
Abstract
Investigations of apolipoprotein E (APOE) gene, the major genetic risk modifier for Alzheimer’s disease (AD), have yielded significant insights into the pathogenic mechanism. Among the three common coding variants, APOE*ε4 increases, whereas APOE*ε2 decreases the risk of late-onset AD compared with APOE*ε3. Despite increased understanding of the detrimental effect of APOE*ε4, it remains unclear how APOE*ε2 confers protection against AD. Accumulating evidence suggests that APOE*ε2 protects against AD through both amyloid-β (Aβ)-dependent and independent mechanisms. In addition, APOE*ε2 has been identified as a longevity gene, suggesting a systemic effect of APOE*ε2 on the aging process. However, APOE*ε2 is not entirely benign; APOE*ε2 carriers exhibit increased risk of certain cerebrovascular diseases and neurological disorders. Here, we review evidence from both human and animal studies demonstrating the protective effect of APOE*ε2 against AD and propose a working model depicting potential underlying mechanisms. Finally, we discuss potential therapeutic strategies designed to leverage the protective effect of APOE2 to treat AD.
Similar content being viewed by others
Background
Apolipoprotein E (APOE), as an apolipoprotein mediating lipid metabolism in circulation and the brain, is the strongest genetic risk modifier of late-onset Alzheimer’s disease (LOAD, referred to as AD in this review) [1,2,3,4]. Among the three common coding variants of APOE, APOE*ε4 increases, whereas APOE*ε2 decreases, the risk of AD compared with the most common APOE*ε3 allele [5, 6]. The mechanism underlying the protective effect of APOE*ε2 against AD remains unclear. Human studies show that APOE*ε2 is associated with reduced Aβ deposition in the brains of non-demented aged individuals and AD patients [7,8,9,10,11], suggesting that APOE*ε2 reduces AD risk at least partially through Aβ-dependent pathways. APOE*ε2 may also protect against AD through Aβ-independent pathways. Supporting this, APOE*ε2/2 and APOE*ε2/3 individuals (referred to as APOE*ε2 carriers in this review) are more likely to be cognitively intact compared with APOE*ε3/3 homozygotes among individuals with minimal Aβ pathology [12]. In addition, studies show that APOE*ε2 protects against cognitive impairment in individuals over 90 years of age who have high levels of Aβ in the brain [13, 14]. In vitro and in vivo studies suggest multiple potential pathways through which APOE2 confers protection independently of Aβ pathology. These pathways likely involve the neuroprotective effect of APOE2 and the regulatory roles of APOE2 in lipid metabolism and synaptic functions [15,16,17,18].
Although APOE*ε2 has also been associated with longevity [19,20,21,22,23], which might be independent of its protective role against AD [24, 25], it is not entirely benign. APOE*ε2 is associated with an increased risk of cerebral amyloid angiopathy (CAA) which often co-exists with AD pathology and is a major cause of hemorrhagic stroke [26, 27]. APOE*ε2 is also associated with increased risk of certain neurological disorders such as post-traumatic stress disorder (PTSD) [28], age-related macular degeneration (AMD) [29], supranuclear palsy (PSP), and argyrophilic grain disease (AGD) [30, 31]. In this review, we summarize recent progress in APOE*ε2 research and propose a hypothetical working model depicting the protective effect of APOE*ε2 against AD. We also discuss potential therapeutic strategies for AD inspired by APOE*ε2-related protective mechanisms.
Main text
Biology of APOE
Human APOE
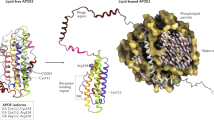
Human APOE is a 34-kDa glycoprotein consisting of 299 amino acids [32], encoded by the APOE gene located on chromosome 19q13.32 [33]. The three allelic variants, namely, APOE*ε2, APOE*ε3, and APOE*ε4, encode three isoforms that differ from each other at two amino acid positions 112 and 158: APOE2 (Cys112; Cys158), APOE3 (Cys112; Arg158), and APOE4 (Arg112; Arg158) [32, 34]. Structurally, APOE has two independently-folded domains referred to as the N-terminal domain and the C-terminal domain [35, 36] (Fig. 1a). These two domains are linked by a flexible loop region that is thrombolytically cleavable [37, 38]. The N-terminal domain contains the receptor-binding site (residues 136-150) [39], whereas the C-terminal domain contains the lipid-binding region (residues 244-272) [4, 40]. Additionally, residues 136-147 in the N-terminal domain and the basic residue Lys233 in the C-terminal domain are required for APOE binding to heparin/heparan sulfate polysaccharide chains of HSPG, another important receptor of APOE [41, 42].

Human APOE. a Human APOE is an O-linked glycoprotein consisting of 299 amino acids. The N-terminal domain (residues 1-167) and the C-terminal domain (residues 206-299) are linked by a flexible hinge region. The receptor binding site (residues 136-150) on the N-terminus overlaps with the heparin binding region (residues 136-147). A second heparin-binding site on the C-terminal domain adjacent to the lipid binding site (residue 244-272) requires K233. Amino acid substitutions at position 112 and 158 result in the three major isoforms: APOE2 (Cys112; Cys158), APOE3 (Cys112; Arg158) and APOE4 (Arg158; Arg158). APOE has other less common isoforms; APOE (V236E) and APOE3 Christchurch (R136S) (blue triangles) are two examples that have also been suggested to protect against AD. b Lipidated APOE-containing lipoprotein particles contain phospholipids and unesterified cholesterol in the shell, and esterified cholesterol and triglycerides form the core. APOE molecules are partially embedded in the phospholipid layer of the particles
In humans, peripheral and central nervous system (CNS) APOE do not cross the blood-brain barrier (BBB), thus forming two independent APOE pools with no APOE-containing lipoprotein exchange [43]. In the periphery, APOE is produced primarily by liver hepatocytes [44], while in the CNS, the majority of APOE derives from astrocytes, microglia, vascular mural cells, and the choroid plexus [45, 46]. Stressed neurons also produce APOE, albeit to a much lesser extent [45, 47]. APOE levels in human plasma follow the APOE genotype rank order of APOE*ε2/2 > APOE*ε2/3 > APOE*ε3/3 (or APOE*ε2/4) > APOE*ε3/4 > APOE*ε4/4 [48,49,50,51]. In contrast, the impact of APOE genotype on CSF APOE levels varies across studies with different quantification methods. While enzyme-linked immunosorbent assay (ELISA)-based measurements show a similar APOE genotype effect to that in plasma [48], mass-spectrometric assays find no such effect [52, 53]. Similar to results from human plasma, cortical APOE levels measured by Western blot and ELISA are highest in APOE*ε2 carriers and lowest in APOE*ε4 carriers [54]. This is consistent with observations from APOE-targeted replacement (APOE-TR) mice in which the murine Apoe gene locus is replaced with human APOE alleles [55], showing that APOE2-TR mice have higher levels of APOE in the interstitial fluid (ISF) and brain lysate than APOE3-TR mice, followed by APOE4-TR mice [56,57,58].
APOE receptors
APOE functions through binding to cell surface receptors, including low-density lipoprotein receptor (LDLR), very low-density lipoprotein receptor (VLDLR), LDLR-related protein 1 (LRP1), APOE receptor 2 (APOER2, also known as LRP8), and heparan sulfate proteoglycans (HSPGs) [59,60,61]. In addition, recent studies show the triggering receptor expressed on myeloid cells 2 (TREM2), which is specifically expressed by microglia in the brain, is a receptor for APOE [62,63,64]. The interaction between APOE and receptors shows isoform-specificity and is affected by APOE lipidation status (Table 1), which is best exemplified by LDLR that recognizes only lipidated APOE [67,68,69], and shows much weaker binding to APOE2 relative to APOE3 and APOE4 [65, 66].
APOE binding to receptors either triggers the uptake of APOE or activates downstream singling cascades involving primarily mitogen-activated protein (MAP) kinases [15,16,17, 61]. The APOE receptor-mediated ligand uptake represents the major mechanism of lipoprotein clearance in the periphery and lipid transport in the CNS [70, 91]. However, the physiological role of APOE-triggered signaling pathways is less clear. In vitro studies show that APOE, regardless of the lipidation status, triggers diverse signaling pathways in neurons, likely through LRP1, to support versatile functions such as neuronal protection and synaptogenesis [15,16,17]. The functional significance of the interaction between APOE and TREM2 remains to be elucidated, although evidence suggests a role in microglia-mediated clearance of Aβ and damaged neurons [64, 87].
Biological functions of APOE
APOE and lipid metabolism
In the periphery, APOE plays a major role in mediating the clearance of triglyceride-rich lipoproteins (chylomicrons, VLDL, and their remnants) by interacting with hepatic APOE receptors [70]. Individuals of different APOE genotypes differ in their plasma lipid profiles. Compared with APOE*ε3/3 homozygotes, APOE*ε3/4 and APOE*ε4/4 individuals (referred to as APOE*ε4 carriers in this review) exhibit higher levels of total cholesterol, LDL, and triglycerides (TGs), and lower levels of HDL, whereas APOE*ε2 carriers have lower levels of total cholesterol and LDL, and higher levels of HDL and TGs in the plasma [92, 93]. The APOE genotype-specific plasma lipid profile is a combinatory result of multiple factors [70, 94]. For example, while impaired binding of APOE2 to LDLR is causally linked to type III hyperlipoproteinemia, characterized by the accumulation of remnants of TG-rich lipoproteins [65, 66, 94, 95], hyperlipidemia is only observed in 5-10% of APOE*ε2/2 homozygotes [94]. The majority of APOE*ε2 carriers have normal or, paradoxically, hypolipemic profile, which is thought to be partially caused by the lower efficiency of lipolytic conversion of APOE2-containing VLDL and IDL to HDL [96,97,98]. Notably, the lipid profile of APOE2-TR mice resembles the small portion of human APOE*ε2 homozygotes who develop hyperlipidemia [99], raising cautions when interpreting results from studies using APOE2-TR mice.
In the CNS, APOE is the major apolipoprotein that transports lipids [91]. CNS APOE is lipidated by cell surface ATP-binding cassette transporters ABCA1 or ABCG1 [100,101,102,103]. Lipidated APOE forms HDL-like particles in size and density containing free cholesterol and phospholipids [104,105,106]. Brain-specific deficiency of Abca1 in mice results in impairments in motor activity and sensorimotor functions, and changes in synaptic structures [107], suggesting a crucial role of APOE-mediated lipid metabolism in the CNS. However, no substantial difference in the brain lipidomics profile has been identified between APOE2-TR, APOE3-TR, and APOE4-TR mice at young and middle-age [108], although aged APOE2-TR mice have lower cortical cholesterol levels than APOE3-TR and APOR4-TR mice [12]. In human AD brains, APOE*ε2 carriers and APOE*ε3/3 homozygotes have similar lipidomics profiles, whereas APOE*ε4 carriers have a significant reduction in ten major lipid classes, including phosphatidylethanolamine, phosphatidic acid, and mitochondrial membrane bilayer-forming phospholipids [109]. Future studies elucidating the role of APOE isoforms in cell type-specific lipid metabolism may aid our understanding of the mechanisms underlying APOE-associated AD risks.
Neurotrophic effect of APOE
The neurotrophic effect of APOE has been well-documented. However, questions remain regarding isoform-specific effects. APOE3, regardless of the lipidation status, promotes neurite outgrowth through a mechanism depending on LRP1, whereas APOE4 has no effect or inhibitory effect [75,76,77,78,79,80]. In addition, APOE3-containing HDL lipoprotein particles protect neurons from apoptosis induced by nutrient depletion at a higher efficiency than APOE4-containing particles, which requires LRP1 as well [16]. APOE also promotes synaptogenesis through mediating cholesterol transport from astrocytes to neurons [110]; however, it is unclear whether the effect is APOE isoform-specific. The neurotrophic effect of APOE2 relative to those of APOE3 and APOE4 has been less studied. Although APOE2-TR mice displayed longer dendritic spines and increased apical dendritic arborization in the cortex at one month of age compared with APOE3-TR mice, the differences have not been observed in older animals [111]. Moreover, there is no difference in dendritic spine density in the hippocampus of APOE2-TR, APOE3-TR, and APOE4-TR mice at different ages [111].
APOE and synaptic functions
Synaptic dysfunction is one of the earliest pathological changes in AD [112, 113]. In vitro data suggest a regulatory role of APOE in synaptic functions. Astrocyte-derived APOE4, but not APOE2 or APOE3, reduces the levels of postsynaptic APOER2, N-methyl-D-aspartate receptor (NMDAR), and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) in cultured neurons by sequestering the receptors in the intracellular compartment [18]. Additionally, lipidated APOE2 enhances, whereas APOE4 suppresses glutamate-induced calcium influx through NMDAR in the presence of Reelin [18]. Lipidated APOE2 also enhances the elimination of synapses by astrocytes more than APOE3 and APOE4 in culture, indicating that APOE2 may protect synaptic functions by reducing senescent synapses and the accumulation of neural debris [114].
