
Abstract
Background
Defects in the glycosylphosphatidylinositol (GPI) biosynthesis pathway can result in a group of congenital disorders of glycosylation known as the inherited GPI deficiencies (IGDs). To date, defects in 22 of the 29 genes in the GPI biosynthesis pathway have been identified in IGDs. The early phase of the biosynthetic pathway assembles the GPI anchor (Synthesis stage) and the late phase transfers the GPI anchor to a nascent peptide in the endoplasmic reticulum (ER) (Transamidase stage), stabilizes the anchor in the ER membrane using fatty acid remodeling and then traffics the GPI-anchored protein to the cell surface (Remodeling stage).
Results
We addressed the hypothesis that disease-associated variants in either the Synthesis stage or Transamidase+Remodeling-stage GPI pathway genes have distinct phenotypic spectra. We reviewed clinical data from 58 publications describing 152 individual patients and encoded the phenotypic information using the Human Phenotype Ontology (HPO). We showed statistically significant differences between the Synthesis and Transamidase+Remodeling Groups in the frequencies of phenotypes in the musculoskeletal system, cleft palate, nose phenotypes, and cognitive disability. Finally, we hypothesized that phenotypic defects in the IGDs are likely to be at least partially related to defective GPI anchoring of their target proteins. Twenty-two of one hundred forty-two proteins that receive a GPI anchor are associated with one or more Mendelian diseases and 12 show some phenotypic overlap with the IGDs, represented by 34 HPO terms. Interestingly, GPC3 and GPC6, members of the glypican family of heparan sulfate proteoglycans bound to the plasma membrane through a covalent GPI linkage, are associated with 25 of these phenotypic abnormalities.
Conclusions
IGDs associated with Synthesis and Transamidase+Remodeling stages of the GPI biosynthesis pathway have significantly different phenotypic spectra. GPC2 and GPC6 genes may represent a GPI target of general disruption to the GPI biosynthesis pathway that contributes to the phenotypes of some IGDs.
Similar content being viewed by others


Introduction
Glycosylphosphatidylinositols (GPIs) are glycolipids that act as membrane anchors of many cell surface proteins. The GPI-anchor biosynthesis pathway covalently attaches the glycolipid to the C-termini of nascent proteins as a post-translational modification [1]. Defects in this pathway represent a relatively new subclass of congenital disorders of glycosylation (CDG) termed inherited GPI deficiencies (IGDs), which are the result of mutations in one of nearly 30 genes that encode portions of the GPI biosynthetic pathway [2].
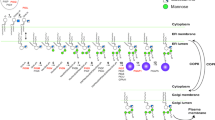
The GPI-anchor biosynthesis can be broken down into a Synthesis and a Transamidase+Remodeling stage (Fig. 1). The first or Synthesis stage results in the stepwise construction of the GPI anchor. The second or Transamidase+Remodeling-stage involves the transfer of a nascent peptide to the GPI anchor by the transamidase complex and results in fatty acid remodeling necessary to stabilize the anchor in the membrane. This happens possibly in conjunction with lipid raft formation before it is trafficked by secretory vesicles to the cell surface [7, 8]. Approximately 150 proteins are GPI anchored, including enzymes, structural molecules, receptors, and regulatory proteins [1]. Misregulation of the GPI-anchored proteins (GPI-AP), which occurs as a result of mutations in the GPI-biosynthesis pathway, leads to the variety of phenotypes observed in IGD.

GPI biosynthesis pathway. Illustrated is the biosynthetic pathway of GPI-AP. In the Synthesis-stage, twenty genes are responsible for synthesizing the GPI anchor (Synthesis Group, genes highlighted in red). The Transamidase+Remodelinglate-stage couples the protein to the GPI anchor and mediates trafficking through the Golgi apparatus to the cell surface (Tranasmidase+Remodeling Group, genes highlighted in blue) [3,4,5,6]. Abbreviations: PI: phosphatidylinositol; EtNP: ethanolamine phosphate; GlcN: D-Glucosamine; GlcNAc: N-Acetyl-D-glucosamine; E.R.:endoplasmic reticulum. Gene symbols: See Tables 1 and 2
IGD disorders are more common than initially recognized. A study of 4293 parent-child developmental disability trios suggested that IGDs alone account for 0.15% of all developmental disorders [9]. The IGD-associated disorders listed in Table 1 result from complete or partial inactivation of these GPI biosynthesis enzymes. The phenotypes that characterize these disorders often include seizures, intellectual disability, coarse facial features, hypotonia, microcephaly, hearing impairment, and joint contractures. The diseases also display phenotypic abnormalities of the skin, heart, urinary system, and skeleton, which are less common features [3]. Reduced surface levels of GPI-APs or abnormal GPI-AP structure are common in IGD [23].
The first disease identified of the IGDs, hyperphosphatasia with mental retardation syndrome (HPMRS), is associated with variants in both Synthesis-stage and Transamidase+Remodeling-stage genes [24,25,26,27,28,29]. Specifically, HPMRS is caused by mutations in one of the four genes necessary for the biosynthesis of the GPI anchor in the endoplasmic reticulum (PIGV, PIGO, PIGW, and PIGY), or two genes necessary for the post-GPI attachment to proteins (PGAP) type 2 (PGAP2) and type 3 (PGAP3) [24, 30]. Multiple congenital anomalies-hypotonia-seizures (MCAHS) syndrome is a related disorder, although patients do not have hyperphosphatasia (persistently elevated alkaline phosphatase). MCAHS1 [31,32,33,34,35] results from inherited PIGN mutations, a critical gene within the GPI biosynthetic pathway [31, 32, 34]. There is considerable phenotypic variability in MCAHS1, probably reflecting the fact that there is residual GPI associated function [22, 33, 36]. Germline PIGA mutations give rise to an X-linked MCAHS2 [14, 37, 38], and somatic mutations in bone marrow cells result in paroxysmal nocturnal hemoglobinuria [39,40,41]. Finally, MCAHS3 syndrome results from the autosomal recessive inheritance of mutations in PIGT [42,43,44,45]. Especially at the severe end of the phenotypic spectrum of HPMRS, there is substantial phenotypic overlap with MCAHS [46]. Additional IGDs are not classified as either HPMRS or MCAHS have been identified. An overview of all IGD described at the time of this writing is provided in Tables 1 and 2.
