
Abstract
Propionic acidemia (PA) is a rare autosomal recessive congenital disease caused by mutations in the PCCA or PCCB genes. Elevated propionylcarnitine, 2-methylcitric acid (2MCA), propionylglycine, glycine and 3-hydroxypropionate can be used to diagnose PA. Early-onset PA can lead to acute deterioration, metabolic acidosis, and hyperammonemia shortly after birth, which can result in high mortality and disability. Late-onset cases of PA have a more heterogeneous clinical spectra, including growth retardation, intellectual disability, seizures, basal ganglia lesions, pancreatitis, cardiomyopathy, arrhythmias, adaptive immune defects, rhabdomyolysis, optic atrophy, hearing loss, premature ovarian failure, and chronic kidney disease. Timely and accurate diagnosis and appropriate treatment are crucial to saving patients’ lives and improving their prognosis. Recently, the number of reported PA cases in China has increased due to advanced diagnostic techniques and increased research attention. However, an overview of PA prevalence in China is lacking. Therefore, this review provides an overview of recent advances in the pathogenesis, diagnostic strategies, and treatment of PA, including epidemiological data on PA in China. The most frequent variants among Chinese PA patients are c.2002G > A in PCCA and c.1301C > T in PCCB, which are often associated with severe clinical symptoms. At present, liver transplantation from a living (heterozygous parental) donor is a better option for treating PA in China, especially for those exhibiting a severe metabolic phenotype and/or end-organ dysfunction. However, a comprehensive risk–benefit analysis should be conducted as an integral part of the decision-making process. This review will provide valuable information for the medical care of Chinese patients with PA.
Similar content being viewed by others


Background
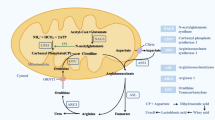
Propionic acidemia (PA) is an autosomal recessive metabolic disorder caused by impaired functioning of propionyl coenzyme A (propionyl-CoA) carboxylase (PCC) in the mitochondria. This results in the metabolic disturbances of propiogenic amino acids (valine, isoleucine, threonine, and methionine), propionate, and odd-chain fatty acids. Patients with PA typically present with vomiting and anorexia, followed by dehydration, weight loss, lethargy, hypothermia, hypotonia, and convulsions. These symptoms can progress rapidly and result in decompensation and poor neurological outcomes. Over time, PA can lead to multiple organ complications, including those affecting the brain, heart, liver, kidney, pancreas, and bone marrow. In severe cases, PA can result in death [1]. Patients with PA typically experience recurrent episodes of hyperammonemia, ketoacidosis, metabolic acidosis, neutropenia, and thrombocytopenia [2]. The clinical presentation is diverse and lacks specificity, with a rapid clinical progression and poor prognosis. Although PA cannot be cured at this time, prompt and proper diagnosis and treatment can stabilize the disease and prevent severe complications.
To gather relevant information for this review, we conducted a literature search from 2010 to 2022 through CNKI and PUBMED databases with the keywords “propionic acidemia”, “organic acidemia”, “metabolic disease”, “newborn screening”, and “China”. Our search yielded a total of 119 papers that met our search criteria and were included in this review.
Etiology and pathogenesis of PA
Structure and function of PCC
PCC is a biotin-dependent carboxylase heterododecamer holoenzyme composed of six α and six β subunits, which is primarily localized in the mitochondria and loosely bound to the inner mitochondrial membrane matrix [3]. Alpha and beta subunits are encoded by the genes PCCA (OMIM232000) and PCCB (OMIM232050), respectively [4]. PCCA gene is located on chromosome 13q32.3 and its encoded α-subunit is 72–80 kDa with three splice isoforms. Classical isoform A (NM_000282.3) is the longest splice isoform encoded by 24 exons containing 728 amino acids. The isoform B is shorter and is encoded by 23 exons containing 702 amino acids. The isoform C is the shortest and is encoded by 23 exons containing 681 amino acids [5]. The PCCB gene is located on chromosome 3q22.3 and its encoded β-subunit is 58 kDa. The β-subunit contains 15 exons and encodes 539 amino acids. α-Subunit decorates the outside of a hexamer nucleus formed from 6 β-subunits. α-Subunit contains the N-terminal biotin-binding domain and the C-terminal biotin carboxylase domain, which are responsible for the formation of carboxy biotin after ATP hydrolysis through interaction. β-Subunit hexamer with a propionyl-CoA binding site and a carboxyltransferase domain is responsible for transferring the carboxyl group to propionyl-CoA [6]. Propionyl-CoA is a metabolite that is produced under physiological conditions from propionate, amino acids (valine, isoleucine, threonine, and methionine), side chain of cholesterol, and odd-chain fatty acids. Propionyl-CoA is carboxylated to methylmalonyl-CoA by PCC in the mitochondria and further isomerized to succinyl-CoA. This process is known as the anaplerosis that replenishes the loss of TCA cycle intermediates.
Pathogenesis of PA
PA is caused by impaired functioning of PCC due to mutations in the PCCA or PCCB genes. Mutations of biallelic sites in either PCCA or PCCB genes or compound heterozygous mutations in two genes result in the loss of PCC activity and lead to abnormal accumulation of propionyl-CoA and its metabolites, such as propionylcarnitine, propionylglycine, 3-hydroxypropionate and 2-methylcitric acid(2MCA). These metabolic disturbances result in a series of biochemical abnormalities and multiple organ complications [7]. Elevated levels of propionyl-CoA can compete with the comparatively lower levels of acetyl-CoA for citrate synthase, leading to the overproduction of 2MCA from propionyl-CoA and oxaloacetate instead of citrate. This excessive 2MCA production in propionic acidemia diverts oxaloacetate away from the tricarboxylic acid (TCA) cycle. 2MCA is reported to be an inhibitor of multiple enzymes involved in TCA cycle, such as citrate synthase, aconitase, isocitrate dehydrogenase, in rat liver mitochondria [8]. The harmful effects on mitochondria induced by 2MCA may play a role in the development of brain damage and neurological complications in patients with PA [9]. High levels of propionyl-CoA and its metabolites are reported to inhibit multiple enzymes including pyruvate dehydrogenase complex and respiratory chain complexes in the TCA cycle, hindering mitochondrial energy production [10]. Additionally, the production of succinyl-CoA from propionyl-CoA is also reduced. The above metabolic perturbations could inhibit TCA cycle flux and impair energy production. Life-threatening acute metabolic decompensations (AMD) are a prominent feature in PA, characterized biochemically by hyperammonemia, metabolic acidosis with a high anion gap, and lactic acidosis [11]. Hyperammonemia is primarily caused by urea cycle disorders. Propionyl-CoA acts as a competitive inhibitor of N-acetylglutamate synthase, which reduces the synthesis of N-acetylglutamate. N-acetylglutamate is an agonist of carbamylphosphate synthase-1, and its reduction can subsequently lead to a decrease in the activity of carbamylphosphate synthase-1 in the urea cycle [12]. Some evidence also suggests the amino acid substrates of the urea cycle, especially citrulline, ornithine and arginine are reduced in patients with PA [13]. During PA decompensations, the body's compensatory mechanisms rely heavily on glutamate/glutamine metabolism to convert α-ketoglutarate. This process results in an excessive generation of ammonia. Another potential mechanism to explain the occurrence of hyperammonemia during PA decompensations is the decrease in glutamine levels [14].
Significantly higher concentrations of the branched-chain amino acids (BCAAs), including leucine (Leu), valine (Val), and isoleucine (Ile), and their intermediate metabolites have been observed during decompensation episodes. This suggests that there may be an increase in protein catabolism. The breakdown of BCAAs acutely increases the amount of circulating toxic metabolites. Due to their acidic nature, these metabolites can rapidly lead to metabolic acidosis by decreasing the pool of bicarbonate in the body [9, 15]. Lactate acidosis may result from the decline of pyruvate dehydrogenase activity, which can lead to the excessive conversion of pyruvate to lactate. Elevated levels of propionate and its metabolites 3-hydroxypropionate and methylcitrate can cause bone marrow suppression, which may lead to anemia, granulocytopenia, and thrombocytopenia [16]. In addition, severe anemia in PA could be ascribed to low levels of BCAA in plasma, which could be a result of a low natural protein intake and high demands for protein synthesis [17]. Ketoacidosis is often reported in PA with metabolic decompensation and its underlying mechanism remains unclear. Studies on the PA mouse model have suggested that propionate overload can stimulate ketone production by increasing fatty acid oxidation in the liver via the lowering of malonyl-CoA [18]. Furthermore, the accumulation of propionyl-CoA can interfere with glycine cleavage by reducing H protein production, leading to hyperglycinemia [19]. Hepatic encephalopathy resulting from hyperammonemia may be a major cause of neurological damage in individuals with PA.
