
Abstract
Background
Skeletal deformity is characterized by an abnormal anatomical structure of bone and cartilage. In our previous studies, we have found that a substantial proportion of patients with skeletal deformity could be explained by monogenic disorders. More recently, complex phenotypes caused by more than one genetic defect (i.e., dual molecular diagnosis) have also been reported in skeletal deformities and may complicate the diagnostic odyssey of patients. In this study, we report the molecular and phenotypic characteristics of patients with dual molecular diagnosis and variable skeletal deformities.
Results
From 1108 patients who underwent exome sequencing, we identified eight probands with dual molecular diagnosis and variable skeletal deformities. All eight patients had dual diagnosis consisting of two autosomal dominant diseases. A total of 16 variants in 12 genes were identified, 5 of which were of de novo origin. Patients with dual molecular diagnosis presented blended phenotypes of two genetic diseases. Mendelian disorders occurred more than once include Osteogenesis Imperfecta Type I (COL1A1, MIM:166200), Neurofibromatosis, Type I (NF1, MIM:162200) and Marfan Syndrome (FBN1, MIM:154700).
Conclusions
This study demonstrated the complicated skeletal phenotypes associated with dual molecular diagnosis. Exome sequencing represents a powerful tool to detect such complex conditions.
Similar content being viewed by others

Background
Skeletal deformity is characterized by an abnormal anatomical structure of the bone and cartilage [1]. Genetic factors are essential for the pathogenesis of skeletal deformities [2]. The tenth version of the Nosology and Classification of Genetic Skeletal Disorders included 461 different diseases which can be classified into 42 groups based on their clinical, radiographic, and/or molecular findings [3]. In previous studies, we found that a substantial proportion of cases with early-onset scoliosis could be explained by monogenic disorders such as achondroplasia (MIM: 100800), Freeman–Sheldon syndrome (FSS) (MIM:193700), and spondyloepimetaphyseal dysplasia (MIM: 602557) [4,5,6]. In addition to monogenic conditions, complex phenotypes caused by two genetic disorders (i.e., dual molecular diagnosis) have also been reported in skeletal deformities. For example, a fetus with complex joint dislocations and congenital scoliosis was identified to be double heterozygote for putatively pathogenic FBN1 and FBN2 variants [7]. Tang et al. identified a patient with pathogenic variants in both FBN1 and PTPN11, which caused blended phenotypes of Marfan syndrome (MIM:154700) and LEOPARD syndrome (MIM:151100) [8].
The co-existence of two Mendelian conditions challenges the diagnosis and clinical management in patients with skeletal deformities. The precise diagnosis of such conditions needs comprehensive genetic testing tools such as exome sequencing (ES) [9,10,11]. Thus far, systematic investigations of dual molecular diagnosis have been performed in neurodevelopmental disorders [12, 13], genetic muscle diseases [14], and endocrine dysfunction [13]. However, the phenotypic characteristics of dual diagnosis in patients with skeletal deformities are still less studied.
Here, we report eight cases with dual molecular diagnosis from the Deciphering disorders Involving Scoliosis and COmorbidities (DISCO) study. We describe the phenotypic characteristics of these patients and clinical relevance for the molecular diagnoses.
Results
Patients with dual molecular diagnosis and a variety of skeletal deformities
From 1108 patients who underwent exome sequencing in the DISCO study, we identified eight probands with dual molecular diagnosis and a variety of skeletal deformities (Table 1). All eight probands have dual diagnosis of two autosomal dominant (AD) diseases. A total of 16 pathogenic variants in 12 genes were identified, 5 of which were de novo. The frequently observed molecular diagnoses (observed in more than one patient) include Osteogenesis Imperfecta Type I (COL1A1, MIM:166200), Neurofibromatosis, Type I (NF1, MIM:162200) and Marfan Syndrome (FBN1, MIM:154700).
The complex clinical features of patients with dual molecular diagnosis
Patients with dual molecular diagnosis presented blended phenotypes of two genetic diseases, which significantly complicated their diagnostic odysseys. Here we report the detailed clinical characteristics of these patients.
