibilities and explore the molecular mechanisms underlying BL thickening in PD. Here, we discuss the changes and functions of four major components of the BL in PD individually.
Collagen IV is increased in PD
Most studies find increased collagen IV level in PD brains. For example, enhanced expression of COL4A2 has been observed in the brains of α-synucleinA30P mice [320]. Echoed with this finding, collagen IV accumulation is also found in postmortem brains from PD patients [241, 318]. Although one study finds no alterations in total length and density of collagen IV+ blood vessels in PD patients, collagen IV intensity is not reported in this study [319]. Since many other genes are associated with fPD, it is important to determine if collagen IV displays similar changes in other PD mouse models in the future.
Although the functional significance of collagen IV upregulation in PD is unknown, there are several hypotheses. One possibility is that enhanced collagen IV may contribute to PD pathogenesis by inducing ER stress. In support of this hypothesis, abnormal expression of collagen IV has been reported to increase ER stress [321], and a strong association of altered Golgi morphology and COL4A2 upregulation is observed in PD models [320]. Another possibility is that upregulation of collagen IV may alter BL morphology and function, and thus affect BBB integrity and brain influx/efflux function. In addition, collagen IV may also affect PD pathogenesis by regulating α-synuclein aggregation in the brain. Future studies should focus on testing these hypotheses and determining the exact role of collagen IV in PD.
Laminin alteration in PD is unknown
There are no studies examining how laminin alters in PD brains. Future research should fill this gap of knowledge by determining how each individual laminin isoform changes in PD brains using subunit-specific antibodies.
Accumulating evidence suggests that laminin exerts a neuroprotective role in PD. First, laminin has been shown to act as a neurite outgrowth-promoting factor for neurons in vitro [322]. Next, laminin-HSPG complex is able to enhance neurite outgrowth in vitro [323]. Similarly, peptide nanofibers with HS mimetic and laminin-derived epitopes significantly promote neurite outgrowth in vitro [324]. Consistent with these in vitro studies, the KDI domain of laminin-γ1 has been shown to protect rat dopaminergic neurons from 6-hydroxydopamine-induced toxicity [325], highlighting a beneficial role of γ1-containing laminins in PD. In addition, HS-mimetic and laminin-mimetic peptide amphiphile nanofibers substantially enhance dopamine and tyrosine hydroxylase levels, reduce cleaved Cas-3 level, and improve function and tissue integrity in the 6-hydroxydopamine-induced PD model in rats [326], indicating a therapeutic potential of laminin and HS in PD. It is unclear, however, whether this beneficial effect is from laminin, HS, or both.
In addition, it remains unknown which laminin isoforms mediate the neuroprotective function in PD. There is evidence suggesting that laminin-511 exerts a beneficial role in PD. Specifically, laminin-511 has been shown to promote the survival and differentiation of midbrain dopaminergic neurons by inducing miR-130a to suppress PTEN (phosphatase and tensin homolog) [327, 328]. Since laminin-511 is mainly produced by BMECs [142, 191], which undergo degeneration in PD [319], it is hypothesized that BMEC degeneration-induced loss of laminin-511 significantly contributes to PD. This hypothesis, however, needs further investigation.
Nidogen alteration in PD is unknown
There are no reports on how nidogen changes in PD brains. This is an interesting area for future research.
The function of nidogen in PD is unknown. This is predominantly due to the mutual compensation between two nidogen isoforms [165,166,167,168, 170] and embryonic lethality of the double knockouts [65, 171, 172]. It is interesting to see if nidogen single knockout mice develop any phenotype in PD background. In addition, mice with conditional knockout of both nidogen isoforms may enable investigation of nidogen’s function in PD pathogenesis in a cell-specific manner. Future research should focus on generating these mutant mice.
HSPG alteration in PD is unknown
Like laminin and nidogen, how HSPG changes in PD brains remains unknown. This important question needs to be answered in future studies.
The function of HSPGs in PD remains unclear. However, there are reports showing that laminin-HSPG complex exerts a beneficial role in PD. For instance, laminin-HSPG complex is able to enhance neurite outgrowth in vitro [323]. In addition, not only do peptide nanofibers with HS mimetic and laminin-derived epitopes induce neurite outgrowth in vitro [324], they also reduce brain injury and enhance functional recovery in a rat model of PD [326]. These results suggest a neuroprotective role of HS and laminin in PD. Whether this beneficial effect is from HS, laminin or both, however, remains elusive. Since HSPGs are absent in Lewy bodies and do not regulate the fibrillization & stabilization of Lewy bodies [329], it is more likely that the above-described neuroprotective function is from laminin rather than HSPGs. This possibility, however, needs further investigation.
Basal lamina and amyotrophic lateral sclerosis
ALS, first described in 1869, is a progressive nervous system disease clinically characterized by muscular weakness and paralysis. It affects approximately 2 in every 100,000 people, and ~16,000 Americans are living with ALS at any time [330]. Unfortunately, there are no effective disease-modifying therapies at present. The typical pathology of ALS is motor neuron degeneration in the spinal cord, motor cortex, and brain stem. Like AD and PD, the vast majority of ALS cases are sporadic (sALS), while only ~10% are familial (fALS). fALS is genetically linked to at least 15 genes, including SOD1 (Cu/Zn superoxide dismutase), TARDBP (TAR DNA-binding protein), FUS (fused in sarcoma), ANG (angiogenin precursor), and OPTN (optineurin) [331]. Approximately 20% of fALS cases are caused by a missense mutation in SOD1 [332]. The mutant SOD1 forms intracellular aggregates, which alter gene expression and protein interactions, leading to motor neuron death via toxic hydroxyl radicals [333]. In addition, mounting evidence suggests that BBB integrity is disrupted in both sALS and fALS. For example, serum protein leakage, tight junction protein reduction, pericyte loss, astrocytic endfeet detachment and degeneration, and BL component changes have been reported independently in ALS in both humans [3, 45, 47, 48, 334,335,336] and mice [40,41,42,43,44,45]. Since BBB disruption occurs before motor neuron degeneration and neuroinflammation [42, 43, 45], it is believed that BBB breakdown actively contributes to ALS. Given the critical role of BL in BBB maintenance, we hypothesize that BL defects may contribute to ALS pathogenesis. Here, we summarize (Table 5) and illustrate (Fig. 3) how the BL and its major components change in ALS, and discuss their functional significance.
The BL is thickened in ALS
Abnormalities in the BL and its surrounding structures are observed in ALS. First, BL thickening and multiple layers of BL and BMECs are detected in the spinal cord and brain stem in the SOD1G93A mouse model of ALS [40]. In addition, reduced pericyte density, retracted astrocytic endfeet, and extracellular edema are also found in these mutants [40, 344]. In sALS patients, the BL is exposed to plasma proteins through detached BMECs, which leads to accumulation of fibrin and collagen IV within the BL [48]. This BL abnormality may contribute to pericyte degeneration and astrocyte defects. Future research should address if similar BL defects are observed in ALS patients.
Although a correlation between BL defects and ALS severity is observed, it remains unclear if BL changes are a cause or consequence of ALS. Based on that BL thickening and duplication occur at early symptomatic stage in SOD1G93A mice [40], it is speculated that these BL changes may be a defense mechanism to BMEC degeneration. In addition, these BL defects may also contribute to ALS pathogenesis via affecting BBB integrity and possibly the meningeal lymphatic/glymphatic pathway. It has been shown that BMEC detachment exposes the BL to plasma proteins, leading to BL thickening and enhanced vascular permeability [40, 334]. These possibilities should be assessed in future studies.
Collagen IV alteration in ALS is controversial
Controversial results exist on how collagen IV changes in ALS. On one hand, increased collagen IV is observed in the spinal cords of SOD1G93A mice at 18 weeks of age [45]. Collagen IV-expressing microglia start to appear in the anterior horn in these mice at 15 weeks of age [45]. Similarly, collagen accumulation has been found in capillary BL in the medulla and spinal cords of ALS patients [48]. On the other hand, decreased perivascular collagen IV has been reported in ALS patients [45, 337]. Additionally, reduced or unaltered collagen IV levels are observed in non-neural tissues in ALS patients. For example, it has been reported that collagen IV is decreased in the skin and serum [338], while unaffected in the muscle of ALS patients [339]. This discrepancy may be explained by different experimental protocols, species, regions, and/or cell types. Based on that collagen IV is mostly increased in glial cells but reduced in other cells in ALS [45], it is speculated that there is a compensatory upregulation of collagen IV in glial cells in response to reduced collagen IV or other components of the BL in ALS. This hypothesis, however, needs further investigation.
The significant alteration of collagen IV in neural tissue suggests an important role of collagen IV in ALS. There are at least two different interpretations. First, the enhanced expression of collagen IV could be caused by upregulation in glial cells. In this case, collagen IV may exert a neuroprotective role to alleviate ALS injury. Alternatively, the high level of collagen IV in glial cells could be caused by increased uptake. In this case, collagen IV may exert a detrimental role to aggravate ALS injury. Future studies should focus on distinguishing these two possibilities and elucidating the function of collagen IV in ALS pathogenesis.
Laminin alteration in ALS is controversial
Inconsistent findings exist on how laminin changes in ALS. Using a laminin-α1/β1 antibody, it has been reported that laminin level is reduced in the spinal cords of symptomatic SOD1G93A mice [41]. Similarly, laminin-α2 and -β2 are decreased in muscle BL, laminin-α4 is absent in limb muscles but not extraocular muscles in ALS patients [342], while pan-laminin is unaltered in muscles in ALS patients [339]. In contrast to these findings, laminin γ1 is strongly stained in astrocytes in white matter along the cervical and thoracic spinal cords in ALS patients, which correlates with disease severity [340]. Echoed with this result, increased laminin-111 expression has been reported in the skin of ALS patients [341]. This disparity can be explained by different laminin antibodies. Since pericyte deficits [50] and BMEC/astrocyte degeneration [40] are observed in ALS, changes in pericytic, endothelial, and/or astrocytic laminins are also expected. Subunit-specific laminin antibodies should be used to elucidate how each individual laminin isoform changes in ALS in future studies.
Although the function of laminin in ALS pathogenesis remains largely unknown, its substantial alteration in ALS suggests a crucial role of laminin in this disorder. Given that laminin-γ1 is significantly upregulated in astrocytes of ALS patients [340], and that the KDI domain of laminin-γ1 is neuroprotective in both glutamate-induced excitotoxicity [345] and 6-hydroxydopamine-induced neuronal death [325], it is speculated that laminin-γ1 may play a beneficial role in ALS [340]. This hypothesis, however, needs further investigation. In addition, the functions of other laminin subunits/isoforms in ALS should also be determined in future studies.
