
Abstract
Background
Congenital anomalies are the leading cause of perinatal, neonatal and infant mortality in developed countries. Large long-term follow-up studies investigating survival beyond the first year of life in children with rare congenital anomalies are costly and sufficiently large standardized cohorts are difficult to obtain due to the rarity of some anomalies. This study aimed to investigate the survival up to 10 years of age of children born with a rare structural congenital anomaly in the period 1995–2014 in Western Europe.
Methods
Live births from thirteen EUROCAT (European network for the epidemiological surveillance of congenital anomalies) population-based registries were linked to mortality records. Survival for 12,685 live births with one of the 31 investigated rare structural congenital anomalies (CAs) was estimated at 1 week, 4 weeks and 1, 5 and 10 years of age within each registry and combined across Europe using random effects meta-analyses. Differences between registries were evaluated for the eight rare CAs with at least 500 live births.
Results
Amongst the investigated CAs, arhinencephaly/holoprosencephaly had the lowest survival at all ages (58.1%, 95% Confidence Interval (CI): 44.3–76.2% at 1 week; 47.4%, CI: 36.4–61.6% at 1 year; 35.6%, CI: 22.2–56.9% at 10 years). Overall, children with rare CAs of the digestive system had the highest survival (> 95% at 1 week, > 84% at 10 years). Most deaths occurred within the first four weeks of life, resulting in a 10-year survival conditional on surviving 4 weeks of over 95% for 17 out of 31 rare CAs. A moderate variability in survival between participating registries was observed for the eight selected rare CAs.
Conclusions
Pooling standardised data across 13 European CA registries and the linkage to mortality data enabled reliable survival estimates to be obtained at five ages up to ten years. Such estimates are useful for clinical practice and parental counselling.
Similar content being viewed by others

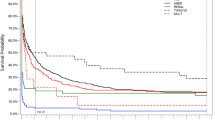

Background
Congenital anomalies (CA) affect approximately 3% of births in Europe and in the United States [1, 2]. CAs, including structural defects, chromosomal anomalies, and genetic syndromes, are the leading cause of perinatal, neonatal and infant mortality in developed countries [3,4,5].
Advances in neonatal and paediatric care have led to an overall improvement in survival of children with CAs beyond infancy [6, 7]. However, large long-term follow-up studies investigating survival beyond the first year of life in children with rare CAs are costly and time-consuming; therefore, such research is scarce and little is known about the long-term outcomes of children born with certain rare CAs. Published results mainly refer to case series or hospital cohorts often estimating mortality at a point in time and very rarely starting from birth [8,9,10,11,12,13,14]. Due to the rarity of some CAs, sufficiently large standardized cohorts are difficult to obtain and the only way to accurately study survival in children with these CAs is to pool data across several registries and link cases to mortality databases [15, 16]. Pooling CA data from registries across Europe using standardized definitions and classification of CAs provides the opportunity to produce reliable survival estimates for children with rare CAs and a rich dataset for future research.
This study aimed to investigate the survival up to 10 years of age of children born with a rare structural CA in the period 1995–2014 using data from 13 EUROCAT (European network for the epidemiological surveillance of CAs) registries. The study is part of the EUROlinkCAT project that linked data of live born children with CAs to mortality data sources and other electronic administrative, healthcare and education databases to investigate the survival, morbidity and educational outcomes up to 10 years of age of European children born with a major CA [17].
Methods
Design and population
This was a European, population-based linkage cohort study. The cohort included all live births with rare structural CAs collected and validated by population-based CA registries which are members of EUROCAT (https://eu-rd-platform.jrc.ec.europa.eu/eurocat_en) [18,19,20].
All liveborn children with a major CA born between 1st January 1995 and 31st December 2014 recorded in the 13 registries of nine Western European countries were linked to mortality records up to the child’s 10th birthday or to 31st December 2015 (whichever was earlier), so that all live births had information on at least the first year’s survival (Table 1). Full details on the linkage methods are reported elsewhere [21,22,23]. A major CA is defined as an anomaly that require surgical treatment, have serious adverse effects on health or development, or have significant cosmetic impact [24].
