
Abstract
Background
Duchenne muscular dystrophy (DMD) is a severe rare progressive inherited neuromuscular disorder, leading to loss of ambulation (LOA) and premature mortality. The standard of care for patients with DMD has been treatment with corticosteroids for the past decade; however a synthesis of contemporary data describing the clinical course of DMD is lacking. The objective was to summarize age at key clinical milestones (loss of ambulation, scoliosis, ventilation, cardiomyopathy, and mortality) in the corticosteroid-treatment-era.
Methods
A systematic review was conducted using MEDLINE and EMBASE. The percentage experiencing key clinical milestones, and the mean or median age at those milestones, was synthesized from studies from North American populations, published between 2007 and 2018.
Results
From 5637 abstracts, 29 studies were included. Estimates of the percentage experiencing key clinical milestones, and age at those milestones, showed heterogeneity. Up to 30% of patients lost ambulation by age 10 years, and up to 90% by 15 years of age. The mean age at scoliosis onset was approximately 14 years. Ventilatory support began from 15 to 18 years, and up to half of patients required ventilation by 20 years of age. Registry-based estimates suggest that 70% had evidence of cardiomyopathy by 15 years and almost all by 20 years of age. Finally, mortality rates up to 16% by age 20 years were reported; among those surviving to adulthood mortality was up to 60% by age 30 years.
Conclusions
Contemporary natural history studies from North America report that LOA on average occurs in the early teens, need for ventilation and cardiomyopathy in the late teens, and death in the third or fourth decade of life. Variability in rates may be due to differences in study design, treatment with corticosteroids or other disease-modifying agents, variations in clinical practices, and dystrophin mutations. Despite challenges in synthesizing estimates, these findings help characterize disease progression among contemporary North American DMD patients.
Similar content being viewed by others


Background
Duchenne muscular dystrophy (DMD) is a rare, progressive, life-limiting neuromuscular disorder [1] occurring in 15.9 to 19.5 per 100,000 live male births [2,3,4]. It is caused by mutations in the dystrophin gene [2, 5]; lack of dystrophin compromises muscle structure and integrity, leading to progressive muscular degeneration [6, 7]. Patients with DMD are typically identified in early childhood with symptoms including delays in motor milestones and frequent falls [8]. Over time, these patients experience progressive functional impairments leading to loss of ambulation (LOA), pulmonary insufficiency, cardiomyopathy, and early mortality [2, 5, 9].
Although there is presently no cure for DMD, advancements to the standard of care, including the introduction of systemic corticosteroids in the 1990s, have helped slow disease progression and improve survival [10,11,12]. However, the impact of these changes in standard of care across the full range of clinically-relevant disease progression milestones experienced by those with DMD has not been fully characterized. In 2017, Ryder et al. published a systematic review examining the epidemiology, burden, and treatment of DMD; however this review focused only on studies published between 2011 and 2015 [6]. Other reviews focused on the prevalence of DMD [13] or the impact of surgery on pulmonary decline [14]. While robust outcomes data are available from large cohort studies including the Cooperative International Neuromuscular Research Group (CINRG) [15], Duchenne Registry [16], and Centers for Disease Control and Prevention’s Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet) [17], a synthesis of data from recent studies is lacking [18]. The objective of this systematic review was to characterize the clinical course of DMD in the era of corticosteroid treatment in North America.
Methods
A comprehensive search of the Medline/Medline In-Process and EMBASE databases was performed (see Additional file 1: Table S1 for search strategy), the design of which was guided by the study-specific PECOS (Population, Exposures, Comparators, Outcomes, Study design) criteria (Table 1). Studies published in English between database inception (1946) and November 2018 that reported estimates of the age at occurrence of key clinical milestones occur among males with DMD were selected. To focus on more generalizable outcomes from a more homogeneous set of patients, the review targeted observational studies from North America (or international studies including North America patients) that aimed to estimate the frequency of key clinical events from large (n > 50) samples of DMD patients treated with corticosteroids. Animal studies, or studies that included patients with other muscular dystrophies, were excluded.
Outcomes of interest that describe the clinical course of DMD included LOA, scoliosis, need for ventilatory support (stratified by any ventilation/type unspecified, non-invasive ventilation [NIV] or invasive ventilation [IV]), pulmonary dysfunction, cardiomyopathy, and mortality. Relevant measures included the mean or median age at the outcome of interest, or the percentage experiencing the outcome over time or at a particular time (t). Scores on assessments of ambulatory, pulmonary, or cardiac function over a minimum of one year of follow-up were also included (Table 1). Two reviewers independently screened abstracts and potentially eligible full-text articles for inclusion, and any discrepancies were resolved through discussion to achieve consensus.
Data were extracted by two researchers; study characteristics extracted included authors, year, study duration, objective(s) and design, sample size, and inclusion and exclusion criteria. Patient characteristics included details of corticosteroid treatment and baseline demographics. Cohorts were classified as ‘corticosteroid-treated’ if all patients were so treated, ‘mixed corticosteroid use’ if the sample represented a mix of corticosteroid-treated and -untreated patients, and ‘likely corticosteroid-treated’ if the study was published after 2005 and did not state the sample was untreated. Available data on use of cardioprotective medications, such as angiotensin-converting enzyme (ACE) inhibitors, were also extracted where available.
For continuous variables, the mean, median, standard deviation (SD), confidence interval (CI), interquartile ranges (IQR), and range was extracted whenever available. For dichotomous and categorical variables, the number of patients and proportion was extracted. For studies reporting on the mean or median age at the outcome, the range of estimates was tabulated. The percentage of the sample who experienced the outcome at time of reporting was also described (where available). Data on the percentage experiencing the outcome at specific time points or over time were described using Kaplan–Meier (KM) curves, as well as presented as point estimates at time t by the original authors. Where available, scores on functional and clinical measures of interest over time were plotted using line graphs.
The strength of the available evidence was assessed using the STrengthening the Reporting of Observational studies in Epidemiology (STROBE) Statement for observational studies and non-randomized clinical trials [19].