Case 1
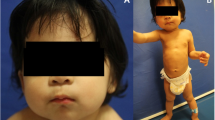
Patient SCO2003P1972 was a 7-year-old boy with early-onset scoliosis (Fig. 1a, Additional file 1: Fig. S1A, B). At 2 years old, he was diagnosed with congenital dislocation of hip joint (Table 1) and underwent a surgical reduction. A mild scoliosis was found during the hospitalization. At 7 years old, he was identified to have congenital scoliosis with segmentation failure of T10-L1 (Fig. 1a, Table 1). Physical examination showed ocular hypertelorism and intellectual disability (Fig. 1a, Table 1). ES revealed a pathogenic heterozygous nonsense variant c.2649G > A (p.Trp883Ter) in FBN1 (Table 1), which is associated with Marfan syndrome (MIM:154700) [4]. Consistently, the proband presented Marfan syndrome-related phenotypes including mitral valve prolapse, mild arachnodactyly and scoliosis. This variant was inherited from his mother, who had severe scoliosis, arachnodactyly and dolichostenomelia (Fig. 1a). Through further analysis of the exome data, a de novo variant in POGZ c.1180_1181del (p.Met394ValfsTer9) (Table 1) was found in the proband. This variant was previously reported in patients with White–Sutton syndrome (WHSUS) (MIM:616364). WHSUS is characterized by intellectual disability, ocular abnormalities and brachydactyly [15], which largely overlapped with the phenotypes of this patient. Therefore, the complex phenotypes of this patient could be explained by a combined effect of variants in POGZ and FBN1.

Representative clinical photographs of 6 patients with skeletal deformity. a Patient SCO2003P1972. b Patient SCO1908P0067. c Patient PCT2007P0019. d Patient SSS1910P0094. e Patient SSS2010P0110. f Patient RDD2001P0005
Case 2
In case 2, the proband (SCO1908P0067) was referred to the clinic at the age of 15 years because of scoliosis (Fig. 1b, Table 1). He also presented pectus carinatum (Table 1). He experienced fracture at the age of 10 and 13. ES identified two pathogenic variants, including a de novo variant in COL1A1 (c.1081C > T, p.Arg361Ter) and a maternally inherited variant in FBN1 (c.1453C > T, p.Arg485Cys) (Table 1). These two reported variants led to dual molecular diagnosis consisting of osteogenesis imperfecta type I and Marfan syndrome [16, 17]. Although patients with osteogenesis imperfecta type I often present with short stature [18], the patient was tall (Height: 185 cm), which might be related with his second diagnosis of Marfan syndrome. Although the FBN1 variant in our patient has been reported and considered as a pathogenic variant [17], no other features of Marfan Syndrome such as dolichostenomelia, arachnodactyly, joint laxity, velvety skin, ectopia lentis and cardiovascular manifestations were identified. This patient exemplified the agonistic effects of two Mendelian disorders on one trait (height in this case).
Case 3
Patient PCT2007P0019 was an 8-year-old girl. The proband had right tibia fracture at the age of 2 years. At around the age of 6 years, the patient developed scoliosis and razorback deformity, with her right shoulder slightly lower than left. The scoliosis progressed in the next 2 years (Additional file 1: Fig. S1D, E). A series of blended clinical phenotypes, such as blue sclera, cafe-au-Lait macules were observed as well (Fig. 1c, Table 1). Her father and younger brother also presented with blue sclera but did not have history of bone fracture. ES identified a pathogenic variant in NF1 (c.2307del, p.Thr770LeufsTer21) and another pathogenic variant in COL1A1 (c.2028 + 4A > G) (Table 1), both transmitted from her father. The NF1 variant could lead to a frameshift of NF1 (Table 1) and thus a loss-of-function effect, which is associated with neurofibromatosis type I and could explain the cafe-au-lait macules in this patient (Fig. 1c, Table 1). The COL1A1 splicing variant (c.2028 + 4A > G) has been previously described to cause osteogenesis imperfecta [19], which could explain the recurrent bone fracture history in this patient. Interestingly, both neurofibromatosis type I and osteogenesis imperfecta could lead to scoliosis with incomplete penetrance [18, 20]. Therefore, the scoliotic phenotype in this patient might be caused by the synergistic effects of the COL1A1 and the NF1 variant.
Case 4
In our previous study, we reported a 6-year-old boy (SSS2008P0037) with short stature and craniofacial deformities, including depressed nasal bridge and long philtrum [21] (Table 1). Then the patient was detected to be double heterozygote for putatively pathogenic ANKRD11 (c.2508dup, p.Leu837ThrfsTer81) and COL11A1 (c.1180_1181del, p.Met394ValfsTer9) variants on ES (Table 1). The ANKRD11 and COL11A1 variants were confirmed as de novo and paternal status, respectively (Table 1). Clinical findings such as short stature and long philtrum were consistent with both KBG syndrome (MIM: 148050) and Marshall syndrome (MIM: 154780). In addition, the depressed nose in this patient is more likely to be associated with Marshall syndrome. The phenotypes of the patient together with the reported phenotypes of KBG syndrome and Marshall syndrome indicated overlapping clinical features in this case.