Nidogen alteration in ALS is unknown
There are currently no studies examining how nidogen changes in ALS. This question should be answered in future research.
The functional significance of nidogen in ALS is unknown, mainly due to mutual compensation between nidogen-1/2 and embryonic lethality of the double knockout mice. Similarly, nidogen single knockout mice should be crossed into the ALS background to determine if loss of either isoform affects ALS pathology. In addition, mice with conditional deletion of both nidogen isoforms simultaneously may enable investigation of nidogen’s function in ALS in a cell-specific manner. Future research should focus on generating these genetic tools.
HSPG alteration in ALS
Agrin expression is reduced in ALS
Agrin has been found to substantially reduced in both spinal cords [43] and neuromuscular junctions [343] in symptomatic but not pre-symptomatic SOD1G93A animals. It remains unclear how agrin changes in spinal cords from ALS patients, although no alteration is detected in neuromuscular junctions [339]. This disparity may be explained by distinct experimental methods and/or different species.
The correlation between agrin reduction and ALS symptoms in SOD1G93A mice suggests that agrin may play an essential role in the onset of symptoms. Given that agrin regulates AChR clustering and other features of postsynaptic membranes in the muscle [346], it is hypothesized that agrin may modulate symptomatic onset by affecting receptor clustering and/or postsynaptic function. This hypothesis, however, needs further investigation.
Perlecan alteration in ALS is unknown
Currently, there are no studies examining perlecan alteration in ALS or the functional significance of these changes. These important questions need to be answered in future studies.
Perspectives and conclusions
Thanks to advances in genetic and biochemical techniques, significant progress has been made in BL research and neurodegenerative disorders. There are, however, several key challenges that need to be addressed and several key questions that need to be answered in future research.
First, the BL is not unique to blood vessels in the CNS. Located at the abluminal side of endothelial cells, the BL covers the entire vasculature. However, due to the contribution of astrocyte-derived ECM proteins, CNS BL has different composition and possibly distinct functions compared to peripheral BL. It should be noted that most studies reporting BL alterations in neurodegenerative disorders examined BL in the CNS. Whether and how peripheral BL changes in these conditions, however, remain unknown. If peripheral BL does change in neurodegenerative disorders, what is the timeline and how is it compared to that of other hallmarks of the diseases (e.g. BBB breakdown)? Answers to these questions will determine if BL changes specifically influence BBB function and if BL changes are early events in the pathogenesis of neurodegenerative disorders.
Second, controversial findings exist on how BL and its major components change in neurodegenerative disorders. This is mainly caused by the use of different antibodies and detection methods. For example, many previous studies used pan-laminin antibodies, which make it impossible to determine the changes of individual laminin isoforms. To address this issue, subunit-specific antibodies and standard protocols should be used in future studies.
Next, the BL is difficult to study due to its unique features. As a high organized structure composed of ECM proteins, the BL exhibits low solubility, high crosslinking, and heavy glycosylation [56, 106, 129, 347,348,349]. The BL is more resistant to detergent-based dissolving/extraction methods compared to other proteins. It is thus challenging to accurately determine the composition of the BL. Innovative techniques that enable separation of the BL from other cellular components and subsequent unbiased assays, such as decellularization followed by mass spectrometry, would address this issue. For example, various decellularization protocols have been developed and employed to isolate the kidney, heart, and liver BL in mice [350,351,352,353]. This approach has enabled direct visualization of the BL in situ and subsequent analyses [354]. Successful decellularization of CNS tissues will allow accurate assessment of BL biochemical properties (e.g. BL composition) in various neurodegenerative disorders and substantially move the field forward.
Fourth, the functions of BL and its major components in neurodegenerative disorders are largely unknown. One major challenge is embryonic lethality of many knockout mice, which prevents loss-of-function studies in adulthood. Another challenge is functional compensation among different isoforms, which leads to a grossly normal phenotype in the single knockouts. Embryonic lethality may be overcome by using conditional knockout mice. Mutual compensation can be addressed by using compound knockout mice, in which multiple isoforms are abrogated simultaneously. Generating these tools will allow us to define the functional significance of the BL and its major components in various neurodegenerative disorders.
Fifth, the causes of many neurological disorders are still unclear. Although transgenic animals replicate many features of the diseases, it should be noted that genetic models only represent familial cases, and most cases of neurodegenerative diseases are sporadic rather than familial. In addition, since multiple genes are linked to AD, PD, or ALS, there are multiple animal models for these diseases. To ensure unbiased and strong conclusions, findings from one animal model should be validated in other animal models and postmortem human samples (both familial and sporadic).
Sixth, targeting the BL therapeutically is challenging. Although BBB impairment is observed in almost all neurodegenerative disorders, only small molecules can cross the compromised BBB in the early phase of these diseases. Given that most BL components are large ECM proteins, intact recombinant proteins are usually ineffective as therapeutics. Short fragments from these proteins, such as key signaling domains, may have a therapeutic potential. This approach requires a thorough understanding of the biochemical properties of major BL components. Alternatively, endogenous levels of BL components may be targeted. This can be achieved by modulating their synthesis and/or degradation using either small-molecular compounds or virus-based approaches. Currently, there are no known small molecules that are able to regulate the expression and/or metabolism of ECM proteins. Although there are safety concerns, virus-based approaches can regulate ECM protein levels in a cell-specific manner. This allows manipulation of certain isoforms of ECM proteins specifically, which may reduce unwanted off-target effects. These therapeutic options should be explored in future studies.
Availability of data and materials
Not applicable.
Abbreviations
- Aβ:
-
β-amyloid
- ACH:
-
amyloid cascade hypothesis
- AD:
-
Alzheimer’s disease
- ALS:
-
amyotrophic lateral sclerosis
- ANG:
-
angiogenin precursor
- APP:
-
amyloid precursor protein
- AQP4:
-
aquaporin-4
- BBB:
-
blood-brain barrier
- BM:
-
basement membrane
- BMECs:
-
brain microvascular endothelial cells
- CAA:
-
cerebral amyloid angiopathy
- CSF:
-
cerebrospinal fluid
- ECM:
-
extracellular matrix
- FUS:
-
fused in sarcoma
- GBA:
-
glucocerebrosidase
- HS:
-
heparan sulfate
- HSPGs:
-
heparan sulfate proteoglycans
- ICH:
-
intracerebral hemorrhage
- ISF:
-
interstitial fluid
- LRP1:
-
low-density receptor-related protein 1
- LRRK2:
-
leucine rich repeat kinase 2
- NFTs:
-
neurofibrillary tangles
- OPTN:
-
optineurin
- PD:
-
Parkinson’s disease
- PSEN1/2:
-
presenilin-1/2
- SNCA:
-
α-synuclein
- SOD1:
-
Cu/Zn superoxide dismutase
- SPs:
-
senile plaques
- TARDBP:
-
TAR DNA-binding protein
- VPS35:
-
vacuolar protein sorting 35 homolog
- SMCs:
-
smooth muscle cells
References
Alzheimer's Association. Alzheimer's disease facts and figures. Alzheimers Dement. 2016;2016(12):459–509.
Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133–50.
Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol Rev. 2019;99:21–78.
Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201.
Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer's disease. Biochim Biophys Acta. 1862;2016:887–900.
Montine TJ, Koroshetz WJ, Babcock D, Dickson DW, Galpern WR, Glymour MM, et al. Recommendations of the Alzheimer's disease-related dementias conference. Neurology. 2014;83:851–60.
Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296–302.
Montagne A, Nation DA, Pa J, Sweeney MD, Toga AW, Zlokovic BV. Brain imaging of neurovascular dysfunction in Alzheimer's disease. Acta neuropathologica. 2016;131:687–707.
Kisler K, Nelson AR, Montagne A, Zlokovic BV. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci. 2017;18:419–34.
Snyder HM, Corriveau RA, Craft S, Faber JE, Greenberg SM, Knopman D, et al. Vascular contributions to cognitive impairment and dementia including Alzheimer's disease. Alzheimers Dement: J Alzheimers Assoc. 2015;11:710–7.
Sweeney MD, Sagare AP, Zlokovic BV. Cerebrospinal fluid biomarkers of neurovascular dysfunction in mild dementia and Alzheimer's disease. J Cereb Blood Flow Metab. 2015;35:1055–68.
Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Perez JM, Evans AC. Alzheimer's Disease Neuroimaging I: Early role of vascular dysregulation on late-onset Alzheimer's disease based on multifactorial data-driven analysis. Nat Commun. 2016;7:11934.
van de Haar HJ, Burgmans S, Jansen JF, van Osch MJ, van Buchem MA, Muller M, et al. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology. 2016;281:527–35.
van de Haar HJ, Jansen JFA, Jeukens C, Burgmans S, van Buchem MA, Muller M, et al. Subtle blood-brain barrier leakage rate and spatial extent: Considerations for dynamic contrast-enhanced MRI. Med Phys. 2017;44:4112–25.
van de Haar HJ, Jansen JFA, van Osch MJP, van Buchem MA, Muller M, Wong SM, et al. Neurovascular unit impairment in early Alzheimer's disease measured with magnetic resonance imaging. Neurobiol Aging. 2016;45:190–6.
Brundel M, Heringa SM, de Bresser J, Koek HL, Zwanenburg JJ, Jaap Kappelle L, et al. High prevalence of cerebral microbleeds at 7Tesla MRI in patients with early Alzheimer's disease. J Alzheimers Dis. 2012;31:259–63.
Heringa SM, Reijmer YD, Leemans A, Koek HL, Kappelle LJ, Biessels GJ. Utrecht Vascular Cognitive Impairment Study G: Multiple microbleeds are related to cerebral network disruptions in patients with early Alzheimer's disease. J Alzheimers Dis. 2014;38:211–21.
Yates PA, Desmond PM, Phal PM, Steward C, Szoeke C, Salvado O, et al. Incidence of cerebral microbleeds in preclinical Alzheimer disease. Neurology. 2014;82:1266–73.
Poliakova T, Levin O, Arablinskiy A, Vasenina E, Zerr I. Cerebral microbleeds in early Alzheimer's disease. J Neurol. 2016;263:1961–8.
Shams S, Martola J, Granberg T, Li X, Shams M, Fereshtehnejad SM, et al. Cerebral microbleeds: different prevalence, topography, and risk factors depending on dementia diagnosis-the Karolinska Imaging Dementia Study. AJNR Am J Neuroradiol. 2015;36:661–6.
Shams S, Wahlund LO. Cerebral microbleeds as a biomarker in Alzheimer's disease? A review in the field. Biomark Med. 2016;10:9–18.
Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12:723–38.
Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and Dysfunction of the Blood-Brain Barrier. Cell. 2015;163:1064–78.
Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, Winkler EA, et al. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer's disease. J Cereb Blood Flow Metab. 2016;36:216–27.
O'Brien JT, Thomas A. Vascular dementia. Lancet. 2015;386:1698–706.
Akiguchi I, Tomimoto H, Suenaga T, Wakita H, Budka H. Blood-brain barrier dysfunction in Binswanger's disease; an immunohistochemical study. Acta neuropathol. 1998;95:78–84.
Farrall AJ, Wardlaw JM. Blood-brain barrier: ageing and microvascular disease--systematic review and meta-analysis. Neurobiol Aging. 2009;30:337–52.
Wallin A, Blennow K, Rosengren L. Cerebrospinal fluid markers of pathogenetic processes in vascular dementia, with special reference to the subcortical subtype. Alzheimer Dis Assoc Disord. 1999;13(Suppl 3):S102–5.
Matsuda K, Tashiro K, Hayashi Y, Monji A, Yoshida I, Mitsuyama Y. Measurement of laminins in the cerebrospinal fluid obtained from patients with Alzheimer's disease and vascular dementia using a modified enzyme-linked immunosorbent assay. Dement Geriatr Cogn Disord. 2002;14:113–22.
Nation DA, Sweeney MD, Montagne A, Sagare AP, D'Orazio LM, Pachicano M, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25:270–6.
Hanyu H, Asano T, Tanaka Y, Iwamoto T, Takasaki M, Abe K. Increased blood-brain barrier permeability in white matter lesions of Binswanger's disease evaluated by contrast-enhanced MRI. Dement Geriatr Cogn Disord. 2002;14:1–6.
Wardlaw JM, Doubal F, Armitage P, Chappell F, Carpenter T, Munoz Maniega S, et al. Lacunar stroke is associated with diffuse blood-brain barrier dysfunction. Ann Neurol. 2009;65:194–202.
Taheri S, Gasparovic C, Huisa BN, Adair JC, Edmonds E, Prestopnik J, et al. Blood-brain barrier permeability abnormalities in vascular cognitive impairment. Stroke; J Cereb Circ. 2011;42:2158–63.
Barcia C, Bautista V, Sanchez-Bahillo A, Fernandez-Villalba E, Faucheux B, Y Poza MP, et al. Changes in vascularization in substantia nigra pars compacta of monkeys rendered parkinsonian. J Neural Transm (Vienna). 2005;112:1237–48.
Rite I, Machado A, Cano J, Venero JL. Blood-brain barrier disruption induces in vivo degeneration of nigral dopaminergic neurons. J Neurochem. 2007;101:1567–82.
Chao YX, He BP, Tay SS. Mesenchymal stem cell transplantation attenuates blood brain barrier damage and neuroinflammation and protects dopaminergic neurons against MPTP toxicity in the substantia nigra in a model of Parkinson's disease. J Neuroimmunol. 2009;216:39–50.
Gray MT, Woulfe JM. Striatal blood-brain barrier permeability in Parkinson's disease. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab. 2015;35:747–50.
Kortekaas R, Leenders KL, van Oostrom JC, Vaalburg W, Bart J, Willemsen AT, et al. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol. 2005;57:176–9.
Al-Bachari S, Naish JH, Parker GJM, Emsley HCA, Parkes LM. Blood-brain barrier leakage is increased in Parkinson's disease. Front Physiol. 2020;11:593026.
Garbuzova-Davis S, Haller E, Saporta S, Kolomey I, Nicosia SV, Sanberg PR. Ultrastructure of blood-brain barrier and blood-spinal cord barrier in SOD1 mice modeling ALS. Brain Res. 2007;1157:126–37.
Garbuzova-Davis S, Saporta S, Haller E, Kolomey I, Bennett SP, Potter H, et al. Evidence of compromised blood-spinal cord barrier in early and late symptomatic SOD1 mice modeling ALS. PloS One. 2007;2:e1205.
Zhong Z, Deane R, Ali Z, Parisi M, Shapovalov Y, O'Banion MK, et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci. 2008;11:420–2.
Nicaise C, Mitrecic D, Demetter P, De Decker R, Authelet M, Boom A, et al. Impaired blood-brain and blood-spinal cord barriers in mutant SOD1-linked ALS rat. Brain Res. 2009;1301:152–62.
Nicaise C, Soyfoo MS, Authelet M, De Decker R, Bataveljic D, Delporte C, et al. Aquaporin-4 overexpression in rat ALS model. Anat Rec (Hoboken). 2009;292:207–13.
Miyazaki K, Ohta Y, Nagai M, Morimoto N, Kurata T, Takehisa Y, et al. Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J Neurosc Res. 2011;89:718–28.
Waters S, Swanson MEV, Dieriks BV, Zhang YB, Grimsey NL, Murray HC, et al. Blood-spinal cord barrier leakage is independent of motor neuron pathology in ALS. Acta Neuropathol Commun. 2021;9:144.
Henkel JS, Beers DR, Wen S, Bowser R, Appel SH. Decreased mRNA expression of tight junction proteins in lumbar spinal cords of patients with ALS. Neurology. 2009;72:1614–6.
Garbuzova-Davis S, Hernandez-Ontiveros DG, Rodrigues MC, Haller E, Frisina-Deyo A, Mirtyl S, et al. Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res. 2012;1469:114–28.
Garbuzova-Davis S, Sanberg PR. Blood-CNS Barrier Impairment in ALS patients versus an animal model. Front Cell Neurosci. 2014;8:21.
Winkler EA, Sengillo JD, Sullivan JS, Henkel JS, Appel SH, Zlokovic BV. Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125:111–20.
Vu LT, Bowser R. Fluid-based biomarkers for amyotrophic lateral sclerosis. Neurotherapeutics. 2017;14:119–34.
De Reuck J, Deramecourt V, Cordonnier C, Auger F, Durieux N, Bordet R, et al. Detection of microbleeds in post-mortem brains of patients with frontotemporal lobar degeneration: a 7.0-Tesla magnetic resonance imaging study with neuropathological correlates. Eur J Neurol. 2012;19:1355–60.
De Reuck J, Deramecourt V, Cordonnier C, Leys D, Pasquier F, Maurage CA. Prevalence of cerebrovascular lesions in patients with Lewy body dementia: a neuropathological study. Clin Neurol Neurosurg. 2013;115:1094–7.
Sjogren M, Folkesson S, Blennow K, Tarkowski E. Increased intrathecal inflammatory activity in frontotemporal dementia: pathophysiological implications. J Neurol Neurosurg Psychiatry. 2004;75:1107–11.
Llorens F, Schmitz M, Gloeckner SF, Kaerst L, Hermann P, Schmidt C, et al. Increased albumin CSF/serum ratio in dementia with Lewy bodies. J Neurol Sci. 2015;358:398–403.
Xu L, Nirwane A, Yao Y. Basement membrane and blood-brain barrier. Stroke Vasc Neurol. 2019;4:78–82.
Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–6.
Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–61.
Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–27.
Montagne A, Nikolakopoulou AM, Zhao Z, Sagare AP, Si G, Lazic D, et al. Pericyte degeneration causes white matter dysfunction in the mouse central nervous system. Nat Med. 2018;24:326–37.
Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosc. 2011;14:1398–405.
Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215.
Yao Y. Laminin: loss-of-function studies. Cell Mol Life Sci. 2017;74:1095–115.
Poschl E, Schlotzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131:1619–28.
Bader BL, Smyth N, Nedbal S, Miosge N, Baranowsky A, Mokkapati S, et al. Compound genetic ablation of nidogen 1 and 2 causes basement membrane defects and perinatal lethality in mice. Molecular and cellular biology. 2005;25:6846–56.
Hawkes CA, Hartig W, Kacza J, Schliebs R, Weller RO, Nicoll JA, et al. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 2011;121:431–43.
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–7.
Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–48.
Buratti E. Functional significance of TDP-43 mutations in disease. Adv Genet. 2015;91:1–53.
Al-Chalabi A, van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol. 2017;13:96–104.
Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4:147ra111.
Iliff JJ, Lee H, Yu M, Feng T, Logan J, Nedergaard M, et al. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Invest. 2013;123:1299–309.
Rasmussen MK, Mestre H, Nedergaard M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018;17:1016–24.
Mestre H, Tithof J, Du T, Song W, Peng W, Sweeney AM, et al. Flow of cerebrospinal fluid is driven by arterial pulsations and is reduced in hypertension. Nat Commun. 2018;9:4878.
Albargothy NJ, Johnston DA, MacGregor-Sharp M, Weller RO, Verma A, Hawkes CA, et al. Convective influx/glymphatic system: tracers injected into the CSF enter and leave the brain along separate periarterial basement membrane pathways. Acta Neuropathol. 2018;136:139–52.
Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D, et al. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol. 2014;76:845–61.
Mestre H, Mori Y, Nedergaard M. The Brain's Glymphatic System: Current Controversies. Trends Neurosci. 2020;43:458–66.
Mestre H, Du T, Sweeney AM, Liu G, Samson AJ, Peng W, et al. Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science. 2020;367:eaax7171.
Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–7.
Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, et al. Clearance of Alzheimer's amyloid-ss (1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–99.
Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–44.
Wu Z, Guo H, Chow N, Sallstrom J, Bell RD, Deane R, et al. Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat Med. 2005;11:959–65.
Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, et al. Transport pathways for clearance of human Alzheimer's amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab. 2007;27:909–18.
Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, et al. Zlokovic BV: apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Investigation. 2008;118:4002–13.
Zhao Z, Sagare AP, Ma Q, Halliday MR, Kong P, Kisler K, et al. Central role for PICALM in amyloid-beta blood-brain barrier transcytosis and clearance. Nat Neurosci. 2015;18:978–87.
Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, Perlmutter D, et al. GLUT1 reductions exacerbate Alzheimer's disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci. 2015;18:521–30.
Nelson AR, Sagare AP, Zlokovic BV. Role of clusterin in the brain vascular clearance of amyloid-beta. Proc Natl Acad Sci U S A. 2017;114:8681–2.
Mastorakos P, McGavern D. The anatomy and immunology of vasculature in the central nervous system. Sci Immunol. 2019;4:eaav0492.
Alves de Lima K, Rustenhoven J, Kipnis J. Meningeal Immunity and Its Function in Maintenance of the Central Nervous System in Health and Disease. Annu Rev Immunol. 2020;38:597–620.