In vivo, young adult APOE2-TR, APOE3-TR, and APOE4-TR mice have similar levels of postsynaptic density protein 95 (PSD-95) in the cortex and hippocampus [12]. However, electrophysiological studies show comparable or lower LTP amplitudes in APOE2-TR mice compared with APOE3-TR mice [115, 116]. The absence of an APOE isoform effect on synaptic functions in young APOE-TR mice is not surprising given comparable cognitive performance between APOE2-TR, APOE3-TR, and APOE4-TR mice, and between humans of different APOE genotypes at young ages (< 60 years old) [12, 117, 118]. Since the protective effect of APOE2 against cognitive decline is most prominent in the elderly [12, 119, 120], one would assume a better synaptic function in aged APOE2-TR mice compared with APOE3-TR and APOE4-TR mice of the similar age. Indeed, one study show that aged APOE4-TR mice display poorer spatial memory acquisition, whereas APOE2-TR mice exhibit better spatial memory retention than APOE3-TR mice [12].
APOE and innate immunity
Innate immunity plays a crucial role in AD pathogenesis [121,122,123,124,125]. The involvement of APOE in AD-associated immune response is evident in recent transcriptomics studies [89, 90, 126,127,128]. In amyloid mouse models, APOE upregulation is a major molecular signature of the subtype of microglia known as disease-associated microglia (DAM) [89, 90, 126]. The acquisition of the neurodegenerative phenotype (MGnD) of DAM is driven by Aβ plaques via a TREM2-dependent pathway [89, 90]. Trem2 knockout abolishes Aβ-driven upregulation of Apoe and reduces plaque-associated APOE protein in amyloid mouse models [89, 129]. Consistent with these findings, microglial APOE is also upregulated in pathologically-confirmed human AD brains [126, 127].
APOE likely modulates microglial function in an isoform-dependent manner through TREM2-mediated pathways [130]. However, it remains unclear how APOE isoforms differentially regulate the immune response, in particular in AD pathogenesis. Evidence from studies of lipopolysaccharide (LPS)-induced immune reactivity shows a greater response associated with APOE*ε4 [131,132,133,134]. However, there are conflicting results regarding the regulatory role of APOE*ε2 in innate immunity. Although one study showed that microglial culture derived from APOE2-TR mice display reduced immune response upon LPS treatment than that derived from APOE3-TR mice [134], others found no such difference [131]. Moreover, APOE2-TR and APOE3-TR mice show comparable cytokine release and glial activation after intracerebroventricular LPS injection [135]. As LPS treatment induces acute immune responses, which does not capture the AD-related conditions, future studies on APOE isoform-specific role in innate immunity should be carried out with AD mouse models bearing amyloid and/or tau pathology.
APOE and blood-brain barrier integrity
BBB breakdown is present in multiple neurodegenerative diseases, including AD [136]. Animal studies show that APOE*ε4 correlates with decreased BBB integrity [137] and slower BBB repair after brain injury [138], which is consistent with the observation in humans that aged APOE*ε4 carriers have increased BBB permeability compared with APOE*ε3 homozygotes, irrespective of the cognitive status [139]. Moreover, the association between APOE*ε4 and BBB breakdown in humans is independent of Aβ and tau pathologies [139], but appears to be caused by functional changes of pericytes [137, 139, 140]. However, whether APOE*ε2 also affects BBB integrity in humans and animal models remains elusive.
Protective effect of APOE*ε2
APOE*ε2 and brain structure
Progressive cortical thinning and volume loss occur along the AD trajectory, namely, from cognitively normal to mild cognitive impairment (MCI) to AD [141,142,143,144,145,146]. However, it remains unclear whether APOE*ε2 reduces AD risk by preserving the cortical structure. Evidence from imaging studies shows no structural difference in cortices between APOE*ε2 carriers and APOE*ε3/3 homozygotes in children and young adolescents [147,148,149]. However, studies of adults yield conflicting results. Although some investigators report that APOE*ε2 is associated with increased cortical thickness and lower atrophy rate in sub-regions of the temporal lobe relative to APOE*ε3/3 homozygotes in non-demented aged people [150,151,152], others find no such difference [153, 154]. Nevertheless, APOE*ε2 carriers appear to have better preserved cortical structures than non-carriers among MCI and AD patients [152, 154], a finding that requires validation in larger cohorts.
APOE*ε2 and cognition
A plausible explanation of the protective effect of APOE*ε2 against AD may be that APOE*ε2 carriers have better baseline cognition, which sets a higher threshold for cognitive impairment. However, efforts to identify the beneficial effects of APOE*ε2 on cognition in young to middle-aged non-demented individuals have generated mixed results. Although one study reported that non-demented, middle-aged APOE*ε2 carriers perform slightly better in cognitive domains including episodic memory and executive functions [155], APOE exerts no effect on intelligence quotient (IQ), memory and school attainment tests in children and college students [156, 157]. Likewise, another study on a community-based cohort in Australia failed to identify APOE*ε2 effects on a battery of cognitive tests in non-demented individuals aged 20 to 60+ [118].
In contrast to observations from young subjects, the cognitive effect of APOE*ε2 in non-demented aged people is more consistent across studies. APOE*ε2 carriers outperform non-carriers in memory tests, visuospatial measures, and global cognition in cross-sectional studies [158,159,160]. Moreover, longitudinal studies show that APOE*ε2 carriers have lower rate of age-related decline in global cognition [12, 161], episodic memory [119], executive function [120], and verbal learning ability [162]. Interestingly, the protective effect of APOE*ε2 on cognition is more prominent in females than in males [12, 163].
APOE*ε2 and longevity
APOE*ε2 has been well-associated with longevity. Cauley and colleagues first reported a higher allele frequency of APOE*ε2 and a lower allele frequency of APOE*ε4 in the elderly than those middle-aged [164]. Although their study focused exclusively on females, similar observations have been reported in French male centenarians [165]. These results have been further validated by cross-sectional case-control studies [166,167,168] and longitudinal studies [24, 25]. The association between the APOE gene locus and longevity has also been confirmed by several case-control genome-wide association studies (GWAS) [19,20,21,22,23].
Despite ample evidence supporting the APOE allele-specific effect on longevity, the mechanisms driving the effect remain unknown. Although APOE*ε2 may increase longevity by protecting against AD [169], evidence also suggests a beneficial effect of APOE*ε2 on survival among cognitively normal individuals [24, 25]. Likewise, although dementia is likely the major cause of death among seniors of APOE*ε4 carriers [25], APOE*ε4 also mediates a detrimental effect on survival in non-demented aged people [24]. Furthermore, evidence shows that non-sex-specific cancer reduces life expectancy in APOE*ε4 carriers more than in non-carriers [170].
APOE*ε2 protects against AD: the clinical evidence
The protective effect of APOE*ε2 against AD was first uncovered in 1994 when the APOE*ε2 allele was found to be underrepresented in AD patients [171, 172]. Compared to APOE*ε3/3 homozygotes, the risk of AD in APOE*ε2 carriers is approximately 50% less [5, 6]. Moreover, AD patients who are APOE*ε2 carriers exhibit slower cognitive decline compared with non-carriers [173]. APOE*ε2 also protects against AD in Down’s syndrome (DS) patients whose amyloid-beta precursor protein (APP) gene is triplicated [174]. Amongst DS individuals, APOE*ε2 carriers have reduced risk and delayed age at onset of AD [175,176,177].
How demographic factors such as gender, race, and age may modify the protective effect of APOE*ε2 against AD has been investigated. For example, APOE*ε2 appears to be more protective in females than in males [178], but equally protective across ethnicities [5]. Although the effect of APOE*ε4 on AD risk peaks at age 60-69, individuals of different age groups are equally protected by APOE*ε2 [6, 179] (Fig. 2a). Furthermore, APOE*ε2 carriers appear to benefit more from cognitive-enhancing life experiences, such as education and reading, regarding their roles in reducing AD risk than non-carriers [180].

APOE*ε2 protects against AD. a Age-stratified odds ratio for AD risk (with APOE*ε3/3 as reference group) in individuals of different APOE genotypes. APOE*ε2/2 and APOE*ε2/3 individuals have reduced risk of AD (OR < 1) compared to APOE*ε3/3 individuals and the protective effect sizes are similar in different age groups. In contrast, APOE*ε4 carriers and APOE*ε2/4 individuals, have increased risk of AD (OR > 1) and the effect size varies among different age groups. b Kaplan-Meier curves showing the percentage of pathologically confirmed AD cases in individuals of different APOE genotypes. APOE*ε2 carriers are less likely to be pathologically diagnosed as AD. The protective effect is more prominent in APOE*ε2/2 homozygotes. a A reproduction of published data by Genin, et al., Mol Psychiatry. 2011 Sep;16(9):903-7, with permission. b A visual adaptation of a figure from Reiman et al., Nat Commun. 2020 Feb 3;11(1):667, with permission
APOE*ε2 protects against AD: the pathological evidence
APOE*ε2 reduces Aβ pathology in humans
The protective effect of APOE*ε2 is more pronounced in pathologically confirmed AD than clinically diagnosed AD [10] (Fig. 2b). Postmortem AD brains from APOE*ε2 carriers have lower densities of Aβ containing neuritic plaques than those from APOE*ε3/3 individuals [7,8,9], suggesting a slower antemortem Aβ deposition in APOE*ε2 carriers. Supporting this, positron emission tomography (PET) imaging in non-demented individuals shows that brain amyloid accumulates at a lower rate in APOE*ε2 carriers than in APOE*ε3/3 homozygotes during aging [11]. Moreover, APOE*ε2 carriers have an older age of amyloid positivity onset than non-carriers [11]. CSF Aβ42 is a widely-used biomarker for AD [181]. Reduced Aβ42 levels in the CSF correlate well with increased Aβ load in the brain shown by amyloid PET imaging [182, 183] or autopsy [184]. Consistent with the imaging study, APOE*ε2 is also associated with higher levels of CSF Aβ42 in middle-aged to aged individuals, irrespective of the cognitive and neurodegeneration status of the subjects [185,186,187].
APOE*ε2 affects not only the global Aβ load but also the region-specific Aβ deposition. Multimodal neuroimaging in non-demented individuals shows reduced amyloid load in the precuneus in APOE*ε2 carriers compared with APOE*ε3/3 homozygotes [188]. Moreover, the precuneal Aβ burden in APOE*ε2/3 individuals does not increase significantly with age, contrasting to non-carriers [188]. Interestingly, despite ample evidence supporting the protective effect of APOE*ε2 against Aβ deposition, studies show that non-demented APOE*ε2 carriers over 90 years of age (oldest old) have a higher burden of neuritic plaques relative to non-carriers [13, 189], raising the possibility that APOE*ε2 carriers are more resilient to Aβ pathology than non-carriers so that the oldest old individuals can survive better from Aβ toxicity and have cognitive functions preserved. The protective role of APOE*ε2 against Aβ-associated toxicity is discussed in detail below.
APOE*ε2 and Aβ aggregation in animal models
How APOE2 affects Aβ deposition has been investigated through crossing 5xFAD mice with APOE-TR mice (denoted as EFAD mice) [190]. One group found that E2FAD mice have similar levels of Aβ42 in the hippocampus at different ages and higher levels of total Aβ42 in the cortex at six months of age compared with E3FAD mice [191]. The lack of protective effect of APOE2 against hippocampal Aβ deposition in animal models has also been shown in a model of PDAPP transgenic mice crossed with APOE-TR mice (denoted as PDAPP/TRE mice) [192]. However, PDAPP/TRE2 animals show lower Aβ load measured by immunohistochemistry in the cortex than PDAPP/TRE3 animals at 18 months of age [192]. The impact of APOE on Aβ pathology has also been investigated through viral-mediated overexpression of human APOE in amyloid mouse models expressing murine Apoe. In PDAPP mice, lentiviral-mediated overexpression of APOE*ε2 reduces hippocampal Aβ levels more than APOE*ε3 and APOE*ε4 overexpression [193]. Consistently, Aβ40 and Aβ42 levels in both soluble and insoluble fractions of the brain lysate are reduced with APOE*ε2, but not APOE*ε3 or APOE*ε4 gene delivery in APP/ PS1 mice [194]. Taken together, these studies suggest that the effect of APOE2 on amyloid pathology in animal models can be affected by age, brain region of interest, the strain of amyloid mouse models, and the presence of murine Apoe.
APOE*ε2 and Aβ production
An imbalance between Aβ production and clearance is considered a crucial event initiating the amyloid cascade in AD [195]. Whether APOE2 impacts Aβ deposition in humans by affecting Aβ production remains inconclusive. Although APOE has a negligible effect on APP processing [196,197,198], there are conflicting results regarding the role of APOE isoforms in APP transcription. One recent study showed that both lipidated and non-lipidated APOE upregulates APP expression in human neurons derived from embryonic stem cells (ESC) or human induced pluripotent stem cells (iPSCs) through the DLK → MKK7 → ERK1/2 signaling pathway. The effect is most prominent for APOE4, followed by APOE3, and then APOE2 [17, 199]. However, the described APOE isoform-specific role in APP transcription conflicts with a transcriptomic study showing that APOE2-TR, APOE3-TR, and APOE4-TR mice have similar levels of endogenous murine App in the brain (the result can be found through the searchable web interface: https://www.epaad.org/blog/index.php/gene-expresssion-database/) [200].