Case 5
This patient (SSS1910P0094) was an 8-year-old girl with idiopathic short stature and global developmental delay. She also presented widespread cafe-au-lait macules and mild lumbar scoliosis (Fig. 1d, Table 1). Additionally, her father (162 cm) and mother (140 cm) had short stature. Brain magnetic resonance (MR) showed abnormal signals in bilateral globus pallidus, thalamus, hippocampus and dentate nucleus of cerebellum were observed. Two pathogenic variants (Table 1), including a splicing variant in the NF1 gene (c.6705-1G > A) and a frameshift variant in the GLI2 gene (c.1189del, p.Val397CysfsTer124) were identified. Variants in GLI2 have been shown to cause short stature, abnormal development of brain structures in Culler–Jones syndrome (MIM:615849) [22]. We suggested this patient’s presentation represents a mixture of distinct phenotypes, i.e., Cafe-au-Lait spots for NF type 1 (NF1, MIM:162200) and short stature for Culler–Jones syndrome (MIM:615849).
Case 6
Case 6 (SSS2010P0110) was a 12-year-old girl. She presented mild scoliosis, short stature, low posterior hairline, hyper pigmentation and webbed neck (Fig. 1e). ES identified a pathogenic nonsense variant in TP63 (c.109C > T, p.Arg37Ter) and another pathogenic missense variant in PTPN11 (c.1510A > G, p.Met504Val) (Table 1). Therefore, this patient reveived dual molecular diagnosis of Rapp–Hodgkin syndrome (MIM: 129400) and Noonan syndrome (MIM: 163950). However, this patient has minimal scoliosis and no major documented manifestation of Rapp-Hodgkin syndrome, suggesting a reduced expressivity of the TP63 variant.
Case 7
A 3-year-old patient (RDD2001P0005) presented at birth with widespread interphalangeal joint contractures of the hands (Fig. 1f) and atrial septal defect (ASD) (Table 1). His father also had camptodactyly. ES revealed a paternally inherited heterozygous missense variant (c.3437A > G, p.Tyr1146Cys) in FBN2 and a de novo heterozygous frameshift ANKRD11 variant (c.3024_3025del, p.Lys1009GlyfsTer8) (Table 1). The FBN2 variant causes Beals syndrome (Congenital contractual arachnodactyly) (MIM:121050), which is characterized by arachnodactyly and camptodactyly [23, 24]; The ANKRD11 mutation causes KBG syndrome, which may contribute to the ASD in this patient [25]. Nevertheless, ASD has also been reported in patients with Beals syndrome [26], suggesting the overlapping phenotype associated with the dual molecular diagnosis.
Case 8
This case is a 5-year-old male (P19009402) with complex phenotypes. He presented congenital syndactyly of hand and foot (Additional file 1: Fig. S1F, G), cloverleaf skull, orbital hypertelorism, proptosis and midfacial hypoplasia (Table 1). He also had a history of malignant hyperthermia during general anesthesia. ES identified a de novo missense variant (c.755C > G, p.Ser252Trp) in FGFR2 gene and a paternally inherited frameshift variant in RYR1 gene (c.12788_12793dup, p.Glu4263_Gly4264dup) (Table 1). The FGFR2 variant occurred in a known Apert syndrome hotspot [27]. Apert syndrome was characterized by craniosynostosis, proptosis, midfacial hypoplasia and severe syndactyly of the hands and feet [27], which are concordant with the phenotypes of this patient. Pathogenic RYR1 variants are associated with malignant hyperthermia susceptibility 1(MHS 1) (MIM:145600) [28], which could explain the hyperthermia history in the patient.