In brief, all the registries linked their CA data to vital statistics, except Malta and Valencian Region (Spain), which linked to mortality records (Table 1). Linkage to vital statistics provides information on whether the child was still alive or had died; in contrast, in a mortality database the child was assumed to be alive if no death certificate was present. Careful examination of the accuracy of the linkage was undertaken and birth years in registries during which the linkage quality was judged poor were excluded from this analysis [21]. The study period differed between registries due to different years of EUROCAT membership and because only years with high quality linkage were retained (Table 1).
Investigated anomalies
This study focused on the survival of children with 24 different rare structural CAs, selected within EUROlinkCAT as CAs with a live birth prevalence lower than 1 per 10,000 according to the EUROCAT prevalence tables. In addition, five new congenital anomaly subgroups were defined that were not in the standard EUROCAT prevalence tables, but were assumed to have a prevalence lower than 1 per 10,000. Four of them were subsequently found to have a prevalence slightly above 1 per 10,000 when analysed in the EUROCAT database, but are included in this study. In addition, Hirschsprung’s disease, with a prevalence of 1.64 per 10,000 and pulmonary valve atresia with a prevalence of 1.01 per 10,000 are also included. Table 2 shows the 31 CAs included in this study with their livebirth prevalence estimates, together with the coding of the International Statistical Classification of Diseases and Related Health Problems version 10 (ICD-10) and version 9 (ICD-9) with the British Paediatric Association (BPA) extension used by EUROCAT to identify each anomaly [25, 26].
Statistical analysis
The analyses were based on standardized EUROCAT variables together with a common data model developed for EUROlinkCAT to standardize the local variables obtained from the linkage [17]. Such standardization allowed centrally written syntax scripts to be developed both for checking the quality of data linkage and for analysing the data to be run by all participating registries [17, 21].
To account for censoring of individuals due to emigration or reaching the study end date before reaching the 10th birthday, Kaplan–Meier survival analyses were performed within each registry by running centrally written syntax scripts. The survival estimates with 95% confidence intervals (CI) together with the number at risk (alive at the beginning of each time period) and the number of deaths in each time period for each CA subgroup were uploaded by each registry to the CRR at Ulster University (UK) and then transferred to the research team using a secure web platform. No individual case data were shared.
The Kaplan–Meier survival estimates from each registry were then combined centrally in a random-effects meta-analysis of the survival at five ages separately (1 week, 4 weeks and 1, 5 and 10 years) to estimate the overall survival for each CA.
Similarly, 10-year survival estimates conditional on having survived at 4 weeks calculated for each registry were combined in a random-effects meta-analysis.
Differences between registries were evaluated for eight rare CAs, where each had at least 500 live births at risk, by plotting the forest plot of the meta-analysis of the survival at 5 years and reporting the I2 statistic as a measure of the observed between-registry heterogeneity.
All statistical analyses were performed using Stata16 (StataCorp LP, College Station, TX, USA).
Results
Thirteen European registries from nine countries, covering a population of 6,159,520 births in 1995–2014 were included in the study, with 5 out of 13 registries covering all 20 birth years (Table 1). There was a total of 12,685 liveborn children with one of the 31 rare structural CAs (Table 3).
Prune belly sequence and anophthalmos were the rarest investigated CAs with only 48 and 103 live births respectively and unilateral renal agenesis the most common with 1237 live births (Table 3).
Table 3 shows the survival estimates (with 95% CIs) calculated for each rare CA. As expected, there was considerable variation in survival between individual anomalies.
At 1 week, only children with arhinencephaly/holoprosencephaly and prune belly sequence had survival below 80%, being 58.1% (95% CI: 44.3–76.2) and 76.4% (95% CI: 52.9–100.0) respectively.
Ten-year survival varied from 35.6% (95% CI: 22.2–56.9) for children with arhinencephaly/holoprosencephaly to 99.5% (95% CI: 98.5–100.0) for children with epispadias.
Ten-year survival was below 80% for 17 CAs, which included all nine severe rare congenital heart defects (CHDs). In particular, children with common arterial truncus and aortic atresia/interrupted aortic arch had 10-year survival lower than 65%.