Clinical features and diagnosis
Patients with PA exhibit a diverse range of clinical manifestations that can present from infancy to adulthood. These manifestations are typically categorized as either early-onset (occurring at or before 3 months of age) or late-onset (occurring after 3 months of age) [20]. Early-onset typically affects newborns who were born with normal gestation and delivery. Patients may not exhibit symptoms immediately after birth and can remain asymptomatic for hours, days, or even months. However, they are at high risk of sudden acute metabolic decompensation, which can be life-threatening. Children with early-onset PA often exhibit symptoms such as poor feeding, lethargy, disorders of consciousness, repeated vomiting, convulsions, dyspnea, growth retardation, epilepsy and motor disorders. Late-onset PA can be divided into two types: chronically progressive and intermittent seizure. The intermittent onset type has an acute phase followed by a stable phase. The acute decompensation phase is typically triggered by metabolic stress, such as infection, prolonged or intense physical exercise, injury, surgery and/or general anesthesia, excessive protein intake, and the attack has a neonatal-like onset. In contrast, the stable phase can be associated with various complications [21].
The most common complications associated with PA are cardiac and neurological disorders [22]. Cardiac complications include cardiomyopathy and arrhythmias. Chronic neurological and cognitive complications are frequent in PA, including movement disorders, spastic paresis, intellectual disability and strokes of basal ganglia [23]. Late manifestations of neuropsychological disorders, such as autism and borderline personality traits, have also been reported [24, 25]. Other complications may include recurrent pancreatitis, adaptive immune defects, rhabdomyolysis, optic atrophy, hearing loss, premature ovarian failure, and chronic kidney disease [26,27,28,29]. In China, most of the reported clinical cases are early-onset. Delayed cardiomyopathy and neurological complications have also been reported in individual cases. The characteristics and clinical data of patients we reviwed are presented in Table 3.
Neonatal screening by mass spectrometry is currently an effective tool for early identification and diagnosis of PA. The diagnosis of PA is confirmed by measuring the levels of C3 and the ratio of C3/acetylcarnitine (C2) in blood by liquid chromatography-tandem mass spectrometry (LC–MS/MS). The typical range of values for C3 (propionylcarnitine) is between 0.2 and 4.3 μmol/L. Meanwhile, the C3/C2 (acetylcarnitine) ratio typically falls between 0.03 and 0.2 [61]. In addition, the levels of 3-hydroxypropiona, propionylglycine and 2MCA in urine can also be measured using gas chromatography-mass spectrometry (GC–MS) to aid in diagnosis [20]. When PA is suspected, further examinations should be conducted to confirm the initial biochemical diagnosis. General tests such as blood ammonia, blood glucose, blood gas analysis, and myocardial zymogram analysis, as well as urine tests for ketone bodies and organic acids, can be conducted to aid in diagnosis [30]. In China, patients with early-onset PA are often misdiagnosed with intermittent neurological deterioration and neonatal sepsis [16]. Following standard clinical and analytical procedures is critical for the differential diagnosis. In newborns with clinical distress and suspicion of sepsis, seizures, or organic acidemias must be considered in the differential diagnosis from the outset [21, 22]. PA and methylmalonic acidemia (MMA) are both disorders in the propionyl-CoA metabolic pathway with shared clinical presentations. PA and MMA are not easily distinguished when screening for inherited metabolic disorders in newborns. Both MMA and PA can cause an elevation of C3 in the acylcarnitine spectrum, which is a common metabolic biomarker for both disorders. However, since PA directly causes an increase in C3, the rise of C3 is usually more pronounced with PA than with MMA in theory, as demonstrated in clinical cases [31, 32]. Determination of methylmalonate from dried blood spots (DBS), serum, and urine samples is commonly performed for differential diagnosis of PA and MMA in clinical diagnostics and newborn screening. The biochemical indicators of PA include elevated 2MCA and propionylglycine, and C3, while high urinary methylmalonate is specific to MMA [33]. Higher levels of 2MCA are generally observed in patients with PA than with MMA. Additionally, PA patients with a severe phenotype and significant long-term complications tend to have even higher levels of 2MCA [34]. A retrospective study conducted at a single center revealed that C3 level, C3/C2 ratio, and 2MCA level in the amniotic fluid supernatant are reliable biochemical markers for the diagnosis of PA. This study also suggested that the C3/C2 ratio is the most dependable biochemical marker for the prenatal diagnosis of PA [35]. Table 1 summarizes the various biomarkers used for the differential diagnosis of PA and MMA and can serve as a reference for clinical practice. The diagnosis of PA is confirmed through the analysis of mutations in the PCCA and PCCB genes, as well as the measurement of PCC enzyme activity. Sanger sequencing, quantitative polymerase chain reaction (qPCR), next generation sequencing (NGS) are employed to detect and identify the pathogenic mutations of biallelic sites in PCCA or PCCB genes. In special cases, additional technologies such as cDNA analysis, multiplex ligation-dependent probe amplification (MLPA), or long-read whole-genome sequencing may be necessary to improve the detection rates [36]. Splicing variants need to be confirmed at the mRNA level. The identification of biochemical markers and gene mutations can aid in family genetic counseling and prenatal diagnosis, particularly for families with a proband affected by PA [37].
Epidemiology data of PA in China
The incidence of PA varies across different countries and regions, with global estimates ranging from 1/313,000 to 1/1000. In the United States, the estimated live-birth incidence is 1/105,000–1/130,000 [32, 38] and in Italy 1/166,000 [39]. In a selective and expanded newborn screening, the frequencies of PA in Japan, South Korea, and Germany were 1/41,000, 1/313,000 and 1/250,000, respectively [40]. The birth incidence in the Middle East is generally higher, with rates of 1/20,000–1/45,000 in the United Arab Emirates [41] and 1/28,000 in Saudi Arabia [42]. Some Saudi tribes have even higher rates, ranging from 1/2000 to 1/5000 [43]. The highest birth incidence (1/1000) is among the Greenlandic Inuits [44].
In mainland China, newborn screening by mass spectrometry was first introduced in 2004 [45]. With the wide application of LC–MS/MS and GC/MS, newborn screening for PA has been available and reported in most parts of China. However, the overall prevalence of PA in China remains unknown. We conducted a retrospective study analyzing screening data from different districts of China and collected incidence data of neonatal inherited metabolic diseases from 23 provinces or municipalities over the last decade (from 2010 to 2022). The results are presented in Table 2. Furthermore, we mapped out the provincial-level prevalence of PA in China in Fig. 1, based on the reported cases.

Distribution and incidence of PA in each province/municipality in China. HLJ, Heilongjiang; JL, Jilin; LN, Liaoning; XJ, Xinjiang; IM, Inner Mongolia; BJ, Beijing; TJ, Tianjin, HEB, Hebei; SX, Shanxi; SAX, Shaanxi; NX, Ningxia; GS, Gansu; QH, Qinghai; SD, Shandong; JS, Jiangsu; AH, Anhui; HEN, Henan; SH, Shanghai; HUB, Hubei; CQ, Chongqing; SC, Sichuan; ZJ, Zhejiang; JX, Jiangxi; HUN, Hunan; GZ, Guizhou; YN, Yunnan; FJ, Fujian; TW, Taiwan; GD, Guangdong; GX, Guangxi; HN, Hainan, HK, Hongkong; MC, Macau. Areas without color indicate that those without screening data. The darker the color the higher the incidence
To better analyze the incidence of PA in China, we divided country into two regions based on the Qinling Mountains-Huaihe River line, namely the northern and southern regions. The overall incidence of PA in the southern region of China was found to be higher, ranging from 1/489,148 to 1/515, with the highest in Guangning city, Guangdong Province. Out of 8,238 neonates screened from October 2010 to October 2015 in Guangning, 16 were diagnosed with PA [46]. The lowest incidence was found in Wenzhou city, Zhejiang Province, where 489,148 newborns were screened from October 1, 2013 to December 31, 2018, and the incidence of PA was reported as 1/489,148 [47].
The overall incidence rate of PA in northern China was from 1/283,459 to 1/3,156. Among 850,486 neonates screened in Henan province from January 2013 to August 2019 [48], three cases of PA were detected with an incidence rate of 1/283,459, which was the lowest in northern China. On the other hand, in Yancheng city, Jiangsu Province, 6 cases of PA were diagnosed among 18,988 screened neonates from June 2012 to June 2014, resulting in an incidence rate of 1/3,156, which was the highest in northern China [49].