Discussion
The development of ES has significantly improved diagnostic yield of rare disease. Yang et al. found ES identified the underlying genetic defect in 25% of consecutive patients referred for evaluation of a possible genetic condition and 4.6% patients with blended phenotypes resulting from two single gene defects [10]. Farwell et al. found 11 (7.2%) in 152 probands with a positive or likely positive finding received a dual molecular diagnosis [29]. In a sizable cohort of 7374 patients, Posey et al. identified 2182 independent molecular diagnoses and identified two or more molecular diagnoses in 101 patients (4.9%) [9, 11]. In our study, we observed a relatively lower rate of dual molecular diagnosis in patients with skeletal diseases (≈ 1%), which might be attributed to the lower baseline molecular diagnosis rate as compared with those developmental disorder cohorts [30]. Most probands (5 out of 8) had family members with at least one of the diseases, consistent with the report by Balci et al. [31].
There are two kinds of effect caused by the interaction of two pathogenic variants. One was called “synergistic effect”, implicating that the combination of two mutational genes in patients would lead to more severe phenotypes. For instance, Xe et al. revealed a patient with variants in CSNK2A1 and TRPS1, which resulted in a dual molecular diagnosis of tricho-rhino-phalangeal syndrome type I (TRPS I) and Okur-Chung neurodevelopmental syndrome (OCNDS). These two syndromes are both associated with short stature. Notably, this patient had a shorter stature as compared with other patients diagnosed with one of the diseases [12]. Moreover, Ye et al. reported a familial case with 13 patients affected by osteogenesis imperfecta (OI) type I, short stature and advanced bone age, with or without early-onset osteoarthritis and/or osteochondritis dissecans (SSOAOD). The proband was found to have two variants in COL1A1 and ACAN. After comprehensive analysis of the height within the family, this study discovered a synergistic effect that the patients with two variants present severe short stature [32]. These manifestations resembled clinical presentations of one of our patients (SCO2003P1972). The kid was presented with a segmentation failure of T10-L1 of spinal vertebrae, which was an unusual phenotype of classical Marfan Syndrome. Additionally, a severe thoracolumbar curve (Cobb > 80°) were also observed in the kid. We concluded that the POGZ and FBN1 variants both contributed to the skeletal deformity in this patient. However, in some patients who were diagnosed with dual molecular diagnosis, certain phenotypes may be opposite to that caused by a single genetic mutation. We proposed that these phenomena were caused by an antagonistic effect, as exemplified by the patient’s height in case 2. In addition, some of the cases with dual molecular diagnosis presented major phenotypic manifestation of one genetic disorder and only minor phenotypes of the other, as exemplified by our case 6 and case 8. This suggests that the genetic terminologies ‘variable expressivity’ and ‘incomplete penetrance’ are also applicable to the condition of dual molecular diagnosis.
All the dual diagnosis conditions in our study were identified through ES. However, the high expense of ES hinders it from being either a stand-alone or a first-tier diagnostic approach, especially in developing country. Therefore, selecting the most appropriate molecular diagnostic tool is important when ordering genetic testing. Single-gene testing should be recommended when the clinical features for a patient were typical for a specific disorder and the association between the disorder and a single gene was well-established [33]. For example, TBX6-associated congenital scoliosis was characterized by simple hemi‐/wedge‐shaped vertebrae in the lower half of the spine [34,35,36]. Furthermore, our previous study found that a novel de novo FBN1 variant could explain the Marfanoid–progeroid–lipodystrophy syndrome (MIM:616914) [37]. Under these circumstances, single-gene testing should be ordered. In contrast, in cases with complex phenotypes that cannot be explained by one genetic defect as shown in our examples, ES can be utilized as first line test which could shorten the diagnostic odyssey of the patients [33].
In conclusion, this study revealed the molecular diagnoses and complex diagnostic odyssey of dual molecular diagnosis through analyzing the clinical traits and genetic data of bone deformity in eight patients.
Conclusions
This study revealed the complicated skeletal phenotypes associated with dual molecular diagnosis. Exome sequencing represents a powerful tool to detect these complex conditions.
Methods
Study design
This is a retrospective study which reports the clinical and genetic characteristics of a group of patients with dual molecular diagnosis.
Subjects
Cases with skeletal disorders from the DISCO study (http://www.discostudy.org/) who underwent ES were included. The chief complains include early-onset scoliosis (EOS) (N = 447), short stature (N = 561), and congenital limb malformations (CLM) (N = 100). Deep phenotyping and radiological examinations including X-ray, computed tomography (CT), and magnetic resonance imaging (MRI) were performed on each patient as previously reported [4, 38], (Fan et al., Journal of Genetics and Genomics, 2021, in press). Written informed consent was obtained from every participant; if the participant was younger than 16 years old, written informed consent was obtained from their parents or legal guardians. The study was approved by the institutional review board of PUMCH (JS-2364), Beijing Jishuitan Hospital (201808-09) and the Second Affiliated Hospital of Guangxi Medical University (G-1-1).