Children with prune belly sequence had consistently low survival across all age points, declining from 76.4% (95% CI: 52.9–100.0) at 1 week to 67.0% (95% CI: 43.2–100.0) at 1 year and further to 57.4% (95% CI: 34.4–96.1) at 10 years of age.
In general, children with rare CAs of the digestive and urinary system, with the exception of prune belly sequence, had a relatively high survival at all five age points.
Figure 1 shows the proportion of deaths for each CA at each age by group of CA. For children with arhinencephaly/holoprosencephaly, encephalocele, prune belly sequence, posterior urethral valve, anotia, anophthalmos, and indeterminate sex more than 50% of deaths occurred within the first week. In general, for children with rare CHDs more than 50% of deaths occurred within the first month. Children with anomalies of the digestive or urinary system had a much higher proportion of deaths occurring at later ages.

Proportion of deaths at 1 week (1 w) of age, between 1 and 4 weeks (4 w), between 4 weeks and 1 year (1 y), between 1 and 5 years (5 y), and between 5 and 10 years (10 y) for the rare structural congenital anomalies sorted according to decreasing proportion of deaths at 1 week of age and showed by group
The 10-year conditional survival estimates (i.e. the survival at 10 years of age of children who have survived at 4 weeks), are all above 90% (Table 3), with the exception of children with arthrogryposis multiplex congenita (89.8%) and arhinencephaly/holoprosencephaly (77.8%). For 17 out of 31 CAs, the 10-year conditional survival was higher than 95%.
Figure 2 shows the differences in the survival at 5 years among registries for eight rare CAs with at least 500 live births. The greatest heterogeneity among registries (I2 > 50%) was observed for the subgroups anophthalmos/microphthalmos, anomalies of intestinal fixation and unilateral renal agenesis. A moderate heterogeneity between registries was observed for all the other rare structural CAs, with the exception of choanal atresia for which the survival appeared almost homogeneous across the investigated areas.

Five-year survival estimates (with 95% Confidence Intervals (CI)) by registry (birth year period: 1995–2014) and the I2 statistic as a measure of the observed between-registry heterogeneity calculated by a random effect meta-analysis on eight rare CAs with at least 500 liveborn cases. d = days; w = week, m = months, y = years
Discussion
This study reports the survival of children born with rare structural CAs in western Europe, using population-based data on children born in the period 1995–2014 linked to mortality data.
There are few studies on population-based long-term survival of children with rare structural CAs, therefore it is not possible to make a direct comparison between our results and other published studies. Most of the studies on rare CAs are hospital-based series mainly reporting fatality rates rather than survival estimates in live births.
The rare CAs investigated in this study are heterogeneous and can be associated with different complex conditions, which accounts for the considerable variation in survival observed between individual anomalies. In addition, the proportion of associated anomalies might differ substantially by type of CA: it has been reported that 32–34% of the respiratory and of eye, face and neck CAs most likely occur with other CAs and that CHDs, limb, and genital are the least likely to occur with other CAs (12–13%) [27]. This different proportions may have an impact on survival mainly for the less severe anomalies [28, 29].
Children with arhinencephaly/holoprosencephaly had the lowest survival at all five investigated ages with only 58.1% of children surviving the first week of life. Holoprosencephaly is a brain malformation characterized by four forms (in decreasing order of severity: alobar, semilobar, lobar and middle interhemispheric variant) with the alobar form present approximately in two out of three individuals, thus probably explaining the observed low survival [30].
Children with prune belly sequence also had a low survival in the first week of life (76.4%). Prune belly sequence can be characterized by a wide variability in severity and clinical manifestations, such as abdominal muscle deficiency, lung and renal dysfunction, associated congenital heart defects, and cryptorchidism [31].
Children with arthrogryposis multiplex congenita (AMC), a CA that can be associated with multiple developmental defects and be part of a large number of syndromes with or without central nervous system involvement, also showed one of the lowest survival estimates both in the first week (85.6%) and at 10 years (69.4%). A EUROCAT population-based study on AMC including the birth period 1980–2006 reported that 23% of children had died within the first week of life. The higher mortality observed in that study may have been due to the earlier time period studied or bias arising from missing data on first week survival [29].