A recent review of the phenotypes from 202 IGD patients taken either from the literature or from in-house clinical data chronicled the wide array of phenotypes observed with mutations in each of the GPI-anchor biosynthesis pathway genes, including cognitive impairment, seizures, and congenital malformations [3]. Here, we identified published clinical case studies describing individuals with disease-causing variants in any gene coding for an enzyme in the GPI-anchoring biosynthesis pathway. We then compared phenotypic differences in the Synthesis and Transamidase+Remodeling stages of the GPI-anchoring pathway utilizing the Human Phenotype Ontology (HPO), a standardized vocabulary of phenotypic abnormalities [47]. We demonstrated a number of statistically significant differences in the phenotypic spectrum of diseases in the two groups, suggesting that differential effects on the biochemical function of the GPI pathway may result in different clinical manifestations. We reviewed the phenotypes of diseases caused by defects in individual GPI-anchored proteins and identified a number of candidate GPI-anchored proteins that could cause individual component phenotypes that characterize the IGDs.
Results
In this work, we present a computational analysis to address the question of whether there are differences in the phenotypic spectrum of diseases associated with genes in the Synthesis and Transamidase+Remodeling phases of GPI anchor biosynthesis (Fig. 1). We first performed a comprehensive literature review of all published case reports about individuals diagnosed with diseases caused by variants in a GPI-anchor pathway gene. We then extracted the patient information, mutation(s) information, and all phenotypic data about each patient using terms from the HPO [48,49,50] (Tables 1 and 2). Clinical data from 58 publications were included in this study, comprising a total of 152 individual patients for whom detailed phenotypic descriptions were available, representing IGDs associated with a total of 22 genes involved in the GPI-biosynthesis pathway (Additional file 1: Table S1).
Synthesis vs. Transamidase+Remodeling phenotypes
We divided the GPI biosynthesis pathway into Synthesis and Transamidase+Remodeling stages. Enzymes in the Synthesis Group mediate assembly of the GPI precursor backbone in the endoplasmic reticulum (ER) membrane. Enzymes in the Transamidase+Remodeling Group facilitate the coupling of the GPI to the C-terminus of a newly synthesized protein within the lumen of the ER, cleavage of a C-terminal GPI-addition signal peptide, and enable lipid and carbohydrate side-chain modifications that regulate GPI-AP trafficking from the ER to the plasma membrane [51] (Fig. 1, Tables 1 and 2).
We compared the phenotypic abnormalities in patients with mutations in Synthesis and Transamidase+Remodeling Group genes. Several skeletal phenotypes were significantly more likely to occur in patients with mutations in the Synthesis stage of the biosynthetic pathway (Synthesis Group). The Synthesis Group had a greater occurrence (33% of patients) of Abnormal digit morphology (HPO terms listed using italics). Other phenotypes observed in the Synthesis Group patients were Absent distal phalanges, Aplasia/Hypoplasia of fingers, Short digit, Broad finger and Broad toe, Clubbing, Clinodactyly, and other abnormalities (Table 3). Transamidase+Remodeling Group patients (patients with mutations in the later stage genes) had fewer incidences of Abnormal digit morphology (6.7%, Table 3). For instance, Synthesis Group patients are statistically more likely to have Short digit (24% of the patients), whereas only one individual (< 2%) was indicated as having a Short digit in the Transamidase+Remodeling Group (Table 3).
Moreover, Synthesis Group patients were significantly more likely to have Abnormal muscle morphology, Abnormal tendon morphology, and/or Abnormal joint morphology. This was mainly related to the term Flexion contracture or descendants thereof (Fig. 2). Eighteen of the ninety-three patients in the Synthesis group had a flexion contracture of one or more joints (descendant of Flexion contracture). A “contracture” is a shortening or hardening of the muscle or tendon that leads to the loss of motion of that joint and therefore is listed under the muscle, tendon, and joint hierarchies of the HPO. In addition to flexion contractures, a handful of other types of phenotypes add significance to these parent classes. One Synthesis Group patient [12] displayed Abnormality of the Achilles tendon which is a child of Abnormal tendon morphology. As for Abnormality of joint morphology, a single patient had Axillary pterygia, which is the presence of a cutaneous membrane in the armpit [31]. Additionally, several Synthesis Group patients and a single Transamidase+Remodeling Group patient had Joint hypermobility (Tables 1 and 2). Besides contractures, several other observed phenotypes contributed to the Abnormal muscle morphology phenotype being significantly increased in the Synthesis Group. Such phenotypes include Muscle dystrophy, Camptodactyly, Generalized amyotrophy, Macroglossia, Myopathy, Rimmed vacuoles, Muscle fiber splitting, Skeletal muscle atrophy, Abnormal muscle, and fiber dystrophin expression. Two Transamidase+Remodeling Group patients have noted Abnormal muscle morphology, but this group differs in the types of phenotypes reported (Skeletal muscle atrophy and Increased muscle lipid content).