APOE*ε2 and Aβ clearance and degradation
Brain parenchymal Aβ is eliminated through multiple pathways, including cellular uptake, extracellular enzymatic degradation, CSF absorption, clearance via the BBB, and ISF bulk flow [201]. APOE mediates Aβ elimination from the brain in an isoform-dependent manner in which APOE4 mediates Aβ clearance at a lower efficiency than APOE3 [74, 81, 198]. In contrast, APOE2 tends to mediate Aβ clearance across the BBB at a higher efficiency than APOE3 [81, 198]. APOE2 also regulates cellular uptake and degradation of Aβ. One study showed that macrophages in culture from APOE2-TR mice are more efficient in degrading both soluble and insoluble Aβ than macrophages from APOE3-TR and APOE4-TR mice. The higher efficacy of APOE2-associated Aβ degradation is likely related to the enhanced matrix metalloproteinase-9 activity [202]. Additionally, APOE has been shown to mediate soluble Aβ degradation by microglia at an efficacy order of APOE2 > APOE3 > APOE4 [203].
APOE*ε2 protects against Aβ toxicity
Previous studies have shown that amyloid load correlates poorly with cognitive impairment and AD severity [204]. Instead, soluble oligomeric Aβ is suggested to be more directly linked to the neurotoxicity in AD brains [204, 205]. The regulatory role of APOE isoforms in Aβ oligomerization has been demonstrated by split-luciferase assays showing that immortalized astrocyte or HEK293 cell-derived APOE promotes Aβ oligomerization with a potency order of APOE4 > APOE3 > APOE2 [206]. However, the in vitro observation of reduced Aβ oligomerization associated with APOE2 was not supported by a study of EFAD mice reporting similar levels of oligomeric Aβ in the soluble fraction of the brain lysate in E2FAD and E3FAD mice [191]. In addition to different modeling systems used, a direct comparison of results from these two studies can be challenging due to the dynamic nature and complex composition of Aβ oligomeric species [205, 207]. Future studies using combinatory approaches (e.g., conformation-specific antibody-based assay or mass spectrometry) to quantify oligomeric Aβ in the brain lysate and CSF of human subjects of different APOE genotypes may help address the question of whether APOE2 reduces oligomeric, toxic Aβ species.
APOE2 also appears to exert anti-toxic effects against Aβ. Both lipidated and non-lipidated APOE2 protect the B12 neuronal cell line against Aβ25-35-induced cell death more than APOE3 and APOE4 [208]. Moreover, hippocampal slices prepared from young adult APOE2-TR mice are more resistant to AD brain lysate or Aβ42-induced LTP suppression than slices prepared from APOE3-TR and APOE4-TR animals of the same age [18, 209]. There is also evidence suggesting that APOE2 expression reduces synaptic loss and neuritic dystrophy in amyloid mouse models [194, 210]. Additionally, APOE2 appears to confer protection for other brain cell types, including cultured pericytes [211] and endothelial cells [212], which potentially constitute indirect pathways for neuronal protection.
APOE and Aβ interaction: essential for Aβ deposition?
The essential role of murine APOE in Aβ deposition in animal models has been well-recognized [213]. However, inferring the isoform-specific role of human APOE in Aβ deposition based on studies of murine APOE may be difficult as there is only one APOE isoform in mice, which is structurally and functionally different from human APOE [190, 214]. How human APOE is involved in Aβ deposition is not entirely clear. In vitro studies show that human APOE forms SDS-insoluble complexes with Aβ, irrespective of the lipidation status [215,216,217,218,219]. The complex formation requires the C-terminal lipid-binding domain of APOE [220], and shows APOE isoform-dependency, with lipidated APOE2 binds Aβ at a higher affinity than lipidated APOE3, followed by lipidated APOE4 [219, 221]. Consistently, E2FAD mice have higher levels of SDS-resistant APOE/Aβ complex than E3FAD mice > E4FAD mice in brain lysate [222]. In postmortem human brains, APOE co-deposits with Aβ plaques [216, 223]. Taken together, these studies suggest that APOE-Aβ complex formation can either protect against or promote Aβ deposition, likely in an APOE isoform-specific manner. Interestingly, blocking the interaction between human APOE and Aβ with Aβ12-28P, a synthetic peptide that is homologous to the APOE binding domain of Aβ, reduces brain Aβ levels in APP/PS1 mice crossed with APOE-TR animals [224]. However, Verghese et al. show that APOE has minimal binding with soluble Aβ in human CSF and in the ISF of animal models [225], raising the possibility that Aβ deposition in humans does not require APOE/Aβ complex formation, but instead is affected by a direct seeding effect of APOE on amyloids [226, 227].
APOE*ε2 and neurofibrillary tangles (NFTs)
NFTs containing hyperphosphorylated tau represent another pathological hallmark of AD [228,229,230]. Autopsy studies show reduced NFTs in postmortem AD brains of APOE*ε2 carriers [7,8,9]. Although the mechanism underlying this reduction is poorly understood, the protective effect of APOE*ε2 against AD tau may be partially mediated through its effect on Aβ deposition, as APOE*ε2 negatively correlates with tau pathology only in Aβ positive but not in Aβ negative individuals [231]. Whether and to what extent APOE*ε2 may protect against tau pathology independently of Aβ in AD remains elusive.
Progress in our understanding of tau pathogenesis in AD is hampered by a lack of sophisticated mouse models that mimic human NFT tau [232, 233]. The widely used tau models, including rTg (tauP301L)4510 mice and Tau P301S/PS19 mice, carry the familial frontotemporal lobar degeneration (FTLD) MAPT mutation at the P301 residue, which is not found in AD patients [233]. Thus, results from studies using these models should be interpreted carefully. Bearing this in mind, one study showed that PS19 mice have similar levels of tau pathology and brain atrophy when crossed with APOE2-TR mice versus when crossed with APOE3-TR mice [131]. However, another study found that viral-mediated TauP301L expression induces more tau pathology in APOE2-TR mice than in APOE3-TR mice, suggesting that APOE2 increases the risk of primary tauopathies [30]. Supporting this, APOE*ε2 has been associated with increased risks of PSP and argyrophilic grain disease (AGD) in humans [30, 31]. Future studies to gain mechanistic insights into the impact of APOE isoforms on AD tau require novel animal models that harbor both Aβ and tau pathologies. In addition, the emerging tau PET imaging will permit the exploration of tau pathogenesis in human brains in vivo [234, 235].
How APOE*ε2 protects against AD: a working model
Taken together, APOE*ε2 may protect against AD through multiple, interconnected mechanisms. Based on a growing body of evidence, we propose that hyperlipidation of APOE2 is a central mechanism underlying the protective effect of APOE*ε2 (Fig. 3). Although direct evidence showing increased lipidation of APOE2 relative to APOE3 and APOE4 in human brains is not available, accumulating evidence demonstrates that APOE2 from human CSF [236], immortalized astrocytes [237], as well as primary microglia and astrocyte culture derived from human APOE knock-in mice, are more lipidated than APOE3 and APOE4 [46]. Lipidation substantially impacts APOE binding to receptors and other proteins, such as Aβ [67,68,69, 238], and also affects APOE catabolism, leading to changes in peripheral and CNS APOE levels [239]. Differential lipidation of APOE isoforms potentially contributes to the distinct cognitive and pathological outcomes in humans of different APOE genotypes through both Aβ-independent (e.g., neurotrophic effect, lipid metabolism, synaptic function, and immunomodulation) and Aβ-dependent pathways.

Potential mechanisms underlying APOE2 protective effects against AD. CNS APOE is produced primarily by astrocytes, and also by activated microglia. Newly synthesized APOE is lipidated through cell surface ABCA1 or ABCG1, generating HDL-like lipoprotein particles. In the CNS, APOE2-containing lipoprotein particles are more lipidated than APOE3 and APOE4-containing particles, thus are larger in size. The lipidation of APOE can be modulated by targeting the transcription factors, LXR, and RXR, which regulate the expression of APOE and ABCA1. Lipidated APOE plays a critical role in lipid transport from astrocytes to neurons. Due to hyperlipidation, APOE2-containing lipoprotein particles likely deliver lipids to neurons at a higher efficiency than APOE3 and APOE4. APOE2 may also maintain synaptic plasticity during AD, potentially through interacting with synaptic APOE receptors. During AD pathogenesis, Aβ is produced primarily by neurons through proteolytic processing of APP. APOE regulates Aβ metabolism in an isoform-dependent manner. APOE2 likely mediates Aβ clearance via BBB at a higher efficiency than APOE3 and APOE4. In addition, APOE2 may have a stronger effect in promoting the proteolytic degradation of Aβ by extracellular enzymes. The regulatory roles of APOE in Aβ metabolism may be partially mediated through APOE/Aβ complex formation. APOE*ε2 has also been associated with reduced neurofibrillary tangles in AD patients, though the mechanism is unclear. Additionally, APOE2 may confer protection against AD by affecting the plasma lipid and metabolomics profiles. ACID, intracellular domain of the amyloid-precursor protein; sAPPβ, soluble amyloid precursor protein β
APOE2 may have a greater neurotrophic effect, which maintains neuronal survival and synaptic functions during AD pathogenesis. This is likely achieved by APOE2-mediated lipid metabolism and APOE2-triggered neuroprotective signaling pathways [15,16,17]. In addition, evidence suggests a critical role of APOE in microglial functions during AD pathogenesis [89, 90, 126,127,128]. How APOE2 may regulate the immune response of microglia differently than APOE3 and APOE4 remains unclear. Previous studies have shown that promoting cholesterol efflux reduces the immune response of macrophages [240, 241]. Given that APOE2 is a better cholesterol acceptor than APOE3 and APOE4 [242, 243], one may assume a reduced inflammatory response of microglia associated with APOE2 in AD, which requires further investigation. Additionally, hyperlipidation of APOE2 may contribute to reduced Aβ deposition. Supporting this, Abca1-knockout increases [244], whereas Abca1-overexpression decreases Aβ deposition in PDAPP mouse models [245]. Furthermore, increasing APOE lipidation through pharmacological activation of liver X receptors (LXRs) reduces Aβ deposition in AD transgenic animal models [203, 246, 247]. APOE2 also has been associated with longevity [19,20,21,22,23]. Although the reduced AD risk in APOE*ε2 carriers may contribute to their longer life expectancy, it is also possible that there are unknown anti-aging effects that contribute to their reduced risk of AD through a systemic impact on the whole body. These factors could be APOE2-specific proteins, lipids, and/or metabolites in the plasma [200, 248].
APOE*ε2 and other proteinopathies
APOE*ε2 and TDP-43 proteinopathy
Intracellular TDP-43 inclusion is a shared pathological hallmark of amyotrophic lateral sclerosis (ALS) and FTLD [249]. TDP-43 aggregation is commonly present in hippocampal sclerosis and AD brains [250,251,252,253]. Although clinical evidence shows no correlation between APOE and ALS risk [254], APOE*ε2/2 ALS patients exhibit decreased glucose metabolism in extra-motor areas compared with APOE*ε3/3 homozygote patients, implying an increased risk of cognitive impairment associated with APOE*ε2 in ALS patients [255]. The impact of APOE*ε2 on FTLD risk remains inconclusive, with APOE*ε2 exerting either no effect or an increased risk of FTLD [256,257,258].
Pathologically, APOE*ε4 has been associated with exacerbated TDP-43 proteinopathy in FTLD [259]. There is also evidence showing that APOE*ε4 increases the TDP-43 burden in the brain independently of Aβ and tau load, which mediates the increased risk of hippocampal sclerosis in APOE*ε4 carriers [260]. However, the effect of APOE*ε2 on TDP-43 pathology remains unknown.
APOE*ε2 and α-synuclein proteinopathy
Dementia with Lewy bodies (DLB) and Parkinson’s disease dementia (PDD) are two neurodegenerative diseases collectively known as Lewy body dementia (LBD) [261]. Pathologically, LBD is characterized by cytoplasmic α-synuclein (αSyn) positive inclusions known as Lewy bodies. α-Syn pathology also affects multiple system atrophy (MSA) [262], and is present in over 50% of the pathologically-confirmed AD brains [263]. Although human studies have shown that APOE*ε4 increases the risk of DLB [264, 265], the impact of APOE*ε2 is less clear. Evidence from a Norwegian cohort suggests a reduced risk of DLB in APOE*ε2 carriers [266], but further validation is required. Although the association between APOE and PD has been disproved [267,268,269], evidence shows an increased risk of PDD in APOE*ε2 carriers [270, 271]. Similar to PD, MSA appears to be also exempted from the impact of APOE [272, 273].
Recent studies addressing the effects of APOE isoforms on α-synuclein pathology and related toxicity in vivo have produced interesting findings. αSyn pathology in APOE-TR mice induced by adeno-associated viruses (AAV)-mediated overexpression of human wild type αSyn, or in transgenic mice that overexpress the PD-associated mutant, αSyn (A53T), is exacerbated by APOE4, but not by APOE2 or APOE3 [274, 275]. Although APOE2 protects against αSyn pathology in αSyn (A53T) transgenic mice [274], the protective effect was not observed in the study using the viral-mediated approach [275].