In our study, all children with rare CHDs had 10-year survival lower than 80%. All the rare CHDs here investigated are severe, commonly requiring cardiac surgery in the first year of life. Some of them, such as common arterial truncus, total anomalous pulmonary venous return and single ventricle, are incompatible with survival without a surgery early in infancy. A meta-analysis by Best and Rankin [32] investigating the long-term survival of children born with CHDs reported pooled 5-year survival estimates of 47.4%, 59.8%, 61.2% and 65.6% for common arterial truncus, single ventricle, total anomalous pulmonary venous return and Ebstein’s anomaly respectively, compared to survival of 61.4%, 72.1%, 76.4% and 78.9% estimated in our study. The lower meta-analytic estimates observed by Best and Rankin may be in part attributed to their cohorts encompassing earlier periods (birth years starting in the 1970s–1980s) and the inclusion of diverse study designs, CHD classification, geographical areas and mortality sources.
Children with atresia of bile ducts almost always survive the first week of life (99.3%). The diagnosis is usually given 2–6 weeks after birth [33]. However, survival estimates at 5 and 10 years of age (84.5% and 84.1%, respectively) in children with this severe CA, which is incompatible with life if not operated early in infancy, are still relatively low despite a wider use of liver transplantation for these patients in recent decades. Our survival estimates are comparable with pooled estimates in a systematic review of children with biliary atresia (85% and 82% for 5- and 10-year survival respectively) [6].
For children with Hirschsprung’s disease and anomalies of intestinal fixation, mortality may be due to enterocolitis and acute complications with intestinal ischemia [34].
Mortality of children with anophthalmos and microphthalmos is most likely explained by associated anomalies such as severe cerebral anomalies and/or lethal chromosomal anomalies [35]. Similarly, survival estimates lower than 100% for children with congenital glaucoma can be explained by associated anomalies or genetic diseases [36].
In children with situs inversus, survival of 91.6% was observed at 1 year (89.6% at 10 years) which is probably due to the presence of associated severe CHDs such as transposition of great vessels [37].
In general, for rare CAs, the number of live births is too small to evaluate geographical differences across different registries, but eight investigated anomalies with more than 500 children were considered suitable for a meta-analysis aimed at evaluating regional variations. However, we observed, as expected, a lower precision of the estimates for some registries in some of these rare CAs due to small numbers of events. The results showed a moderate variation in survival between participating registries that is in full agreement with what was reported in a methodological study on the geographical variation in survival showing a high variability only for major subgroups of CAs [22]. The low/moderate heterogeneity observed in our study suggests consistency and generalizability of our results and, as a consequence, accurate survival estimates for the rare CAs investigated.
Study strengths and limitations
The main strength of the study is that the pooling of data from high-quality population-based specialized EUROCAT registries from across Europe resulted in the largest cohort of children born with rare structural CAs at European level to date. This allowed reliable survival estimates up to 10 years of age to be produced. Another strength was the use of standardized approaches in EUROCAT registries (data collection, coding and classification) and in EUROlinkCAT (standardising variables to a common data model) which enabled common syntax scripts to produce standardised analytic results.
A limitation of the study is that isolated cases of each rare CA could not be analysed due to the extremely small sample sizes in the registries. However, as the data is from a population-based cohort, the presence of associated anomalies reflects the expected occurrence of anomalies in future births and therefore the survival rates can be considered an unbiased estimate of predicted survival for children with these rare anomalies. A second limitation is that all registries report cases diagnosed within 1 year of age, but some of the investigated rare CAs (e.g. anomalies of intestinal fixation, unilateral renal agenesis, accessory kidney, situs inversus if not picked up by ultrasound scans) may be diagnosed later and children with these anomalies may not be included in the study; this is particularly true for less severe cases without associated anomalies. For this reason, there might be an overestimate of mortality for some rare CA due to the exclusion of less severe cases diagnosed after infancy. Another limitation is the lack of registries from Central and Eastern Europe. Three EUROCAT member registries from Central and European Europe participated in EUROlinkCAT, but we excluded them from the analysis: two of them were excluded due to low quality data linkage and one due to extremely low survival rates. As other studies have also found higher infant mortality in Eastern Europe [38] it was decided that including only one Eastern European country would not enable us anyway to produce survival estimates referred to the whole Europe. For these reasons, the results of our study should be inteded as representative of Western Europe only.