Example of HPO hierarchy. The hierarchy in the HPO for cleft palate and neighboring phenotypes
Transamidase+Remodeling Group patients, overall, displayed more diverse phenotypic abnormalities that selectively affected this population as opposed to the Synthesis Group (Tables 3 and 4). The most common alterations were in bone and facial development and neurodevelopmental disabilities. The frequency of Abnormality of bone density is significantly higher in Transamidase+Remodeling Group patients as compared to Synthesis Group patients. Osteopenia, a reduction in bone mineral density below normal but not as severe as Osteoporosis, occurred in 22% of the Transamidase+Remodeling Group patients, while only 2% of the patients in the Synthesis Group were reported to have Osteopenia. Osteopenia contributes almost entirely to the significance identified in Reduced bone mineral density (parent term), Abnormality of bone mineral density (grandparent term), Abnormal bone ossification (great-grandparent term), and Abnormal bone structure (great-great-grandparent term) in the Transamidase+Remodeling Group patients. The only additional Abnormal bone structure phenotypes observed were a Thin bony cortex observed in a single patient in the Synthesis Group [52], and two patients were observed to have Reduced bone mineral density and Osteoporosis in one patient in the Transamidase+Remodeling Group [42] (Table 4).
Similarly, patients in the Transamidase+Remodeling Group, predominantly patients with PGAP3 mutations, are significantly more likely to having Macrotia. The Transamidase+Remodeling Group patients were classified as having Macrotia (large ears greater than 2x the standard deviation) 25% of the time, while the incidence was only 2% for the Synthesis Group patients. The vast majority of these Transamidase+Remodeling Group patients were described as specifically having Large fleshy ears, a child of Macrotia (Table 4).
Other facial developmental abnormalities found in Transamidase+Remodeling Group patients were Abnormal hard palate morphology and its child term, Cleft palate. Both phenotypes occurred with significantly higher frequency in the Transamidase+Remodeling Group as compared to the Synthesis Group (29% vs. 6% of patients) (Table 4, Fig. 2). Cleft palate was the predominant phenotype identified 16 Transamidase+Remodeling Group patients (vs. 6 patients in the Synthesis Group), which resulted in both Abnormal hard palate morphology and Cleft palate reaching significance. Additionally, two patients in the Transamidase+Remodeling Group were described as having Median cleft lip and palate, great-grandchild of Cleft palate, which also contributed to the significance of these two phenotypes (Table 4, Fig. 2). Because the term Cleft palate has multiple parents in the HPO, Oral cleft was also identified as selectively enriched in Transamidase+Remodeling Group patients. Besides the phenotypes already mentioned, the significance of the term Oral cleft stemmed from Cleft upper lip and Cleft lip (Table 4, Fig. 2).
Numerous patients in both groups have Abnormality of the nose, but patients in the Transamidase+Remodeling Group were significantly more likely to have alterations in their nose (Synthesis Group = 28% vs. Transamidase+Remodeling Group = 61%). Both groups have Abnormalities of the nose, such as Broad nasal tip and Wide nasal bridge being the most common in each group. While many of the nose abnormalities are present in both groups, Prominent nose was only found in the Transamidase+Remodeling Group (15%). Prominent nose appears to be strongly associated with mutations in PGAP3 and was only reported in those patients. One patient in the Synthesis Group had a Prominent nasal bridge (Table 4).
There are numerous mental and cognitive phenotypes affecting both groups, however, the Remodeling Group appears to have been impacted more often. While both groups have a large percentage of patients with Neurodevelopmental abnormality, 98% of the Transamidase+Remodeling Group were noted with Neurodevelopmental abnormality, as opposed to 73% of the Synthesis Group. More specifically, the Transamidase+Remodeling Group had an increased incidence of Neurodevelopmental delay, Intellectual disability, and Behavioral abnormality (92, 66, and 42%, respectively) while the Synthesis Group had a significantly smaller population with these abnormalities (61, 16, and 15%, respectively) (Table 4).
The division we chose between Synthesis and Transamidase+Remodeling Groups is but one of many possible ways of dividing the GPI pathway, and we reasoned that other partitionings might display other phenotypic differences. In order to explore this, we defined a group consisting of the GPI synthesis genes as well as the transamidase complex genes (Synthesis+Transamidase Group) and compared it with the genes responsible for fatty-acid remodeling (Remodeling Group). The Remodeling group consists of the genes PGAP1, PGAP3, PGAP2, and PGAP5 (a subset of the original Transamidase+Remodeling Group). The Synthesis+Transamidase Group showed enrichment of Urinary tract anomalies. The Remodeling group showed enrichment for some of the same terms as in the Transamidase+Remodeling Group, including Behavioral abnormality, Neurodevelopmental delay, Abnormality of the hard palate, Oral cleft, and Cleft palate. Additionally, the Remodelling group had Decreased head circumference, Altered eye location, Ear and Eyelid morphology abnormalities, Wide nasal bridge, Upper lip abnormalities, and Elevated alkaline phosphatase (Additional file 1: Table S3).
Candidate causal genes for component phenotypes of the IGDs
Mutations in genes that encode enzymes of the GPI biosynthesis pathway result in mistargeting of GPI-APs [53], but the abnormal distribution of GPI-APs in the IGDs has not been characterized in detail. Our hypothesis is that mis-anchoring and, therefore, mistargeting of individual GPI-APs leads to dysfunction of the targeted proteins which in turn leads to some or all of the phenotypic abnormalities observed in the IGDs. A better understanding of the mistargeting of GPI-APs could, therefore, clarify the molecular pathogenesis of the IGDs and shed light on genotype-phenotype correlations.