APOE*ε2 and risks of other neurological disorders
Studies have suggested APOE*ε2 as a risk factor for PTSD, given there is a disproportionately high representation of APOE*ε2 carriers among PTSD patients [28]. Moreover, PTSD patients carrying the APOE*ε2 allele display more severe symptoms [276] and potentially have stronger stress responses than non-carriers [277]. The negative effect of APOE*ε2 on PTSD is also supported by an in vivo animal study showing a slower fear extinction in APOE knock-in mice expressing APOE*ε2 than those expressing other APOE alleles [277].
AMD is the leading cause of vision loss in the elderly [278]. The polymorphism of APOE has been associated with AMD risk [279,280,281]. Opposite to the risk profile of AD, APOE*ε2/2 individuals have increased, whereas APOE*ε4 carriers have decreased risk of AMD compared to APOE*ε3/3 homozygotes [29]. In animals, APOE2-TR mice exhibit increased subretinal accumulation of mononuclear phagocytes (MP), retinal degeneration, and choroidal neovascularization than APOE3-TR and APOE4-TR mice at 12 months of age [282]. The detrimental effect of APOE2 in AMD may be partially caused by the APOE*ε2-associated activation of MPs, as blocking the activity of the innate immunity receptor cluster in MPs reduces AMD pathogenies in aged APOE2-TR animals [282].
APOE*ε2 may also modify the risks of other less common neurological disorders. For example, APOE*ε2 has been associated with a reduced risk of Creutzfeldt-Jakob Disease [283], and increased risks of cerebral palsy [284] and Machado-Joseph Disease [285]. However, the evidence should be examined carefully, given the small sample size of these studies.
Large-scale human studies have disputed the association between APOE and multiple sclerosis (MS) [286, 287], whereas the impact of APOE*ε2 on Huntington disease (HD) remains elusive. Despite an earlier report of a younger age at onset of HD in male APOE*ε2/3 patients [288], the observation has not been replicated by others [289].
APOE*ε2 and cerebrovascular diseases
Cerebral amyloid angiopathy (CAA)
CAA is caused by Aβ deposition in cerebral vessel walls [290]. As a common concurrence in AD, CAA mostly affects small arteries and capillaries in the CNS [291]. Despite APOE*ε2 being protective against Aβ deposition in the brain parenchyma, APOE*ε2 carriers are at higher risk and severity of CAA compared to APOE*ε3/3 individuals [26, 27]. APOE*ε2-associated accumulation of Aβ causes amyloid-laden vessels to undergo vasculopathic changes such as fibrinoid necrosis, leading to vessel rupture and resultant hemorrhages in APOE*ε2 CAA patients [292, 293]. In contrast, APOE*ε4 CAA patients more commonly exhibit microbleeds than hemorrhages [27, 294]. APOE*ε2 and APOE*ε4 impact blood vessels of varying sizes, thereby differentially affecting CAA-related pathological outcomes. For example, APOE*ε4, but not APOE*ε2, has been associated with capillary amyloid angiopathy [27]. The mechanism underlying the difference is unclear but possibly related to the differential APOE receptor expression [295, 296], or isoform-specific impact on different vascular cell types [140, 297].
CAA is a common cause of recurrent lobar intracerebral hemorrhage (ICH) [298, 299]. Although ICH-related stroke is relatively uncommon, it is associated with high mortality and morbidity [300]. The APOE*ε2 allele is associated with an increased risk for hematoma expansion in lobar ICH patients, especially in ICH cases with CAA [299], predisposing patients for subsequent hemorrhages. In agreement with that, ICH recurrence within two years of the first event is 18% higher in APOE*ε2 carriers as compared to APOE*ε3/3 individuals [301]. Additionally, the effect of APOE*ε2 on ICH risk appears to be affected by ethnic background, such that APOE*ε2 imposes a higher risk of ICH for Asian than for European individuals [302].
Stroke
APOE*ε2, like APOE*ε4, is also a genetic risk factor for stroke [303]. Compared with APOE*ε3/3 individuals, APOE*ε2 carriers are at higher risk for cerebral and cortical infarction [304]. Furthermore, APOE*ε2 is associated with higher chances of both ischemic and hemorrhagic stroke recurrence [298, 301, 302, 305]. Notably, the impact of APOE*ε2 on stroke occurrence may be modulated by age, as the stroke risk in APOE*ε2 carriers decreases significantly after age 70 [304].
APOE*ε2-inspired therapeutic strategies
As APOE-targeting strategies for AD treatment have been extensively reviewed elsewhere [4, 306,307,308,309], herein, we focus on the development of therapies inspired by recent APOE*ε2 studies.
Viral-mediated APOE*ε2 overexpression
Given APOE2 protects against AD likely due to its greater neuroprotective functions than that of APOE3 and APOE4 (Fig. 3), introducing APOE2 into the brain of AD patients who lack APOE*ε2 may have therapeutic effects. This idea has been tested with amyloid mouse models expressing murine Apoe. Viral-mediated overexpression of APOE2, but not APOE3 or APOE4 in the brain at the age when Aβ starts to deposit halts Aβ accumulation and reduces Aβ burden [193, 194], which may be attributed to the increased Aβ clearance in APOE2-expressing animals [194]. Moreover, evidence shows that APOE*ε2 gene delivery into amyloid mouse models with APOE4 expression reduces Aβ levels in the brain [310]. However, since APOE2 increases the risk of certain diseases such as CAA [26, 27], stroke [303], PTSD [28], AMD [29], and primary tauopathy [30], the long-term safety of APOE2 overexpression in human brains should be carefully assessed.
Increasing APOE lipidation
As has been discussed, hyperlipidation of APOE2 lipoprotein may be the central mechanism underlying its protective effect. Thus, pharmacological enhancement of APOE lipidation represents an attractive approach for AD treatment [311,312,313]. LXRs are transcriptional factors that form heterodimers with retinoid X receptors (RXRs) to regulate the expression of a battery of genes involved in lipid metabolism, including ABCA1 and APOE [314]. Oral administration of the LXR agonists, such as GW3965 and TO901317, increases the protein level and lipidation of brain APOE in mice [203, 246, 247, 315, 316]. Long-term (one month or longer) treatment with GW3965 or TO901317 during early-stage Aβ deposition reduces brain Aβ load and improves cognitive performances of amyloid transgenic animals [203, 246, 247]. However, conflicting reports exist regarding the treatment effect of LXR agonists when there is already substantial Aβ deposition in the brain. One study reported that although TO901317 administration for seven weeks reduces Aβ deposition in the cortex, it yields no impact on the cognition of APP23 mice [315]. Conversely, other studies show that long-term GW3965 or TO901317 treatment rescues cognitive impairments in different amyloid mouse models without affecting the Aβ burden in the brain [317,318,319].
The potential therapeutic effect of RXR agonists for AD also has been explored, which is best exemplified by the Food and Drug Administration (FDA)-approved drug, Bexarotene. Like LXR agonists, oral administration of Bexarotene upregulates APOE and ABCA1 in mouse brains [320,321,322,323]. Studies show that both short-term and long-term treatment of Bexarotene in amyloid mouse models after Aβ has been deposited in the brain restores cognitive performances of the animals, with or without affecting the brain Aβ load [320, 321, 324]. However, the treatment effect of Bexarotene in either cognition or Aβ pathology in animal models has not been replicated by others [323, 325,326,327,328]. Interestingly, despite conflicting results from amyloid mouse models expressing murine Apoe, there is consistent evidence showing that short-term Bexarotene treatment reverses memory deficit, increases Aβ clearance, and reduces soluble Aβ42 in the hippocampal lysate of amyloid mouse models expressing human APOE isoforms [329, 330].
In humans, Bexarotene treatment increases APOE levels in the CSF [331]. However, the treatment has no impact on brain amyloid load and cognitive functions in AD patients [332]. Moreover, Bexarotene has been reported to cause systemic adverse effects, including hypertriglyceridemia [332], which may limit its potential clinical use in AD patients.
While identifying and testing novel LXR/RXR agonists could be a future direction for AD treatment [333], modulating APOE lipidation by targeting ABCA1 may be a promising alternative option. Overexpression of murine Abca1 under mouse prion promoter reduces Aβ deposition in PDAPP mice brains [245]. In addition, brain ABCA1 is upregulated by genetic deletion of the small non-coding microRNA (miRNA), miR-33 [334]. Intracerebroventricular infusion of anti-miR-33 oligonucleotides reduces cortical Aβ40 levels in 3-month-old APP/PS1 mice [334]. The activity of ABCA1 may also be enhanced by the APOE mimetic peptide CS-6253 [335]. However, whether CS-6253 induces beneficial effects against AD remains to be tested.
Converting APOE*ε4 to APOE*ε2
With the emergence of powerful gene-editing tools such as the CRISPR-Cas system [336,337,338], generating isogenic iPSC lines from one APOE genotype (normally APOE*ε3/3) to other genotypes becomes efficient and cost-effective [339,340,341]. Compared to APOE*ε3 cells, isogenic APOE*ε4 cells show dramatic phenotypic changes, including increased Aβ42 and phosphorylated tau in neurons, impaired Aβ uptake and cholesterol metabolism in astrocyte, and reduced phagocytosis of Aβ in microglia [339, 340]. How isogenic APOE*ε2 cells may be functionally different from APOE*ε3 and APOE*ε4 cells remains unclear. Future studies should address the clinical potential of converting APOE*ε4 to APOE*ε2 in vivo as a treatment option for AD.
Plasma APOE-based therapy
Although it remains controversial whether and how peripheral APOE may contribute to AD pathogenesis [46, 342], evidence suggests that low plasma APOE levels are associated with increased AD and dementia risk, independent of APOE genotype [343, 344]. Moreover, higher levels of APOE in APOC3-free HDL particles in the plasma have been associated with better cognitive performance and reduced risk of dementia in humans [345]. Since APOE*ε2 carriers have higher levels of plasma APOE [48,49,50,51] and HDL [92, 93], whole plasma or plasma APOE-containing lipoprotein particles from APOE*ε2 carriers may hold promise as a therapeutic strategy for AD.
Conclusions
Despite compelling evidence from human studies supporting the protective effect of APOE*ε2 against AD, the underlying mechanisms remain mostly elusive. APOE*ε2 likely confers protection against AD through both Aβ-dependent and independent mechanisms, both of which appear to be underpinned by increased lipidation of APOE2-containing lipoprotein particles (Fig. 3). To validate the mechanisms proposed in this review, more evidence from humans and animal models is required. Interpretation of data from these studies should be context-dependent, with age, sex, and AD pathology being considered. Furthermore, improved understanding of the roles of APOE2 in other diseases, such as cerebrovascular diseases and different proteinopathies, including tau, TDP-43, and α-Syn pathologies, will aid in the comprehensive assessment of safety regarding APOE2-targeted therapeutics for AD.
In addition to APOE2, other APOE variants have been suggested to protect against AD. For example, the APOE (V236E) variant in the APOE3 backbone has been associated with a significant reduction in AD risk [346]. Additionally, the possession of two copies of the APOE3 Christchurch variant (R136S) markedly delayed cognitive decline in a presenilin 1 (PSEN1) mutation carrier, likely by limiting tau accumulation in the brain [86]. Future studies to validate and to understand the mechanisms underlying the protective effect of these variants will shed light on identifying disease-modifying interventions targeting APOE for AD therapies.
Availability of data and materials
Not applicable.
Change history
12 November 2020
The original article has been revised to include ORCIDs for all authors.
Abbreviations
- AAV:
-
Adeno-associated virus
- AD:
-
Alzheimer’s disease
- AGD:
-
Argyrophilic grain disease
- ALS:
-
Amyotrophic lateral sclerosis
- AMD:
-
Age-related macular degeneration
- AMPAR:
-
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- APOE:
-
Apolipoprotein E
- APOER2:
-
APOE receptor 2
- APOE-TR:
-
APOE-targeted replacement
- APP:
-
Amyloid-beta precursor protein
- BBB:
-
Blood-brain barrier
- CAA:
-
Cerebral amyloid
- CNS:
-
Central nervous system
- DAM:
-
Disease-associated microglia
- DLB:
-
Dementia with Lewy bodies
- DS:
-
Down’s syndrome
- ELISA:
-
Enzyme-linked immunosorbent assay
- ESC:
-
Embryonic stem cells
- FDA:
-
Food and Drug Administration
- FTLD:
-
Frontotemporal lobar degeneration
- GWAS:
-
Genome-wide association studies
- HD:
-
Huntington disease
- HSPG:
-
Heparan sulfate proteoglycan
- ICH:
-
Intracerebral hemorrhage
- iPSC:
-
Human induced pluripotent stem cell
- IQ:
-
Intelligence quotient
- ISF:
-
Interstitial fluid
- LBD:
-
Lewy body dementia
- LDLR:
-
Low-density lipoprotein receptor
- LOAD:
-
Late-onset Alzheimer’s disease
- LPS:
-
Lipopolysaccharide
- LRP1:
-
LDLR-related protein 1
- LXR:
-
Liver X receptor
- MAP:
-
Mitogen-activated protein
- MCI:
-
Mild cognitive impairment
- MGnD:
-
Neurodegenerative phenotype
- miRNA:
-
MicroRNA
- MP:
-
Mononuclear phagocytes
- MS:
-
Multiple sclerosis
- MSA:
-
Multiple system atrophy
- NFT:
-
Neurofibrillary tangles
- NMDAR:
-
N-methyl-D-aspartate receptor
- PDD:
-
Parkinson’s disease dementia
- PET:
-
Positron emission tomography
- PSD-95:
-
Postsynaptic density protein 95
- PSEN:
-
Presenilin
- PSP:
-
Supranuclear palsy
- PTSD:
-
Post-traumatic stress disorder
- RXR:
-
Retinoid X receptor
- TREM2:
-
Triggering receptor expressed on myeloid cells 2
- VLDLR:
-
Very low-density lipoprotein receptor
- αSyn:
-
α-synuclein
- βVLDL:
-
β-migrating VLDL
References
Belloy ME, Napolioni V, Greicius MD. A Quarter Century of APOE and Alzheimer's Disease: Progress to Date and the Path Forward. Neuron. 2019;101(5):820–38.