Results
The search strategy identified 5,637 potentially-relevant records; four (< 1%) were removed after de-duplication and 5,213 (92.5%) were excluded on abstract review (Fig. 1). Of the remaining 410 records, 381 were excluded on full-text review, leaving 29 eligible studies. Study designs included single-center or multicenter chart reviews and DMD registries (including 6 publications from CINRG and 4 publications from MD STARnet; Table 2). Available details of corticosteroid treatment (including the age at initiation, follow-up protocols, and frequency of reported side effects) are summarized in Additional file 1: Table S2; however, the level of detail provided varied by study, and few studies examined how variability in parameters such as age at corticosteroid initiation impacted the clinical course of DMD. Available details of treatment with cardioprotective medications are summarized in Additional file 1: Table S3. A summary of the quality of included studies in Additional file 1: Table S4.

PRISMA diagram outlining study inclusion and exclusion. PRISMA Preferred Reporting Items for Systematic Reviews and Meta-Analyses, CS corticosteroid, RTC randomized controlled trial
Loss of ambulation
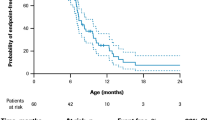
Six studies reported on the mean age at [20,21,22,23,24,25], 10 studies on median age at [26,27,28,29,30,31,32,33,34,35], and 13 studies on the percentage experiencing LOA (Table 2) [20, 22,23,24,25, 28,29,30,31, 33, 34, 36, 37]. Two studies provided subgroup-specific estimates [21, 31]. Among studies of corticosteroid-treated patients, the mean (SD) age at LOA ranged from 9.5 (0.2) years (among 112 patients from MD STARnet) [21] to 12.5 (3.0) years (in 68% of 75 patients from a single-center chart review [22]; Fig. 2a). Estimates were similar from the three studies reporting on mixed corticosteroid use patients; the mean ages at LOA ranged from 9.8 (2.2) years (in 26.6% of 432 Mexican DMD patients) [24] to 10.8 (2.1) years (in 63.2% of 462 patients from MD STARnet) [20]. The earliest mean age at LOA (9.5 years) was observed among patients with ≤ 3 years of corticosteroid treatment, compared with 12.3 years among those with > 3 year corticosteroid use (MD STARnet) [21].

Age at LOA or mortality: a mean/median age at LOA; b LOA over time, c mean/median age at mortality; and d Mortality over time. LOA = loss of ambulation; CS = corticosteroid; LT = long-term; NR = not reported; ST = short term; yrs = years DFZ = deflazacort; NR = not reported; Pred = prednisone; yrs = years; CINRG-DNHS = The Cooperative International Neuromuscular Research Group Duchene Natural History Study; MD STARnet = Muscular Dystrophy Surveillance, Tracking, and Research Network; CM = cardiomyopathy; CPT = cardiopulmonary therapies; Died RF = died from respiratory failure; Died CF = died cardiac failure; Died Oth = died from other causes; IV = invasive ventilation; LVD = left ventricular dysfunction; NIV = non-invasive ventilation; CV = cardiovascular. Notes **Middle value in range of medians. Long follow up = 10–20 years; median follow up = 5.4–7.1 years; short follow up = 1.9–2 years; unknown = not reported
Thirteen estimates from ten studies described median age at LOA (Fig. 2b) [26,27,28,29,30,31,32,33,34,35]. Estimates from 7 studies of corticosteroid-treated samples ranged from 12.0 (11.3–14.0) years (in 63 patients from CINRG) [29] to 16.0 (NR) years (in 765 patients from the Duchenne Registry) [26]. The latter study reported age at LOA by genotype, from 12 years (patients with exon 51 and 53 skip amenable mutations) to 20 years (patients with exon 44 skip amenable mutations). Six studies reported estimates from mixed corticosteroid use samples, and the range was tighter; from 10.0 (range: 4.0–14.0) years (in 67.4% of 85 patients from a single-center chart review) [34] to 12.4 years (in 64.9% of 225 patients from CINRG) [29].
The percentage who experienced LOA increased with time (Fig. 2c) [20, 22,23,24,25, 28,29,30,31, 33, 34, 36, 37], from 12.3% at 10 years (from 223 corticosteroid-treated CINRG patients) [30] to 89.9% at 15 years (from 53 corticosteroid-treated MD STARnet patients) [31]. Estimates from longitudinal studies report that up to 30% of DMD patients lose ambulation by 10 years (CINRG) [28], and 90% by 15 years (MD STARnet) [31]. While these effects were fairly consistent across studies of different sample sizes, mixed corticosteroid use samples tended to have higher rates of LOA at a given age than corticosteroid-treated samples.
Scoliosis
One study reported the mean age at scoliosis [38], 2 studies the median age at scoliosis [31, 35], and 5 studies the percentage with scoliosis by age (Table 2) [22, 30, 31, 35, 37]. How scoliosis was defined varied across studies. In a single-center study of 56 patients, the mean age at spinal surgery was 14.0 years; and 14.5 years in the subset (n = 20) undergoing pulmonary function testing (Fig. 3a) [38]. The median (range) age at scoliosis surgery among a mixed corticosteroid use sample from MD STARnet was 14.6 (10.2–20.2) years (with surgery observed in 52.4% of n = 208) [35]. In the remaining study of 274 corticosteroid-treated patients (also from MD STARnet), the median (range) age (by spinal curvature > 30° or surgery) was 14.2 (12.5–15.6) years among the 107 patients with scoliosis [31]. The percentage with scoliosis increased with increasing age (Fig. 3b) [22, 30, 31, 35, 37]. Results from a longitudinal study from MD STARnet suggest that up to 59% of patients with DMD will have scoliosis by 15 years of age, and up to 72% by 20 years of age [31].