Over 142 human proteins have been identified in UniProt as being GPI-anchored (Additional file 1: Table S2). Of these, 23 (or 16%) of these genes encoding for GPI-APs have been associated with at least one Mendelian disease (a total of 34 Mendelian diseases were identified), and therefore, numerous phenotypes that define these diseases. We did not observe a significant enrichment of Gene Ontology terms for the genes, nor an enrichment of Mammalian Phenotype Ontology terms (including embryonic lethality) among the orthologs of these genes (data not shown). Thirty-four phenotypes in patients with mutations in GPI-anchored genes overlap with the phenotypes of CDG patients (Table 1 and 2). The fact that GPI-biosynthesis gene mutation and GPI-anchored gene mutations may cause overlapping but not identical phenotypes is expected since mutations in the GPI-biosynthesis pathway would likely alter the activity and function of a number of GPI-anchored proteins, and therefore, multiple signaling pathways.
To further delve into the pathways affected by GPI-biosynthesis gene mutations, we investigated the phenotypes that were observed to be more frequent in the Synthesis or Transamidase+Remodeling Groups. In the Synthesis Group, genes associated with the 5 characteristic phenotypes (Table 3) were compared. A total of 102 genes were associated with Mendelian diseases that share each of the five phenotypic features (Additional file 1: Figure S1).
When comparing the genes associated with the Transamidase+Remodeling Group, two genes were associated with 15 of the 16 Transamidase+Remodeling Group enriched phenotypes: fibroblast growth factor receptor tyrosine kinase (FGFR2), and a downstream signaling partner, B-Raf (BRAF) (Additional file 1: Figure S2). FGFR2 and B-Raf are associated with all the Transamidase+Remodeling Group phenotypes except Large fleshy ears. Notably, these genes are associated with the parent term of Large fleshy ears, Macrotia. The exclusion of Large fleshy ears may be due to the fact that patients present with large ears but not Large fleshy ears, or it could be due to the specificity in which physicians are presenting patient data or the detail recorded by curators and researchers. Mutations in FGFR2 are associated with over ten distinct diseases including Pfeiffer syndrome and Crouzon syndrome [16, 54]. Mutations in BRAF are associated with seven diseases including Noonan syndrome type 7 and Cardiofaciocutaneous syndrome.
Although neither FGFR2, a membrane-spanning protein, nor B-Raf has been identified as GPI-APs, FGFR2 has been shown to associate with lipid rafts in oligodendrocytes [55] and osteoblasts [56] and B-Raf translocation occurs more rapidly in the presence of lipid rafts [57]. GPI-APs are associated with lipid rafts [1, 7] suggesting that this may be a key altered pathway for Transamidase+Remodeling-Group-specific mutations (Fig. 3). Several other signaling partners within the FGFR2 pathway are also associated with lipid rafts, including ligand FGF2 [55, 58], and FRS2 [55]. There may be several targets or interactions with GPI-APs and FGFR2 signaling pathways.

Schematic representation of FGFR2 signaling through the Ras/Raf/MAPK pathway. FGFR2 and B-Raf were found 2 associated with 15 of 16 phenotypes over-represented in the Transamidase+Remodeling Group and are in signaling cascades associated with lipid rafts which contain GPI-anchored proteins. *Numerous FGFs activate the FGFR2. Only FGF2 is known to be associated with lipid rafts (purple) [55,56,57,58]
Interestingly, two target proteins, GPC3 and GPC6 are associated with 25 of GPI-AP associated phenotypic abnormalities (Table 5). Both proteins are members of the glypican family of heparan sulfate proteoglycans that are bound to the cytoplasmic surface of the plasma membrane through a covalent GPI linkage. GPC3 can act as an FGFR1 and FGFR2 coreceptor required for the reception and subsequent relay of the FGF9 signals responsible for the control of coronary vascular development [59], suggesting a possible link.
Discussion
The 29 GPI-biosynthesis enzymes are critical for building the GPI backbone, adding the GPI anchor to proteins, and targeting them to subcellular compartments. The first ~ 20 genes in the pathway are dedicated to building the GPI anchor, while the last ~ 9 genes anchor the proteins to the GPI backbone, and further modify the backbone, thereby regulating targeting of the anchored protein (Table 1). We hypothesized that the clinical features of diseases associated with either the Synthesis or the Transamidase+Remodeling stage of the GPI-biosynthesis pathway may be significantly distinct in each group. Although naively one might think that a mutation of any component of the GPI biosynthesis pathway would have identical phenotypic consequences, it is likely that defects in various components of the pathway affect the biochemistry and functions of GPI-anchored proteins in different ways. Our analysis showed 5 phenotypic abnormalities that were significantly more common with mutations in Synthesis pathway genes and 16 abnormalities that were more common with mutations in Transamidase+Remodeling pathway genes.
While our computational analysis is not able to identify the biochemical mechanisms underlying these differences, we can speculate about the range of factors that might be involved. IGDs do not lead to a uniform reduction of GPI-APs on all cells. For example, some patients with mental retardation, autosomal recessive 53 (MRT53) resulting from variants of PIGG have normal GPI expression on granulocytes while fibroblasts show a reduced global level of GPI anchors and of specific GPI-linked markers [60]. Therefore, one potential factor related to different phenotypic spectrums of the IGDs might be related to the distribution and degree of reduction of GPI-APs in different tissues [61].
The biochemical consequences of individual GPIs may be specific to particular tissues or developmental stages and would affect the target proteins, the GPI-APs, differentially, leading to different phenotypic consequences. The Synthesis Group displayed an enrichment of terms related to Flexion contracture, Abnormal digit, and Short digit, while the Synthesis Group showed an overrepresentation of Neurodevelopmental abnormalities, Bone density abnormalities, and additional facial developmental anomalies (Tables 3 and 4), which we speculate suggests a differential effect of mutations in Synthesis and Transamidase+Remodeling genes on the corresponding tissues.