Zhao N, et al. Apolipoprotein E, Receptors, and Modulation of Alzheimer's Disease. Biol Psychiatry. 2018;83(4):347–57.
Guo T, et al. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer's disease. Mol Neurodegener. 2020;15(1):40.
Yamazaki Y, et al. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15(9):501–18.
Farrer LA, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. Jama. 1997;278(16):1349–56.
Genin E, et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry. 2011;16(9):903–7.
Nagy Z, et al. Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer's disease. Neuroscience. 1995;69(3):757–61.
Bennett DA, et al. Neuropathologic intermediate phenotypes enhance association to Alzheimer susceptibility alleles. Neurology. 2009;72(17):1495–503.
Serrano-Pozo A, et al. APOEepsilon2 is associated with milder clinical and pathological Alzheimer disease. Ann Neurol. 2015;77(6):917–29.
Reiman EM, et al. Exceptionally low likelihood of Alzheimer's dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun. 2020;11(1):667.
Jansen WJ, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA. 2015;313(19):1924–38.
Shinohara M, et al. APOE2 eases cognitive decline during Aging: Clinical and preclinical evaluations. Ann Neurol. 2016;79(5):758–74.
Berlau DJ, et al. APOE epsilon2 is associated with intact cognition but increased Alzheimer pathology in the oldest old. Neurology. 2009;72(9):829–34.
Berlau DJ, et al. Dissociation of neuropathologic findings and cognition: case report of an apolipoprotein E epsilon2/epsilon2 genotype. Arch Neurol. 2007;64(8):1193–6.
Hoe HS, Harris DC, Rebeck GW. Multiple pathways of apolipoprotein E signaling in primary neurons. J Neurochem. 2005;93(1):145–55.
Hayashi H, et al. Apolipoprotein E-containing lipoproteins protect neurons from apoptosis via a signaling pathway involving low-density lipoprotein receptor-related protein-1. J Neurosci. 2007;27(8):1933–41.
Huang YA, et al. Differential Signaling Mediated by ApoE2, ApoE3, and ApoE4 in Human Neurons Parallels Alzheimer's Disease Risk. J Neurosci. 2019;39(37):7408–27.
Chen Y, et al. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci U S A. 2010;107(26):12011–6.
Deelen J, et al. Genome-wide association study identifies a single major locus contributing to survival into old age; the APOE locus revisited. Aging Cell. 2011;10(4):686–98.
Nebel A, et al. A genome-wide association study confirms APOE as the major gene influencing survival in long-lived individuals. Mech Ageing Dev. 2011;132(6-7):324–30.
Sebastiani P, et al. Genetic signatures of exceptional longevity in humans. PLoS One. 2012;7(1):e29848.
Deelen J, et al. Genome-wide association meta-analysis of human longevity identifies a novel locus conferring survival beyond 90 years of age. Hum Mol Genet. 2014;23(16):4420–32.
Zeng Y, et al. Novel loci and pathways significantly associated with longevity. Sci Rep. 2016;6:21243.
Corder EH, et al. Apolipoprotein E genotype determines survival in the oldest old (85 years or older) who have good cognition. Arch Neurol. 1996;53(5):418–22.
Rosvall L, et al. APOE-related mortality: effect of dementia, cardiovascular disease and gender. Neurobiol Aging. 2009;30(10):1545–51.
Nelson PT, et al. APOE-epsilon2 and APOE-epsilon4 correlate with increased amyloid accumulation in cerebral vasculature. J Neuropathol Exp Neurol. 2013;72(7):708–15.
Yu L, et al. APOE and cerebral amyloid angiopathy in community-dwelling older persons. Neurobiol Aging. 2015;36(11):2946–53.
Kim TY, et al. Apolipoprotein E gene polymorphism, alcohol use, and their interactions in combat-related posttraumatic stress disorder. Depress Anxiety. 2013;30(12):1194–201.
McKay GJ, et al. Evidence of association of APOE with age-related macular degeneration: a pooled analysis of 15 studies. Hum Mutat. 2011;32(12):1407–16.
Zhao N, et al. APOE ε2 is associated with increased tau pathology in primary tauopathy. Nat Commun. 2018;9(1):4388.
Ghebremedhin E, et al. Argyrophilic grain disease is associated with apolipoprotein E epsilon 2 allele. Acta Neuropathol. 1998;96(3):222–4.
Rall SC, Weisgraber KH, Mahley RW. Human apolipoprotein E. The complete amino acid sequence. J Biol Chem. 1982;257(8):4171–8.
Das HK, et al. Isolation, characterization, and mapping to chromosome 19 of the human apolipoprotein E gene. J Biol Chem. 1985;260(10):6240–7.
Weisgraber KH, Rall SC, Mahley RW. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J Biol Chem. 1981;256(17):9077–83.
Wetterau JR, et al. Human apolipoprotein E3 in aqueous solution. I. Evidence for two structural domains. J Biol Chem. 1988;263(13):6240–8.
Weisgraber KH. Apolipoprotein E: structure-function relationships. Adv Protein Chem. 1994;45:249–302.
Aggerbeck LP, et al. Human apolipoprotein E3 in aqueous solution. II. Properties of the amino- and carboxyl-terminal domains. J Biol Chem. 1988;263(13):6249–58.
Tolar M, et al. Neurotoxicity of the 22 kDa thrombin-cleavage fragment of apolipoprotein E and related synthetic peptides is receptor-mediated. J Neurosci. 1997;17(15):5678–86.
Lalazar A, et al. Site-specific mutagenesis of human apolipoprotein E. Receptor binding activity of variants with single amino acid substitutions. J Biol Chem. 1988;263(8):3542–5.
Dong LM, et al. Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. J Biol Chem. 1994;269(35):22358–65.
Saito H, et al. Characterization of the heparin binding sites in human apolipoprotein E. J Biol Chem. 2003;278(17):14782–7.
Futamura M, et al. Two-step mechanism of binding of apolipoprotein E to heparin: implications for the kinetics of apolipoprotein E-heparan sulfate proteoglycan complex formation on cell surfaces. J Biol Chem. 2005;280(7):5414–22.
Linton MF, et al. Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. J Clin Invest. 1991;88(1):270–81.
Elshourbagy NA, et al. Apolipoprotein E mRNA is abundant in the brain and adrenals, as well as in the liver, and is present in other peripheral tissues of rats and marmosets. Proc Natl Acad Sci U S A. 1985;82(1):203–7.
Xu Q, et al. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 2006;26(19):4985–94.
Huynh T-V, et al. Lack of hepatic apoE does not influence early Aβ deposition: observations from a new APOE knock-in model. Mol Neurodegener. 2019;14(1):37.
Horsburgh K, et al. Influence of apolipoprotein E genotype on neuronal damage and apoE immunoreactivity in human hippocampus following global ischemia. J Neuropathol Exp Neurol. 1999;58(3):227–34.
Cruchaga C, et al. Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer's disease. Hum Mol Genet. 2012;21(20):4558–71.
Khan TA, et al. Apolipoprotein E genotype, cardiovascular biomarkers and risk of stroke: systematic review and meta-analysis of 14,015 stroke cases and pooled analysis of primary biomarker data from up to 60,883 individuals. Int J Epidemiol. 2013;42(2):475–92.
Rezeli M, et al. Quantification of total apolipoprotein E and its specific isoforms in cerebrospinal fluid and blood in Alzheimer’s disease and other neurodegenerative diseases. EuPA Open Proteomics. 2015;8:137–43.
Corsetti JP, et al. Apolipoprotein E levels and apolipoprotein E genotypes in incident cardiovascular disease risk in subjects of the Prevention of Renal and Vascular End-stage disease study. J Clin Lipidol. 2016;10(4):842–50.
Martínez-Morillo E, et al. Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer's disease patients and controls. Acta Neuropathol. 2014;127(5):633–43.
Minta K, et al. Quantification of total apolipoprotein E and its isoforms in cerebrospinal fluid from patients with neurodegenerative diseases. Alzheimers Res Ther. 2020;12(1):19.
Conejero-Goldberg C, et al. APOE2 enhances neuroprotection against Alzheimer's disease through multiple molecular mechanisms. Mol Psychiatry. 2014;19(11):1243–50.
Knouff C, et al. Apo E structure determines VLDL clearance and atherosclerosis risk in mice. J Clin Invest. 1999;103(11):1579–86.
Ramaswamy G, et al. Effect of domain interaction on apolipoprotein E levels in mouse brain. J Neurosci. 2005;25(46):10658–63.
Riddell DR, et al. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J Neurosci. 2008;28(45):11445–53.
Ulrich JD, et al. In vivo measurement of apolipoprotein E from the brain interstitial fluid using microdialysis. Mol Neurodegener. 2013;8:13.
Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(3):a006312.
Lane-Donovan C, Herz J. ApoE, ApoE Receptors, and the Synapse in Alzheimer's Disease. Trends Endocrinol Metab. 2017;28(4):273–84.
Bu G. Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10(5):333–44.
Atagi Y, et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J Biol Chem. 2015;290(43):26043–50.
Bailey CC, DeVaux LB, Farzan M. The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein E. J Biol Chem. 2015;290(43):26033–42.
Yeh FL, et al. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron. 2016;91(2):328–40.
Weisgraber KH, Innerarity TL, Mahley RW. Abnormal lipoprotein receptor-binding activity of the human E apoprotein due to cysteine-arginine interchange at a single site. J Biol Chem. 1982;257(5):2518–21.
Innerarity TL, et al. Normalization of receptor binding of apolipoprotein E2. Evidence for modulation of the binding site conformation. J Biol Chem. 1984;259(11):7261–7.
Ruiz J, et al. The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J Lipid Res. 2005;46(8):1721–31.
Frieden C, Wang H, Ho CMW. A mechanism for lipid binding to apoE and the role of intrinsically disordered regions coupled to domain-domain interactions. Proc Natl Acad Sci U S A. 2017;114(24):6292–7.
Gupta V, et al. Lipid-induced extension of apolipoprotein E helix 4 correlates with low density lipoprotein receptor binding ability. J Biol Chem. 2006;281(51):39294–9.
Mahley RW, Rall SC Jr. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–37.
Kowal RC, et al. Opposing effects of apolipoproteins E and C on lipoprotein binding to low density lipoprotein receptor-related protein. J Biol Chem. 1990;265(18):10771–9.
Sagare A, et al. Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med. 2007;13(9):1029–31.
LaDu MJ, et al. Self-assembly of HEK cell-secreted ApoE particles resembles ApoE enrichment of lipoproteins as a ligand for the LDL receptor-related protein. Biochemistry. 2006;45(2):381–90.
Ma Q, et al. Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-β42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol Neurodegener. 2018;13(1):57.
Nathan BP, et al. Apolipoprotein E4 inhibits, and apolipoprotein E3 promotes neurite outgrowth in cultured adult mouse cortical neurons through the low-density lipoprotein receptor-related protein. Brain Res. 2002;928(1-2):96–105.
Puttfarcken PS, et al. Effect of apolipoprotein E on neurite outgrowth and beta-amyloid-induced toxicity in developing rat primary hippocampal cultures. J Neurochem. 1997;68(2):760–9.
Teter B, et al. Human apolipoprotein E isoform-specific differences in neuronal sprouting in organotypic hippocampal culture. J Neurochem. 1999;73(6):2613–6.
Holtzman DM, et al. Low density lipoprotein receptor-related protein mediates apolipoprotein E-dependent neurite outgrowth in a central nervous system-derived neuronal cell line. Proc Natl Acad Sci U S A. 1995;92(21):9480–4.
Fagan AM, et al. Apolipoprotein E-containing high density lipoprotein promotes neurite outgrowth and is a ligand for the low density lipoprotein receptor-related protein. J Biol Chem. 1996;271(47):30121–5.
Nathan BP, et al. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science. 1994;264(5160):850–2.
Deane R, et al. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118(12):4002–13.
D'Arcangelo G, et al. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;24(2):471–9.
Hiesberger T, et al. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24(2):481–9.
Niu S, et al. Reelin promotes hippocampal dendrite development through the VLDLR/ApoER2-Dab1 pathway. Neuron. 2004;41(1):71–84.
Xian X, et al. Reversal of ApoE4-induced recycling block as a novel prevention approach for Alzheimer's disease. Elife. 2018;7:e40048.
Arboleda-Velasquez JF, et al. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019;25(11):1680–3.
Shi Y, Holtzman DM. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol. 2018;18(12):759–72.