Exome sequencing and variant interpretation
ES was performed on DNA extracted from blood of all 8 probands and their family members. The sequencing data were analyzed and annotated using an in-house developed analytical pipeline, Peking Union Medical college hospital Pipeline (PUMP) [39,40,41]. All variants were presumed to be pathogenic were subjected to Sanger sequencing.
Identification of dual molecular diagnosis
Patients with more than one molecular diagnosis from the included patients were selected for analyses. Each molecular diagnosis was manually curated based on the pathogenicity of the variants and the Mendelian expectations for inheritance mode. The pathogenicity of the variants was evaluated according to the American College of Medical Genetics and Genomics (ACMG) guidelines [42]. The Mendelian expectations for inheritance mode include autosomal dominant (AD) inheritance, autosomal recessive (AR) inheritance and X-linked dominant/recessive (XLD/XLR) inheritance. For AD/XLD traits, one heterozygous pathogenic/likely pathogenic variant is sufficient to establish a molecular diagnosis. For AR/XLR traits, one homozygous, one hemizygous or one pair of compound heterozygous pathogenic/likely pathogenic variants are required for a molecular diagnosis.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- COL1A1 :
-
Collagen, type I, alpha-1
- NF1 :
-
Neurofibromatosis, type I
- FBN1 :
-
Fibrilin-1
- FBN2 :
-
Fibrillin-2
- PTPN11 :
-
Protein tyrosine phosphatase, non-receptor type 11 gene
- ES:
-
Exome sequencing
- EOS:
-
Early-onset scoliosis
- WHSUS :
-
White–Sutton syndrome
- Gli2 :
-
GLI-Kruppel family member 2
- ANKRD11 :
-
Ankyrin repeat domain-containing protein 11
- FGFR2 :
-
Fibroblast growth factor receptor 2
- RYR1 :
-
Ryanodine receptor 1
- CSNK2A1 :
-
Casein kinase II, alpha subunit
- TRPS1 :
-
Trichorhinophalangeal syndrome type I
References
Spranger JW, Brill PW, Hall C, Nishimura G, Superti-Furga A, Unger S. Bone dysplasias: an atlas of genetic disorders of skeletal development. Oxford: Oxford University Press; 2018.
Rodriguez Celin M, Moosa S, Fano V. Uncommon IFITM5 mutation associated with severe skeletal deformity in osteogenesis imperfecta. Ann Hum Genet. 2018;82(6):477–81.
Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos S, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A. 2019;179(12):2393–419.
Zhao S, Zhang Y, Chen W, Li W, Wang S, Wang L, et al. Diagnostic yield and clinical impact of exome sequencing in early-onset scoliosis (EOS). J Med Genet. 2021;58(1):41–7.
Toydemir RM, Rutherford A, Whitby FG, Jorde LB, Carey JC, Bamshad MJ. Mutations in embryonic myosin heavy chain (MYH3) cause Freeman–Sheldon syndrome and Sheldon–Hall syndrome. Nat Genet. 2006;38(5):561–5.
Egunsola AT, Bae Y, Jiang MM, Liu DS, Chen-Evenson Y, Bertin T, et al. Loss of DDRGK1 modulates SOX9 ubiquitination in spondyloepimetaphyseal dysplasia. J Clin Investig. 2017;127(4):1475–84.
Aggarwal S, Das Bhowmik A, Tandon A, Dalal A. Exome sequencing reveals blended phenotype of double heterozygous FBN1 and FBN2 variants in a fetus. Eur J Med Genet. 2018;61(7):399–402.
Tang S, Hoshida H, Kamisago M, Yagi H, Momma K, Matsuoka R. Phenotype-genotype correlation in a patient with co-occurrence of Marfan and LEOPARD syndromes. Am J Med Genet A. 2009;149A(10):2216–9.
Posey JE, Rosenfeld JA, James RA, Bainbridge M, Niu Z, Wang X, et al. Molecular diagnostic experience of whole-exome sequencing in adult patients. Genet Med. 2016;18(7):678–85.
Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369(16):1502–11.
Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. 2017;376(1):21–31.