Our survey included 15 regions in the south and 8 regions in the north based on available reports. However, cases of PA reported in other regions of China without regional incidence data were not included in this review. For example, from 2007 to 2010, 113 cases of PA were diagnosed from 5,931 children screened in major hospitals in Hebei province. Additionally, PA is a common organic acidemia found in Shijiazhuang [50]. Table 2. displays the epidemiological data of PA in China, indicating a significant regional difference in PA incidence across the country. The variation in incidence might be due to differences in genetic and ethnic backgrounds, as well as the total number of people screened. Moreover, the dates of newborn screening differ among various regions. For instance, in Zhejiang Province, the number of newborns screened for PA reached 1,861,262 from January 2009 to December 2016 [51], while the screening data of Sichuan Province was only collected from November 2017 to December 2018 and the number of newborns screened was 39,648 in this area [52]. In addition, various regions have different newborn screening rates conducted at disparate times. For example, Qingdao reported a screening rate of 79.0% in 2016 [53], while Yancheng reported a screening rate of 29.0% from January to December in 2012 [49].
Correlation between genotypes and phenotypes of PA in China
The correlation between gene mutations and clinical phenotype in PA patients has become a hot topic due to the increase in reported PA cases in recent years. To contribute to this field, we conducted a study analyzing the PCC mutations in 61 Chinese patients with PA reported in literature since 2014 and examined the phenotype-genotype correlations.
PCCA and PCCB variants in Chinese PA patients
Among 61 patients, 25 (41.0%) patients harbored PCCA variants including three pairs of siblings and 36 (59.0%) harbored PCCB variants including two pairs of siblings. There were 48 variants in PCCA and 66 variants in PCCB. Seventeen (27.9%) patients were diagnosed with newborn screening or prenatal diagnosis. Of the remaining 49 patients, 35 had an early-onset (≤ 3 months) disease and 7 had a late-onset (> 3 months) disease. Following the guidelines developed by the American College of Medical Genetics and Genomics (ACMG), we classified the gene mutations into five levels: pathogenic, likely pathogenic, benign, likely benign, and uncertain significance. We also listed the CADD pathogenicity score, and the mutation significance cut-off score was set at 20 (Table 3.).
The frequency of gene mutation types was calculated and shown in Fig. 2. The most common variants found in the reported Chinese PA patients were c.2002G > A in PCCA and c.1301C > T in PCCB, with frequencies of 25.0% (12/48 alleles) and 18.2% (12/66 alleles), respectively. Missense mutations has been reported to account for approximately 50% of the variations and are the most frequent type in PCCA and PCCB [54]. Our analysis also reveals that missense mutations are the most common mutant form of PCCA and PCCB, accounting for 46.0% (23/50) and 51.4% (37/72), respectively. The distribution of PCCA gene mutations is more dispersed in all exons except for exons 1, 5, 8, 17, 18, 20, 23, and 24, while the PCCB gene mutations are mainly on exons 1, 3, 12, and 13. In other countries, gene mutations of PCCA and PCCB are mostly found in exons 12, 13, 18, 19 and exons 6, 11, 12, 15, respectively [44, 55,56,57,58].

The frequency of PCCA and PCCB gene mutation types. A The distribution of PCCA variants in 25 Chinese patients diagnosed with PA. B The distribution of PCCB variants in 36 Chinese patients diagnosed with PA
Genotype–phenotype correlations in Chinese PA patients
We examined reported cases of PA in Chinese patients to determine whether there are correlations between genotypes and clinical phenotypes. We identified 50 PCCA gene mutation sites in 25 patients (48 alleles were detected). The most common PCCA variant was c.2002G > A, with a frequency of 25.0% (12/48 alleles). The second most common variant was c.229C > T, accounting for 8.3% (4/48 alleles). Additionally, c.1850 T > C, c.1288C > T, c.1426C > T, c.1746G > C, exon3-4del, c.1845 + 1G > A, and c.446delA occurred at a frequency of 4.2% (2/48 alleles), while all other mutation sites were reported only once in our dataset. Homozygous variants with higher frequencies are often accompanied by a certain clinical phenotype. The c.2002G > A mutation, which results in a missense substitution of glycine by arginine at codon 668 of the PCCA protein (p.Gly668Arg), is one such variant. The mutant protein, which is defective in biotinylation, has been found to have only 1.4% residual activity compared to the wild-type PCCA protein [5]. A case study [59] reported that twin siblings with PA were homozygous for the PCCA c.2002G > A mutation, yet they presented slightly different clinical symptoms. The firstborn daughter developed the disease early and became more severe. She had respiratory failure, granulocytopenia, and cardiac damage with a faster disease progression. Although the second son had disorders of consciousness, feeding difficulties, hyperammonemia, and metabolic acidosis, his symptoms were relatively mild and progressed slowly. The elder daughter died after being discharged on the same day and the second son died one month after discharge. Hu et al. also reported a case of this mutation, in which the patient died at three years and nine months of age [60]. The two patients registered in Xuzhou [61] were found to have compound heterozygous mutations (PCCA c.2002G > A/c.2040G > A and c.2002G > A/c.131delinsATT). They exhibited growth retardation and the onset of disease started in early infancy. Zhou et al. [62] reported a patient with late-onset PA due to c.2002G > A homozygous mutation. This patient was admitted to hospital at the age of two years and six months for PA-associated cardiomyopathy. Despite strict dietary control and drug treatment, the child experienced several acute metabolic acidosis decompensation events and suffered severe growth and development delay which greatly affected her quality of life. Preoperative echocardiography suggested a mild left ventricular dilation with a reduced overall motion amplitude of the left ventricle and a decreased left ventricular ejection fraction. To prevent further deterioration of her cardiac, neurologic, and other systems' function or the development of irreversible complications, liver transplantation was performed. The patient had a normal function of transplanted liver and cardiac function was restored based on echocardiographic test at 13.8 months after her liver transplant. The quality of life was greatly improved, although the growth delay was not improved. A patient with c.2002G > A/c.1288C > T compound heterozygous mutation was reported by Yang et al. The patient had seizures and possible cardiomyopathy and eventually died of cardiac arrest at the age of 6.5 months [7]. These cases suggest that the clinical phenotype associated with c.2002G > A is more severe and may develop cardiomyopathy. Furthermore, c.2002G > A is one of the hot spots of PCCA gene mutation in China, which is consistent with the findings by Liu et al. [63] According to ClinVar, PCCA c.229C > T (p.Arg77Trp) results in a non-conservative amino acid change located in the biotin carboxylation domain and biotin carboxylase-like, N-terminal domain of the encoded protein sequence. In Taiwan, a PA patient with compound heterozygous mutations (PCCA c.1262A > C/c.229C > T) exhibited normal to very mild developmental delay [64]. One of 82 PA patients reported in the literature was homozygous for the PCCA c.229C > T mutation and exhibited convulsions and lethargy at the age of 5 years [60]. However, during the next three years, the child had no recurrence and had moderate academic performance. The height and weight of this child were in the normal range, suggesting the clinical phenotype of c.229C > T mutation is mild in these cases.
Among 36 patients with 72 PCCB gene mutation sites (only 66 were detected), c.1301C > T was the most common PCCB variant with a frequency of 18.2% (12/66 alleles), followed by c.838dupC (frequency of 12.1%, 8/66 alleles), c.1087 T > C (frequency of 6.1%, 4/66 alleles), and c.1316A > G (frequency of 6.1%, 4/66 alleles). The c.1301C > T mutation, which is located on exon13 of the PCCB gene, is a missense mutation (p.Ala434Val). A case of c.1301C > T homozygous mutation was reported in a child from mainland China [65], who presented with severe and early-onset clinical manifestations, including poor feeding, hyperglycinemia, hyperammonemia, metabolic acidosis, early recurrent infections, and developmental delay. Although he was positively treated such as sodium bicarbonate IV to correct acidosis, l-carnitine supplement, BCAA restriction and protein intake reduction, intellectual development was still delayed. The genotypes of 10 PA patients in Taiwan were analyzed by Chiu et al. [64]. Half of PCCB allele mutations reported in Taiwan are c.1301C > T(p.A434V) which is a common mutation in this region and leads to low enzyme activity, presenting the classic phenotype of PA [66]. Therefore, c.1301C > T mutation is associated with an early-onset and severe clinical symptoms.
Liu et al. reported a case of a child with a homozygous c.838dupC mutation in the PCCB gene, which caused a significant alteration in the spatial conformation of the subunit of the mutant PCCB protein [16]. The mutant causes the premature termination with the loss of 250 amino acids in the peptide chain of the PCCB protein. Despite active treatment, the child's disorders of consciousness gradually worsened, and he died of pneumonia, indicating that the c.838dupC mutation most likely contributes to severe pathogenicity. Three patients with PCCB c.1316 A > G mutation were diagnosed in the Jining region, Shandong province and were suspected to be a common mutation in Jining City [67]. According to Hu and others [60], one child with a c.1316 A > G homozygous mutation in the PCCB gene did not have another acute onset after the first onset at nine months of age. Two cases of c.1316 A > G had normal intelligence. One had an acute onset at only 20 months of age, and the other had no clinical onset. Therefore the clinical phenotype of PCCB c.1316A > G mutation is likely mild.