The analyses of the Synthesis vs. Transamidase+Remodeling Groups, as well as the Synthesis+Transamidase vs. Remodelling groups, clearly demonstrate that the phenotypic abnormalities observed with mutations of genes involved in the GPI-biosynthesis pathway are not consistent across all of the genes. We limited our analysis to two comparisons and showed statistically significant differences in each case. As larger numbers of case reports become available, it may be possible to identify significant differences on a finer scale, involving smaller groups of genes, individual genes, or even specific variants. Understanding these differences may be helpful for precision management or even treatment of the GPI diseases in the future.
Methods
Patient selection
An extensive literature search was conducted using Clinvar [62], Pubmed, OMIM [63], and references for each GPI-anchoring gene to identify case studies. Searches were initiated using the terms “GPI”, “glycosylphosphatidylinositol”, and “mutation”, allowing citations in each publication to be looked up. Variants associated with each disease-related gene in the GPI pathway were examined in ClinVar to identify relevant case reports containing phenotypic descriptions. All case studies of individuals with pathogenic mutations in the GPI-anchoring synthesis pathway were included as long as they met the following criteria: an identifiable mutation was presented, phenotypic information about the patient was included, and the patient was not already included in another paper (See Tables 1, 2, Additional file 1: Table S4). Patient identifiers, phenotypes, and genetic variants were recorded.
Biocuration
Biocuration was performed with an in-house and freely available Java desktop tool for the curation of case reports called HpoCaseAnnotator (https://github.com/monarch-initiative/HpoCaseAnnotator). The tool was used to enter and track PMID, variant information, patient ID, and phenotype(s) associated with each patient. HpoCaseAnnotator offers a concept recognition tool and other convenient functions to streamline HPO-based phenotype annotation. HpoCaseAnnotator calls VariantValidator [64] to check the HGVS syntax and chromosomal locations of variants.
Analysis of the distribution of phenotypes in the selected groups
The biocurated case reports were used to analyze the distribution of phenotypic features within selected groups. Patients were classified into either Synthesis and Transamidase+Remodeling Groups or Synthesis+Transamidase and Remodeling Groups according to the gene mutation identified (Table 1, Additional file 1: Table S1, Additional file 1: Table S4). For each HPO term appearing in any proband record, the software counts the number of patients in each group annotated with that term. These counts are propagated upward in the HPO hierarchy so that a patient annotated with term T is included in the count for any term that subsumes T (i.e., for ancestors of T in the ontology). For instance, if a patient is annotated to Flexion contracture of the 2nd finger (HP:0009537), then implicitly the patient is also annotated to Flexion contracture of finger (HP:0012785) and Abnormal 2nd finger morphology (HP:0004100), as well as all the ancestor terms on the path to the root of the ontology. Using a χ2 test with one degree of freedom, the incidence of each phenotype term was compared between the two groups. Comparisons with insufficient data (expected value below 5 in any cell of the 2 × 2 contingency table) were omitted. A Bonferroni correction for multiple comparisons was applied to achieve α ≤ 0.05. The analysis was implemented as a Java application (code available at https://github.com/monarch-initiative/phenoCompare, release v1.0.0).
Availability of data and materials
All data and materials are included in this published article and its supplementary information files. Information about the analysis tools can be found in the HpoCaseAnnotator Repository (https://github.com/monarch-initiative/HpoCaseAnnotator) or in phenoCompare Repository (https://github.com/monarch-initiative/phenoCompare).
Abbreviations
- BRAF:
-
B-Raf
- CDG:
-
Congenital disorders of glycosylation
- EIEE:
-
Early Infantile Epileptic Encephalopathy
- ER:
-
Endoplasmic reticulum
- EtNP:
-
Phosphatidylinositol; ethanolamine phosphate
- FGFR2:
-
Fibroblast growth factor receptor tyrosine kinase
- GlcN:
-
D-Glucosamine
- GlcNAc:
-
N-Acetyl-D-glucosamine
- GPI:
-
Glycophosphatidylinositol
- GPI-AP:
-
GPI-anchored proteins
- HPMRS:
-
Hyperphosphatasia with mental retardation
- HPO:
-
Human phenotype ontology
- IGD:
-
Inherited GPI deficiencies
- MRT53:
-
Mental retardation, autosomal recessive 53
- PGAP:
-
Post-GPI attachment to proteins
- PNH2:
-
Paroxysmal nocturnal hemoglobinuria 2
References
Kinoshita T, Fujita M. Biosynthesis of GPI-anchored proteins: special emphasis on GPI lipid remodeling. J Lipid Res. 2016;57:6–24.
Ng BG, Freeze HH. Perspectives on glycosylation and its congenital disorders. Trends Genet. 2018;34:466–76.
Bellai-Dussault K, Nguyen TTM, Baratang NV, Jimenez-Cruz DA, Campeau PM. Clinical variability in inherited glycosylphosphatidylinositol deficiency disorders. Clin Genet. 2019;95:112–21.
Maeda Y, Kinoshita T. Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Prog Lipid Res. 2011;50:411–24.
Mayor S, Rao M. Rafts: scale-dependent, active lipid organization at the cell surface. Traffic. 2004;5:231–40.
de la Morena-Barrio ME, Hernández-Caselles T, Corral J, García-López R, Martínez-Martínez I, Pérez-Dueñas B, Altisent C, Sevivas T, Kristensen SR, Guillén-Navarro E, Miñano A, Vicente V, Jaeken J, Lozano ML. GPI-anchor and GPI-anchored protein expression in PMM2-CDG patients. Orphanet J Rare Dis. 2013;8:170.