Xu S, Lian Q, Wu J, Li L, Song J. Dual molecular diagnosis of tricho-rhino-phalangeal syndrome type I and Okur–Chung neurodevelopmental syndrome in one Chinese patient: a case report. BMC Med Genet. 2020;21(1):158.
Jehee FS, de Oliveira VT, Gurgel-Giannetti J, Pietra RX, Rubatino FVM, Carobin NV, et al. Dual molecular diagnosis contributes to atypical Prader–Willi phenotype in monozygotic twins. Am J Med Genet A. 2017;173(9):2451–5.
Hannah-Shmouni F, Al-Shahoumi R, Brady LI, Wu L, Frei J, Tarnopolsky MA. Dual molecular diagnoses in a neurometabolic specialty clinic. Am J Med Genet A. 2020;185:766–73.
Assia Batzir N, Posey JE, Song X, Akdemir ZC, Rosenfeld JA, Brown CW, et al. Phenotypic expansion of POGZ-related intellectual disability syndrome (White–Sutton syndrome). Am J Med Genet A. 2020;182(1):38–52.
Zhytnik L, Maasalu K, Pashenko A, Khmyzov S, Reimann E, Prans E, et al. COL1A1/2 pathogenic variants and phenotype characteristics in Ukrainian osteogenesis imperfecta patients. Front Genet. 2019;10:722.
Overwater E, Efrat R, Barge-Schaapveld D, Lakeman P, Weiss MM, Maugeri A, et al. Autosomal dominant Marfan syndrome caused by a previously reported recessive FBN1 variant. Mol Genet Genomic Med. 2019;7(2):e00518.
Kang H, Aryal ACS, Marini JC. Osteogenesis imperfecta: new genes reveal novel mechanisms in bone dysplasia. Transl Res. 2017;181:27–48.
Swinnen FK, Coucke PJ, De Paepe AM, Symoens S, Malfait F, Gentile FV, et al. Osteogenesis Imperfecta: the audiological phenotype lacks correlation with the genotype. Orphanet J Rare Dis. 2011;6:88.
Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nat Rev Dis Primers. 2017;3:17004.
Fan X, Zhao S, Yu C, Wu D, Yan Z, Fan L, et al. Exome sequencing reveals genetic architecture in patients with isolated or syndromic short stature. J Genet Genom. 2021;48(5):396–402.
Valenza F, Cittaro D, Stupka E, Biancolini D, Patricelli MG, Bonanomi D, et al. A novel truncating variant of GLI2 associated with Culler–Jones syndrome impairs Hedgehog signalling. PLoS ONE. 2019;14(1):e0210097.
Lavillaureix A, Heide S, Chantot-Bastaraud S, Marey I, Keren B, Grigorescu R, et al. Mosaic intragenic deletion of FBN2 and severe congenital contractural arachnodactyly. Clin Genet. 2017;92(5):556–8.
Callewaert BL, Loeys BL, Ficcadenti A, Vermeer S, Landgren M, Kroes HY, et al. Comprehensive clinical and molecular assessment of 32 probands with congenital contractural arachnodactyly: report of 14 novel mutations and review of the literature. Hum Mutat. 2009;30(3):334–41.
Morel Swols D, Foster J 2nd, Tekin M. KBG syndrome. Orphanet J Rare Dis. 2017;12(1):183.
Macnab AJ, D’Orsogna L, Cole DE, Baguley PE, Adderley RJ, Patterson MW. Cardiac anomalies complicating congenital contractural arachnodactyly. Arch Dis Child. 1991;66(10 Spec No):1143–6.
Slaney SF, Oldridge M, Hurst JA, Moriss-Kay GM, Hall CM, Poole MD, et al. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am J Hum Genet. 1996;58(5):923–32.
Brandom BW. The genetics of malignant hyperthermia. Anesthesiol Clin North Am. 2005;23(4):615–9.
Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. 2015;17(7):578–86.
Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, van Kogelenberg M, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2015;385(9975):1305–14.
Balci TB, Hartley T, Xi Y, Dyment DA, Beaulieu CL, Bernier FP, et al. Debunking Occam’s razor: diagnosing multiple genetic diseases in families by whole-exome sequencing. Clin Genet. 2017;92(3):281–9.
Ye X, Fang D, He Y, Yan H, Qiu W, Sun Y. Dual diagnosis of osteogenesis imperfecta (OI) and short stature and advanced bone age with or without early-onset osteoarthritis and/or osteochondritis dissecans (SSOAOD) reveals a cumulative effect on stature caused by mutations in COL1A1 and ACAN genes. Eur J Med Genet. 2020;63(12):104074.