Balin BJ, Broadwell RD. Salcman M, el-Kalliny M: Avenues for entry of peripherally administered protein to the central nervous system in mouse, rat, and squirrel monkey. J Comp Neurol. 1986;251:260–80.
Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci. 2019;22:1021–35.
Herz J, Filiano AJ, Smith A, Yogev N, Kipnis J. Myeloid Cells in the Central Nervous System. Immunity. 2017;46:943–56.
Norris GT, Kipnis J. Immune cells and CNS physiology: Microglia and beyond. J Exp Med. 2019;216:60–70.
Kipnis J. Multifaceted interactions between adaptive immunity and the central nervous system. Science. 2016;353:766–71.
Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity. 2018;48:380–95 e386.
Rua R, McGavern DB. Advances in Meningeal Immunity. Trends Mol Med. 2018;24:542–59.
Korin B, Ben-Shaanan TL, Schiller M, Dubovik T, Azulay-Debby H, Boshnak NT, et al. High-dimensional, single-cell characterization of the brain's immune compartment. Nat Neurosci. 2017;20:1300–9.
Coles JA, Myburgh E, Brewer JM, McMenamin PG. Where are we? The anatomy of the murine cortical meninges revisited for intravital imaging, immunology, and clearance of waste from the brain. Prog Neurobiol. 2017;156:107–48.
Nayak D, Zinselmeyer BH, Corps KN, McGavern DB. In vivo dynamics of innate immune sentinels in the CNS. Intravital. 2012;1:95–106.
Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol. 2017;18:123–31.
Abbott NJ, Pizzo ME, Preston JE, Janigro D, Thorne RG. The role of brain barriers in fluid movement in the CNS: is there a `glymphatic’ system. Acta Neuropathol. 2018;135:387–407.
Hannocks MJ, Pizzo ME, Huppert J, Deshpande T, Abbott NJ, Thorne RG, et al. Molecular characterization of perivascular drainage pathways in the murine brain. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab. 2018;38:669–86.
Nikolakopoulou AM, Montagne A, Kisler K, Dai Z, Wang Y, Huuskonen MT, et al. Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat Neurosci. 2019;22:1089–98.
Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–6.
Kisler K, Nikolakopoulou AM, Sweeney MD, Lazic D, Zhao Z, Zlokovic BV. Acute Ablation of Cortical Pericytes Leads to Rapid Neurovascular Uncoupling. Front Cell Neurosci. 2020;14:27.
Nirwane A, Yao Y. Laminins and their receptors in the CNS. Biol Rev Camb Philos Soc. 2019;94:283–306.
Ahn JH, Cho H, Kim JH, Kim SH, Ham JS, Park I, et al. Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature. 2019;572:62–6.
Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–41.
Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015;212:991–9.
Antila S, Karaman S, Nurmi H, Airavaara M, Voutilainen MH, Mathivet T, et al. Development and plasticity of meningeal lymphatic vessels. J Exp Med. 2017;214:3645–67.
Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015;11:457–70.
Mesquita DAK, Queiroz EF, Oliveira MA, Cunha C, Maia FM, Correa RV. The Old One Technique in a New Style: Developing Procedural Skills in Paracentesis in a Low Cost Simulator Model. Arq Gastroenterol. 2018;55:375–9.
Da Mesquita S, Fu Z, Kipnis J. The Meningeal Lymphatic System: A New Player in Neurophysiology. Neuron. 2018;100:375–88.
Louveau A, Herz J, Alme MN, Salvador AF, Dong MQ, Viar KE, et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci. 2018;21:1380–91.
Patel TK, Habimana-Griffin L, Gao X, Xu B, Achilefu S, Alitalo K, et al. Dural lymphatics regulate clearance of extracellular tau from the CNS. Mol Neurodegener. 2019;14:11.
Nedergaard M. Neuroscience. Garbage truck of the brain. Science. 2013;340:1529–30.
Mestre H, Hablitz LM, Xavier AL, Feng W, Zou W, Pu T, et al. Aquaporin-4-dependent glymphatic solute transport in the rodent brain. eLife. 2018;7:e40070.
Hirose M, Asano M, Watanabe-Matsumoto S, Yamanaka K, Abe Y, Yasui M, et al. Stagnation of glymphatic interstitial fluid flow and delay in waste clearance in the SOD1-G93A mouse model of ALS. Neurosci Res. 2021;171:74–82.
Smith AJ, Yao X, Dix JA, Jin BJ, Verkman AS. Test of the `glymphatic’ hypothesis demonstrates diffusive and aquaporin-4-independent solute transport in rodent brain parenchyma. eLife. 2017;6:e27679.
Asgari N, Berg CT, Morch MT, Khorooshi R, Owens T. Cerebrospinal fluid aquaporin-4-immunoglobulin G disrupts blood brain barrier. Ann Clin Transl Neurol. 2015;2:857–63.
Asgari M, de Zelicourt D, Kurtcuoglu V. Glymphatic solute transport does not require bulk flow. Sci Rep. 2016;6:38635.
Jin BJ, Smith AJ, Verkman AS. Spatial model of convective solute transport in brain extracellular space does not support a “glymphatic” mechanism. J Gen Physiol. 2016;148:489–501.
Holter KE, Kehlet B, Devor A, Sejnowski TJ, Dale AM, Omholt SW, et al. Interstitial solute transport in 3D reconstructed neuropil occurs by diffusion rather than bulk flow. Proc Natl Acad Sci U S A. 2017;114:9894–9.
Spector R, Robert Snodgrass S, Johanson CE. A balanced view of the cerebrospinal fluid composition and functions: Focus on adult humans. Exp Neurol. 2015;273:57–68.
Hladky SB, Barrand MA. Mechanisms of fluid movement into, through and out of the brain: evaluation of the evidence. Fluids Barriers CNS. 2014;11:26.
Plog BA, Nedergaard M. The Glymphatic System in Central Nervous System Health and Disease: Past, Present, and Future. Annu Rev Pathol. 2018;13:379–94.
Jessen NA, Munk AS, Lundgaard I, Nedergaard M. The Glymphatic System: A Beginner's Guide. Neurochem Res. 2015;40:2583–99.
Yao Y. Basement membrane and stroke. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab. 2019;39:3–19.
Kang M, Yao Y. Basement Membrane Changes in Ischemic Stroke. Stroke. 2020;51:1344–52.
Morris AW, Sharp MM, Albargothy NJ, Fernandes R, Hawkes CA, Verma A, et al. Vascular basement membranes as pathways for the passage of fluid into and out of the brain. Acta Neuropathol. 2016;131:725–36.
Rauch SM, Huen K, Miller MC, Chaudry H, Lau M, Sanes JR, et al. Changes in brain beta-amyloid deposition and aquaporin 4 levels in response to altered agrin expression in mice. J Neuropathol Exp Neurol. 2011;70:1124–37.
Yao Y, Chen ZL, Norris EH, Strickland S. Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat Commun. 2014;5:3413.
Gautam J, Cao Y, Yao Y. Pericytic Laminin Maintains Blood-Brain Barrier Integrity in an Age-Dependent Manner. Transl Stroke Res. 2020;11:228–42.
Gautam J, Zhang X, Yao Y. The role of pericytic laminin in blood brain barrier integrity maintenance. Sci Rep. 2016;6:36450.
Da Mesquita S, Louveau A, Vaccari A, Smirnov I, Cornelison RC, Kingsmore KM, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer's disease. Nature. 2018;560:185–91.
Ding XB, Wang XX, Xia DH, Liu H, Tian HY, Fu Y, et al. Impaired meningeal lymphatic drainage in patients with idiopathic Parkinson's disease. Nat Med. 2021;27:411–8.
Zou W, Pu T, Feng W, Lu M, Zheng Y, Du R, et al. Blocking meningeal lymphatic drainage aggravates Parkinson's disease-like pathology in mice overexpressing mutated alpha-synuclein. Transl Neurodegener. 2019;8:7.
Vracko R, Strandness DE Jr. Basal lamina of abdominal skeletal muscle capillaries in diabetics and nondiabetics. Circulation. 1967;35:690–700.
Vracko R, Benditt EP. Basal lamina: the scaffold for orderly cell replacement. Observations on regeneration of injured skeletal muscle fibers and capillaries. J Cell Biol. 1972;55:406–19.
Vracko R, Benditt EP. Capillary basal lamina thickening. Its relationship to endothelial cell death and replacement. J Cell Biol. 1970;47:281–5.
Vracko R. Basal lamina scaffold-anatomy and significance for maintenance of orderly tissue structure. Am J Pathol. 1974;77:314–46.
Sixt M, Engelhardt B, Pausch F, Hallmann R, Wendler O, Sorokin LM. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol. 2001;153:933–46.
Hallmann R, Horn N, Selg M, Wendler O, Pausch F, Sorokin LM. Expression and function of laminins in the embryonic and mature vasculature. Physiol Rev. 2005;85:979–1000.
Owens T, Bechmann I, Engelhardt B. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol. 2008;67:1113–21.
Paulsson M. Basement membrane proteins: structure, assembly, and cellular interactions. Crit Rev Biochem Mol Biol. 1992;27:93–127.
Jeanne M, Labelle-Dumais C, Jorgensen J, Kauffman WB, Mancini GM, Favor J, et al. COL4A2 mutations impair COL4A1 and COL4A2 secretion and cause hemorrhagic stroke. Am J Hum Genet. 2012;90:91–101.
Favor J, Gloeckner CJ, Janik D, Klempt M, Neuhauser-Klaus A, Pretsch W, et al. Type IV procollagen missense mutations associated with defects of the eye, vascular stability, the brain, kidney function and embryonic or postnatal viability in the mouse, Mus musculus: an extension of the Col4a1 allelic series and the identification of the first two Col4a2 mutant alleles. Genetics. 2007;175:725–36.
Kuo DS, Labelle-Dumais C, Mao M, Jeanne M, Kauffman WB, Allen J, et al. Allelic heterogeneity contributes to variability in ocular dysgenesis, myopathy and brain malformations caused by Col4a1 and Col4a2 mutations. Hum Mol Genet. 2014;23:1709–22.
Gould DB, Phalan FC, van Mil SE, Sundberg JP, Vahedi K, Massin P, et al. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med. 2006;354:1489–96.
Gould DB, Phalan FC, Breedveld GJ, van Mil SE, Smith RS, Schimenti JC, et al. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308:1167–71.
Jeanne M, Jorgensen J, Gould DB. Molecular and Genetic Analyses of Collagen Type IV Mutant Mouse Models of Spontaneous Intracerebral Hemorrhage Identify Mechanisms for Stroke Prevention. Circulation. 2015;131:1555–65.