The analysis above suggests that certain gene mutations are relatively common among Chinese patients with PA, and their associated with certain phenotypes. However, for less frequent mutations, the relationship between genotype and phenotype may be less clear, possibly due to genetic heterogeneity in both PCCA and PCCB mutations. There are several challenges in correlating genotype with phenotype in patients with PA. One of these challenges is the presence of compound heterozygous mutations in many patients, which can further complicate the relationship between genotype and phenotype. In addition, determining the severity of the condition in patients with mutations in both the PCCA and PCCB genes, which encode for the PCC holoenzyme, is particularly difficult. Finally, the interaction and influence of different genes on the development and progression of the disease are not well understood [43].
Therapeutic interventions for PA
Diet control and drug administration are common clinical treatments for PA. Organ transplantation becomes an option when the disease progresses and does not respond to diet control and drug interventions. Liver transplantation (LT) is an effective surgery since the liver can efficiently metabolize propionyl-CoA and its metabolites. Other organ transplants, such as heart and kidney [68], have also been reported when complication in these organs become severe.
PA is typically treated based on the phase of the disease that the patient is experiencing. The acute and chronic phases have different dietary and medication requirements, which are taken into consideration during treatment in the clinic. However, the primary aim of treatment for PA is to prevent protein catabolism and maintain biochemical control, growth, and development through adequate calorie intake. Long-term dietary management of PA involves restricting natural protein and supplementing with medical formulas that are enriched with Leu but free of Val, Ile, Met, and Thr to reduce the concentration of elevated metabolites [69]. Recent studies have suggested that the medical formula used for PA patients may contribute to growth retardation, despite high total protein intake. This is due to enhanced oxidation of Val and Ile in the presence of abundant Leu, known as BCAA antagonism. This imbalance makes both Val and Ile less available for anabolism, resulting in an unfavorable outcome for growth [70]. Therefore, Saleemani and colleagues have suggested that optimal protein synthesis can be achieved by reducing Leu intake and maintaining Ile and Val at the minimal level of PA recommendations. A BCAA ratio of 1:0.26:0.28 (Leu:Ile:Val) to 1:0.35:0.4 (Leu:Ile:Val) was associated with optimal protein synthesis [71]. A retrospective cohort study in Dutch evaluated both longitudinal dietary treatment and clinical course of MMA and PA patients. The study found that one-fourth of MMA and PA patients had a natural protein prescription that exceeded the recommended daily allowances (RDA). Additionally, many patients received additional amino acid mixtures (AAM) protein prescription despite already meeting the RDA for natural protein. In patients with early-onset PA, a higher natural protein prescription was associated with more frequent AMD. Therefore, it is recommended to exercise caution when prescribing AAM and to reduce protein prescriptions in patients, especially for those severely affected, who have already been given protein above RDA [72].
The current drug development for PA primarily focuses on the harmful biomarkers of the disease. l-carnitine supplement is another common treatment for PA patients by converting propionyl-CoA to propionylcarnitine which can be excreted through urine. Coenzmy Q10, an antioxidative nutritional supplement, may help prevent the chronic complications associated with mitochondrial dysfunction in PA. In a prospective study, seven patients with PA received supplements of CoQ10 in the form of ubiquinol (10 mg/kg/day for 6 months). Supplementation with ubiquinol normalized plasma CoQ10 concentrations in six patients who had shown a reduction. Furthermore, urinary citrate levels markedly increased, along with an elevation in the citrate/methylcitrate ratio [73]. Antibiotics such as metronidazole (MTZ) are often prescribed to patients with PA to inhibit the production of propionate by the intestinal microbiome. However, two patients with severe neonatal onset PA who were on chronic MTZ therapy developed axonal peripheral neuropathy. As a result, their peripheral nerve function was closely monitored during the course of MTZ treatment [74]. Arginine and sodium benzoate are used to reduce ammonia [75]. N-carbamylglutamate (NCG) has been found to be effective not only in the acute phase, but also significantly reduce ammonia levels during the chronic phase. However, additional research is needed to determine the optimal dosage of NCG [76]. Clinical studies have shown that carglumic acid is effective in reducing plasma ammonia levels in patients with PA and mitigating the frequency of hyperammonemia episodes. Additionally, carglumic acid is well-tolerated in long-term treatment [77]. Recent studies have demonstrated that a novel small molecule, HST5040 (2,2-dimethylbutyric acid), can reduce the ratio of C3/C2 and propionyl-CoA in primary hepatocytes of patients with PA in a dose-dependent manner by redistributing free and conjugated CoA pools. These findings suggest that HST5040 may be a promising drug candidate for the treatment of PA [78]. Furthermore, two allosteric pantothenate kinase activators, PZ-3022 and BBP-671, have been found to improve mitochondrial function in a mouse model of PA by improving intracellular C3:C2-CoA and plasma C3:C2-carnitine ratios and restoring liver CoA pool and acteyl-CoA to wild-type amounts in males and females [79, 80].
Liver transplantation (LT) is currently the most common surgical intervention for PA. The enzymatic activity of PCC can be restored by implanting a normal liver, which improves the metabolic stability of PA [81]. The study by Barshes et al. [82] showed that the survival rate of 12 patients with PA who received LT reached 72.2% within one year. Therefore, LT can be considered for PA patient whose condition exacerbates even with strict dietary restrictions and other medical treatments. However, LT improves certain complications but cannot completely cure PA [83]. There is still a risk of recurrence and death after LT, and ongoing follow-up treatment remains necessary.
In China, the treatment regimen for PA follows the international guideline of PA. Patients with PA should adhere to a strict and individualized nutritional intervention and receive long-term medications such as carnitine, MTZ, and NCG. In the event of severe hyperammonemia or acidosis, prompt treatment with peritoneal or blood dialysis is necessary [84]. LT becomes the first option when metabolic decompensation occurs frequently even after strict dietary restriction and drug therapy [63]. In a study by Zeng et al., six Chinese patients with PA underwent living donor liver transplantation (LDLT) due to frequent metabolic decompensation and one patient with PA underwent LDLT for prophylactic treatment. All the recipients were alive with 100% allograft survival. This study also demonstrated that hepatic expressions of PCCA and PCCB was consistent at the protein level in both heterozygous donor and the healthy donor for the first time. Liver supply from relatives carrying heterozygous genes is relatively easy to obtain and less burdensome for patients’ families [85] (Additional file 1). For children with mild PA, prophylaxis is recommended for patients under one year of age [86]. It is undeniable that patients with PA may still develop complications even after LT, such as renal failure, hepatic artery thrombosis [86, 87]. However, LT can largely prevent metabolic decompensation, achieve protein diet liberalization, improve neurodevelopmental delay to some extent, and may even treat cardiomyopathy [85].
It is worth noting that extrahepatic tissues lacking PCC continue to produce toxic metabolites after LT, which means that LT cannot provide a complete cure for PA. At a tertiary center, a total of 14 children underwent LT, and three of them died after LT. Among the 11 survivors, two experienced metabolic stroke but made a full recovery, and three developed mild cardiomyopathy after LT [88]. In some cases, PA complications may continue to progress even after a successful LT. For example, there was a case, who presented with a fatal metabolic stroke 11 years after undergoing a successful LT [89]. Two case were reported to develop recurrent cardiomyopathy following LT [90]. Several issues need to be addressed when considering LT for patients with PA: (1) minimizing LT complications such as rejection, hepatic artery thrombosis, cytomegalovirus/Epstein-Barr virus infection, and biliary complications; (2) determining the optimal timing for LT, and considering prophylactic LT; and (3) assessing long-term safety and feasibility of protein intake liberalization after LT.
Emerging therapies
Hepatocyte transplantation and hepatic progenitor cell transplantation have also been investigated as emerging treatments for urea cycle defects, including PA, in clinical trials [91, 92]. Gene therapy is emerging as a promising therapeutic approach for patients with PA, although it still faces challenges [93]. The first gene therapy product, AAV9-hPCCA (NCATS-BL0746), using adeno-associated virus serotype 9 to deliver human PCCA, was granted a rare pediatric disease designation by the U.S. Food and Drug Administration (FDA) for clinical trials on PCCA-related PA [94].
Recently, an enzyme replacement approach to treat PA has been reported. This approach involves the administration of a combination of two mRNAs encoding human PCCA and PCCB encapsulated in biodegradable lipid nanoparticles (LNPs), to a murine model with a hypomorphic phenotype, Pcca(− / −)(p.A138T). The treatment resulted in the production of functional PCC enzyme in the liver and a reduction in primary disease-associated toxins in a dose-dependent manner in both 3- and 6-month repeat-dose studies in mice with PA [95].