Finally, due to the small sample sizes occurring in each registry it was not possible to investigate any association between survival and socio-economic status that might provide useful information for making inferences about the quality of care provided to children born with CAs.
Conclusions
This multi-centre population-based European study provided a sufficiently large, standardized cohort to produce reliable survival estimates of children with rare structural anomalies up to 10 years of age born in the period 1995–2014. There was considerable variation in survival for children with the different anomalies, with only moderate variability between registries. For the majority of anomalies, more than 50% of deaths occurred within the first month and the 10-year survival conditional on surviving the first four weeks of life was above 95%. Having reliable information on long-term survival of children born with specific CAs is of major importance for the health professionals involved in counseling parents, especially when facing a prenatal diagnosis of a rare CA.
Availability of data and materials
The data that support the findings of this study are available from the participating registries of congenital anomalies, but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors for scientifically valid requests and with permission of the participating registries of congenital anomalies.
Abbreviations
- AMC:
-
Arthrogryposis multiplex congenita
- CAs:
-
Congenital anomalies
- CHDs:
-
Congenital heart defects
- CI:
-
Confidence interval
- CRR:
-
Central Results Repository
- ICD-10:
-
International Classification of Diseases version 10
References
Morbidity and Mortality Weekly Report, Centers for Disease Control and Prevention, January 11, 2008/57(01);1–5.
EUROCAT website: https://eu-rd-platform.jrc.ec.europa.eu/eurocat/eurocat-data/prevalence_en. Accessed 29 June 2021.
Boyle B, Addor M-C, Arriola L, Barisic I, Bianchi F, Csáky-Szunyogh M, de Walle HEK, Dias CM, Draper E, Gatt M, Garne E, Haeusler M, Källén K, Latos-Bielenska A, McDonnell B, Mullaney C, Nelen V, Neville AJ, O’Mahony M, Queisser-Wahrendorf A, Randrianaivo H, Rankin J, Rissmann A, Ritvanen A, Rounding C, Tucker D, Verellen-Dumoulin C, Wellesley D, Wreyford B, Zymak-Zakutnia N, Dolk H. Estimating global burden of disease due to congenital anomaly: an analysis of European data. Arch Dis Child Fetal Neonatal Ed. 2018;103:F22–8.
Heron M. Deaths: leading causes for 2017. Natl Vital Stat Rep. 2019;68(6):1–77.
World Health Organization The Global Health Observatory. World health data platform. Distribution of causes of death among children aged < 5 years (%). 2017. https://www.who.int/data/gho/data/indicators/indicator-details/GHO/distribution-of-causes-of-death-among-children-aged-5-years-(-). Accessed 20 Apr 2021.
Glinianaia SV, Morris JK, Best KE, Santoro M, Coi A, Armaroli A, Rankin J. Long-term survival of children born with congenital anomalies: a systematic review and meta-analysis of population-based studies. PLoS Med. 2020;17(9):e1003356.
Tennant PWG, Pearce MS, Bythell M, Rankin J. 20-Year survival of children born with congenital anomalies: a population-based study. Lancet. 2010;375(9715):649–56.
Routh JC, Huang L, Retik AB, Nelson CP. Contemporary epidemiology and characterization of newborn males with prune belly syndrome. Urology. 2010;76(1):44–8.
Kaliaperumal C, Ndoro S, Mandiwanza T, Reidy F, McAuliffe F, Caird J, Crimmins D. Holoprosencephaly: antenatal and postnatal diagnosis and outcome. Childs Nerv Syst. 2016;32(5):801–9.
Domadia S, Kumar SR, Votava-Smith JK, Pruetz JD. Neonatal outcomes in total anomalous pulmonary venous return: the role of prenatal diagnosis and pulmonary venous obstruction. Pediatr Cardiol. 2018;39(7):1346–54.
Herbst KW, Tomlinson P, Lockwood G, Mosha MH, Wang Z, D’Alessandri-Silva C. Survival and kidney outcomes of children with an early diagnosis of posterior urethral valves. Clin J Am Soc Nephrol. 2019;14(11):1572–80.