Ulrich JD, et al. ApoE facilitates the microglial response to amyloid plaque pathology. J Exp Med. 2018;215(4):1047–58.
Keren-Shaul H, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer's Disease. Cell. 2017;169(7):1276–1290.e17.
Krasemann S, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity. 2017;47(3):566–581.e9.
Mahley RW. Central Nervous System Lipoproteins: ApoE and Regulation of Cholesterol Metabolism. Arterioscler Thromb Vasc Biol. 2016;36(7):1305–15.
Rasmussen KL. Plasma levels of apolipoprotein E, APOE genotype and risk of dementia and ischemic heart disease: A review. Atherosclerosis. 2016;255:145–55.
Wolters FJ, et al. The impact of APOE genotype on survival: Results of 38,537 participants from six population-based cohorts (E2-CHARGE). PLoS One. 2019;14(7):e0219668.
Mahley RW, Huang Y, Rall SC Jr. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia). Questions, quandaries, and paradoxes. J Lipid Res. 1999;40(11):1933–49.
de Beer F, et al. Expression of type III hyperlipoproteinemia in apolipoprotein E2 (Arg158 --> Cys) homozygotes is associated with hyperinsulinemia. Arterioscler Thromb Vasc Biol. 2002;22(2):294–9.
Huang Y, et al. Apolipoprotein E2 reduces the low density lipoprotein level in transgenic mice by impairing lipoprotein lipase-mediated lipolysis of triglyceride-rich lipoproteins. J Biol Chem. 1998;273(28):17483–90.
Chait A, et al. Type-III Hyperlipoproteinaemia ("remnant removal disease"). Insight into the pathogenetic mechanism. Lancet. 1977;1(8023):1176–8.
Chait A, et al. Impaired very low density lipoprotein and triglyceride removal in broad beta disease: comparison with endogenous hypertriglyceridemia. Metabolism. 1978;27(9):1055–66.
Lewandowski CT, Maldonado Weng J, LaDu MJ. Alzheimer's disease pathology in APOE transgenic mouse models: The Who, What, When, Where, Why, and How. Neurobiol Dis. 2020;139:104811.
Karten B, et al. Expression of ABCG1, but not ABCA1, correlates with cholesterol release by cerebellar astroglia. J Biol Chem. 2006;281(7):4049–57.
Kim WS, et al. Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein E discs and suppression of amyloid-beta peptide generation. J Biol Chem. 2007;282(5):2851–61.
Wahrle SE, et al. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem. 2004;279(39):40987–93.
Krimbou L, et al. Molecular interactions between apoE and ABCA1: impact on apoE lipidation. J Lipid Res. 2004;45(5):839–48.
Sun Y, et al. Glial fibrillary acidic protein-apolipoprotein E (apoE) transgenic mice: astrocyte-specific expression and differing biological effects of astrocyte-secreted apoE3 and apoE4 lipoproteins. J Neurosci. 1998;18(9):3261–72.
Fagan AM, et al. Unique lipoproteins secreted by primary astrocytes from wild type, apoE (−/−), and human apoE transgenic mice. J Biol Chem. 1999;274(42):30001–7.
LaDu MJ, et al. Nascent astrocyte particles differ from lipoproteins in CSF. J Neurochem. 1998;70(5):2070–81.
Karasinska JM, et al. Specific loss of brain ABCA1 increases brain cholesterol uptake and influences neuronal structure and function. J Neurosci. 2009;29(11):3579–89.
Sharman MJ, et al. Profiling brain and plasma lipids in human APOE epsilon2, epsilon3, and epsilon4 knock-in mice using electrospray ionization mass spectrometry. J Alzheimers Dis. 2010;20(1):105–11.
Lefterov I, et al. APOE2 orchestrated differences in transcriptomic and lipidomic profiles of postmortem AD brain. Alzheimers Res Ther. 2019;11(1):113.
Mauch DH, et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294(5545):1354–7.
Dumanis SB, et al. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J Neurosci. 2009;29(48):15317–22.
Chen Y, Fu AKY, Ip NY. Synaptic dysfunction in Alzheimer's disease: Mechanisms and therapeutic strategies. Pharmacol Ther. 2019;195:186–98.
Chakroborty S, et al. Reduced presynaptic vesicle stores mediate cellular and network plasticity defects in an early-stage mouse model of Alzheimer's disease. Mol Neurodegener. 2019;14(1):7.
Chung WS, et al. Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc Natl Acad Sci U S A. 2016;113(36):10186–91.
Korwek KM, et al. ApoE isoform-dependent changes in hippocampal synaptic function. Mol Neurodegener. 2009;4:21.
Trommer BL, et al. ApoE isoform affects LTP in human targeted replacement mice. Neuroreport. 2004;15(17):2655–8.
Weissberger GH, et al. Meta-analysis of cognitive ability differences by apolipoprotein e genotype in young humans. Neurosci Biobehav Rev. 2018;94:49–58.
Bunce D, et al. APOE genotype and cognitive change in young, middle-aged, and older adults living in the community. J Gerontol A Biol Sci Med Sci. 2014;69(4):379–86.
Rajan KB, et al. Apolipoprotein E Genotypes, Age, Race, and Cognitive Decline in a Population Sample. J Am Geriatr Soc. 2019;67(4):734–40.
Reas ET, et al. Effects of APOE on cognitive aging in community-dwelling older adults. Neuropsychology. 2019;33(3):406–16.
Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16(6):358–72.
Brown GC, St George-Hyslop PH. Deciphering microglial diversity in Alzheimer's disease. Science. 2017;356(6343):1123–4.
Wang Y, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell. 2015;160(6):1061–71.
Pandey RS, et al. Genetic perturbations of disease risk genes in mice capture transcriptomic signatures of late-onset Alzheimer's disease. Mol Neurodegener. 2019;14(1):50.
Gratuze M, Leyns CEG, Holtzman DM. New insights into the role of TREM2 in Alzheimer's disease. Mol Neurodegener. 2018;13(1):66.
Zhou Y, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer's disease. Nature medicine. 2020;26(1):131–42.
Mathys H, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570(7761):332–7.
Sala Frigerio C, et al. The Major Risk Factors for Alzheimer's Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019;27(4):1293–1306.e6.
Parhizkar S, et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci. 2019;22(2):191–204.
Fitz NF, et al. Trem2 deficiency differentially affects phenotype and transcriptome of human APOE3 and APOE4 mice. Mol Neurodegener. 2020;15(1):41.
Shi Y, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549(7673):523–7.
Gale SC, et al. APOε4 is associated with enhanced in vivo innate immune responses in human subjects. J Allergy Clin Immunol. 2014;134(1):127–34.
Vitek MP, Brown CM, Colton CA. APOE genotype-specific differences in the innate immune response. Neurobiol Aging. 2009;30(9):1350–60.
Maezawa I, et al. Neurotoxicity from innate immune response is greatest with targeted replacement of E4 allele of apolipoprotein E gene and is mediated by microglial p38MAPK. Faseb j. 2006;20(6):797–9.
Zhu Y, et al. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia. 2012;60(4):559–69.
Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14(3):133–50.
Bell RD, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485(7399):512–6.
Main BS, et al. Apolipoprotein E4 impairs spontaneous blood brain barrier repair following traumatic brain injury. Mol Neurodegener. 2018;13(1):17.
Montagne A, et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature. 2020;581(7806):71–6.
Yamazaki Y, et al. ApoE (Apolipoprotein E) in Brain Pericytes Regulates Endothelial Function in an Isoform-Dependent Manner by Modulating Basement Membrane Components. Arterioscler Thromb Vasc Biol. 2020;40(1):128–44.
Singh V, et al. Spatial patterns of cortical thinning in mild cognitive impairment and Alzheimer's disease. Brain. 2006;129(Pt 11):2885–93.
Lerch JP, et al. Focal decline of cortical thickness in Alzheimer's disease identified by computational neuroanatomy. Cereb Cortex. 2005;15(7):995–1001.
Julkunen V, et al. Differences in cortical thickness in healthy controls, subjects with mild cognitive impairment, and Alzheimer's disease patients: a longitudinal study. J Alzheimers Dis. 2010;21(4):1141–51.
Pegueroles J, et al. Longitudinal brain structural changes in preclinical Alzheimer's disease. Alzheimers Dement. 2017;13(5):499–509.
Jack CR Jr, et al. MR-based hippocampal volumetry in the diagnosis of Alzheimer's disease. Neurology. 1992;42(1):183–8.
Killiany RJ, et al. Temporal lobe regions on magnetic resonance imaging identify patients with early Alzheimer's disease. Arch Neurol. 1993;50(9):949–54.
Shaw P, et al. Cortical morphology in children and adolescents with different apolipoprotein E gene polymorphisms: an observational study. Lancet Neurol. 2007;6(6):494–500.
Chang L, et al. Gray matter maturation and cognition in children with different APOE epsilon genotypes. Neurology. 2016;87(6):585–94.
Khan W, et al. No differences in hippocampal volume between carriers and non-carriers of the ApoE ε4 and ε2 alleles in young healthy adolescents. J Alzheimers Dis. 2014;40(1):37–43.
Fennema-Notestine C, et al. Presence of ApoE ε4 allele associated with thinner frontal cortex in middle age. J Alzheimers Dis. 2011;26(Suppl 3):49–60.
Fan M, et al. Cortical thickness is associated with different apolipoprotein E genotypes in healthy elderly adults. Neurosci Lett. 2010;479(3):332–6.
Hostage CA, et al. Dissecting the gene dose-effects of the APOE ε4 and ε2 alleles on hippocampal volumes in aging and Alzheimer's disease. PLoS One. 2013;8(2):e54483.
den Heijer T, et al. Hippocampal, amygdalar, and global brain atrophy in different apolipoprotein E genotypes. Neurology. 2002;59(5):746–8.
Liu Y, et al. APOE ε2 allele is associated with larger regional cortical thicknesses and volumes. Dement Geriatr Cogn Disord. 2010;30(3):229–37.
Sinclair LI, Pleydell-Pearce CW, Day INM. Possible positive effect of the APOE epsilon2 allele on cognition in early to mid-adult life. Neurobiol Learn Mem. 2017;146:37–46.
Taylor AE, et al. IQ, educational attainment, memory and plasma lipids: associations with apolipoprotein E genotype in 5995 children. Biol Psychiatry. 2011;70(2):152–8.
Chen XF, et al. Demographic and Lifestyle Characteristics, but Not Apolipoprotein E Genotype, Are Associated with Intelligence among Young Chinese College Students. PLoS One. 2015;10(11):e0143157.
Helkala EL, et al. The association of apolipoprotein E polymorphism with memory: a population based study. Neurosci Lett. 1995;191(3):141–4.
Chey J, Kim JW, Cho HY. Effects of apolipoprotein E phenotypes on the neuropsychological functions of community-dwelling elderly individuals without dementia. Neurosci Lett. 2000;289(3):230–4.
Kang JH, et al. Apolipoprotein E, cardiovascular disease and cognitive function in aging women. Neurobiol Aging. 2005;26(4):475–84.
Blair CK, et al. APOE genotype and cognitive decline in a middle-aged cohort. Neurology. 2005;64(2):268–76.
Helkala EL, et al. Memory functions in human subjects with different apolipoprotein E phenotypes during a 3-year population-based follow-up study. Neurosci Lett. 1996;204(3):177–80.
Hyman BT, et al. Apolipoprotein E and cognitive change in an elderly population. Ann Neurol. 1996;40(1):55–66.
Cauley JA, et al. Apo E allele frequencies in younger (age 42-50) vs older (age 65-90) women. Genet Epidemiol. 1993;10(1):27–34.
Schachter F, et al. Genetic associations with human longevity at the APOE and ACE loci. Nat Genet. 1994;6(1):29–32.
Hirose N, et al. Tokyo Centenarian Study. 4. Apolipoprotein E phenotype in Japanese centenarians living in the Tokyo Metropolitan area. Nihon Ronen Igakkai Zasshi. 1997;34(4):267–72.
Jian-Gang Z, et al. Apolipoprotein E and longevity among Han Chinese population. Mech Ageing Dev. 1998;104(2):159–67.
Frisoni GB, et al. Longevity and the epsilon2 allele of apolipoprotein E: the Finnish Centenarians Study. J Gerontol A Biol Sci Med Sci. 2001;56(2):M75–8.
Wolfson C, et al. A reevaluation of the duration of survival after the onset of dementia. N Engl J Med. 2001;344(15):1111–6.
Kulminski AM, et al. Age, gender, and cancer but not neurodegenerative and cardiovascular diseases strongly modulate systemic effect of the Apolipoprotein E4 allele on lifespan. PLoS Genet. 2014;10(1):e1004141.
Smith AD, et al. Protective effect of apo epsilon 2 in Alzheimer's disease. Oxford Project to Investigate Memory and Ageing (OPTIMA). Lancet. 1994;344(8920):473–4.
West HL, William Rebeck G, Hyman BT. Frequency of the apolipoprotein E ε2 allele is diminished in sporadic Alzheimer disease. Neurosci Lett. 1994;175(1-2):46–8.
Martins CA, et al. APOE alleles predict the rate of cognitive decline in Alzheimer disease: a nonlinear model. Neurology. 2005;65(12):1888–93.