Conclusion
PA is a genetic disease with a poor prognosis that requires early detection and treatment for efficient preventive management. LC–MS/MS and GC–MS are essential in early clinical screening and diagnosis. More attention should be paid to the specific effects of PCCA and PCCB mutations on the organ complications for the precise treatment strategy. Common mutations in China, such as PCCA c.2002G > A and PCCB c.1301C > T, require further clinical studies, as well as ancestry analysis and haplotype mapping, to determine if they are hot spot mutations in the overall Chinese population. However, we would like to note that the availability of data for ancestry analysis and haplotype mapping for these variants is currently unknown, and as such, we cannot determine if they are hot spot mutations in the Chinese population. The mainstay of nutrition therapy is a low protein intake while ensuring essential requirements of amino acids, Ile, Val, Met, and Thr. Medical formula is recommended to supplement the RDA not met by natural protein, and plasma amino acid levels should be monitored closely. Further research is needed to determine the optimal BCAA ratio in patients with PA to optimize their total protein synthesis. Selection of therapeutic drugs for hyperammonemia should also be carefully examined due to different mechanisms of hyperammonemia occurring in PA. Although patients with PA have been reported in most regions of China, neonatal screening for PA is mainly concentrated in certain areas, which limits the research results' selection and bias. Therefore, obtaining accurate information and data nationwide based on the application of neonatal screening strategies will provide a scientific basis for the treatment of PA.
Availability of data and materials
The datasets used and/or analyzed during the current study can be obtained from the corresponding author upon reasonable request.
Abbreviations
- PA:
-
Propionic acidemia
- propionyl-CoA:
-
Propionyl coenzyme A
- PCC:
-
Propionyl carboxylase
- AMD:
-
Acute metabolic decompensations
- BCAA:
-
Branched-chain amino acid
- LC–MS/MS:
-
Liquid chromatography-tandem mass spectrometry
- GC/MS:
-
Gas chromatography-mass spectrometry
- 2MCA:
-
2-Methylcitric acid
- C3:
-
Propionylcarnitine
- C2:
-
Acetylcarnitine
- MMA:
-
Methylmalonic acidemia
- qPCR:
-
Quantitative polymerase chain reaction
- NGS:
-
Next generation sequencing
- NCG:
-
N-carbamylglutamate
- LT:
-
Liver transplantation
- LDLT:
-
Living donor liver transplantation
References
Pena L, Burton BK. Survey of health status and complications among propionic acidemia patients. Am J Med Genet A. 2012;158A(7):1641–6.
Rodriguez-Gonzalez M, Perez-Reviriego AA, Castellano-Martinez A, Cascales-Poyatos HM. Cardiac complications in patients with propionic acidemia. J Rare Dis Res Treat. 2018;3(3):13–21.
Frenkel EP, Kitchens RL. Intracellular localization of hepatic propionyl-CoA carboxylase and methylmalonyl-CoA mutase in humans and normal and vitamin B12 deficient rats. Br J Haematol. 1975;31(4):501–13.
Wongkittichote P, Ah Mew N, Chapman KA. Propionyl-CoA carboxylase—a review. Mol Genet Metab. 2017;122(4):145–52.
Clavero S, Martínez MA, Pérez B, Pérez-Cerdá C, Ugarte M, Desviat LR. Functional characterization of PCCA mutations causing propionic acidemia. Biochim Biophys Acta. 2002;1588(2):119–25.
Pérez-Cerdá C, Clavero S, Pérez B, Rodríguez-Pombo P, Desviat LR, Ugarte M. Functional analysis of PCCB mutations causing propionic acidemia based on expression studies in deficient human skin fibroblasts. Biochim Biophys Acta. 2003;1638(1):43–9.
Yang Q, Xu H, Luo J, Li M, Yi S, Zhang Q, Geng G, Feng S, Fan X. Case reports: three novel variants in PCCA and PCCB genes in Chinese patients with propionic acidemia. BMC Med Genet. 2020;21(1):72.
Cheema-Dhadli S, Leznoff CC, Halperin ML. Effect of 2-methylcitrate on citrate metabolism: implications for the management of patients with propionic acidemia and methylmalonic aciduria. Pediatr Res. 1975;9(12):905–8.
Stanescu S, Belanger-Quintana A, Fernandez-Felix BM, Ruiz-Sala P, Del Valle M, Garcia F, Arrieta F, Martinez-Pardo M. Interorgan amino acid interchange in propionic acidemia: the missing key to understanding its physiopathology. Amino Acids. 2022;54(5):777–86.
Schwab MA, Sauer SW, Okun JG, Nijtmans LG, Rodenburg RJ, van den Heuvel LP, Dröse S, Brandt U, Hoffmann GF, Ter Laak H, Kölker S, Smeitink JA. Secondary mitochondrial dysfunction in propionic aciduria: a pathogenic role for endogenous mitochondrial toxins. Biochem J. 2006;398(1):107–12.
Haijes HA, Jans JJM, van der Ham M, van Hasselt PM, Verhoeven-Duif NM. Understanding acute metabolic decompensation in propionic and methylmalonic acidemias: a deep metabolic phenotyping approach. Orphanet J Rare Dis. 2020;15(1):68.
Coude FX, Sweetman L, Nyhan WL. Inhibition by propionyl-coenzyme A of N-acetylglutamate synthetase in rat liver mitochondria. A possible explanation for hyperammonemia in propionic and methylmalonic acidemia. J Clin Invest. 1979;64(6):1544–51.
Scholl-Bürgi S, Sass JO, Zschocke J, Karall D. Amino acid metabolism in patients with propionic acidaemia. J Inherit Metab Dis. 2012;35(1):65–70.
Filipowicz HR, Ernst SL, Ashurst CL, Pasquali M, Longo N. Metabolic changes associated with hyperammonemia in patients with propionic acidemia. Mol Genet Metab. 2006;88(2):123–30.
Haijes HA, van Hasselt PM, Jans JJM, Verhoeven-Duif NM. Pathophysiology of propionic and methylmalonic acidemias. Part 2: Treatment strategies. J Inherit Metab Dis. 2019;42(5):745–61.
Shu L, Mingyong L, Jinqun L, Nuan C, Himei O, Weihong Z, Xunjie X, Liying C, Jianhui J. Severe anemia, malnutrition, hypotonia, and aggravation of the conscious disturbance, methyl malonic acidemia or propionic acidemia ? Chinese J Appl Clin Pediatr. 2017;32(20):1575–9.
Stanescu S, Belanger-Quintana A, Fernandez-Felix BM, Arrieta F, Quintero V, Maldonado MS, Alcaide P, Martínez-Pardo M. Severe anemia in patients with Propionic acidemia is associated with branched-chain amino acid imbalance. Orphanet J Rare Dis. 2021;16(1):226.
He W, Wang Y, Xie EJ, Barry MA, Zhang GF. Metabolic perturbations mediated by propionyl-CoA accumulation in organs of mouse model of propionic acidemia. Mol Genet Metab. 2021;134(3):257–66.
Hayasaka K, Narisawa K, Satoh T, Tateda H, Metoki K, Tada K, Hiraga K, Aoki T, Kawakami T, Akamatsu H, Matsuo N. Glycine cleavage system in ketotic hyperglycinemia: a reduction of H-protein activity. Pediatr Res. 1982;16(1):5–7.
Yi L, Yanling Y. Screening, diagnosis, and treatment of propionic acidemia. Chinese Clin J Pract Pediatr. 2019;20:1531–4.
Baumgartner MR, Hörster F, Dionisi-Vici C, Haliloglu G, Karall D, Chapman KA, Huemer M, Hochuli M, Assoun M, Ballhausen D, Burlina A, Fowler B, Grünert SC, Grünewald S, Honzik T, Merinero B, Pérez-Cerdá C, Scholl-Bürgi S, Skovby F, Wijburg F, MacDonald A, Martinelli D, Sass JO, Valayannopoulos V, Chakrapani A. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis. 2014;9:130.
Fraser JL, Venditti CP. Methylmalonic and propionic acidemias: clinical management update. Curr Opin Pediatr. 2016;28(6):682–93.
Kör D, Şeker-Yılmaz B, Bulut FD, Kılavuz S, Öktem M, Ceylaner S, Yıldızdaş D, Önenli-Mungan N. Clinical features of 27 Turkish Propionic acidemia patients with 12 novel mutations. Turk J Pediatr. 2019;61(3):330–6.
Cotrina ML, Ferreiras S, Schneider P. High prevalence of self-reported autism spectrum disorder in the propionic acidemia registry. JIMD Rep. 2019;51(1):70–5.
Tarrada A, Frismand-Kryloff S, Hingray C. Functional neurologic disorders in an adult with propionic acidemia: a case report. BMC Psychiatry. 2021;21(1):587.