Occurrence of other key clinical milestones: a Mean/median age at scoliosis; b Percentage with scoliosis over time; c Mean/median age at respiratory support; d Percentage on respiratory support over time; e Mean/median age at cardiomyopathy; f Percentage with cardiomyopathy over time. 6MWD = 6 min walk distance; PEF = peak expiratory flow; FVC = forced vital capacity; SF = shortening fraction; LVED = left ventricular end-diastolic dimension; EF = ejection fraction. Notes: **Scoliosis includes both severe scoliosis and spinal surgery
Pulmonary function and need for ventilatory support
Four studies reported the mean or median age at ventilation [33, 35, 39, 40], 3 studies reported the percentage needing ventilation by age [30, 33, 35], 4 studies reported the age at transitioning to key pulmonary functional milestones [30, 41,42,43], and 2 studies reported pulmonary function over time (Table 2) [42, 43].
In terms of age at need for ventilation, one multicenter chart review of 324 mixed corticosteroid-treated DMD patients reported a median age at ‘any ventilation’ of 15 years (Fig. 3c) [33]. Three studies reported the age at NIV to range from a median (IQR) age of 18.0 (9.4–26.8) years (in 47.6% of 208 mixed corticosteroid-treated patients on nasal NIV from MD STARnet) [35], to a mean of 22.3 (4.7) years (in 39.3% of 275 likely-corticosteroid-treated patients receiving continuous NIV in a single-center chart review) [39]. Two studies reported age at IV; a single-center chart review reporting a mean (SD) age of 18.6 (2.3) years (in 9.1% of 275 likely-corticosteroid-treated patients with continuous tracheostomy mechanical ventilation) [39], and an MD STARnet study reporting a median (IQR) age of 19.1 (13.4–27.0) years (in 21.2% in 208 mixed-corticosteroid-treated patients with tracheostomy) [35].
The percentage of patients requiring ventilation tended to increase over time, with variability in estimates observed due to type of ventilation (Fig. 3d) [30, 33, 35]. By 20 years of age, 27.2% (n = 88) of mixed corticosteroid use patients in a multicenter chart review required ‘any ventilation’ [33]. Two studies describing NIV reported estimates of 21.2% (among 44 corticosteroid-treated patients from MD STARnet) [35], and 39.6% (among 21 mixed corticosteroid-treated patients from CINRG) [30] by 20 years. The MD STARnet study also reported that 47.6% of patients with mixed corticosteroid use were on IV by 20 years [35].
Absolute measures of pulmonary function generally show relatively preserved function until adolescence, which declines with increasing age (Fig. 4a, c). Two studies reported absolute and percent predicted peak expiratory flow (PEF). A substantial decline in PEF was observed among 330 corticosteroid-treated CINRG patients, from 243.7 L/min (age = 17 years) to 76.1 L/min (age = 29 years). Trends were similar among 60 mixed corticosteroid-treated patients from a single-center chart review (from 269.4 L/min [age = 18 years] to 67.9 L/min [age = 24 years]) [42]. Estimates of percent predicted PEF show loss of function relative to age-matched healthy controls; the magnitude increases with age (Fig. 4a), reaching a low of 11.8% by age 29 years in the CINRG study. Those same two studies also reported FVC (L) over time (Fig. 4d) which demonstrated an initial increase in function followed by progressive decline after approximately 15 years; the percent predicted FVC showed loss of function relative to age-matched controls with increasing age, to 10.4% at 29 years of age (Fig. 4c) [42, 43].

Measures of functional status over time: a–d pulmonary function measures; e–g cardiac function measures. PEF = peak expiratory flow; FVC = forced vital capacity; SF = shortening fraction; LVED = left ventricular end-diastolic dimension; EF = ejection fraction. Notes a = HR in the upper quartile (> 96 BPM), b = HR in the lower quartile (≤ 96 BPM), c = Left ventricular dysfunction, d = No Left ventricular dysfunction
Four studies reported the age at transitioning to key pulmonary milestones; specifically, reaching FVC < 1L, FVC < 30% or PEF < 30% [30, 41,42,43]. FVC < 1L was first reported at 20 years of age in the 60 mixed corticosteroid-treated patients from a single-center chart review [42] and 23 years of age in a CINRG study of 330 corticosteroid-treated patients [30]. Mean (SD) ages at FVC < 30% and PEF < 30% were similar from a CINRG study of 223 mixed corticosteroid-treated patients (FVC < 30%: 24.0 (1.5) years, and PEF < 30%: 24.9 (0.8) years); the same CINRG study also reported that 50% progressed to FVC < 30% or PEF < 30% by 25 years of age [41]. Estimates of the percentage with severe pulmonary dysfunction (FVC < 50%) at 20 years of age ranged from 13.6% [43] to 29.7% [30]. Finally, among 330 corticosteroid-treated patients from CINRG, among those with LOA at < 10 years, the median age at FVC < 1L was 18.1 years, vs 20.1 years among those with LOA between 10–13 years of age, and 24.4 years among patients with LOA at ≥ 13 years [30].
Cardiac function and cardiomyopathy
Seven studies reported the mean or median age at diagnosis [20, 25, 26, 31, 34, 35, 44] and 9 studies reported the percentage of patients with cardiomyopathy [20, 25, 31, 34, 35, 37, 44,45,46]; 3 studies reported changes in cardiac function over time (Table 2) [45,46,47].
Of the 7 studies reporting the age at cardiomyopathy, 5 described samples not selected using cardiovascular-risk-related criteria (Fig. 3e) [20, 31, 34, 35, 44]. The mean (SD) age at cardiomyopathy ranged from 12.7 (3.0) years (in 37.0% of 67 corticosteroid-treated patients from a multicenter chart review) [44] to 15.8 (range: 9–29) years (in 48.2% of 85 patients of mixed corticosteroid-treatment status from a single-center chart review) [34]. Estimates of median (IQR) age at cardiomyopathy ranged from 14.9 (4.9) years (in 69.7% of 208 mixed corticosteroid-treated patients from MD STARnet) [35] to 18.0 (CI: 6.9–18.5) years (in 39.0% of 218 corticosteroid-treated patients from MD STARnet) [31]. The reported age at cardiomyopathy was lower among two studies reporting on mixed corticosteroid-treated samples either treated with cardiopulmonary therapy (median 18.0 (7.0–27.3) years, in 70.2% of 57 patients) [46], or with LV dysfunction (mean, 15.4 (8–27) years, in 32.5% of 77 patients) [25].