Menezes MJ, McClenahan FK, Leiton CV, Aranmolate A, Shan X, Colognato H. The Extracellular Matrix Protein Laminin alpha2 Regulates the Maturation and Function of the Blood-Brain Barrier. J Neurosci: Off J Soc Neurosci. 2014;34:15260–80.
Thyboll J, Kortesmaa J, Cao R, Soininen R, Wang L, Iivanainen A, et al. Deletion of the laminin alpha4 chain leads to impaired microvessel maturation. Mol Cell Biol. 2002;22:1194–202.
Nguyen NM, Miner JH, Pierce RA, Senior RM. Laminin alpha 5 is required for lobar septation and visceral pleural basement membrane formation in the developing mouse lung. Dev Biol. 2002;246:231–44.
Miner JH, Cunningham J, Sanes JR. Roles for laminin in embryogenesis: exencephaly, syndactyly, and placentopathy in mice lacking the laminin alpha5 chain. J Cell Biol. 1998;143:1713–23.
Coles EG, Gammill LS, Miner JH, Bronner-Fraser M. Abnormalities in neural crest cell migration in laminin alpha5 mutant mice. Dev Biol. 2006;289:218–28.
Song J, Lokmic Z, Lammermann T, Rolf J, Wu C, Zhang X, et al. Extracellular matrix of secondary lymphoid organs impacts on B-cell fate and survival. Proc Natl Acad Sci U S A. 2013;110:E2915–24.
Song J, Zhang X, Buscher K, Wang Y, Wang H, Di Russo J, et al. Endothelial Basement Membrane Laminin 511 Contributes to Endothelial Junctional Tightness and Thereby Inhibits Leukocyte Transmigration. Cell Rep. 2017;18:1256–69.
Gautam J, Miner JH, Yao Y. Loss of Endothelial Laminin alpha5 Exacerbates Hemorrhagic Brain Injury. Transl Stroke Res. 2019;10:705–18.
Miner JH, Li C, Mudd JL, Go G, Sutherland AE. Compositional and structural requirements for laminin and basement membranes during mouse embryo implantation and gastrulation. Development. 2004;131:2247–56.
Smyth N, Vatansever HS, Meyer M, Frie C, Paulsson M, Edgar D. The targeted deletion of the LAMC1 gene. Ann N Y Acad Sci. 1998;857:283–6.
Smyth N, Vatansever HS, Murray P, Meyer M, Frie C, Paulsson M, et al. Absence of basement membranes after targeting the LAMC1 gene results in embryonic lethality due to failure of endoderm differentiation. J Cell Biol. 1999;144:151–60.
Chen ZL, Yao Y, Norris EH, Kruyer A, Jno-Charles O, Akhmerov A, et al. Ablation of astrocytic laminin impairs vascular smooth muscle cell function and leads to hemorrhagic stroke. J Cell Biol. 2013;202:381–95.
Yao Y, Norris EH, Strickland S. The cellular origin of laminin determines its role in blood pressure regulation. Cell Mol Life Sci. 2015;72:999–1008.
Kang SH, Kramer JM. Nidogen is nonessential and not required for normal type IV collagen localization in Caenorhabditis elegans. Mol Biol Cell. 2000;11:3911–23.
Murshed M, Smyth N, Miosge N, Karolat J, Krieg T, Paulsson M, et al. The absence of nidogen 1 does not affect murine basement membrane formation. Mol Cell Biol. 2000;20:7007–12.
Dong L, Chen Y, Lewis M, Hsieh JC, Reing J, Chaillet JR, et al. Neurologic defects and selective disruption of basement membranes in mice lacking entactin-1/nidogen-1. Laboratory investigation. J Tech Methods Pathol. 2002;82:1617–30.
May CA. Distribution of nidogen in the murine eye and ocular phenotype of the nidogen-1 knockout mouse. ISRN Ophthalmol. 2012;2012:378641.
Miosge N, Sasaki T, Timpl R. Evidence of nidogen-2 compensation for nidogen-1 deficiency in transgenic mice. Matrix Biol. 2002;21:611–21.
Schymeinsky J, Nedbal S, Miosge N, Poschl E, Rao C, Beier DR, et al. Gene structure and functional analysis of the mouse nidogen-2 gene: nidogen-2 is not essential for basement membrane formation in mice. Mol Cell Biol. 2002;22:6820–30.
Bose K, Nischt R, Page A, Bader BL, Paulsson M, Smyth N. Loss of nidogen-1 and -2 results in syndactyly and changes in limb development. J Biol Chem. 2006;281:39620–9.
Mokkapati S, Baranowsky A, Mirancea N, Smyth N, Breitkreutz D, Nischt R. Basement membranes in skin are differently affected by lack of nidogen 1 and 2. J Invest Dermatol. 2008;128:2259–67.
Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, et al. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–35.
Arikawa-Hirasawa E, Watanabe H, Takami H, Hassell JR, Yamada Y. Perlecan is essential for cartilage and cephalic development. Nat Genet. 1999;23:354–8.
Costell M, Gustafsson E, Aszodi A, Morgelin M, Bloch W, Hunziker E, et al. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. 1999;147:1109–22.
Rossi M, Morita H, Sormunen R, Airenne S, Kreivi M, Wang L, et al. Heparan sulfate chains of perlecan are indispensable in the lens capsule but not in the kidney. EMBO J. 2003;22:236–45.
Rodgers KD, Sasaki T, Aszodi A, Jacenko O. Reduced perlecan in mice results in chondrodysplasia resembling Schwartz-Jampel syndrome. Hum Mol Genet. 2007;16:515–28.
Nakamura K, Ikeuchi T, Nara K, Rhodes CS, Zhang P, Chiba Y, et al. Perlecan regulates pericyte dynamics in the maintenance and repair of the blood-brain barrier. J Cell Biol. 2019;218:3506–25.
Webersinke G, Bauer H, Amberger A, Zach O, Bauer HC. Comparison of gene expression of extracellular matrix molecules in brain microvascular endothelial cells and astrocytes. Biochem Biophys Res Commun. 1992;189:877–84.
Tilling T, Engelbertz C, Decker S, Korte D, Huwel S, Galla HJ. Expression and adhesive properties of basement membrane proteins in cerebral capillary endothelial cell cultures. Cell Tissue Res. 2002;310:19–29.
Stratman AN, Malotte KM, Mahan RD, Davis MJ, Davis GE. Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood. 2009;114:5091–101.
Kose N, Asashima T, Muta M, Iizasa H, Sai Y, Terasaki T, et al. Altered expression of basement membrane-related molecules in rat brain pericyte, endothelial, and astrocyte cell lines after transforming growth factor-beta1 treatment. Drug Metab Pharmacokinet. 2007;22:255–66.
Allt G, Lawrenson JG. Pericytes: cell biology and pathology. Cells Tissues Organs. 2001;169:1–11.
Hudson BG, Reeders ST, Tryggvason K. Type IV collagen: structure, gene organization, and role in human diseases. Molecular basis of Goodpasture and Alport syndromes and diffuse leiomyomatosis. J Biol Chem. 1993;268:26033–6.
Filie JD, Burbelo PD, Kozak CA. Genetic mapping of the alpha 1 and alpha 2 (IV) collagen genes to mouse chromosome 8. Mamm Genome. 1995;6:487.
Sado Y, Kagawa M, Naito I, Ueki Y, Seki T, Momota R, et al. Organization and expression of basement membrane collagen IV genes and their roles in human disorders. J Biochem. 1998;123:767–76.
Khoshnoodi J, Pedchenko V, Hudson BG. Mammalian collagen IV. Microsc Res Tech. 2008;71:357–70.
Vanacore RM, Shanmugasundararaj S, Friedman DB, Bondar O, Hudson BG, Sundaramoorthy M. The alpha1.alpha2 network of collagen IV. Reinforced stabilization of the noncollagenous domain-1 by noncovalent forces and the absence of Met-Lys cross-links. J Biol Chem. 2004;279:44723–30.
McKee KK, Harrison D, Capizzi S, Yurchenco PD. Role of laminin terminal globular domains in basement membrane assembly. J Biol Chem. 2007;282:21437–47.
Hohenester E, Yurchenco PD. Laminins in basement membrane assembly. Cell adhesion & migration. 2013;7:56–63.
Sorokin LM, Pausch F, Frieser M, Kroger S, Ohage E, Deutzmann R. Developmental regulation of the laminin alpha5 chain suggests a role in epithelial and endothelial cell maturation. Dev Biol. 1997;189:285–300.
Jucker M, Tian M, Norton DD, Sherman C, Kusiak JW. Laminin alpha 2 is a component of brain capillary basement membrane: reduced expression in dystrophic dy mice. Neuroscience. 1996;71:1153–61.
Vanlandewijck M, He L, Mae MA, Andrae J, Ando K, Del Gaudio F, et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature. 2018;554:475–80.
Wu C, Ivars F, Anderson P, Hallmann R, Vestweber D, Nilsson P, et al. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med. 2009;15:519–27.
Fox JW, Mayer U, Nischt R, Aumailley M, Reinhardt D, Wiedemann H, et al. Recombinant nidogen consists of three globular domains and mediates binding of laminin to collagen type IV. EMBO J. 1991;10:3137–46.
Bezakova G, Ruegg MA. New insights into the roles of agrin. Nat Rev Mol Cell Biol. 2003;4:295–308.
Nitkin RM, Smith MA, Magill C, Fallon JR, Yao YM, Wallace BG, et al. Identification of agrin, a synaptic organizing protein from Torpedo electric organ. J Cell Biol. 1987;105:2471–8.
McMahan UJ. The agrin hypothesis. Cold Spring Harb Symp Quant Biol. 1990;55:407–18.
Denzer AJ, Brandenberger R, Gesemann M, Chiquet M, Ruegg MA. Agrin binds to the nerve-muscle basal lamina via laminin. J Cell Biol. 1997;137:671–83.
Groffen AJ, Buskens CA, van Kuppevelt TH, Veerkamp JH, Monnens LA, van den Heuvel LP. Primary structure and high expression of human agrin in basement membranes of adult lung and kidney. Eur J Biochem/FEBS. 1998;254:123–8.
Jury EC, Kabouridis PS. New role for Agrin in T cells and its potential importance in immune system regulation. Arthritis Res Ther. 2010;12:205.
Barber AJ, Lieth E. Agrin accumulates in the brain microvascular basal lamina during development of the blood-brain barrier. Dev Dyn: Off Publ Am Assoc Anatomists. 1997;208:62–74.