Abel JS, Berg C, Geipel A, Gembruch U, Herberg U, Breuer J, Brockmeier K, Gottschalk I. Prenatal diagnosis, associated findings and postnatal outcome of fetuses with truncus arteriosus communis (TAC). Arch Gynecol Obstet. 2021;304(6):1455–66.
Global PaedSurg Research Collaboration. Mortality from gastrointestinal congenital anomalies at 264 hospitals in 74 low-income, middle-income, and high-income countries: a multicentre, international, prospective cohort study. Lancet. 2021;398(10297):325–39.
Protzenko T, Dos Santos Gomes Junior SC, Bellas A, Salomão JFM. Hydrocephalus and occipital encephaloceles: presentation of a series and review of the literature. Childs Nerv Syst. 2021;37(11):3437–45.
Copeland GE, Kirby RS. Using birth defects registry data to evaluate infant and childhood mortality associated with birth defects: an alternative to traditional mortality assessment using underlying cause of death statistics. Birth Defects Res A Clin Mol Teratol. 2007;79(11):792–7.
Castilla EE, Mastroiacovo P. Very rare defects: what can we learn? Am J Med Genet C Semin Med Genet. 2011;157C(4):252–61.
Morris JK, Garne E, Loane M, Barisic I, Densem J, Latos-Bieleńska A, Neville A, Pierini A, Rankin J, Rissmann A, de Walle H, Tan J, Given JE, Claridge H, EUROlinkCAT Consortium. EUROlinkCAT protocol for a European population-based data linkage study investigating the survival, morbidity and education of children with congenital anomalies. BMJ Open. 2021;11:e047859.
Boyd PA, Haeusler M, Barisic I, Loane M, Garne E, Dolk H. Paper 1: the EUROCAT network-organization and processes. Birth Defects Res A Clin Mol Teratol. 2011;91(Suppl 1):S2-15.
Tucker FD, Morris JK, JRC Management Committee, Neville A, Garne E, Kinsner-Ovaskainen A, Lanzoni M, Loane MA, Martin S, Nicholl C, Rankin J, Rissmann AK. EUROCAT: an update on its functions and activities. J Community Genet. 2018;9(4):407–10.
Kinsner-Ovaskainen A, Lanzoni M, Garne E, Loane M, Morris J, Neville A, Nicholl C, Rankin J, Rissmann A, Tucker D, Martin S. A sustainable solution for the activities of the European network for surveillance of congenital anomalies: EUROCAT as part of the EU Platform on Rare Diseases Registration. Eur J Med Genet. 2018;61(9):513–7.
Loane M, Given JE, Tan J, Reid A, Akhmedzhanova D, Astolfi G, Barišić I, Bertille N, Bonet LB, Cavero-Carbonell C, Mokoroa Carollo O, Coi A, Densem J, Draper E, Garne E, Gatt M, Glinianaia SV, Heino A, Den Hond E, Jordan S, Khoshnood B, Kiuru-Kuhlefelt S, Klungsøyr K, Lelong N, Lutke LR, Neville AJ, Ostapchuk L, Puccini A, Rissmann A, Santoro M, Scanlon I, Thys G, Tucker D, Urhoj S-K, de Walle HEK, Wellesley D, Zurriaga O, Morris JK. Linking a European cohort of children born with congenital anomalies to vital statistics and mortality records: a EUROlinkCAT study. PLoS ONE. 2021;16(8):e0256535.
Santoro M, Coi A, Pierini1A, Rankin J, Glinianaia SV, Tan J, Reid A, Garne E, Loane M, Given J, Aizpurua A, Astolfi G, Barisic I, Cavero-Carbonell C, de Walle HEK, Den Hond1 E, García-Villodre L, Gatt M, Gissler M, Jordan S, Khoshnood B, Kiuru-Kuhlefelt S, Klungsøyr K, Lelong N, Lutke LR, Mokoroa O, Nelen V, Neville AJ, Odak L, Rissmann A, Scanlon I, Kjaer Urhoj S, Wellesley D, Wertelecki W, Yevtushok L, Morris JK. Temporal and geographical variations in survival of children born with congenital anomalies in Europe: a EUROlinkCAT study. Paediatr Perinat Epidemiol 2021 (submitted).