The percentage with cardiomyopathy was higher with increasing age (Fig. 3f) [20, 25, 31, 34, 35, 37, 44,45,46]; this effect was consistent across studies of different sample sizes. At 15 years of age, the percentage with cardiomyopathy ranged from 23.3% (among 218 patients who initiated corticosteroids after 5 years of age) [31] to 69.7% (among 208 mixed corticosteroid-treated patients) [35]; both estimates were from MD STARnet. By 20 years of age, the percentage with cardiomyopathy ranged from 68.2% (of 85 mixed corticosteroid-treated patients from a single-center chart review) [34] to 92.8% (of 47 patients who initiated corticosteroids before 5 years of age from MD STARnet) [31]. By age 25, the percentage with cardiomyopathy ranged from 87.6% (of 85 mixed corticosteroid-treated patients from a single-center chart review) [34] to 100% (291 corticosteroid-treated patients from MD STARnet) [20].
Measures of cardiac function show preserved function until adolescence and then decline with age (Fig. 4e–g) [45,46,47]. In a long-term observational study of 63 DMD patients treated with cardiopulmonary therapies and corticosteroids, the ejection fraction decreased to 53% by 20 years of age [45]. That study and two other single-center studies also reported worsening of cardiac function by left ventricular end diastolic diameter (LVED) and shortening fraction (SF) among corticosteroid-treated patients with DMD [45,46,47].
Mortality
Eight studies reported the mean age [24, 32, 35, 39, 40, 44,45,46] and 1 study the median age at mortality [35]; 9 studies reported case fatality by age (Table 2) [24, 30, 32, 34, 35, 40, 44, 46, 48].
Of the 8 studies reporting mean (SD) age at mortality [24, 32, 35, 39, 40, 44,45,46], 4 were reflective of the overall DMD population [24, 32, 44, 45] one was from a sample of DMD patients with cardiomyopathy [46], and 3 were from samples of patients who were non-ambulatory or on ventilation [35, 39, 40]. From studies of the overall population, the mean (SD) age at mortality ranged from 18.1 (3.8) years (in 11% of 101 mixed corticosteroid-treated patients from a multicenter chart review) [44] to 20.0 (15–31) years (in 13% of 437 mixed corticosteroid-treated patients from an administrative database study; Fig. 2d) [32]. In the single study that described outcomes among DMD patients with cardiomyopathy, the mean (SD) age at mortality was 26.0 (6.8) years (in 47.4% of 57 mixed corticosteroid-treated patients from a single-center chart review; Fig. 2d). The mean (SD) age at mortality among DMD patients who were non-ambulatory or on ventilation ranged from 25.8 (7.8) years (in 17 likely-corticosteroid-treated patients from a single-center chart review, who died from causes other than respiratory or cardiac dysfunction) [40] to 31.4 (5.7) years (in 14 likely-corticosteroid-treated patients from that single-center chart review, with death due to respiratory complications; Fig. 2d) [40]. The median (IQR) age at mortality among DMD patients who were non-ambulatory or on ventilation was 21.5 (3.8) years (in 28.3% of 208 mixed corticosteroid-treated patients from MD STARnet; Fig. 2d) [35].
In terms of the proportion surviving over time, up to 16.2% mortality was reported by age 20 years (Fig. 2e) [24]. Estimates of survival after 20 years are available only from studies enrolling adult patients with DMD; and these reported rates of 44.2% to 56.8% mortality by age 30 years (Fig. 2e) [40].
Discussion
A comprehensive systematic review was conducted to identify estimates of the age at key clinical milestones, and trajectories on relevant functional measures over time, among studies including North American patients with DMD. Age at LOA was the most widely reported with estimates available from many large studies; these tended to range from 10 to 14 years of age [27, 34]. However, robust data on the timing of the onset of scoliosis-, cardiac-, pulmonary- and ventilation-related outcomes were less frequently presented, particularly from large longitudinal studies. While reported estimates of the mean age at diagnosis of scoliosis were fairly consistent across studies (at 14–15 years of age), how scoliosis was classified differed widely [31, 35, 38]. Pulmonary function in DMD patients declines with age from the mid-teens [30, 41], and while most have severe pulmonary dysfunction by 25 years [30], the mean age at initiation of ventilatory support ranged from 15 to 22 years depending on the type of ventilation considered and treatment center [33, 39]. Data on age at mortality in DMD were also variable, and estimates were impacted by the inclusion criteria of the individual studies; for example estimates of mortality among those with cardiomyopathy or on ventilation were drawn from populations surviving to adulthood [40, 46]. In addition to selection criteria, factors impacting the timing of key clinical milestones include corticosteroid regimen [31] and disease genotype [26]. The findings of this review help summarize the likely timing of disease progression milestones for North American patients with DMD, and also highlight potential heterogeneity in timing observed both within and across study populations.