Shchelochkov OA, Manoli I, Sloan JL, Ferry S, Pass A, Van Ryzin C, Myles J, Schoenfeld M, McGuire P, Rosing DR, Levin MD, Kopp JB, Venditti CP. Chronic kidney disease in propionic acidemia. Genet Med. 2019;21(12):2830–5.
Kido J, Matsumoto S, Sawada T, Endo F, Nakamura K. Rhabdomyolysis in organic acidemia patients manifesting with metabolic decompensation. Hemodial Int. 2019;23(4):E115–9.
Grotto S, Sudrié-Arnaud B, Drouin-Garraud V, Nafeh-Bizet C, Chadefaux-Vekemans B, Gobin S, Bekri S, Tebani A. Dilated cardiomyopathy and premature ovarian failure unveiling propionic aciduria. Clin Chem. 2018;64(4):752–4.
Altun I, Kiykim A, Zubarioglu T, Burtecene N, Hopurcuoglu D, Topcu B, Cansever MS, Kiykim E, Cokugras HC, Aktuglu Zeybek AC. Altered immune response in organic acidemia. Pediatr Int. 2022;64(1): e15082.
Xiuyun X, Yaru D, Jun W, Fengqiu Y, Zhengli D, Zhiqin L. Molecular pathology report of late-onset propionic acidemia in adults: one case report and literature review. Chin J Contemp Neurol Neurosurg. 2021;21(04):289–95.
Han L, Gao X, Ye J, Qiu W, Gu X. Application of tandem mass spectrometry in diagnosis of organic acidemias. Chinese J Pediatr. 2005;05:325–30.
Chace DH, DiPerna JC, Kalas TA, Johnson RW, Naylor EW. Rapid diagnosis of methylmalonic and propionic acidemias: quantitative tandem mass spectrometric analysis of propionylcarnitine in filter-paper blood specimens obtained from newborns. Clin Chem. 2001;47(11):2040–4.
Longo N, Sass JO, Jurecka A, Vockley J. Biomarkers for drug development in propionic and methylmalonic acidemias. J Inherit Metab Dis. 2022;45(2):132–43.
Maines E, Catesini G, Boenzi S, Mosca A, Candusso M, Dello Strologo L, Martinelli D, Maiorana A, Liguori A, Olivieri G, Taurisano R, Piemonte F, Rizzo C, Spada M, Dionisi-Vici C. Plasma methylcitric acid and its correlations with other disease biomarkers: the impact in the follow up of patients with propionic and methylmalonic acidemia. J Inherit Metab Dis. 2020;43(6):1173–85.
Dai M, Xiao B, Zhang H, Ye J, Qiu W, Zhu H, Wang L, Liang L, Zhan X, Ji W, Wang Y, Yu Y, Gu X, Han L. Biochemical and genetic approaches to the prenatal diagnosis of propionic acidemia in 78 pregnancies. Orphanet J Rare Dis. 2020;15(1):276.
Wang H, Meng L, Li W, Du J, Tan Y, Gong F, Lu G, Lin G, Zhang Q. Combinations of exonic deletions and rare mutations lead to misdiagnosis of propionic acidemia. Clin Chim Acta. 2020;502:153–8.
Wang HR, Liu YQ, He XL, Sun J, Zeng FW, Yan CB, Li H, Gao SY, Yang Y. A novel delins (c.773_819+47delinsAA) mutation of the PCCA gene associated with neonatal-onset propionic acidemia: a case report. BMC Med Genet. 2020;21(1):166.
Couce ML, Castiñeiras DE, Bóveda MD, Baña A, Cocho JA, Iglesias AJ, Colón C, Alonso-Fernández JR, Fraga JM. Evaluation and long-term follow-up of infants with inborn errors of metabolism identified in an expanded screening programme. Mol Genet Metab. 2011;104(4):470–5.
Dionisi-Vici C, Rizzo C, Burlina AB, Caruso U, Sabetta G, Uziel G, Abeni D. Inborn errors of metabolism in the Italian pediatric population: a national retrospective survey. J Pediatr. 2002;140(3):321–7.
Shibata N, Hasegawa Y, Yamada K, Kobayashi H, Purevsuren J, Yang Y, Dung VC, Khanh NN, Verma IC, Bijarnia-Mahay S, Lee DH, Niu DM, Hoffmann GF, Shigematsu Y, Fukao T, Fukuda S, Taketani T, Yamaguchi S. Diversity in the incidence and spectrum of organic acidemias, fatty acid oxidation disorders, and amino acid disorders in Asian countries: Selective screening vs. expanded newborn screening. Mol Genet Metab Rep. 2018;16:5–10.
Al-Shamsi A, Hertecant JL, Al-Hamad S, Souid AK, Al-Jasmi F. Mutation spectrum and birth prevalence of inborn errors of metabolism among emiratis: a study from Tawam hospital metabolic center, United Arab Emirates. Sultan Qaboos Univ Med J. 2014;14(1):e42–9.
Rashed MS. Clinical applications of tandem mass spectrometry: ten years of diagnosis and screening for inherited metabolic diseases. J Chromatogr B Biomed Sci Appl. 2001;758(1):27–48.
Zayed H. Propionic acidemia in the Arab World. Gene. 2015;564(2):119–24.
Ravn K, Chloupkova M, Christensen E, Brandt NJ, Simonsen H, Kraus JP, Nielsen IM, Skovby F, Schwartz M. High incidence of propionic acidemia in greenland is due to a prevalent mutation, 1540insCCC, in the gene for the beta-subunit of propionyl CoA carboxylase. Am J Hum Genet. 2000;67(1):203–6.
Shi XT, Cai J, Wang YY, Tu WJ, Wang WP, Gong LM, Wang DW, Ye YT, Fang SG, Jing PW. Newborn screening for inborn errors of metabolism in mainland china: 30 years of experience. JIMD Rep. 2012;6:79–83.
Yanping PYGZFST. Clinical analysis and screening results of newborngenetic metabolic disease in Guangning region. Inner Mongolia J Tradit Chinese Med. 2016;35(03):105–6.
Tianhui Wu, Yurun Z. Neonatal screening for inherited metabolic diseases in Wenzhou Area from 2014 to 2018. Zhejiang Med Educ. 2020;19(05):61–3.
Shengju Ma, Dehua Z, Kun Ma, Min Ni, Liwen W, Yunjia O, Lei Z, Xiaoan Z. Retrospective analysis on screening of neonates inherited metabolic diseases from 2013 to 2019 in Henan Province. Laboratory Med Clin. 2020;17(14):1965–8.
Lu Wenying Wu, Jian LL, Jie Q. Screening study of neonatal genetic metabolic diseases in Yancheng from 2012 to 2014. Lab Med Clin. 2015;12(05):669–70.
Zhang Wei. Analysis of the screening results of inherited metabolic disorders part of area in HeBei Province from 2007 to 2010. Hebei Medical University. 2011.
Hong F, Huang X, Zhang Y, Yang J, Tong F, Mao H, Huang X, Zhou X, Yang R, Zhao Z. Screening for newborn organic aciduria in Zhejiang Province: prevalence, outcome and follow-up. J Zhejiang Univ (Med Sci). 2017;46(03):240–7.
Zhang Jiaguo Su, Xingyue LT, Qi Hu, Jingyao Z, Zhang Yu. Analysis of screening for neonatal inherited metabolic disorders by tandem mass spectrometry in parts of Sichuan Province. Chinese J Child Health Care. 2020;28(07):809–12.
Dongpo S, Wenjie Li, Jinfeng Lv, Caijuan W. Analysis of tandem mass spectrometry detection results in Qingdao region from 2012 to 2016. Maternal Child Health Care China. 2018;33(02):388–90.
Desviat LR, Sanchez-Alcudia R, Pérez B, Pérez-Cerdá C, Navarrete R, Vijzelaar R, Ugarte M. High frequency of large genomic deletions in the PCCA gene causing propionic acidemia. Mol Genet Metab. 2009;96(4):171–6.
Yang X, Sakamoto O, Matsubara Y, Kure S, Suzuki Y, Aoki Y, Yamaguchi S, Takahashi Y, Nishikubo T, Kawaguchi C, Yoshioka A, Kimura T, Hayasaka K, Kohno Y, Iinuma K, Ohura T. Mutation spectrum of the PCCA and PCCB genes in Japanese patients with propionic acidemia. Mol Genet Metab. 2004;81(4):335–42.
Campeau E, Dupuis L, Leclerc D, Gravel RA. Detection of a normally rare transcript in propionic acidemia patients with mRNA destabilizing mutations in the PCCA gene. Hum Mol Genet. 1999;8(1):107–13.