Sangiorgio V, Pitto M, Palestini P, Masserini M. GPI-anchored proteins and lipid rafts. Ital J Biochem. 2004;53:98–111.
Wang Y, Murakami Y, Yasui T, Wakana S, Kikutani H, Kinoshita T, Maeda Y. Significance of glycosylphosphatidylinositol-anchored protein enrichment in lipid rafts for the control of autoimmunity. J Biol Chem. 2013;288:25490–9.
Pagnamenta AT, Murakami Y, Taylor JM, Anzilotti C, Howard MF, Miller V, Johnson DS, Tadros S, Mansour S, Temple IK, Firth R, Rosser E, Harrison RE, Kerr B, Popitsch N, Study DDD, Kinoshita T, Taylor JC, Kini U. Analysis of exome data for 4293 trios suggests GPI-anchor biogenesis defects are a rare cause of developmental disorders. Eur J Hum Genet. 2017;25:669–79.
Dancourt J, Vuillaumier-Barrot S, de Baulny HO, Sfaello I, Barnier A, le Bizec C, Dupre T, Durand G, Seta N, Moore SEH. A new intronic mutation in the DPM1 gene is associated with a milder form of CDG Ie in two French siblings. Pediatr Res. 2006;59:835–9.
Barone R, Aiello C, Race V, Morava E, Foulquier F, Riemersma M, Passarelli C, Concolino D, Carella M, Santorelli F, Vleugels W, Mercuri E, Garozzo D, Sturiale L, Messina S, Jaeken J, Fiumara A, Wevers RA, Bertini E, Matthijs G, Lefeber DJ. DPM2-CDG: a muscular dystrophy-dystroglycanopathy syndrome with severe epilepsy. Ann Neurol. 2012;72:550–8.
Lefeber DJ, Schönberger J, Morava E, Guillard M, Huyben KM, Verrijp K, Grafakou O, Evangeliou A, Preijers FW, Manta P, Yildiz J, Grünewald S, Spilioti M, van den Elzen C, Klein D, Hess D, Ashida H, Hofsteenge J, Maeda Y, van den Heuvel L, Lammens M, Lehle L, Wevers RA. Deficiency of Dol-P-man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am J Hum Genet. 2009;85:76–86.
Schenk B, Imbach T, Frank CG, Grubenmann CE, Raymond GV, Hurvitz H, Raas-Rotschild A, Luder AS, Jaeken J, Berger EG, Matthijs G, Hennet T, Aebi M. MPDU1 mutations underlie a novel human congenital disorder of glycosylation, designated type if. J Clin Invest. 2001;108:1687–95.
Kato M, Saitsu H, Murakami Y, Kikuchi K, Watanabe S, Iai M, Miya K, Matsuura R, Takayama R, Ohba C, Nakashima M, Tsurusaki Y, Miyake N, Hamano S, Osaka H, Hayasaka K, Kinoshita T, Matsumoto N. PIGA mutations cause earlyonset epileptic encephalopathies and distinctive features. Neurology. 2014;82(18):1587.
Edvardson S, Murakami Y, Nguyen TTM, Shahrour M, St-Denis A, Shaag A, Damseh N, Le Deist F, Bryceson Y, Abu-Libdeh B, Campeau PM, Kinoshita T, Elpeleg O. Mutations in the phosphatidylinositol glycan C (PIGC) gene are associated with epilepsy and intellectual disability. J Med Genet. 2017;54:196–201.
Rai R, Iwanaga J, Dupont G, Oskouian RJ, Loukas M, Oakes WJ, Tubbs RS. Pfeiffer type 2 syndrome: review with updates on its genetics and molecular biology. Childs Nerv Syst. 2019;1:1–5. Published Online First: 21 June 2019. https://doi.org/10.1007/s00381-019-04244-7.
Pagnamenta AT, Murakami Y, Anzilotti C, Titheradge H, Oates AJ, Morton J, Study DDD, Kinoshita T, Kini U, Taylor JC. A homozygous variant disrupting the PIGH start-codon is associated with developmental delay, epilepsy, and microcephaly. Hum Mutat. 2018;39:822–6.
Fujiwara I, Murakami Y, Niihori T, Kanno J, Hakoda A, Sakamoto O, Okamoto N, Funayama R, Nagashima T, Nakayama K, Kinoshita T, Kure S, Matsubara Y, Aoki Y. Mutations in PIGL in a patient with Mabry syndrome. Am J Med Genet A. 2015;167A:777–85.
Ng BG, Hackmann K, Jones MA, Eroshkin AM, He P, Wiliams R, Bhide S, Cantagrel V, Gleeson JG, Paller AS, Schnur RE, Tinschert S, Zunich J, Hegde MR, Freeze HH. Mutations in the glycosylphosphatidylinositol gene PIGL cause CHIME syndrome. Am J Hum Genet. 2012;90:685–8.
Nicklas JA, Carter EW, Albertini RJ. Both PIGA and PIGL mutations cause GPI-a deficient isolates in the Tk6 cell line. Environ Mol Mutagen. 2015;56:663–73.
Almeida AM, Murakami Y, Layton DM, Hillmen P, Sellick GS, Maeda Y, Richards S, Patterson S, Kotsianidis I, Mollica L, Crawford DH, Baker A, Ferguson M, Roberts I, Houlston R, Kinoshita T, Karadimitris A. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat Med. 2006;12:846–51.
Johnstone DL, Nguyen T-T-M, Murakami Y, Kernohan KD, Tétreault M, Goldsmith C, Doja A, Wagner JD, Huang L, Hartley T, St-Denis A, le Deist F, Majewski J, Bulman DE. Care4Rare Canada consortium, Kinoshita T, Dyment DA, boycott KM, Campeau PM. Compound heterozygous mutations in the gene PIGP are associated with early infantile epileptic encephalopathy. Hum Mol Genet. 2017;26:1706–15.