Estimates of time to key clinical milestones in this review included data from studies from the large North American registries (e.g. CINRG and MD STARnet), and findings are consistent with those from large observational studies and registries from outside of North America. The Translational Research in Europe—Assessment and Treatment of Neuromuscular Diseases (TREAT-NMD) network of DMD registries have published studies documenting the clinical course of patients with DMD [49,50,51,52]. In a large survey of over 1500 DMD patients that characterized the impact of corticosteroid use, mean estimates of age at LOA ranged from 10.1 (non-corticosteroid-treated patients) to 11.4 (corticosteroid-treated) years [49]. An analysis of over 5000 patients also from TREAT-NMD reported age at LOA of 13 years among corticosteroid-treated patients, and that up to 50% of patients required ventilation by 20 years of age [50]
That longitudinal data describing survival specifically among North American DMD patients are few, was one of the major gaps identified in this review. However, mortality rates from included studies were consistent with findings of two important studies on mortality in dystrophin gene-related muscular dystrophy, which did not meet the inclusion criteria for the current review as they also included patients with Becker muscular dystrophy (BMD). The first study, which was based on vital statistics, estimated that 71% of mortality among those with BMD/DMD occurred between the ages of 15 and 29 years; the authors assumed it was most likely related to DMD [53]. The second study, from MD STARnet, estimated mortality in almost 60% of that cohort by age 25 years, with most deaths occurring among those aged 20 to 25 years [54]. Further follow-up from existing large DMD cohorts will help improve contemporary estimates of the timing of key clinical milestones.
Accurately estimating the time of onset of gradually progressive manifestations of DMD can be difficult, and this along with changes in practice patterns and symptom detection, contribute to observed variability in estimates. For example, many studies reporting on scoliosis classify outcomes based on surgery, however with changing treatment patterns [55] the utility of surgery as a proxy for clinically-significant scoliosis will decrease. Similarly, recommended strategies for ventilation vary among clinical centers [39, 56, 57], and practice is changing (in particular for how IV is used) [58], which will impact the comparability of estimates of the timing of respiratory decline across studies from different periods. Finally for cardiomyopathy, with advancements in screening tools [59, 60] as well as evidence of benefits to early treatment [61], it is likely that initial signs will now be detected earlier, which would result in an apparent decrease in the mean age at cardiomyopathy over the coming years.
There are several additional factors impacting the timing of key clinical milestones that require consideration. To capture the impact of corticosteroids in the management of DMD, only studies including patients from the corticosteroid treatment era were included. While details of corticosteroid treatment regimens were extracted and reviewed, there were important limitations that precluded analyzing outcomes according to regimen. First, details on the timing of initiation, duration, type, and dose varied within and between studies. Only a small number of studies reporting on LOA presented results according to agent; but the remainder of the studies for that outcome, and all of the studies for other outcomes of interest, did not stratify by corticosteroid regimen. However, variations in corticosteroid treatment patterns (in terms of duration and dosing) may have affected the timing when patients reached LOA [28, 31, 62, 63], and other important clinical milestones [20, 31, 46, 62,63,64,65]. Evidence on the impact of early initiation of corticosteroids (e.g. before age 6 years) remains mixed [31, 66]; more work is needed to disentangle the potential confounding effect of disease severity and the potential risk for adverse effects of corticosteroid treatment on outcomes in real-world studies. Treatment with ACE inhibitors has also been shown to impact the clinical course of DMD by delaying the onset of cardiomyopathy; however, the use of ACE inhibitors remains variable [20, 67]. While it might be anticipated that studies describing later cohorts would show delayed onset of milestones that define the clinical course of DMD, the interplay between treatment advances and the impact of earlier diagnostics would make these relationships less apparent. Finally, other genetic modifiers may also play a role in the timing of DMD progression [26, 29, 50]; however, outcomes according to genotype are infrequently reported outside of treatment trials [68, 69]. The move from biomarkers to precise genetic diagnosis may also impact the apparent clinical course [70].
Variability in methodology and data sources may also have affected estimates. Data from the CINRG and MD STARnet registries, both large well-documented US cohorts that comprehensively collect longitudinal data on the clinical course of DMD, were used in ten studies within this review. Outside of those, most observational studies and treatment trials do not follow patients for a sufficient time to describe changes across the range of key clinical milestones [21, 30]. Other challenges for studying disease progression in rare diseases include small sample sizes which can amplify the impact of heterogeneity in diseases with varied clinical courses; data presented from convenience samples and case series may not be generalizable, and the impact of selection biases on outcomes (particularly for diseases with high early fatality among more severe cases) can be substantial [71, 72]. The numerous outcome measures used to assess progression in DMD also make comparisons difficult, a limitation recently acknowledged in a workshop held by the DMD research community [73]. Finally, there are useful measures for characterizing DMD progression that were infrequently reported in the studies of this review, such as the North Star Ambulatory Assessment or upper arm function, which are important in understanding patient functional status and ability to participate in activities of daily living.
Some limitations to the published data warrant mention. First, while time to event data using KM curves were presented in some studies, many reported the mean age at an occurrence where the entire sample had not experienced the event at the time of study reporting. As such, these values can be interpreted as the lower limit for when key clinical milestones will occur in DMD. Second, some measures may only be administered to individuals who still have some functional capacity (e.g. tests of ambulation), and patients unable to complete the test would have been excluded. This type of survival bias would result in an inflation of apparent functional status for cohorts as a whole. Third, mean scores on functional tests may reflect the inclusion criteria of each study, rather than the underlying distribution of scores on that functional test among the DMD population. Fourth, because of heterogeneity in designs employed, measures selected, and populations included across studies, meta-analysis was judged to be infeasible [74, 75]; as a result, overall summary estimates of the time to key clinical milestones were not calculable.
Conclusions
This is the first systematic review of published estimates of the frequency and timing of important milestones that characterize the clinical course of DMD in the corticosteroid era. This review has also leant insight into a number of challenges in the interpretation and comparison of estimates of outcomes to characterize the clinical course of DMD. Additional studies on the ages at occurrence of other important DMD clinical milestones, and the relationships between short-term and long-term outcomes, will be valuable in the continuation of knowledge regarding disease progression in DMD.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ACE:
-
Angiotensin-converting enzyme
- BMD:
-
Becker muscular dystrophy
- CI:
-
Confidence interval
- CINRG:
-
Cooperative International Neuromuscular Research Group
- DMD:
-
Duchenne muscular dystrophy
- FVC:
-
Forced vital capacity
- IQR:
-
Interquartile ranges
- IV:
-
Invasive ventilation
- KM:
-
Kaplan–Meier
- LOA:
-
Loss of ambulation
- LV:
-
Left ventricle
- LVED:
-
Left ventricular end diastolic diameter
- MD STARNet:
-
Muscular Dystrophy Surveillance, Tracking, and Research Network
- NIV:
-
Non-invasive ventilation
- PEF:
-
Peak expiratory flow
- SD:
-
Standard deviation
- SF:
-
Shortening fraction
- STROBE:
-
STrengthening the Reporting of Observational studies in Epidemiology
- TREAT-NMD:
-
Translational Research in Europe—Assessment and Treatment of Neuromuscular Diseases
References
Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17(3):251–67.