Pérez B, Desviat LR, Rodríguez-Pombo P, Clavero S, Navarrete R, Perez-Cerdá C, Ugarte M. Propionic acidemia: identification of twenty-four novel mutations in Europe and North America. Mol Genet Metab. 2003;78(1):59–67.
Kim SN, Ryu KH, Lee EH, Kim JS, Hahn SH. Molecular analysis of PCCB gene in Korean patients with propionic acidemia. Mol Genet Metab. 2002;77(3):209–16.
Ying Z, Fang Z, Yang Y. Diagnosis and treatment of propionic academia: a case report of a pair of twin siblings. J Int Reproduc Health/Family Plan. 2020;39(02):138–40.
Hu Y, Han L, Ye J, Qiu W, Zhang H, Liang L, Ji W, Xu F, Chen T, Chen S, Gu X. Treatment and follow-up of 82 children with propionic acidemia. Chinese J Perinatal Med. 2021;24(02):105–12.
Xiuli W, Lei P, Jijie L, Yuan F. Newborn screening and gene mutations of patients with propionic acidemia in Xuzhou City. Chinese J Neonatol. 2019;02:87–92.
Guangpeng Z, Zhijun Z, Liying S, Wei Lin Qu, Wei ZZ, Ying L, Yule T, Jun W. Case report of living donor liver transplantation for pediatric propionic acidemia combined with dilated cardiomyopathy. Chinese J Appl Clin Pediatr. 2021;36(23):1828–31.
Liu Y, Chen Z, Dong H, Ding Y, He R, Kang L, Li D, Shen M, Jin Y, Zhang Y, Song J, Tian Y, Cao Y, Liang D, Yang Y. Analysis of the relationship between phenotypes and genotypes in 60 Chinese patients with propionic acidemia: a fourteen-year experience at a tertiary hospital. Orphanet J Rare Dis. 2022;17(1):135.
Chiu YH, Liu YN, Liao WL, Chang YC, Lin SP, Hsu CC, Chiu PC, Niu DM, Wang CH, Ke YY, Chien YH, Hsiao KJ, Liu TT. Two frequent mutations associated with the classic form of propionic acidemia in Taiwan. Biochem Genet. 2014;52(9–10):415–29.
Na T, Guiling Mo, Honghong Z, Aiping W, Jianqin Qi, Yang Y, Meiyuan S. Clinical characteristics and gene mutation analysis of the two families with propionaemia. Chinese Pediatr Emerg Med. 2016;23(06):418–21.
Chen Y, Lin X, Lin Q, Zeng Y, Qiu X, Liu G, Zhu W. Gene diagnosis and pedigree analysis of two Han ethnicity families with propionic acidemia in Fujian. Medicine. 2021;100(10): e24161.
Yang C, Zhou C, Xu P, Jin X, Liu W, Wang W, Huang C, Jiang M, Chen X. Newborn screening and diagnosis of inborn errors of metabolism: a 5-year study in an Eastern Chinese population. Clin Chim Acta. 2020;502:133–8.
Yap S, Vara R, Morais A. Post-transplantation outcomes in patients with PA or MMA: a review of the literature. Adv Ther. 2020;37(5):1866–96.
Mobarak A, Stockler S, Salvarinova R, Van Karnebeek C, Horvath G. Long term follow-up of the dietary intake in propionic acidemia. Mol Genet Metab Rep. 2021;27: 100757.
Manoli I, Myles JG, Sloan JL, Shchelochkov OA, Venditti CP. A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 1: isolated methylmalonic acidemias. Genet Med. 2016;18(4):386–95.
Saleemani H, Horvath G, Stockler-Ipsiroglu S, Elango R. Determining ideal balance among branched-chain amino acids in medical formula for propionic acidemia: a proof of concept study in healthy children. Mol Genet Metab. 2022;135(1):56–62.
Molema F, Haijes HA, Janssen MC, Bosch AM, van Spronsen FJ, Mulder MF, Verhoeven-Duif NM, Jans JJM, van der Ploeg AT, Wagenmakers MA, Rubio-Gozalbo ME, Brouwers MCGJ, de Vries MC, Fuchs S, Langendonk JG, Rizopoulos D, van Hasselt PM, Williams M. High protein prescription in methylmalonic and propionic acidemia patients and its negative association with long-term outcome. Clin Nutr. 2021;40(5):3622–30.
Stanescu S, Belanger-Quintana A, Fernández-Felix BM, Ruiz-Sala P, Alcaide P, Arrieta F, Martínez-Pardo M. Plasma CoQ10 status in patients with propionic acidaemia and possible benefit of treatment with Ubiquinol. Antioxidants. 2022;11(8):1588.
Diodato D, Olivieri G, Pro S, Maiorani D, Martinelli D, Deodato F, Taurisano R, Di Capua M, Dionisi-Vici C. Axonal peripheral neuropathy in propionic acidemia: a severe side effect of long-term metronidazole therapy. Neurology. 2018;91(12):565–7.
Liang L, Han L. Diagnosis and treatment and screening of organic acidaemia. J Develop Med (Electronic Version). 2020;8(01):15–9.
Kiykim E, Oguz O, Duman C, Zubarioglu T, Cansever MS, Zeybek ACA. Long-term N-carbamylglutamate treatment of hyperammonemia in patients with classic organic acidemias. Mol Genet Metab Rep. 2021;26: 100715.
Alfadhel M, Nashabat M, Saleh M, Elamin M, Alfares A, Al Othaim A, Umair M, Ahmed H, Ababneh F, Al Mutairi F, Eyaid W, Alswaid A, Alohali L, Faqeih E, Almannai M, Aljeraisy M, Albdah B, Hussein MA, Rahbeeni Z, Alasmari A. Long-term effectiveness of carglumic acid in patients with propionic acidemia (PA) and methylmalonic acidemia (MMA): a randomized clinical trial. Orphanet J Rare Dis. 2021;16(1):422.
Armstrong AJ, Henke BR, Collado MS, Taylor JM, Pourtaheri TD, Dillberger JE, Roper TD, Wamhoff BR, Olson MW, Figler RA, Hoang SA, Reardon JE, Johns BA. Identification of 2,2-dimethylbutanoic acid (HST5040), a clinical development candidate for the treatment of propionic acidemia and methylmalonic acidemia. J Med Chem. 2021;64(8):5037–48.
Subramanian C, Frank MW, Tangallapally R, Yun MK, Edwards A, White SW, Lee RE, Rock CO, Jackowski S. Pantothenate kinase activation relieves coenzyme A sequestration and improves mitochondrial function in mice with propionic acidemia. Sci Transl Med. 2021;13(611):eabf5965.
Subramanian C, Frank MW, Tangallapally R, Yun MK, White SW, Lee RE, Rock CO, Jackowski S. Relief of CoA sequestration and restoration of mitochondrial function in a mouse model of propionic acidemia. J Inherit Metab Dis. 2023;46(1):28–42.
Zhou GP, Jiang YZ, Wu SS, Kong YY, Sun LY, Zhu ZJ. Liver transplantation for propionic acidemia: evidence from a systematic review and meta-analysis. Transplantation. 2021;105(10):2272–82.
Barshes NR, Vanatta JM, Patel AJ, Carter BA, O’Mahony CA, Karpen SJ, Goss JA. Evaluation and management of patients with propionic acidemia undergoing liver transplantation: a comprehensive review. Pediatr Transplant. 2006;10(7):773–81.
Zeng F, Tingfang S, Chen C. Nursing experience of children with propionic acidemia after intravital liver transplantation. J Nurs Rehabilit. 2021;20(08):63–5.
Rice GM, Steiner RD. Inborn errors of metabolism (metabolic disorders). Pediatr Rev. 2016;37(1):3–15.
Zeng ZG, Zhou GP, Wei L, Qu W, Liu Y, Tan YL, Wang J, Sun LY, Zhu ZJ. Therapeutic potential of living donor liver transplantation from heterozygous carrier donors in children with propionic acidemia. Orphanet J Rare Dis. 2022;17(1):62.
Huiyi C, Ping W, Qiang X. Progress in liver transplantation for methylmalonic acemia and propionic acidemia. Int J Digest Dis. 2020;40(02):75–86.
Charbit-Henrion F, Lacaille F, McKiernan P, Girard M, de Lonlay P, Valayannopoulos V, Ottolenghi C, Chakrapani A, Preece M, Sharif K, Chardot C, Hubert P, Dupic L. Early and late complications after liver transplantation for propionic acidemia in children: a two centers study. Am J Transplant. 2015;15(3):786–91.
Curnock R, Heaton ND, Vilca-Melendez H, Dhawan A, Hadzic N, Vara R. Liver transplantation in children with propionic acidemia: medium-term outcomes. Liver Transpl. 2020;26(3):419–30.