Farach-Carson MC, Carson DD. Perlecan--a multifunctional extracellular proteoglycan scaffold. Glycobiology. 2007;17:897–905.
Costell M, Sasaki T, Mann K, Yamada Y, Timpl R. Structural characterization of recombinant domain II of the basement membrane proteoglycan perlecan. FEBS Lett. 1996;396:127–31.
Dolan M, Horchar T, Rigatti B, Hassell JR. Identification of sites in domain I of perlecan that regulate heparan sulfate synthesis. J Biol Chem. 1997;272:4316–22.
Hopf M, Gohring W, Mann K, Timpl R. Mapping of binding sites for nidogens, fibulin-2, fibronectin and heparin to different IG modules of perlecan. J Mol Biol. 2001;311:529–41.
Hawkes CA, Gatherer M, Sharp MM, Dorr A, Yuen HM, Kalaria R, et al. Regional differences in the morphological and functional effects of aging on cerebral basement membranes and perivascular drainage of amyloid-beta from the mouse brain. Aging Cell. 2013;12:224–36.
Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer's disease. Prog Neurobiol. 2001;64:575–611.
Kunze A, Abari E, Semkova I, Paulsson M, Hartmann U. Deposition of nidogens and other basement membrane proteins in the young and aging mouse retina. Ophthalmic Res. 2010;43:108–12.
Uspenskaia O, Liebetrau M, Herms J, Danek A, Hamann GF. Aging is associated with increased collagen type IV accumulation in the basal lamina of human cerebral microvessels. BMC Neurosci. 2004;5:37.
Candiello J, Cole GJ, Halfter W. Age-dependent changes in the structure, composition and biophysical properties of a human basement membrane. Matrix Biol: J Int Soc Matrix Biol. 2010;29:402–10.
Keable A, Fenna K, Yuen HM, Johnston DA, Smyth NR, Smith C, et al. Deposition of amyloid beta in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim Biophys Acta. 1862;2016:1037–46.
Norden DM, Godbout JP. Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol. 2013;39:19–34.
Niraula A, Sheridan JF, Godbout JP. Microglia priming with Aging and stress. Neuropsychopharmacology. 2017;42:318–33.
Jucker M, Bialobok P, Hagg T, Ingram DK. Laminin immunohistochemistry in brain is dependent on method of tissue fixation. Brain Res. 1992;586:166–70.
Lane CA, Hardy J, Schott JM. Alzheimer's disease. Eur J Neurol. 2018;25:59–70.
Jost BC, Grossberg GT. The natural history of Alzheimer's disease: a brain bank study. J Am Geriatr Soc. 1995;43:1248–55.
Thomsen MS, Routhe LJ, Moos T. The vascular basement membrane in the healthy and pathological brain. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab. 2017;37:3300–17.
Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol. 2011;10:241–52.
Roher AE, Cribbs DH, Kim RC, Maarouf CL, Whiteside CM, Kokjohn TA, et al. Bapineuzumab alters abeta composition: implications for the amyloid cascade hypothesis and anti-amyloid immunotherapy. PLoS One. 2013;8:e59735.
Armstrong RA. A critical analysis of the ‘amyloid cascade hypothesis’. Folia Neuropathol. 2014;52:211–25.
Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–5.
Podlisny MB, Tolan DR, Selkoe DJ. Homology of the amyloid beta protein precursor in monkey and human supports a primate model for beta amyloidosis in Alzheimer's disease. Am J Pathol. 1991;138:1423–35.
Lee HG, Casadesus G, Zhu X, Takeda A, Perry G, Smith MA. Challenging the amyloid cascade hypothesis: senile plaques and amyloid-beta as protective adaptations to Alzheimer disease. Ann N Y Acad Sci. 2004;1019:1–4.
De Strooper B, Karran E. The cellular phase of Alzheimer's disease. Cell. 2016;164:603–15.
Price JL, McKeel DW Jr, Buckles VD, Roe CM, Xiong C, Grundman M, et al. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging. 2009;30:1026–36.
Hyman BT, Marzloff K, Arriagada PV. The lack of accumulation of senile plaques or amyloid burden in Alzheimer's disease suggests a dynamic balance between amyloid deposition and resolution. J Neuropathol Exp Neurol. 1993;52:594–600.
Wisniewski T, Ghiso J, Frangione B. Alzheimer's disease and soluble A beta. Neurobiol Aging. 1994;15:143–52.
Nalivaeva NN, Turner AJ. The amyloid precursor protein: a biochemical enigma in brain development, function and disease. FEBS Lett. 2013;587:2046–54.
Zlokovic BV. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 2005;28:202–8.
Merlini M, Meyer EP, Ulmann-Schuler A, Nitsch RM. Vascular beta-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAbeta mice. Acta Neuropathol. 2011;122:293–311.
Mehta DC, Short JL, Nicolazzo JA. Altered brain uptake of therapeutics in a triple transgenic mouse model of Alzheimer's disease. Pharm Res. 2013;30:2868–79.
Gama Sosa MA, Gasperi RD, Rocher AB, Wang AC, Janssen WG, Flores T, et al. Age-related vascular pathology in transgenic mice expressing presenilin 1-associated familial Alzheimer's disease mutations. Am J Pathol. 2010;176:353–68.
Bourasset F, Ouellet M, Tremblay C, Julien C, Do TM, Oddo S, et al. Reduction of the cerebrovascular volume in a transgenic mouse model of Alzheimer's disease. Neuropharmacology. 2009;56:808–13.
Lepelletier FX, Mann DM, Robinson AC, Pinteaux E, Boutin H. Early changes in extracellular matrix in Alzheimer's disease. Neuropathol Appl Neurobiol. 2017;43:167–82.
Serot JM, Bene MC, Foliguet B, Faure GC. Altered choroid plexus basement membrane and epithelium in late-onset Alzheimer's disease: an ultrastructural study. Ann N Y Acad Sci. 1997;826:507–9.
Zarow C, Barron E, Chui HC, Perlmutter LS. Vascular basement membrane pathology and Alzheimer's disease. Ann N Y Acad Sci. 1997;826:147–60.
Claudio L. Ultrastructural features of the blood-brain barrier in biopsy tissue from Alzheimer's disease patients. Acta Neuropathol. 1996;91:6–14.
Perlmutter LS, Chui HC. Microangiopathy, the vascular basement membrane and Alzheimer's disease: a review. Brain Res Bull. 1990;24:677–86.
Mancardi GL, Perdelli F, Rivano C, Leonardi A, Bugiani O. Thickening of the basement membrane of cortical capillaries in Alzheimer's disease. Acta Neuropathol. 1980;49:79–83.
Farkas E, De Jong GI, de Vos RA, Jansen Steur EN, Luiten PG. Pathological features of cerebral cortical capillaries are doubled in Alzheimer's disease and Parkinson's disease. Acta Neuropathol. 2000;100:395–402.
Perlmutter LS. Microvascular pathology and vascular basement membrane components in Alzheimer's disease. Mol Neurobiol. 1994;9:33–40.
Zipser BD, Johanson CE, Gonzalez L, Berzin TM, Tavares R, Hulette CM, et al. Microvascular injury and blood-brain barrier leakage in Alzheimer's disease. Neurobiol Aging. 2007;28:977–86.
Hawkes CA, Sullivan PM, Hands S, Weller RO, Nicoll JA, Carare RO. Disruption of arterial perivascular drainage of amyloid-beta from the brains of mice expressing the human APOE epsilon4 allele. PloS One. 2012;7:e41636.
Kurata T, Miyazaki K, Kozuki M, Morimoto N, Ohta Y, Ikeda Y, et al. Progressive neurovascular disturbances in the cerebral cortex of Alzheimer's disease-model mice: protection by atorvastatin and pitavastatin. Neuroscience. 2011;197:358–68.
Kalaria RN, Pax AB. Increased collagen content of cerebral microvessels in Alzheimer's disease. Brain Res. 1995;705:349–52.
Magaki S, Tang Z, Tung S, Williams CK, Lo D, Yong WH, et al. The effects of cerebral amyloid angiopathy on integrity of the blood-brain barrier. Neurobiol Aging. 2018;70:70–7.
Murtomaki S, Risteli J, Risteli L, Koivisto UM, Johansson S, Liesi P. Laminin and its neurite outgrowth-promoting domain in the brain in Alzheimer's disease and Down's syndrome patients. J Neurosci Res. 1992;32:261–73.
Palu E, Liesi P. Differential distribution of laminins in Alzheimer disease and normal human brain tissue. J Neurosci Res. 2002;69:243–56.
Berzin TM, Zipser BD, Rafii MS, Kuo-Leblanc V, Yancopoulos GD, Glass DJ, et al. Agrin and microvascular damage in Alzheimer's disease. Neurobiol Aging. 2000;21:349–55.
Storck SE, Meister S, Nahrath J, Meissner JN, Schubert N, Di Spiezio A, et al. Endothelial LRP1 transports amyloid-beta (1-42) across the blood-brain barrier. J Clin Invest. 2016;126:123–36.
Silverberg GD, Messier AA, Miller MC, Machan JT, Majmudar SS, Stopa EG, et al. Amyloid efflux transporter expression at the blood-brain barrier declines in normal aging. J Neuropathol Exp Neurol. 2010;69:1034–43.
Osgood D, Miller MC, Messier AA, Gonzalez L, Silverberg GD. Aging alters mRNA expression of amyloid transporter genes at the blood-brain barrier. Neurobiol Aging. 2017;57:178–85.
Donahue JE, Flaherty SL, Johanson CE, Duncan JA 3rd, Silverberg GD, Miller MC, et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer's disease. Acta Neuropathol. 2006;112:405–15.
Nikolakopoulou AM, Wang Y, Ma Q, Sagare AP, Montagne A, Huuskonen MT, et al. Endothelial LRP1 protects against neurodegeneration by blocking cyclophilin A. J Exp Med. 2021;218:e20202207.
Peng W, Achariyar TM, Li B, Liao Y, Mestre H, Hitomi E, et al. Suppression of glymphatic fluid transport in a mouse model of Alzheimer's disease. Neurobiol Dis. 2016;93:215–25.
de Leon MJ, Li Y, Okamura N, Tsui WH, Saint-Louis LA, Glodzik L, et al. Cerebrospinal Fluid Clearance in Alzheimer Disease Measured with Dynamic PET. J Nucl Med. 2017;58:1471–6.
Cai Z, Qiao PF, Wan CQ, Cai M, Zhou NK, Li Q. Role of Blood-Brain Barrier in Alzheimer's Disease. J Alzheimers Dis. 2018;63:1223–34.