Xue Y, Ankala A, Wilcox WR, Hegde MR. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. Genet Med. 2015;17(6):444–51.
Wu N, Ming X, Xiao J, Wu Z, Chen X, Shinawi M, et al. TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N Engl J Med. 2015;372(4):341–50.
Liu J, Wu N, Deciphering Disorders Involving Scoliosis and COmorbidities (DISCO) study, Yang N, Takeda K, et al. TBX6-associated congenital scoliosis (TACS) as a clinically distinguishable subtype of congenital scoliosis: further evidence supporting the compound inheritance and TBX6 gene dosage model. Genet Med. 2019;21(7):1548–58.
Chen W, Lin J, Wang L, Li X, Zhao S, Liu J, et al. TBX6 missense variants expand the mutational spectrum in a non-Mendelian inheritance disease. Hum Mutat. 2020;41(1):182–95.
Lin M, Liu Z, Liu G, Zhao S, Li C, Chen W, et al. Genetic and molecular mechanism for distinct clinical phenotypes conveyed by allelic truncating mutations implicated in FBN1. Mol Genet Genomic Med. 2020;8(1):e1023.
Tian W, Huang Y, Sun L, Guo Y, Zhao S, Lin M, et al. Phenotypic and genetic spectrum of isolated macrodactyly: somatic mosaicism of PIK3CA and AKT1 oncogenic variants. Orphanet J Rare Dis. 2020;15(1):288.
Wang K, Zhao S, Liu B, Zhang Q, Li Y, Liu J, et al. Perturbations of BMP/TGF-beta and VEGF/VEGFR signalling pathways in non-syndromic sporadic brain arteriovenous malformations (BAVM). J Med Genet. 2018;55(10):675–84.
Wang K, Zhao S, Zhang Q, Yuan J, Liu J, Ding X, et al. Whole-exome sequencing reveals known and novel variants in a cohort of intracranial vertebral-basilar artery dissection (IVAD). J Hum Genet. 2018;63(11):1119–28.
Chen N, Zhao S, Jolly A, Wang L, Pan H, Yuan J, et al. Perturbations of genes essential for Mullerian duct and Wolffian duct development in Mayer–Rokitansky–Kuster–Hauser syndrome. Am J Hum Genet. 2021;108:337–45.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Acknowledgements
We would like to thank all the individuals involved in the study for their participation. We thank the nurses and doctors from the Department of Orthopedic Surgery of Peking Union Medical College Hospital, the Second Affiliated Hospital of Guangxi Medical University and Beijing Jishuitan Hospital for assistance with patient enrollment. We thank geneseeq inc. for exome sequencing technical support. We thank for ekitech ltd. (Beijing) for big data management and genetic data analysis.
Funding
This research was funded in part by the Beijing Natural Science Foundation (JQ20032 to N.W., 7191007 to Z.W.), Capital’s Funds for Health Improvement and Research (2020-4-40114 to N.W.), National Natural Science Foundation of China (82072391 to N.W., 81930068 and 81772299 to Z.W., 81772301 and 81972132 to G.Q., 81972037 to J.Z.), Tsinghua University-Peking Union Medical College Hospital Initiative Scientific Research Program, Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (No. 2019PT320025), Center for Rare Diseases Research, Chinese Academy of Medical Sciences (Grant No. 2016ZX310174-4).
Author information
Authors and Affiliations
Consortia
Contributions
NW directed the project. NW, JZ and GQ designed the study. LL, LS, YC and MW were involved in data management and statistical analysis and wrote the first draft of the manuscript. CY, YH, HD and SC gathered detailed clinical information for the study. XF, WT and SZ analyzed the data. ZW, GQ, JZ and NW supervised the data analysis, and reviewed and commented on all drafts. All other authors coordinated the study and critically revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the institutional review board of PUMCH (JS-2364), Jishuitan Hospital (201808-09) and the Second Affiliated Hospital of Guangxi Medical University (G-1-1). All participants gave informed consent.
Consent for publication
Consent for publication was obtained from all participants.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary clinical photographs of patients in our study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Liu, L., Sun, L., Chen, Y. et al. Delineation of dual molecular diagnosis in patients with skeletal deformity. Orphanet J Rare Dis 17, 139 (2022). https://doi.org/10.1186/s13023-022-02293-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-022-02293-x