Sivananthan S, Hadžić N, Dhawan A, Heaton ND, Vara R. Fatal metabolic stroke in a child with propionic acidemia 11 years post liver transplant. Am J Transplant. 2021;21(4):1637–40.
Hejazi Y, Hijazi ZM, Al-Saloos H, Omran TB. The re-occurrence of dilated cardiomyopathy in propionic acidemia after liver transplantation requiring heart transplant, first case from Middle East. Cardiol Young. 2022;16:1–4.
Zinan Yu, Zhang Yu, Xinwen H. European guidelines for the diagnosis and treatment of methylmalonic acidemia and propionic acidemia. Chinese J Emerg Med. 2019;5:560–2.
Dhawan A, Mitry RR, Hughes RD. Hepatocyte transplantation for liver-based metabolic disorders. J Inherit Metab Dis. 2006;29(2–3):431–5.
Maestro S, Weber ND, Zabaleta N, Aldabe R, Gonzalez-Aseguinolaza G. Novel vectors and approaches for gene therapy in liver diseases. JHEP Rep. 2021;3(4): 100300.
Lomash RM, Shchelochkov O, Chandler RJ, Venditti CP, Pariser AR, Ottinger EA, NIH PaVe-GT Team. Successfully Navigating Food and Drug Administration Orphan Drug and Rare Pediatric Disease Designations for AAV9-hPCCA Gene Therapy: The National Institutes of Health Platform Vector Gene Therapy Experience. Hum Gene Ther. 2023;34(5–6):217–227.
Jiang L, Park JS, Yin L, Laureano R, Jacquinet E, Yang J, Liang S, Frassetto A, Zhuo J, Yan X, Zhu X, Fortucci S, Hoar K, Mihai C, Tunkey C, Presnyak V, Benenato KE, Lukacs CM, Martini PGV, Guey LT. Dual mRNA therapy restores metabolic function in long-term studies in mice with propionic acidemia. Nat Commun. 2020;11(1):5339.
Tu W. Sereening of high-risk children and patients from neonatal intensive care units for inborn errors of metabolism using quid chromatography-trandem mass spectrometry(LC-MS/MS). Peking Union Medical College, 2010.
Huanhuan Xu, Kaiqi Z, Chunhong N. Analysis of screening of 48 kinds of inherit metabolic disease in newborns from 91 406 cases in kaifeng area. Chinese J Birth Health Heredity. 2018;26(08):67–8.
Sun M, Huang P, Cheng S, Wang W, Jiangning, Wu Y. Screening study of neonatal genetic and metabolic diseases in Shaanxi Province. Chinese Foreign Med Res. 2016;14(11):54–5.
Han F, Han L, Ji W, Xu F, Ye J, Qiu W, Zhang H, Gu X. Prenatal diagnosis of propionic acidemia by amniotic fluid metabolites analysis using mass spectrometry. Chinese J Reprod Contracep. 2017;37(11):918–22.
Zhong J, Fu Q, Lin Y. Systematic analysis of the incidence and disease spectrum of organic academia newborn screening results in Quanzhou, Fujian Province. Screening results of neonatal organic accidemia in Quanzhou, Fujian Province. J Mod Lab Med. 2019;34(05):52–5.
Huiming Y, Zhengjun J, Jing L, Yanghui Z, Hua T, Hui Xi, Jing C, Junqun F, Donghua X, Hua W. Tandem mass spectrometry screening of 565182 newborns for inherited metabolic disease in Hunan province. Chinese J Appl Clin Pediatr. 2019;20:1541–5.
Yuyan S, Jian Li, Gang X. Screening results and follow-up study of neonatal organic acidemia from 79 205 cases in Huaihua. Chinese J Birth Health Heredity. 2018;26(10):76–8.
Pingming G, Yiheng D, Deqin J, Jianwei H, Shuitang Z, Xiangming Z. Selective screening of genetic metabolic defects in neonatal period—clinical analysis of 5 cases. Chinese J Contemporary Pediatr. 2007;06:605–7.
Jincai W. Study on the incidence of neonatal genetic and metabolic diseases in Puning city. Chinese Foreign Med Res. 2012;10(35):47–9.
Wanxiu Z. Screening and follow-up analysis of neonatal genetic metabolic diseases by tandem mass spectrometry technology. Contemporary Med. 2014;20(17):148–9.
Tang C, Tan M, Xie T, Tang F, Liu S, Wei Q, Liu J, Huang Y. Screening for neonatal inherited metabolic disorders by tandem mass spectrometry in Guangzhou. J Zhejiang Univ (Med Sci). 2021;50(04):463–71.
Liu Xiaoyu Wu, Wei SL. Analysis of tandem mass spectrometry screening and follow-up results of neonatal genetic metabolic disorders in Meizhou. Chinese Youjiang Med J. 2021;49(11):834–8.
Manfang X, Haijie S. Gene mutation for inherited metabolic diseases among the newborn population of Hainan minority areas. China Trop Med. 2020;20(09):804–8.
Wang T, Ma J, Zhang Q, Gao A, Wang Q, Li H, Xiang J, Wang B. Expanded newborn screening for inborn errors of metabolism by tandem mass spectrometry in Suzhou, China: disease spectrum, prevalence, genetic characteristics in a chinese population. Front Genet. 2019;10:1052.
Wang Y, Sun Y, Jiang T. Diagnosis of delayed propionic acidemia and identification of one novel mutation. Chinese J Obstetr Gynecol Pediatr (Electron Edition). 2018;14(01):68–72.
Yan Y, Yanqiu L, Jia C, Haiyan L. Analysis of a pedigree with propionic acidemia by trio whole exome sequencing. Chinese J Med Genet. 2020;37(07):751–4.
Xing X, Liu S, Li F, Ma H, Luo Y. Analysis of pathogenic gene mutations in a child with propionic acidemia. In: Proceedings of the 9th national academic exchange conference on genetic disease diagnosis and antenatal diagnosis and new technology seminar on prenatal diagnosis and medical genetics. 2014:128–9.
Yang C, Zhou C, Wan Q, Zhou Y, Chen X, Xu P. Positive cases of organic acidemia were screened in Jining genetic mutational features. Chinese J Birth Health Heredity. 2022;30(05):885–90.
Ao ZZ, Wang J, Li S, Ma Y, Miao T, Liang Z, Shi C, Gu X, Xiao X, Hao H. Application of tandem mass spectrometry combined with second-generation sequencing in the screening and analysis of 20,000 neonatal genetic diseases. Chinese J Appl Clin Pediatr. 2020;35(24):1881–5.
Meixian Wu, Wenhua Q, Guang C. One case of craniocerebral MR imaging of neonatal propionic acidemia. Chinese J Neonatol. 2017;32(03):228.
Peiying Y, Yun S, Dingyuan Ma, Yanyun W, Zhilei Z, Wei C, Tao J. Phenotypes and pathogenic variations in two cases of propionic acidemia. Chinese J Perinatal Med. 2021;24(02):120–5.
Qigang Z, Guanglai F, Shu Z, Yuefang L, Wenjie Z, Qiong P. Identification of two novel variants in the PCCB gene in a pedigree affected with propionic acidemia. Chinese J Med Genet. 2021;38(03):251–4.
Qiong G, Jun Li, Guinan Li. One case of the PCCB gene mutation. J Chinese Phys. 2017;19(12):1907–8.
Yang X, Li D, Tu C, He W, Meng L, Tan YQ, Lu G, Du J, Zhang Q. Novel variants of the PCCB gene in Chinese patients with propionic acidemia. Clin Chim Acta. 2021;519:18–25.
Acknowledgements
We thank Chenrui Niu for making a map of China using the analysis and mapping software Arcmap for this literature and thank for the support of innovative myocardial infarction team at Jining medical university.
Funding
This work is supported by the College Students' Innovation Training Program of Jining Medical University (Y.X.Z. No.cx2021047), NSFC cultivation project of Jining Medical University (Y.W. No.JYP2019KJ05), Shandong Medical Science and Technology Development Project (Y.W. No.2018WS459) and the award from Propionic Acidemia Foundation to G.F.Z.
Author information
Authors and Affiliations
Contributions
YXZ performed the data analysis, drafted and revised the manuscript. CWP, LFW and JWW contributed to the production of forms and the writing of this article. ST Chen contributed to the literature search and the writing of this article. JWW contributed to the production of pictures and the writing of this article. CGW and XXC contributed to the ACMG classification. ZHT was involved in writing the article. SHZ, GFZ and YW designed and supervised the research study. All authors have given approval to the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent to publish.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Demographics, preoperative characteristics, and operative findings of PA patients who underwent liver transplant.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Y., Peng, C., Wang, L. et al. Prevalence of propionic acidemia in China. Orphanet J Rare Dis 18, 281 (2023). https://doi.org/10.1186/s13023-023-02898-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-023-02898-w