Wein N, Alfano L, Flanigan KM. Genetics and emerging treatments for Duchenne and Becker muscular dystrophy. Pediatr Clin North Am. 2015;62(3):723–42.
Mendell JR, Shilling C, Leslie ND, Flanigan KM, al-Dahhak R, Gastier-Foster J, et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. 2012;71(3):304–13.
Moat SJ, Bradley DM, Salmon R, Clarke A, Hartley L. Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK). Eur J Hum Genet. 2013;21(10):1049.
Yiu EM, Kornberg AJ. Duchenne muscular dystrophy. J Paediatr Child Health. 2015;51(8):759–64.
Ryder S, Leadley RM, Armstrong N, Westwood M, De Kock S, Butt T, et al. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J Rare Dis. 2017;12(1):79.
Medicine; UNLo. Genetics home reference: DMD gene 2017. https://ghr.nlm.nih.gov/gene/DMD.
Ryder S, Leadley R, Armstrong N, Westwood M, de Kock S, Butt T, et al. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J Rare Dis. 2017;12(1):79.
Mirski KT, Crawford TO. Motor and cognitive delay in Duchenne muscular dystrophy: implication for early diagnosis. J Pediatr. 2014;165(5):1008–10.
Birnkrant J, Bennett D, Noritz G, Harrington M, Birnkrant D. Prolonged survival and end-of-life care among end-stage patients with Duchenne muscular dystrophy (DMD). Chest. 2011;140(4):1054A.
Birnkrant DJ, Ararat E, Mhanna MJ. Cardiac phenotype determines survival in Duchenne muscular dystrophy. Pediatr Pulmonol. 2016;51(1):70–6.
Andrews JG, Wahl RA. Duchenne and Becker muscular dystrophy in adolescents: current perspectives. Adolesc Health Med Ther. 2018;9:53–63.
Mah JK, Korngut L, Dykeman J, Day L, Pringsheim T, Jette N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord. 2014;24(6):482–91.
Roberto R, Fritz A, Hagar Y, Boice B, Skalsky A, Hwang H, et al. The natural history of cardiac and pulmonary function decline in patients with Duchenne muscular dystrophy. Spine. 2011;36(15):E1009–17.
McDonald CM, Henricson EK, Abresch RT, Han JJ, Escolar DM, Florence JM, et al. The cooperative international neuromuscular research group Duchenne natural history study—a longitudinal investigation in the era of glucocorticoid therapy: design of protocol and the methods used. Muscle Nerve. 2013;48(1):32–54.
Parent Project Muscular Dystrophy. The Duchenne registry. 2019. https://www.duchenneregistry.org/.
Centers for Disease Control and Prevention. Muscular Dystrophy Research and Tracking. 2019. https://www.cdc.gov/ncbddd/musculardystrophy/research.html.
Jewell NP. Natural history of diseases: statistical designs and issues. Clin Pharmacol Ther. 2016;100(4):353–61.
von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP, et al. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet. 2007;370(9596):1453–7.
Barber BJ, Andrews JG, Lu Z, West NA, Meaney FJ, Price ET, et al. Oral corticosteroids and onset of cardiomyopathy in Duchenne muscular dystrophy. J Pediatr. 2013;163(4):1080-4.e1.
Kim S, Campbell KA, Fox DJ, Matthews DJ, Valdez R. STARnet MD. Corticosteroid treatments in males with Duchenne muscular dystrophy: treatment duration and time to loss of ambulation. J Child Neurol. 2015;30(10):1275–80.
King WM, Ruttencutter R, Nagaraja HN, Matkovic V, Landoll J, Hoyle C, et al. Orthopedic outcomes of long-term daily corticosteroid treatment in Duchenne muscular dystrophy. Neurology. 2007;68(19):1607–13.
Labove L, Jaworski M, Nguyen CTE. Duchenne muscular dystrophy: do boys with a shorter stature maintain ambulation longer? J Neuromuscul Dis. 2018;5(Supplement 1):S142–3.
Lopez-Hernandez LB, Gomez-Diaz B, Escobar-Cedillo RE, Gama-Moreno O, Camacho-Molina A, Soto-Valdes DM, et al. Duchenne muscular dystrophy in a developing country: challenges in management and genetic counseling. Genet Couns. 2014;25(2):129–41.
Posner AD, Soslow JH, Burnette WB, Bian A, Shintani A, Sawyer DB, et al. The correlation of skeletal and cardiac muscle dysfunction in Duchenne muscular dystrophy. J Neuromuscul Dis. 2016;3(1):91–9.
Wang RT, Barthelemy F, Martin AS, Douine ED, Eskin A, Lucas A, et al. DMD genotype correlations from the Duchenne Registry: endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype. Hum Mutat. 2018;39(9):1193–202.
Bello L, Morgenroth LP, Gordish-Dressman H, Hoffman EP, McDonald CM, Cirak S. DMD genotypes and loss of ambulation in the CINRG Duchenne Natural History Study. Neurology. 2016;87(4):401–9.
Bello L, Gordish-Dressman H, Morgenroth LP, Henricson EK, Duong T, Hoffman EP, et al. Prednisone/prednisolone and deflazacort regimens in the CINRG Duchenne Natural History Study. Neurology. 2015;85(12):1048–55.