Makrythanasis P, Kato M, Zaki MS, Saitsu H, Nakamura K, Santoni FA, Miyatake S, Nakashima M, Issa MY, Guipponi M, Letourneau A, Logan CV, Roberts N, Parry DA, Johnson CA, Matsumoto N, Hamamy H, Sheridan E, Kinoshita T, Antonarakis SE, Murakami Y. Pathogenic variants in PIGG cause intellectual disability with seizures and Hypotonia. Am J Hum Genet. 2016;98:615–26.
Mabry CC, Bautista A, Kirk RF, Dubilier LD, Braunstein H, Koepke JA. Familial hyperphosphatase with mental retardation, seizures, and neurologic deficits. J Pediatr. 1970;77:74–85.
Gomes WJ, Hunter JL. Mental retardation, cataracts, and unexplained hyperphosphatasia. Arch Dis Child. 1970;45:726–7.
Kruse K, Hanefeld F, Kohlschütter A, Rosskamp R, Gross-Selbeck G. Hyperphosphatasia with mental retardation. J Pediatr. 1988;112:436–9.
Rabe P, Haverkamp F, Emons D. Syndrome of developmental retardation, facial and skeletal anomalies, and hyperphosphatasia in two sisters: nosology and genetics of the coffin-Siris syndrome. American J Med Gen. 1991;41(3):350 https://onlinelibrary.wiley.com/doi/abs/10.1002/ajmg.1320410317.
Thompson MD, Nezarati MM, Gillessen-Kaesbach G, Meinecke P, Mendoza-Londono R, Mendoza R, Mornet E, Brun-Heath I, Squarcioni CP, Legeai-Mallet L, Munnich A, Cole DEC. Hyperphosphatasia with seizures, neurologic deficit, and characteristic facial features: five new patients with Mabry syndrome. Am J Med Genet A. 2010;152A:1661–9.
Thompson MD, Killoran A, Percy ME, Nezarati M, Cole DEC, Hwang PA. Hyperphosphatasia with neurologic deficit: a pyridoxine-responsive seizure disorder? Pediatr Neurol. 2006;34:303–7.
Cole DEC, Thompson MD. Neurogenetic aspects of Hyperphosphatasia in Mabry syndrome. Subcell Biochem. 2015;76:343–61.
Brady PD, Moerman P, De Catte L, Deprest J, Devriendt K, Vermeesch JR. Exome sequencing identifies a recessive PIGN splice site mutation as a cause of syndromic congenital diaphragmatic hernia. Eur J Med Genet. 2014;57:487–93.
Maydan G, Noyman I, Har-Zahav A, Neriah ZB, Pasmanik-Chor M, Yeheskel A, Albin-Kaplanski A, Maya I, Magal N, Birk E, Simon AJ, Halevy A, Rechavi G, Shohat M, Straussberg R, Basel-Vanagaite L. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet. 2011;48:383–9.
McInerney-Leo AM, Harris JE, Gattas M, Peach EE, Sinnott S, Dudding-Byth T, Rajagopalan S, Barnett CP, Anderson LK, Wheeler L, Brown MA, Leo PJ, Wicking C, Duncan EL. Fryns syndrome associated with recessive mutations in PIGN in two separate families. Hum Mutat. 2016;37:695–702.
Ohba_PIGNmutations cause congenital anomalies, developmental delay, hypotonia, epilepsy, and progressive cerebellar atrophy _ SpringerLink.pdf. https://link.springer.com/article/
Fleming L, Lemmon M, Beck N, Johnson M, Mu W, Murdock D, Bodurtha J, Hoover-Fong J, Cohn R, Bosemani T, Barañano K, Hamosh A. Genotype-phenotype correlation of congenital anomalies in multiple congenital anomalies hypotonia seizures syndrome (MCAHS1)/PIGN-related epilepsy. Am J Med Genet A. 2016;170A:77–86.
Thompson MD, Cole DE. Recessive PIGN mutations in Fryns syndrome: evidence for genetic heterogeneity. Hum Mutat. 2016;37:621.
Fauth C, Steindl K, Toutain A, Farrell S, Witsch-Baumgartner M, Karall D, Joset P, Böhm S, Baumer A, Maier O, Zschocke J, Weksberg R, Marshall CR, Rauch A. A recurrent germline mutation in the PIGA gene causes Simpson-Golabi-Behmel syndrome type 2. Am J Med Genet A. 2016;170A:392–402.
Johnston JJ, Gropman AL, Sapp JC, Teer JK, Martin JM, Liu CF, Yuan X, Ye Z, Cheng L, Brodsky RA, Biesecker LG. The phenotype of a germline mutation in PIGA: the gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am J Hum Genet. 2012;90:295–300.
Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, Fujita T, Takahashi M, Kitani T, Kinoshita T. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703–11.
Nafa K, Bessler M, Deeg HJ, Luzzatto L. New somatic mutation in the PIG-A gene emerges at relapse of paroxysmal nocturnal hemoglobinuria. Blood. 1998;92:3422–7.
Ware RE, Rosse WF, Howard TA. Mutations within the Piga gene in patients with paroxysmal nocturnal hemoglobinuria. Blood. 1994;83:2418–22.
Nakashima M, Kashii H, Murakami Y, Kato M, Tsurusaki Y, Miyake N, Kubota M, Kinoshita T, Saitsu H, Matsumoto N. Novel compound heterozygous PIGT mutations caused multiple congenital anomalies-hypotonia-seizures syndrome 3. Neurogenetics. 2014;15:193–200.