Kovac A, Zilkova M, Deli MA, Zilka N, Novak M. Human truncated tau is using a different mechanism from amyloid-beta to damage the blood-brain barrier. J Alzheimers Dis. 2009;18:897–906.
Majerova P, Michalicova A, Cente M, Hanes J, Vegh J, Kittel A, et al. Trafficking of immune cells across the blood-brain barrier is modulated by neurofibrillary pathology in tauopathies. PloS One. 2019;14:e0217216.
Forman MS, Lal D, Zhang B, Dabir DV, Swanson E, Lee VM, et al. Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. J Neurosc: Off J Soc Neurosci. 2005;25:3539–50.
Blair LJ, Frauen HD, Zhang B, Nordhues BA, Bijan S, Lin YC, et al. Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol Commun. 2015;3:8.
Doherty CP, O'Keefe E, Wallace E, Loftus T, Keaney J, Kealy J, et al. Blood-Brain Barrier Dysfunction as a Hallmark Pathology in Chronic Traumatic Encephalopathy. J Neuropathol Exp Neurol. 2016;75:656–62.
Roy S, Ha J, Trudeau K, Beglova E. Vascular basement membrane thickening in diabetic retinopathy. Curr Eye Res. 2010;35:1045–56.
Weisgraber KH, Rall SC Jr, Mahley RW, Milne RW, Marcel YL, Sparrow JT. Human apolipoprotein E. Determination of the heparin binding sites of apolipoprotein E3. J Biol Chem. 1986;261:2068–76.
Huang DY, Weisgraber KH, Strittmatter WJ, Matthew WD. Interaction of apolipoprotein E with laminin increases neuronal adhesion and alters neurite morphology. Exp Neurol. 1995;136:251–7.
Singh-Bains MK, Linke V, Austria MDR, Tan AYS, Scotter EL, Mehrabi NF, et al. Altered microglia and neurovasculature in the Alzheimer's disease cerebellum. Neurobiol Dis. 2019;132:104589.
Narindrasorasak S, Altman RA, Gonzalez-DeWhitt P, Greenberg BD, Kisilevsky R. An interaction between basement membrane and Alzheimer amyloid precursor proteins suggests a role in the pathogenesis of Alzheimer’s disease. Lab Invest. 1995;72:272–82.
Li XF, Thinakaran G, Sisodia SS, Yu FS. Amyloid precursor-like protein 2 promotes cell migration toward fibronectin and collagen IV. J Biol Chem. 1999;274:27249–56.
Kiuchi Y, Isobe Y, Fukushima K. Type IV collagen prevents amyloid beta-protein fibril formation. Life Sci. 2002;70:1555–64.
Kiuchi Y, Isobe Y, Fukushima K, Kimura M. Disassembly of amyloid beta-protein fibril by basement membrane components. Life Sci. 2002;70:2421–31.
Bronfman FC, Garrido J, Alvarez A, Morgan C, Inestrosa NC. Laminin inhibits amyloid-beta-peptide fibrillation. Neurosci Lett. 1996;218:201–3.
Bronfman FC, Alvarez A, Morgan C, Inestrosa NC. Laminin blocks the assembly of wild-type A beta and the Dutch variant peptide into Alzheimer's fibrils. Amyloid. 1998;5:16–23.
Monji A, Tashiro K, Yoshida I, Hayashi Y, Tashiro N. Laminin inhibits A beta 40 fibril formation promoted by apolipoprotein E4 in vitro. Brain Res. 1998;796:171–5.
Castillo GM, Lukito W, Peskind E, Raskind M, Kirschner DA, Yee AG, et al. Laminin inhibition of beta-amyloid protein (Abeta) fibrillogenesis and identification of an Abeta binding site localized to the globular domain repeats on the laminin a chain. J Neurosci Res. 2000;62:451–62.
Narindrasorasak S, Lowery DE, Altman RA, Gonzalez-DeWhitt PA, Greenberg BD, Kisilevsky R. Characterization of high affinity binding between laminin and Alzheimer's disease amyloid precursor proteins. Lab Investigation; J Tech Methods Pathol. 1992;67:643–52.
Zekonyte J, Sakai K, Nicoll JA, Weller RO, Carare RO. Quantification of molecular interactions between ApoE, amyloid-beta (Abeta) and laminin: Relevance to accumulation of Abeta in Alzheimer's disease. Biochim Biophys Acta. 1862;2016:1047–53.
Shimizu H, Ghazizadeh M, Sato S, Oguro T, Kawanami O. Interaction between beta-amyloid protein and heparan sulfate proteoglycans from the cerebral capillary basement membrane in Alzheimer's disease. J Clin Neurosci. 2009;16:277–82.
Perlmutter LS, Chui HC, Saperia D, Athanikar J. Microangiopathy and the colocalization of heparan sulfate proteoglycan with amyloid in senile plaques of Alzheimer's disease. Brain Res. 1990;508:13–9.
Cotman SL, Halfter W, Cole GJ. Agrin binds to beta-amyloid (Abeta), accelerates abeta fibril formation, and is localized to Abeta deposits in Alzheimer's disease brain. Mol Cell Neurosci. 2000;15:183–98.
Lorente-Gea L, Garcia B, Martin C, Quiros LM, Fernandez-Vega I. Heparan sulfate proteoglycans and heparanases in Alzheimer's disease: current outlook and potential therapeutic targets. Neural Regen Res. 2017;12:914–5.
Buee L, Ding W, Anderson JP, Narindrasorasak S, Kisilevsky R, Boyle NJ, et al. Binding of vascular heparan sulfate proteoglycan to Alzheimer's amyloid precursor protein is mediated in part by the N-terminal region of A4 peptide. Brain Res. 1993;627:199–204.
Lindahl B, Westling C, Gimenez-Gallego G, Lindahl U, Salmivirta M. Common binding sites for beta-amyloid fibrils and fibroblast growth factor-2 in heparan sulfate from human cerebral cortex. J Biol Chem. 1999;274:30631–5.
Castillo GM, Lukito W, Wight TN, Snow AD. The sulfate moieties of glycosaminoglycans are critical for the enhancement of beta-amyloid protein fibril formation. J Neurochem. 1999;72:1681–7.
Snow AD, Sekiguchi RT, Nochlin D, Kalaria RN, Kimata K. Heparan sulfate proteoglycan in diffuse plaques of hippocampus but not of cerebellum in Alzheimer's disease brain. Am J Pathol. 1994;144:337–47.
Gupta-Bansal R, Frederickson RC, Brunden KR. Proteoglycan-mediated inhibition of A beta proteolysis. A potential cause of senile plaque accumulation. J Biol Chem. 1995;270:18666–71.
Bame KJ, Danda J, Hassall A, Tumova S. Abeta (1-40) prevents heparanase-catalyzed degradation of heparan sulfate glycosaminoglycans and proteoglycans in vitro. A role for heparan sulfate proteoglycan turnover in Alzheimer's disease. J Biol Chem. 1997;272:17005–11.
Garcia B, Martin C, Garcia-Suarez O, Muniz-Alonso B, Ordiales H, Fernandez-Menendez S, et al. Upregulated Expression of Heparanase and Heparanase 2 in the Brains of Alzheimer's Disease. J Alzheimers Dis. 2017;58:185–92.
Jendresen CB, Cui H, Zhang X, Vlodavsky I, Nilsson LN, Li JP. Overexpression of heparanase lowers the amyloid burden in amyloid-beta precursor protein transgenic mice. J Biol Chem. 2015;290:5053–64.
Donahue JE, Berzin TM, Rafii MS, Glass DJ, Yancopoulos GD, Fallon JR, et al. Agrin in Alzheimer's disease: altered solubility and abnormal distribution within microvasculature and brain parenchyma. Proc Natl Acad Sci U S A. 1999;96:6468–72.
Verbeek MM, Otte-Holler I, van den Born J, van den Heuvel LP, David G, Wesseling P, et al. Agrin is a major heparan sulfate proteoglycan accumulating in Alzheimer's disease brain. Am J Pathol. 1999;155:2115–25.
van Horssen J, Otte-Holler I, David G, Maat-Schieman ML, van den Heuvel LP, Wesseling P, et al. Heparan sulfate proteoglycan expression in cerebrovascular amyloid beta deposits in Alzheimer's disease and hereditary cerebral hemorrhage with amyloidosis (Dutch) brains. Acta Neuropathol. 2001;102:604–14.
Xu Z, Xiao N, Chen Y, Huang H, Marshall C, Gao J, et al. Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Abeta accumulation and memory deficits. Mol Neurodegener. 2015;10:58.
Murlidharan G, Crowther A, Reardon RA, Song J, Asokan A. Glymphatic fluid transport controls paravascular clearance of AAV vectors from the brain. JCI Insight. 2016;1:e88034.
Achariyar TM, Li B, Peng W, Verghese PB, Shi Y, McConnell E, et al. Glymphatic distribution of CSF-derived apoE into brain is isoform specific and suppressed during sleep deprivation. Mol Neurodegener. 2016;11:74.
Xia M, Yang L, Sun G, Qi S, Li B. Mechanism of depression as a risk factor in the development of Alzheimer's disease: the function of AQP4 and the glymphatic system. Psychopharmacology (Berl). 2017;234:365–79.
Timmer NM, Herbert MK, Kleinovink JW, Kiliaan AJ, De Waal RM, Verbeek MM. Limited expression of heparan sulphate proteoglycans associated with Abeta deposits in the APPswe/PS1dE9 mouse model for Alzheimer's disease. Neuropathol Appl Neurobiol. 2010;36:478–86.
Snow AD, Mar H, Nochlin D, Kimata K, Kato M, Suzuki S, et al. The presence of heparan sulfate proteoglycans in the neuritic plaques and congophilic angiopathy in Alzheimer's disease. Am J Pathol. 1988;133:456–63.
Wright S, Parham C, Lee B, Clarke D, Auckland L, Johnston J, et al. Perlecan domain V inhibits alpha2 integrin-mediated amyloid-beta neurotoxicity. Neurobiol Aging. 2012;33:1379–88.
Parham CL, Shaw C, Auckland LD, Dickeson SK, Griswold-Prenner I, Bix G. Perlecan Domain V Inhibits Amyloid-beta Induced Activation of the alpha2beta1 Integrin-Mediated Neurotoxic Signaling Cascade. J Alzheimers Dis. 2016;54:1629–47.
Parham C, Auckland L, Rachwal J, Clarke D, Bix G. Perlecan domain V inhibits amyloid-beta induced brain endothelial cell toxicity and restores angiogenic function. J Alzheimers Dis. 2014;38:415–23.
Parkinson J. An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci. 2002;14:223–36 discussion 222.