Lott IT, Head E. Dementia in Down syndrome: unique insights for Alzheimer disease research. Nat Rev Neurol. 2019;15(3):135–47.
Tyrrell J, et al. A protective effect of apolipoprotein E e2 allele on dementia in Down's syndrome. Biol Psychiatry. 1998;43(6):397–400.
Lai F, et al. APOE genotype and gender effects on Alzheimer disease in 100 adults with Down syndrome. Neurology. 1999;53(2):331–6.
Lambert JC, et al. Analysis of the APOE alleles impact in Down's syndrome. Neurosci Lett. 1996;220(1):57–60.
Neu SC, et al. Apolipoprotein E Genotype and Sex Risk Factors for Alzheimer Disease: A Meta-analysis. JAMA Neurol. 2017;74(10):1178–89.
Saddiki H, et al. Age and the association between apolipoprotein E genotype and Alzheimer disease: A cerebrospinal fluid biomarker-based case-control study. PLoS Med. 2020;17(8):e1003289.
Pettigrew C, et al. Relationship of cognitive reserve and APOE status to the emergence of clinical symptoms in preclinical Alzheimer's disease. Cogn Neurosci. 2013;4(3-4):136–42.
Hampel H, et al. Biomarkers for Alzheimer's disease: academic, industry and regulatory perspectives. Nat Rev Drug Discov. 2010;9(7):560–74.
Palmqvist S, et al. Detailed comparison of amyloid PET and CSF biomarkers for identifying early Alzheimer disease. Neurology. 2015;85(14):1240–9.
Müller EG, et al. Amyloid-β PET-Correlation with cerebrospinal fluid biomarkers and prediction of Alzheimer’s disease diagnosis in a memory clinic. PLoS One. 2019;14(8):e0221365.
Strozyk D, et al. CSF Abeta 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology. 2003;60(4):652–6.
Morris JC, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67(1):122–31.
Hohman TJ, et al. APOE allele frequencies in suspected non-amyloid pathophysiology (SNAP) and the prodromal stages of Alzheimer's Disease. PLoS One. 2017;12(11):e0188501.
Buckley RF, et al. Sex Differences in the Association of Global Amyloid and Regional Tau Deposition Measured by Positron Emission Tomography in Clinically Normal Older Adults. JAMA Neurol. 2019;76(5):542–51.
Grothe MJ, et al. Multimodal characterization of older APOE2 carriers reveals selective reduction of amyloid load. Neurology. 2017;88(6):569–76.
Berlau DJ, et al. Neocortical beta-amyloid area, but not CERAD plaque stage, is associated with dementia status and Apolipoprotein E (APOE) genotype in the oldest old. Alzheimer's & Dementia. 2010;6(4):S125–6.
Tai LM, et al. EFAD transgenic mice as a human APOE relevant preclinical model of Alzheimer's disease. J Lipid Res. 2017;58(9):1733–55.
Youmans KL, et al. APOE4-specific changes in Aβ accumulation in a new transgenic mouse model of Alzheimer disease. J Biol Chem. 2012;287(50):41774–86.
Bales KR, et al. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J Neurosci. 2009;29(21):6771–9.
Dodart JC, et al. Gene delivery of human apolipoprotein E alters brain Abeta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2005;102(4):1211–6.
Hudry E, et al. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci Transl Med. 2013;5(212):212ra161.
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8(6):595–608.
Biere AL, et al. Co-expression of beta-amyloid precursor protein (betaAPP) and apolipoprotein E in cell culture: analysis of betaAPP processing. Neurobiol Dis. 1995;2(3):177–87.
Irizarry MC, et al. Apolipoprotein E modulates gamma-secretase cleavage of the amyloid precursor protein. J Neurochem. 2004;90(5):1132–43.
Castellano JM, et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011;3(89):89ra57.
Huang YA, et al. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell. 2017;168(3):427–41. e21.
Zhao N, et al. Alzheimer's Risk Factors Age, APOE Genotype, and Sex Drive Distinct Molecular Pathways. Neuron. 2020;106(5):727–742.e6.
Tarasoff-Conway JM, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015;11(8):457–70.
Zhao L, et al. Macrophage-mediated degradation of beta-amyloid via an apolipoprotein E isoform-dependent mechanism. J Neurosci. 2009;29(11):3603–12.
Jiang Q, et al. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58(5):681–93.
Panza F, et al. A critical appraisal of amyloid-beta-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2019;15(2):73–88.
Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci. 2012;15(3):349–57.
Hashimoto T, et al. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid beta peptide. J Neurosci. 2012;32(43):15181–92.
Lee SJ, et al. Towards an understanding of amyloid-β oligomers: characterization, toxicity mechanisms, and inhibitors. Chem Soc Rev. 2017;46(2):310–23.
Miyata M, Smith JD. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat Genet. 1996;14(1):55–61.
Trommer BL, et al. ApoE isoform-specific effects on LTP: blockade by oligomeric amyloid-beta1-42. Neurobiol Dis. 2005;18(1):75–82.
Lanz TA, Carter DB, Merchant KM. Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol Dis. 2003;13(3):246–53.
Wilhelmus MM, et al. Apolipoprotein E genotype regulates amyloid-beta cytotoxicity. J Neurosci. 2005;25(14):3621–7.
Folin M, et al. Apolipoprotein-E modulates the cytotoxic effect of beta-amyloid on rat brain endothelium in an isoform-dependent specific manner. Int J Mol Med. 2006;17(5):821–6.
Kanekiyo T, Xu H, Bu G. ApoE and Abeta in Alzheimer's disease: accidental encounters or partners? Neuron. 2014;81(4):740–54.
Balu D, et al. The role of APOE in transgenic mouse models of AD. Neurosci Lett. 2019;707:134285.
Strittmatter WJ, et al. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(17):8098–102.
Strittmatter WJ, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(5):1977–81.
LaDu MJ, et al. Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem. 1994;269(38):23403–6.
LaDu MJ, et al. Purification of apolipoprotein E attenuates isoform-specific binding to beta-amyloid. J Biol Chem. 1995;270(16):9039–42.
Yang DS, et al. Characterization of the binding of amyloid-beta peptide to cell culture-derived native apolipoprotein E2, E3, and E4 isoforms and to isoforms from human plasma. J Neurochem. 1997;68(2):721–5.
Pillot T, et al. Beta-amyloid peptide interacts specifically with the carboxy-terminal domain of human apolipoprotein E: relevance to Alzheimer's disease. J Neurochem. 1999;72(1):230–7.
Aleshkov S, Abraham CR, Zannis VI. Interaction of nascent ApoE2, ApoE3, and ApoE4 isoforms expressed in mammalian cells with amyloid peptide beta (1-40). Relevance to Alzheimer's disease. Biochemistry. 1997;36(34):10571–80.
Tai LM, et al. Levels of soluble apolipoprotein E/amyloid-β (Aβ) complex are reduced and oligomeric Aβ increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples. J Biol Chem. 2013;288(8):5914–26.
Namba Y, et al. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991;541(1):163–6.
Pankiewicz JE, et al. Blocking the apoE/Aβ interaction ameliorates Aβ-related pathology in APOE ε2 and ε4 targeted replacement Alzheimer model mice. Acta Neuropathol Commun. 2014;2:75.
Verghese PB, et al. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc Natl Acad Sci U S A. 2013;110(19):E1807–16.
Liu CC, et al. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron. 2017;96(5):1024–1032.e3.
Huynh TV, et al. Age-Dependent Effects of apoE Reduction Using Antisense Oligonucleotides in a Model of β-amyloidosis. Neuron. 2017;96(5):1013–1023.e4.
van der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid-beta-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci. 2020;21(1):21–35.
Busche MA, Hyman BT. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat Neurosci. 2020;23(10):1183–93.
DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener. 2019;14(1):32.
Farfel JM, et al. Association of APOE with tau-tangle pathology with and without beta-amyloid. Neurobiol Aging. 2016;37:19–25.
Jankowsky JL, Zheng H. Practical considerations for choosing a mouse model of Alzheimer's disease. Mol Neurodegener. 2017;12(1):89.
Götz J, Bodea LG, Goedert M. Rodent models for Alzheimer disease. Nat Rev Neurosci. 2018;19(10):583–98.
Saint-Aubert L, et al. Tau PET imaging: present and future directions. Mol Neurodegener. 2017;12(1):19.
Leuzy A, et al. Tau PET imaging in neurodegenerative tauopathies-still a challenge. Mol Psychiatry. 2019;24(8):1112–34.
Heinsinger NM, Gachechiladze MA, Rebeck GW. Apolipoprotein E Genotype Affects Size of ApoE Complexes in Cerebrospinal Fluid. J Neuropathol Exp Neurol. 2016;75(10):918–24.
Morikawa M, et al. Production and characterization of astrocyte-derived human apolipoprotein E isoforms from immortalized astrocytes and their interactions with amyloid-beta. Neurobiol Dis. 2005;19(1-2):66–76.
Tokuda T, et al. Lipidation of apolipoprotein E influences its isoform-specific interaction with Alzheimer's amyloid beta peptides. Biochem J. 2000;348(Pt 2):359–65.
Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. 2009;63(3):287–303.
Yvan-Charvet L, et al. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest. 2007;117(12):3900–8.
Yvan-Charvet L, et al. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118(18):1837–47.
Michikawa M, et al. Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J Neurochem. 2000;74(3):1008–16.
Hara M, et al. Isoform-dependent cholesterol efflux from macrophages by apolipoprotein E is modulated by cell surface proteoglycans. Arterioscler Thromb Vasc Biol. 2003;23(2):269–74.
Wahrle SE, et al. Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J Biol Chem. 2005;280(52):43236–42.
Wahrle SE, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008;118(2):671–82.
Donkin JJ, et al. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem. 2010;285(44):34144–54.
Lefterov I, et al. Expression profiling in APP23 mouse brain: inhibition of Abeta amyloidosis and inflammation in response to LXR agonist treatment. Mol Neurodegener. 2007;2:20.
Karjalainen JP, et al. The effect of apolipoprotein E polymorphism on serum metabolome - a population-based 10-year follow-up study. Sci Rep. 2019;9(1):458.
Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416–38.
Josephs KA, et al. TAR DNA-binding protein 43 and pathological subtype of Alzheimer's disease impact clinical features. Ann Neurol. 2015;78(5):697–709.
Amador-Ortiz C, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007;61(5):435–45.
James BD, et al. TDP-43 stage, mixed pathologies, and clinical Alzheimer's-type dementia. Brain. 2016;139(11):2983–93.
Nelson PT, et al. Hippocampal sclerosis in advanced age: clinical and pathological features. Brain. 2011;134(Pt 5):1506–18.
Govone F, et al. Lack of association between APOE gene polymorphisms and amyotrophic lateral sclerosis: a comprehensive meta-analysis. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(7-8):551–6.
Canosa A, et al. Correlation between Apolipoprotein E genotype and brain metabolism in amyotrophic lateral sclerosis. Eur J Neurol. 2019;26(2):306–12.
Verpillat P, et al. Apolipoprotein E gene in frontotemporal dementia: an association study and meta-analysis. Eur J Hum Genet. 2002;10(7):399–405.
Rubino E, et al. Apolipoprotein E polymorphisms in frontotemporal lobar degeneration: a meta-analysis. Alzheimers Dement. 2013;9(6):706–13.
Mishra A, et al. Gene-based association studies report genetic links for clinical subtypes of frontotemporal dementia. Brain. 2017;140(5):1437–46.
Vossel KA, et al. ApoE and TDP-43 neuropathology in two siblings with familial FTLD-motor neuron disease. Neurocase. 2013;19(3):295–301.
Yang HS, et al. Evaluation of TDP-43 proteinopathy and hippocampal sclerosis in relation to APOE epsilon4 haplotype status: a community-based cohort study. Lancet Neurol. 2018;17(9):773–81.
Outeiro TF, et al. Dementia with Lewy bodies: an update and outlook. Mol Neurodegener. 2019;14(1):5.
Schweighauser M, et al. Structures of α-synuclein filaments from multiple system atrophy. Nature. 2020;585(7825):464–9.
Twohig D, Nielsen HM. α-synuclein in the pathophysiology of Alzheimer's disease. Mol Neurodegener. 2019;14(1):23.
Tsuang D, et al. APOE epsilon4 increases risk for dementia in pure synucleinopathies. JAMA Neurol. 2013;70(2):223–8.
Dickson DW, et al. APOE epsilon4 is associated with severity of Lewy body pathology independent of Alzheimer pathology. Neurology. 2018;91(12):e1182–95.
Berge G, et al. Apolipoprotein E epsilon2 genotype delays onset of dementia with Lewy bodies in a Norwegian cohort. J Neurol Neurosurg Psychiatry. 2014;85(11):1227–31.
Lill CM, et al. Comprehensive research synopsis and systematic meta-analyses in Parkinson's disease genetics: The PDGene database. PLoS Genet. 2012;8(3):e1002548.
Nalls MA, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet. 2014;46(9):989–93.
Federoff M, et al. A large study reveals no association between APOE and Parkinson's disease. Neurobiol Dis. 2012;46(2):389–92.
de Lau LM, et al. Prognosis of Parkinson disease: risk of dementia and mortality: the Rotterdam Study. Arch Neurol. 2005;62(8):1265–9.