Glinianaia SV, Rankin J, Pierini A, Coi, A, Santoro M. Tan J, Reid A, Ester G, Loane M, Given J, Cavero-Carbonell C, de Walle HEK, Gatt M, Gissler M, Heino A, Khoshnood B, Klungsøyr K, Lelong N, Neville A, Thayer D, Tucker D, Urhøj S, Wellesley D, Zurriaga O, Morris JK. Ten-year survival of children with congenital anomalies: a European Cohort Study. Pediatrics. 2022;149(3):e2021053793. https://doi.org/10.1542/peds.2021-053793.
EUROCAT Special Report: Congenital Anomalies are a Major Group of Mainly Rare Diseases, 2012. https://eu-rd-platform.jrc.ec.europa.eu/sites/default/files/eurocat-pub-docs/Special-Report-Major-Group-of-Mainly-Rare-Diseases.pdf.
World Health Organization. Congenital malformations, deformations and chromosomal abnormalities (Q00-Q99). In: International statistical classification of diseases and related health problems: 10th revision. Geneva, Switzerland: World Health Organization; 2010.
EUROCAT. Chapter 3.3: EUROCAT Subgroups of Congenital Anomalies (version 23.09.2020). In: EUROCAT Guide 1.4: Instruction for the registration of congenital anomalies (Last update version 01/12/2020). Newtownabbey, UK: EUROCAT Central Registry, University of Ulster; 2013.
Calzolari E, Barisic I, Loane M, Morris J, Wellesley D, Dolk H, Addor MC, Arriola L, Bianchi F, Neville AJ, Budd JL, Klungsoyr K, Khoshnood B, McDonnell B, Nelen V, Queisser-Luft A, Rankin J, Rissmann A, Rounding C, Tucker D, Verellen-Dumoulin C, de Walle H, Garne E. Epidemiology of multiple congenital anomalies in Europe: a EUROCAT population-based registry study. Birth Defects Res A Clin Mol Teratol. 2014;100(4):270–6.
Morris JK, Wellesley DG, Barisic I, Addor MC, Bergman JEH, Braz P, Cavero-Carbonell C, Draper ES, Gatt M, Haeusler M, Klungsoyr K, Kurinczuk JJ, Lelong N, Luyt K, Lynch C, O’Mahony MT, Mokoroa O, Nelen V, Neville AJ, Pierini A, Randrianaivo H, Rankin J, Rissmann A, Rouget F, Schaub B, Tucker DF, Verellen-Dumoulin C, Wiesel A, Zymak-Zakutnia N, Lanzoni M, Garne E. Epidemiology of congenital cerebral anomalies in Europe: a multicentre, population-based EUROCAT study. Arch Dis Child. 2019;104(12):1181–7.
Hoff JM, Loane M, Gilhus NE, Rasmussen S, Daltveit AK. Arthrogryposis multiplex congenita: an epidemiologic study of nearly 9 million births in 24 EUROCAT registers. Eur J Obstet Gynecol Reprod Biol. 2011;159(2):347–50.
Solomon BD, Gropman A, Muenke M. Holoprosencephaly overview. Gene Reviews. 2013.
Lopes RI, Baker LA, Dénes FT. Modern management of and update on prune belly syndrome. J Pediatr Urol. 2021;S1477–5131(21):00217–25.
Best KE, Rankin J. Long-term survival of individuals born with congenital heart disease: a systematic review and meta-analysis. J Am Heart Assoc. 2016;5:e002846.
Chardot C. Biliary atresia. Orphanet J Rare Dis. 2006;1:28.
Roorda D, Oosterlaan J, van Heurn E, Derikx JPM. Risk factors for enterocolitis in patients with Hirschsprung disease: a retrospective observational study. J Pediatr Surg. 2021;56(10):1791–8.
Stoll C, Dott B, Alembik Y, Roth MP. Associated malformations among infants with anophthalmia and microphthalmia. Birth Defects Res A Clin Mol Teratol. 2012;94(3):147–52.