Obeso JA, Stamelou M, Goetz CG, Poewe W, Lang AE, Weintraub D, et al. Past, present, and future of Parkinson's disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov Disord. 2017;32:1264–310.
Lang AE. A critical appraisal of the premotor symptoms of Parkinson's disease: potential usefulness in early diagnosis and design of neuroprotective trials. Mov Disord. 2011;26:775–83.
Klein C, Westenberger A. Genetics of Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a008888.
Ascherio A, Schwarzschild MA. The epidemiology of Parkinson's disease: risk factors and prevention. Lancet Neurol. 2016;15:1257–72.
Lees AJ, Hardy J, Revesz T. Parkinson's disease. Lancet. 2009;373:2055–66.
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–7.
Fearnley JM, Lees AJ. Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain. 1991;114(Pt 5):2283–301.
Sui YT, Bullock KM, Erickson MA, Zhang J, Banks WA. Alpha synuclein is transported into and out of the brain by the blood-brain barrier. Peptides. 2014;62:197–202.
Tokuda T, Salem SA, Allsop D, Mizuno T, Nakagawa M, Qureshi MM, et al. Decreased alpha-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson's disease. Biochem Biophys Res Commun. 2006;349:162–6.
Mollenhauer B, Cullen V, Kahn I, Krastins B, Outeiro TF, Pepivani I, et al. Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp Neurol. 2008;213:315–25.
Hong Z, Shi M, Chung KA, Quinn JF, Peskind ER, Galasko D, et al. DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson's disease. Brain. 2010;133:713–26.
Wilhelmus MM, Bol JG, Van Haastert ES, Rozemuller AJ, Bu G, Drukarch B, et al. Apolipoprotein E and LRP1 Increase Early in Parkinson's Disease Pathogenesis. Am J Pathol. 2011;179:2152–6.
Kim SY, Buckwalter M, Soreq H, Vezzani A, Kaufer D. Blood-brain barrier dysfunction-induced inflammatory signaling in brain pathology and epileptogenesis. Epilepsia. 2012;53(Suppl 6):37–44.
Li Y, Li Y, Pang S, Huang W, Zhang A, Hawley RG, et al. Novel and functional ABCB1 gene variant in sporadic Parkinson's disease. Neurosci Lett. 2014;566:61–6.
Booth HDE, Hirst WD, Wade-Martins R. The Role of Astrocyte Dysfunction in Parkinson's Disease Pathogenesis. Trends Neurosci. 2017;40:358–70.
Bertrand E, Lewandowska E, Stepien T, Szpak GM, Pasennik E, Modzelewska J. Amyloid angiopathy in idiopathic Parkinson's disease. Immunohistochemical and ultrastructural study. Folia Neuropathol. 2008;46:255–70.
Yang P, Pavlovic D, Waldvogel H, Dragunow M, Synek B, Turner C, et al. String Vessel Formation is Increased in the Brain of Parkinson Disease. J Parkinsons Dis. 2015;5:821–36.
Paiva I, Jain G, Lazaro DF, Jercic KG, Hentrich T, Kerimoglu C, et al. Alpha-synuclein deregulates the expression of COL4A2 and impairs ER-Golgi function. Neurobiol Dis. 2018;119:121–35.
Firtina Z, Danysh BP, Bai X, Gould DB, Kobayashi T, Duncan MK. Abnormal expression of collagen IV in lens activates unfolded protein response resulting in cataract. J Biol Chem. 2009;284:35872–84.
Manthorpe M, Engvall E, Ruoslahti E, Longo FM, Davis GE, Varon S. Laminin promotes neuritic regeneration from cultured peripheral and central neurons. J Cell Biol. 1983;97:1882–90.
Riopelle RJ, Dow KE. Functional interactions of neuronal heparan sulphate proteoglycans with laminin. Brain Res. 1990;525:92–100.
Mammadov B, Mammadov R, Guler MO, Tekinay AB. Cooperative effect of heparan sulfate and laminin mimetic peptide nanofibers on the promotion of neurite outgrowth. Acta Biomater. 2012;8:2077–86.
Vaananen AJ, Rauhala P, Tuominen RK, Liesi P. KDI tripeptide of gamma1 laminin protects rat dopaminergic neurons from 6-OHDA induced toxicity. J Neurosci Res. 2006;84:655–65.
Sever M, Turkyilmaz M, Sevinc C, Cakir A, Ocalan B, Cansev M, et al. Regenerative effects of peptide nanofibers in an experimental model of Parkinson's disease. Acta Biomater. 2016;46:79–90.
Zhang D, Yang S, Toledo EM, Gyllborg D, Salto C, Carlos Villaescusa J, et al. Niche-derived laminin-511 promotes midbrain dopaminergic neuron survival and differentiation through YAP. Sci Signal. 2017;10:eaal4165.
Stiles B, Groszer M, Wang S, Jiao J, Wu H. PTENless means more. Dev Biol. 2004;273:175–84.
van Horssen J, de Vos RA, Steur EN, David G, Wesseling P, de Waal RM, et al. Absence of heparan sulfate proteoglycans in Lewy bodies and Lewy neurites in Parkinson's disease brains. J Alzheimers Dis. 2004;6:469–74.
Mehta P, Kaye W, Raymond J, Punjani R, Larson T, Cohen J, et al. Prevalence of Amyotrophic Lateral Sclerosis - United States, 2015. MMWR Morb Mortal Wkly Rep. 2018;67:1285–9.
Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011;7:603–15.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62.
Kaur SJ, McKeown SR, Rashid S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene. 2016;577:109–18.
Donnenfeld H, Kascsak RJ, Bartfeld H. Deposits of IgG and C3 in the spinal cord and motor cortex of ALS patients. J Neuroimmunol. 1984;6:51–7.
Kwan JY, Jeong SY, Van Gelderen P, Deng HX, Quezado MM, Danielian LE, et al. Iron accumulation in deep cortical layers accounts for MRI signal abnormalities in ALS: correlating 7 tesla MRI and pathology. PLoS One. 2012;7:e35241.
Yamadera M, Fujimura H, Inoue K, Toyooka K, Mori C, Hirano H, et al. Microvascular disturbance with decreased pericyte coverage is prominent in the ventral horn of patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:393–401.
Ono S, Imai T, Munakata S, Takahashi K, Kanda F, Hashimoto K, et al. Collagen abnormalities in the spinal cord from patients with amyotrophic lateral sclerosis. J Neurol Sci. 1998;160:140–7.
Ono S, Imai T, Takahashi K, Jinnai K, Yamano T, Nagao K, et al. Decreased type IV collagen of skin and serum in patients with amyotrophic lateral sclerosis. Neurology. 1998;51:114–20.
Rao JS, Hantai D, Festoff BW. Thrombospondin, a platelet alpha-granule and matrix glycoprotein, is increased in muscle basement membrane of patients with amyotrophic lateral sclerosis. J Neurol Sci. 1992;113:99–107.
Wiksten M, Vaananen A, Liesi P. Selective overexpression of gamma1 laminin in astrocytes in amyotrophic lateral sclerosis indicates an involvement in ALS pathology. J Neurosci Res. 2007;85:2045–58.
Ono S, Imai T, Shimizu N, Nagao K. Increased expression of laminin 1 in the skin of amyotrophic lateral sclerosis. Eur Neurol. 2000;43:215–20.
Liu JX, Brannstrom T, Andersen PM, Pedrosa-Domellof F. Different impact of ALS on laminin isoforms in human extraocular muscles versus limb muscles. Invest Ophthalmol Vis Sci. 2011;52:4842–52.
Dobrowolny G, Giacinti C, Pelosi L, Nicoletti C, Winn N, Barberi L, et al. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J Cell Biol. 2005;168:193–9.
Coatti GC, Frangini M, Valadares MC, Gomes JP, Lima NO, Cavacana N, et al. Pericytes Extend Survival of ALS SOD1 Mice and Induce the Expression of Antioxidant Enzymes in the Murine Model and in IPSCs Derived Neuronal Cells from an ALS Patient. Stem Cell Rev Rep. 2017;13:686–98.
Wiksten M, Vaananen A, Liebkind R, Rauhala P, Liesi P. Soluble KDI domain of gamma1 laminin protects adult hippocampus from excitotoxicity of kainic acid. J Neurosci Res. 2004;78:411–9.
McConville J, Vincent A. Diseases of the neuromuscular junction. Curr Opin Pharmacol. 2002;2:296–301.
Baeten KM, Akassoglou K. Extracellular matrix and matrix receptors in blood-brain barrier formation and stroke. Dev Neurobiol. 2011;71:1018–39.
Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–9.
Yurchenco PD. Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harb Perspect Biol. 2011;3:a004911.
Segel M, Neumann B, Hill MFE, Weber IP, Viscomi C, Zhao C, et al. Niche stiffness underlies the ageing of central nervous system progenitor cells. Nature. 2019;573:130–4.
Granato AEC, da Cruz EF, Rodrigues-Junior DM, Mosini AC, Ulrich H, Rodrigues BVM, et al. A novel decellularization method to produce brain scaffolds. Tissue Cell. 2020;67:101412.
Gilbert TW, Wognum S, Joyce EM, Freytes DO, Sacks MS, Badylak SF. Collagen fiber alignment and biaxial mechanical behavior of porcine urinary bladder derived extracellular matrix. Biomaterials. 2008;29:4775–82.
Gaudet C, Marganski WA, Kim S, Brown CT, Gunderia V, Dembo M, et al. Influence of type I collagen surface density on fibroblast spreading, motility, and contractility. Biophys J. 2003;85:3329–35.
Gessel M, Spraggins JM, Voziyan P, Hudson BG, Caprioli RM. Decellularization of intact tissue enables MALDI imaging mass spectrometry analysis of the extracellular matrix. J Mass Spectrom. 2015;50:1288–93.
Funding
This work was partially supported by R01HL146574 (to YY), RF1AG065345 (to YY), R21AG073862 (to YY), R21AG064422 (to YY), and American Heart Association Scientist Development Grant 16SDG29320001 (to YY).
Author information
Authors and Affiliations
Contributions
BN searched the literature and wrote the manuscript. GB contributed to the writing of the perlecan sections and co-edited the manuscript. YY drew the diagrams in the figures, supervised the writing, and co-edited the manuscript. All authors have read and agreed on the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
All authors agreed to publish.
Competing interests
The authors declared no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Nguyen, B., Bix, G. & Yao, Y. Basal lamina changes in neurodegenerative disorders. Mol Neurodegeneration 16, 81 (2021). https://doi.org/10.1186/s13024-021-00502-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-021-00502-y