Bello L, Kesari A, Gordish-Dressman H, Cnaan A, Morgenroth LP, Punetha J, et al. Genetic modifiers of ambulation in the cooperative international Neuromuscular research group Duchenne natural history study. Ann Neurol. 2015;77(4):684–96.
McDonald CM, Henricson EK, Abresch RT, Duong T, Joyce NC, Hu F, et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. The Lancet. 2018;391(10119):451–61.
Kim S, Zhu Y, Romitti PA, Fox DJ, Sheehan DW, Valdez R, et al. Associations between timing of corticosteroid treatment initiation and clinical outcomes in Duchenne muscular dystrophy. Neuromuscul Disord. 2017;27(8):730–7.
Deshpande SR, Wittlieb-Weber CA, Gambetta KE, Bock MJ, Lal AK, Conway JL, et al. Duchenne muscular dystrophy-related acute heart failure: multicenter study of hospitalizations and outcomes. J Heart Lung Transpl. 2018;37(4 Supplement 1):S388–9.
Gambetta K, Wittlieb-Weber C, Bock M, Villa C, Johnson J, Lal A, et al. Impact of genotype on boys with duchenne muscular dystrophy. Journal of Heart and Lung Transplantation. 2018;37 (4 Supplement 1):S122.
McKane M, Soslow JH, Xu M, Saville BR, Slaughter JC, Burnette WB, et al. Does body mass index predict premature cardiomyopathy onset for Duchenne muscular dystrophy? J Child Neurol. 2017;32(5):499–504.
Pandya S, James KA, Westfield C, Thomas S, Fox DJ, Ciafaloni E, et al. Health profile of a cohort of adults with Duchenne muscular dystrophy. Muscle Nerve. 2018;58(2):219–23.
Barnard AM, Willcocks RJ, Finanger EL, Daniels MJ, Triplett WT, Rooney WD, et al. Skeletal muscle magnetic resonance biomarkers correlate with function and sentinel events in Duchenne muscular dystrophy. PLoS ONE. 2018;13(3):e0194283.
Wong BL, Rybalsky I, Shellenbarger KC, Tian C, McMahon MA, Rutter MM, et al. Long-term outcome of interdisciplinary management of patients with Duchenne muscular dystrophy receiving daily glucocorticoid treatment. J Pediatr. 2017;182:296-303.e1.
Velasco MV, Colin AA, Zurakowski D, Darras BT, Shapiro F. Posterior spinal fusion for scoliosis in Duchenne muscular dystrophy diminishes the rate of respiratory decline. Spine. 2007;32(4):459–65.
Bach JR, Tran J, Durante S. Cost and physician effort analysis of invasive vs. noninvasive respiratory management of Duchenne muscular dystrophy. Am J Phys Med Rehabil. 2015;94(6):474–82.
Bach JR, Martinez D. Duchenne muscular dystrophy: continuous noninvasive ventilatory support prolongs survival. Respir Care. 2011;56(6):744–50.
Henricson E, McDonald C, Gordish-Dressman H, Abresch T, Cnaan A. Steroid use delays but does not prevent loss of pulmonary function in patients with Duchene muscular dystrophy (DMD). Dev Med Child Neurol. 2017;59(Supplement 4):30.
Mayer OH, Finkel RS, Rummey C, Benton MJ, Glanzman AM, Flickinger J, et al. Characterization of pulmonary function in Duchenne muscular dystrophy. Pediatr Pulmonol. 2015;50(5):487–94.
McDonald CM, Gordish-Dressman H, Henricson EK, Duong T, Joyce NC, Jhawar S, et al. Longitudinal pulmonary function testing outcome measures in Duchenne muscular dystrophy: long-term natural history with and without glucocorticoids. Neuromuscul Disord. 2018;27:S115–6.
Van Dorn CS, Puchalski MD, Weng HY, Bleyl SB, Butterfield RJ, Williams RV. DMD mutation and LTBP4 haplotype do not predict onset of left ventricular dysfunction in Duchenne muscular dystrophy. Cardiol Young. 2018;28(7):910–5.
Schram G, Fournier A, Leduc H, Dahdah N, Therien J, Vanasse M, et al. All-cause mortality and cardiovascular outcomes with prophylactic steroid therapy in Duchenne muscular dystrophy. J Am Coll Cardiol. 2013;61(9):948–54.
Wang M, Birnkrant DJ, Super DM, Jacobs IB, Bahler RC. Progressive left ventricular dysfunction and long-term outcomes in patients with Duchenne muscular dystrophy receiving cardiopulmonary therapies. Open Heart. 2018;5(1):e000783.
Thomas TO, Morgan TM, Burnette WB, Markham LW. Correlation of heart rate and cardiac dysfunction in Duchenne muscular dystrophy. Pediatr Cardiol. 2012;33(7):1175–9.
Connolly AM, Florence JM, Zaidman CM, Golumbek PT, Mendell JR, Flanigan KM, et al. Clinical trial readiness in non-ambulatory boys and men with Duchenne muscular dystrophy: MDA-DMD network follow-up. Muscle Nerve. 2016;54(4):681–9.
Vry J, Gramsch K, Rodger S, Thompson R, Steffensen BF, Rahbek J, et al. European cross-sectional survey of current care practices for Duchenne muscular dystrophy reveals regional and age-dependent differences. J Neuromuscul Dis. 2016;3(4):517–27.
Koeks Z, Bladen CL, Salgado D, Van Zwet E, Pogoryelova O, McMacken G, et al. Clinical outcomes in Duchenne muscular dystrophy: a study of 5345 patients from the TREAT-NMD DMD global database. J Neuromuscul Dis. 2017;4(4):293–306.
Bladen CL, Rafferty K, Straub V, Monges S, Moresco A, Dawkins H, et al. The TREAT-NMD Duchenne muscular dystrophy registries: conception, design, and utilization by industry and academia. Hum Mutat. 2013;34(11):1449–57.
Thangarajh M, Hendriksen J, McDermott MP, Martens W, Hart KA, Griggs RC, et al. Relationships between DMD mutations and neurodevelopment in dystrophinopathy. Neurology. 2019;93(17):e1597–604.