Pedersen KB, Kappelgaard P, Kessel L, Sandfeld L, Zibrandtsen N, Bach-Holm D. Primary congenital glaucoma in Denmark, 1977–2016. Acta Ophthalmol. 2020;98(2):182–9.
Frescura C, Thiene G. The spectrum of congenital heart disease with transposition of the great arteries from the cardiac registry of the University of Padua. Front Pediatr. 2016;4:84.
Pitt MJ, Morris JK. European trends in mortality in children with congenital anomalies: 2000–2015. Birth Defects Res. 2021;113(12):958–67.
Acknowledgements
We are very grateful to the whole EUROlinkCAT Working Group for their contribution to the project (data linkage and standardization, running syntax scripts): Dr Nicole Siemensma-Mühlenberg (University Medical Center Groningen, Groningen, The Netherlands); Óscar Zurriaga, Ana Ruiz Palacio, Sandra Moreno Marro and Laia Barrachina Bonet (Foundation for the Promotion of Health and Biomedical Research in the Valencian Region, Valencia, Spain); Anna Heino and Tuuli Puroharju (THL Finnish Institute for Health and Welfare. Helsinki, Finland); Drs Gianni Astolfi, Aurora Puccini, Annarita Armaroli (Center for Clinical and Epidemiological Research, University of Ferrara, Ferrara, Italy); Nathalie Bertille and Babak Khoshnood (INSERM, Paris, France); Professor Elizabeth Draper (University of Leicester, Leicester, United Kingdom); Professor Jenny Kurinczuk (University of Oxford, Oxford, United Kingdom). We also thank Mr Hugh Claridge (Population Health Research Institute, St George’s, University of London, London, United Kingdom) for the project management.
Funding
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under Grant Agreement No. 733001 (Jan 2017–May 2022) https://ec.europa.eu/programmes/horizon2020/en). The funder had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, or review; and decision to submit the manuscript for publication. The views presented here are those of the authors only and the European Commission is not responsible for any use that may be made of the information presented here.
Author information
Authors and Affiliations
Contributions
AC conceptualized and designed the study, developed the study methods and the statistical analysis plan, interpreted the results, drafted the initial manuscript, and critically reviewed the manuscript. MS and SG conceptualized and designed the study, developed the study methods and the statistical analysis plan, interpreted the results, and critically reviewed the manuscript. AP and JR conceptualized and designed the study, obtained funding for the study developed the study methods and the statistical analysis plan, interpreted the results, and critically reviewed manuscript. JT wrote the analysis programs, contributed to statistical analysis and to the development of the study methods and the statistical analysis plan, interpreted the results, and critically reviewed the manuscript. A-KR wrote the analysis programs, contributed to statistical analysis, interpreted the results and critically reviewed the manuscript. EG contributed to obtaining funding, to the development of the study methods, including data standardization and data linkage, to interpretation of the results, and critically reviewed the manuscript. ML contributed to obtaining funding, was responsible for data standardization and management of data linkage by the participating data providers, interpreted the results, and critically reviewed the manuscript. JG contributed to the development of study methods, including data standardization and data linkage, to the interpretation of the results, and critically reviewed the manuscript. EB, CC-C, HEKdW, MG, LG-V, MG, SJ, SK-K, KK, NL, RLL, AJN, MR, IS, SKU, and DW were responsible for data linkage and standardization for their registries’ data and running centrally written syntax scripts for local analyses, and critically reviewed the manuscript for important intellectual content. JKM conceptualized and designed the study, obtained funding, developed study methods, including data standardization and linkage, supervised writing analysis programs, performed statistical analysis, supervised the work, and critically reviewed and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The EUROCAT registries all have ethical and governance clearances and other permissions required according to their national guidelines for routine surveillance, data collection and transmission of anonymised data to a central database. Additional permissions to link their data to mortality or vital statistics and to transmit anonymous aggregate data and analytic results to a Central Results Repository (CRR) were obtained by each registry.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Coi, A., Santoro, M., Pierini, A. et al. Survival of children with rare structural congenital anomalies: a multi-registry cohort study. Orphanet J Rare Dis 17, 142 (2022). https://doi.org/10.1186/s13023-022-02292-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-022-02292-y