Harhangi BS, et al. APOE and the risk of PD with or without dementia in a population-based study. Neurology. 2000;54(6):1272–6.
Morris HR, et al. Multiple system atrophy/progressive supranuclear palsy: alpha-Synuclein, synphilin, tau, and APOE. Neurology. 2000;55(12):1918–20.
Ogaki K, et al. Multiple system atrophy and apolipoprotein E. Mov Disord. 2018;33(4):647–50.
Davis AA, et al. APOE genotype regulates pathology and disease progression in synucleinopathy. Sci Transl Med. 2020;12(529):eaay3069.
Zhao N, et al. APOE4 exacerbates alpha-synuclein pathology and related toxicity independent of amyloid. Sci Transl Med. 2020;12(529):eaay1809.
Freeman T, et al. Neuropsychiatric associations of apolipoprotein E alleles in subjects with combat-related posttraumatic stress disorder. J Neuropsychiatry Clin Neurosci. 2005;17(4):541–3.
Johnson LA, et al. ApoE2 Exaggerates PTSD-Related Behavioral, Cognitive, and Neuroendocrine Alterations. Neuropsychopharmacology. 2015;40(10):2443–53.
Mitchell P, et al. Age-related macular degeneration. Lancet. 2018;392(10153):1147–59.
Klaver CC, et al. Genetic association of apolipoprotein E with age-related macular degeneration. Am J Hum Genet. 1998;63(1):200–6.
Fritsche LG, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet. 2016;48(2):134–43.
Black JR, Clark SJ. Age-related macular degeneration: genome-wide association studies to translation. Genet Med. 2016;18(4):283–9.
Levy O, et al. APOE Isoforms Control Pathogenic Subretinal Inflammation in Age-Related Macular Degeneration. J Neurosci. 2015;35(40):13568–76.
Pickering-Brown SM, et al. Allelic variations in apolipoprotein E and prion protein genotype related to plaque formation and age of onset in sporadic Creutzfeldt-Jakob disease. Neurosci Lett. 1995;187(2):127–9.
Blackman JA. Apolipoprotein E genotype and cerebral palsy. Dev Med Child Neurol. 2010;52(7):600.
Bettencourt C, et al. The APOE epsilon2 allele increases the risk of earlier age at onset in Machado-Joseph disease. Arch Neurol. 2011;68(12):1580–3.
Xuan C, et al. No association between APOE epsilon 4 allele and multiple sclerosis susceptibility: a meta-analysis from 5472 cases and 4727 controls. J Neurol Sci. 2011;308(1-2):110–6.
Lill CM, et al. Closing the case of APOE in multiple sclerosis: no association with disease risk in over 29 000 subjects. J Med Genet. 2012;49(9):558–62.
Kehoe P, et al. Age of onset in Huntington disease: sex specific influence of apolipoprotein E genotype and normal CAG repeat length. J Med Genet. 1999;36(2):108–11.
Saft C, et al. Apolipoprotein E genotypes do not influence the age of onset in Huntington's disease. J Neurol Neurosurg Psychiatry. 2004;75(12):1692–6.
Revesz T, et al. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol. 2009;118(1):115–30.
Viswanathan A, Greenberg SM. Cerebral amyloid angiopathy in the elderly. Ann Neurol. 2011;70(6):871–80.
McCarron MO, et al. The apolipoprotein E epsilon2 allele and the pathological features in cerebral amyloid angiopathy-related hemorrhage. J Neuropathol Exp Neurol. 1999;58(7):711–8.
Nicoll JA, et al. High frequency of apolipoprotein E epsilon 2 allele in hemorrhage due to cerebral amyloid angiopathy. Ann Neurol. 1997;41(6):716–21.
Charidimou A, et al. Cerebral amyloid angiopathy with and without hemorrhage: evidence for different disease phenotypes. Neurology. 2015;84(12):1206–12.
Gosselet F, et al. Transcriptional profiles of receptors and transporters involved in brain cholesterol homeostasis at the blood-brain barrier: use of an in vitro model. Brain Res. 2009;1249:34–42.
Vanlandewijck M, et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature. 2018;554(7693):475–80.
Zeleny M, et al. Distinct apolipoprotein E isoform preference for inhibition of smooth muscle cell migration and proliferation. Biochemistry. 2002;41(39):11820–3.
Woo D, et al. Genetic and environmental risk factors for intracerebral hemorrhage: preliminary results of a population-based study. Stroke. 2002;33(5):1190–5.
Brouwers HB, et al. Apolipoprotein E genotype is associated with CT angiography spot sign in lobar intracerebral hemorrhage. Stroke. 2012;43(8):2120–5.
Gross BA, Jankowitz BT, Friedlander RM. Cerebral Intraparenchymal Hemorrhage: A Review. JAMA. 2019;321(13):1295–303.
O'Donnell HC, et al. Apolipoprotein E genotype and the risk of recurrent lobar intracerebral hemorrhage. N Engl J Med. 2000;342(4):240–5.
Tzourio C, et al. APOE genotype, ethnicity, and the risk of cerebral hemorrhage. Neurology. 2008;70(16):1322–8.
Schilling S, et al. APOE genotype and MRI markers of cerebrovascular disease: systematic review and meta-analysis. Neurology. 2013;81(3):292–300.
Kokubo Y, et al. Age-dependent association of apolipoprotein E genotypes with stroke subtypes in a Japanese rural population. Stroke. 2000;31(6):1299–306.
Pawlikowska L, et al. Apolipoprotein E epsilon 2 is associated with new hemorrhage risk in brain arteriovenous malformations. Neurosurgery. 2006;58(5):838–43. discussion 838-43.
Yamazaki Y, et al. Apolipoprotein E as a Therapeutic Target in Alzheimer's Disease: A Review of Basic Research and Clinical Evidence. CNS Drugs. 2016;30(9):773–89.
Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148(6):1204–22.
Williams T, Borchelt DR, Chakrabarty P. Therapeutic approaches targeting Apolipoprotein E function in Alzheimer's disease. Mol Neurodegener. 2020;15(1):8.
Cao J, et al. Advances in developing novel therapeutic strategies for Alzheimer's disease. Mol Neurodegener. 2018;13(1):64.
Hu J, et al. Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Abeta metabolism in apoE4-targeted replacement mice. Mol Neurodegener. 2015;10:6.
Koster KP, et al. Rexinoids as Therapeutics for Alzheimer's Disease: Role of APOE. Curr Top Med Chem. 2017;17(6):708–20.
Moutinho M, Landreth GE. Therapeutic potential of nuclear receptor agonists in Alzheimer's disease. J Lipid Res. 2017;58(10):1937–49.
Courtney R, Landreth GE. LXR Regulation of Brain Cholesterol: From Development to Disease. Trends Endocrinol Metab. 2016;27(6):404–14.
Hong C, Tontonoz P. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discov. 2014;13(6):433–44.
Terwel D, et al. Critical role of astroglial apolipoprotein E and liver X receptor-α expression for microglial Aβ phagocytosis. J Neurosci. 2011;31(19):7049–59.
Suon S, et al. Systemic treatment with liver X receptor agonists raises apolipoprotein E, cholesterol, and amyloid-β peptides in the cerebral spinal fluid of rats. Mol Neurodegener. 2010;5:44.
Fitz NF, et al. Improvement of memory deficits and amyloid-β clearance in aged APP23 mice treated with a combination of anti-amyloid-β antibody and LXR agonist. J Alzheimers Dis. 2014;41(2):535–49.
Sandoval-Hernández AG, et al. Role of Liver X Receptor in AD Pathophysiology. PLoS One. 2015;10(12):e0145467.
Vanmierlo T, et al. Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol Aging. 2011;32(7):1262–72.
Muñoz-Cabrera JM, et al. Bexarotene therapy ameliorates behavioral deficits and induces functional and molecular changes in very-old Triple Transgenic Mice model of Alzheimer’s disease. PLoS One. 2019;14(10):e0223578.
Corona AW, et al. ABCA1 is Necessary for Bexarotene-Mediated Clearance of Soluble Amyloid Beta from the Hippocampus of APP/PS1 Mice. J Neuroimmune Pharmacol. 2016;11(1):61–72.
Lefterov I, et al. RNA-sequencing reveals transcriptional up-regulation of Trem2 in response to bexarotene treatment. Neurobiol Dis. 2015;82:132–40.
Price AR, et al. Comment on "ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models". Science. 2013;340(6135):924–d.
Casali BT, Reed-Geaghan EG, Landreth GE. Nuclear receptor agonist-driven modification of inflammation and amyloid pathology enhances and sustains cognitive improvements in a mouse model of Alzheimer's disease. J Neuroinflammation. 2018;15(1):43.
O'Hare E, et al. Lack of support for bexarotene as a treatment for Alzheimer's disease. Neuropharmacology. 2016;100:124–30.
Balducci C, et al. The Continuing Failure of Bexarotene in Alzheimer's Disease Mice. J Alzheimers Dis. 2015;46(2):471–82.
Tesseur I, et al. Comment on "ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models". Science. 2013;340(6135):924–e.
Veeraraghavalu K, et al. Comment on "ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models". Science. 2013;340(6135):924–f.
Tai LM, et al. Amyloid-β pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J Biol Chem. 2014;289(44):30538–55.
Fitz NF, et al. Comment on "ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models". Science. 2013;340(6135):924–c.
Ghosal K, et al. A randomized controlled study to evaluate the effect of bexarotene on amyloid-β and apolipoprotein E metabolism in healthy subjects. Alzheimers Dement (N Y). 2016;2(2):110–20.
Cummings JL, et al. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene Xin moderate Alzheimer's disease. Alzheimers Res Ther. 2016;8:4.
Loren J, et al. Liver X receptor modulators: a review of recently patented compounds (2009 - 2012). Expert Opin Ther Pat. 2013;23(10):1317–35.
Kim J, et al. microRNA-33 Regulates ApoE Lipidation and Amyloid-β Metabolism in the Brain. J Neurosci. 2015;35(44):14717–26.
Hafiane A, Johansson JO, Genest J. ABCA1 Agonist Mimetic Peptide CS-6253 Induces Microparticles Release From Different Cell Types by ABCA1-Efflux-Dependent Mechanism. Can J Cardiol. 2019;35(6):770–81.
Pickar-Oliver A, Gersbach CA. The next generation of CRISPR-Cas technologies and applications. Nat Rev Mol Cell Biol. 2019;20(8):490–507.
Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157(6):1262–78.
So RWL, et al. Application of CRISPR genetic screens to investigate neurological diseases. Mol Neurodegener. 2019;14(1):41.
Lin YT, et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer's Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron. 2018;98(6):1141–1154.e7.
Wang C, et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med. 2018;24(5):647–57.
Schmid B, et al. Generation of a set of isogenic, gene-edited iPSC lines homozygous for all main APOE variants and an APOE knock-out line. Stem Cell Res. 2019;34:101349.
Lane-Donovan C, et al. Genetic Restoration of Plasma ApoE Improves Cognition and Partially Restores Synaptic Defects in ApoE-Deficient Mice. J Neurosci. 2016;36(39):10141–50.
Rasmussen KL, et al. Plasma levels of apolipoprotein E and risk of dementia in the general population. Ann Neurol. 2015;77(2):301–11.
Rasmussen KL, et al. APOE and dementia - resequencing and genotyping in 105,597 individuals. Alzheimers Dement. 2020. https://pubmed.ncbi.nlm.nih.gov/32808727/.
Koch M, et al. Association of Apolipoprotein E in Lipoprotein Subspecies With Risk of Dementia. JAMA Netw Open. 2020;3(7):e209250.
Medway CW, et al. ApoE variant p.V236E is associated with markedly reduced risk of Alzheimer's disease. Mol Neurodegener. 2014;9:11.
Acknowledgements
We thank Dr. Ana Caroline Raulin, Dr. Chia-Chen Liu, Dr. Melissa C. Wren, Mr. Trottier, Zachary A., Dr. Yonghe, Li, Dr. Yuka A. Martens and Dr. Hongmei Li for proofreading of the manuscript.
Funding
APOE-related work in authors’ labs is supported by grants from the National Institutes of Health and Cure Alzheimer’s Fund (to GB), the Coins for Alzheimer’s Research Trust (CART) Foundation (to NZ), and research grants from National Center for Geriatrics and Gerontology, the Hori Sciences and Arts Foundation, and the Yokoyama Foundation for Clinical Pharmacology (to MS).
Author information
Authors and Affiliations
Contributions
ZHL led the writing of the manuscript, devised all the figures and edited the manuscript. MS contributed to writing of the APOE2 and longevity and APOE2 and brain structure sections and co-edited the manuscript. SF wrote the APOE2 and cerebrovascular disease section and contributed to the editing of the manuscript. GB and NZ supervised the writing and co-edited the manuscript. All authors have read and agreed on the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
All authors agreed to publish.
Competing interests
The authors declared no competing of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Z., Shue, F., Zhao, N. et al. APOE2: protective mechanism and therapeutic implications for Alzheimer’s disease. Mol Neurodegeneration 15, 63 (2020). https://doi.org/10.1186/s13024-020-00413-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-020-00413-4