Salzberg DC, Mann JR, McDermott S. Differences in race and ethnicity in muscular dystrophy mortality rates for males under 40 years of age, 2006–2015. Neuroepidemiology. 2018;50(3–4):201–6.
Anonymous. Prevalence of Duchenne/Becker muscular dystrophy among males aged 5–24 years—four states, 2007. Morb Mortal Wkly Rep. 2009; 58(40):1119–22.
Raudenbush BL, Thirukumaran CP, Li Y, Sanders JO, Rubery PT, Mesfin A. Impact of a Comparative Study on the Management of Scoliosis in Duchenne Muscular Dystrophy: Are Corticosteroids Decreasing the Rate of Scoliosis Surgery in the United States? Spine. 2016;41(17):E1030–8.
Jabaley CS, Groff RF, Sharifpour M, Raikhelkar JK, Blum JM. Modes of mechanical ventilation vary between hospitals and intensive care units within a university healthcare system: a retrospective observational study. BMC Res Notes. 2018;11(1):425.
Dellaca RL, Veneroni C, Farre R. Trends in mechanical ventilation: are we ventilating our patients in the best possible way? Breathe (Sheff). 2017;13(2):84–98.
Sheehan DW, Birnkrant DJ, Benditt JO, Eagle M, Finder JD, Kissel J, et al. Respiratory management of the patient with Duchenne muscular dystrophy. Pediatrics. 2018;142(Supplement 2):S62–71.
Brunklaus A, Parish E, Muntoni F, Scuplak S, Tucker SK, Fenton M, et al. The value of cardiac MRI versus echocardiography in the pre-operative assessment of patients with Duchenne muscular dystrophy. Eur J Paediatr Neurol. 2015;19(4):395–401.
Buddhe S, Lewin M, Olson A, Ferguson M, Soriano BD. Comparison of left ventricular function assessment between echocardiography and MRI in Duchenne muscular dystrophy. Pediatr Radiol. 2016;46(10):1399–408.
McNally EM, Kaltman JR, Benson DW, Canter CE, Cripe LH, Duan D, et al. Contemporary cardiac issues in Duchenne muscular dystrophy. Working Group of the National Heart, Lung, and Blood Institute in collaboration with Parent Project Muscular Dystrophy. [Erratum appears in Circulation. 2015 Jun 23;131(25):e539 Note: Groh, William J [added]; PMID: 26099961]. Circulation. 2015;131(18):1590–8.
Moxley RT III, Pandya S, Ciafaloni E, Fox DJ, Campbell K. Change in natural history of Duchenne muscular dystrophy with long-term corticosteroid treatment: implications for management. J Child Neurol. 2010;25(9):1116–29.
Griggs RC, Herr BE, Reha A, Elfring G, Atkinson L, Cwik V, et al. Corticosteroids in Duchenne muscular dystrophy: major variations in practice. Muscle Nerve. 2013;48(1):27–31.
Kim S, Campbell KA, Fox DJ, Matthews DJ, Valdez R. STARnet, MD. Corticosteroid treatments in males with Duchenne muscular dystrophy: treatment duration and time to loss of ambulation. J Child Neurol. 2015;30(10):1275–80.
McDonald CM, Henricson EK, Abresch RT, Duong T, Joyce NC, Hu F, et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet. 2018;391(10119):451–61.
Merlini L, Gennari M, Malaspina E, Cecconi I, Armaroli A, Gnudi S, et al. Early corticosteroid treatment in 4 Duchenne muscular dystrophy patients: 14-year follow-up. Muscle Nerve. 2012;45(6):796–802.
Duboc D, Meune C, Lerebours G, Devaux J-Y, Vaksmann G, Bécane H-M. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol. 2005;45(6):855–7.
Mendell JR, Goemans N, Lowes LP, Alfano LN, Berry K, Shao J, et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol. 2016;79(2):257–71.
McDonald CM, Campbell C, Torricelli RE, Finkel RS, Flanigan KM, Goemans N, et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet. 2017;390(10101):1489–98.
Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet. 2016;53(3):145–51.
Kempf L, Goldsmith JC, Temple R. Challenges of developing and conducting clinical trials in rare disorders. Am J Med Genet A. 2018;176(4):773–83.
Administration FaD. Rare diseases: natural history studies for drug development. Guidance for industry. 2019.
Straub V, Mercuri E. Report on the workshop: meaningful outcome measures for Duchenne muscular dystrophy, London, UK, 30–31 January 2017. Neuromuscul Disord. 2018;28(8):690–701.
Egger M, Smith G, Schneider M. Systematic reviews of observational studies. BMJ Publishing Group; 2010.
Colditz GA, Burdick E, Mosteller F. Heterogeneity in meta-analysis of data from epidemiologic studies: a commentary. Am J Epidemiol. 1995;142(4):371–82.
Funding
Sarepta Therapeutics, Inc.
Author information
Authors and Affiliations
Contributions
All authors contributed to study design, interpretation, and analysis. SMS and AD were responsible for data identification, extraction, and synthesis. SMS was responsible for creating the first draft and all authors read, revised, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not available.
Consent for publication
Not available.
Competing interests
SMS and AMD are employees of Broadstreet HEOR, and MH was at the time of this project; which received funds from Sarepta for this work. RMS and KLG are employees of Sarepta. JM acted as a consultant to Broadstreet HEOR.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1
. Search strategy. Supplementary Table 2. Details of corticosteroid treatment, by study. Supplementary Table 3. Details of ACE inhibitor treatment, by study. Supplementary Table 4. STROBE assessments of included studies.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Szabo, S.M., Salhany, R.M., Deighton, A. et al. The clinical course of Duchenne muscular dystrophy in the corticosteroid treatment era: a systematic literature review. Orphanet J Rare Dis 16, 237 (2021). https://doi.org/10.1186/s13023-021-01862-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-021-01862-w