Kvarnung M, Nilsson D, Lindstrand A, Korenke GC, Chiang SCC, Blennow E, Bergmann M, Stödberg T, Mäkitie O, Anderlid B-M, Bryceson YT, Nordenskjöld M, Nordgren A. A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J Med Genet. 2013;50:521–8.
Lam C, Golas GA, Davids M, Huizing M, Kane MS, Krasnewich DM, Malicdan MCV, Adams DR, Markello TC, Zein WM, Gropman AL, Lodish MB, Stratakis CA, Maric I, Rosenzweig SD, Baker EH, Ferreira CR, Danylchuk NR, Kahler S, Garnica AD, Bradley Schaefer G, Boerkoel CF, Gahl WA, Wolfe LA. Expanding the clinical and molecular characteristics of PIGT-CDG, a disorder of glycosylphosphatidylinositol anchors. Mol Genet Metab. 2015;115:128–40.
Skauli N, Wallace S, Chiang SCC, Barøy T, Holmgren A, Stray-Pedersen A, Bryceson YT, Strømme P, Frengen E, Misceo D. Novel PIGT Variant in two Brothers: expansion of the multiple congenital anomalies-Hypotonia seizures syndrome 3 phenotype. Genes. 2016;7:108. https://doi.org/10.3390/genes7120108.
Knaus A, Pantel JT, Pendziwiat M, Hajjir N, Zhao M, Hsieh T-C, Schubach M, Gurovich Y, Fleischer N, Jäger M, Köhler S, Muhle H, Korff C, Møller RS, Bayat A, Calvas P, Chassaing N, Warren H, Skinner S, Louie R, Evers C, Bohn M, Christen H-J, van den Born M, Obersztyn E, Charzewska A, Endziniene M, Kortüm F, Brown N, Robinson PN, Schelhaas HJ, Weber Y, Helbig I, Mundlos S, Horn D, Krawitz PM. Characterization of glycosylphosphatidylinositol biosynthesis defects by clinical features, flow cytometry, and automated image analysis. Genome Med. 2018;10:3.
Köhler S, Carmody L, Vasilevsky N, JOB J, Danis D, Gourdine J-P, Gargano M, Harris NL, Matentzoglu N, JA MM, Osumi-Sutherland D, Cipriani V, Balhoff JP, Conlin T, Blau H, Baynam G, Palmer R, Gratian D, Dawkins H, Segal M, Jansen AC, Muaz A, Chang WH, Bergerson J, SJF L, Yüksel Z, Beltran S, Freeman AF, Sergouniotis PI, Durkin D, Storm AL, Hanauer M, Brudno M, Bello SM, Sincan M, Rageth K, Wheeler MT, Oegema R, Lourghi H, Della Rocca MG, Thompson R, Castellanos F, Priest J, Cunningham-Rundles C, Hegde A, Lovering RC, Hajek C, Olry A, Notarangelo L, Similuk M, Zhang XA, Gómez-Andrés D, Lochmüller H, Dollfus H, Rosenzweig S, Marwaha S, Rath A, Sullivan K, Smith C, Milner JD, Leroux D, Boerkoel CF, Klion A, Carter MC, Groza T, Smedley D, Haendel MA, Mungall C, Robinson PN. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res. 2019;47(D1):D1018. Published Online First: 22 November 2018. https://doi.org/10.1093/nar/gky1105.
Robinson PN, Köhler S, Bauer S, Seelow D, Horn D, Mundlos S. The human phenotype ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet. 2008;83:610–5.
Köhler S, Vasilevsky NA, Engelstad M, Foster E, McMurry J, Aymé S, Baynam G, Bello SM, Boerkoel CF, Boycott KM, Brudno M, Buske OJ, Chinnery PF, Cipriani V, Connell LE, HJS D, LE DM, Devereau AD, de BBA V, Firth HV, Freson K, Greene D, Hamosh A, Helbig I, Hum C, Jähn JA, James R, Krause R, SJ FL, Lochmüller H, Lyon GJ, Ogishima S, Olry A, Ouwehand WH, Pontikos N, Rath A, Schaefer F, Scott RH, Segal M, Sergouniotis PI, Sever R, Smith CL, Straub V, Thompson R, Turner C, Turro E, MWM V, Vulliamy T, Yu J, von Ziegenweidt J, Zankl A, Züchner S, Zemojtel T, JOB J, Groza T, Smedley D, Mungall CJ, Haendel M, Robinson PN. The Human Phenotype Ontology in 2017. Nucleic Acids Res. 2017;45:D865–76.
Köhler S, Doelken SC, Mungall CJ, Bauer S, Firth HV, Bailleul-Forestier I, GCM B, Brown DL, Brudno M, Campbell J, DR FP, Eppig JT, Jackson AP, Freson K, Girdea M, Helbig I, Hurst JA, Jähn J, Jackson LG, Kelly AM, Ledbetter DH, Mansour S, Martin CL, Moss C, Mumford A, Ouwehand WH, Park S-M, Riggs ER, Scott RH, Sisodiya S, Van Vooren S, Wapner RJ, AOM W, Wright CF, Vulto-van Silfhout AT, de Leeuw N, de BBA V, Washingthon NL, Smith CL, Westerfield M, Schofield P, Ruef BJ, Gkoutos GV, Haendel M, Smedley D, Lewis SE, Robinson PN. The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Res. 2014;42:D966–74.
MAJ F, Hart GW, Kinoshita T. Glycosylphosphatidylinositol Anchors. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, Seeberger PH, editors. Essentials of Glycobiology. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2017.
Swoboda KJ, Margraf RL, Carey JC, Zhou H, Newcomb TM, Coonrod E, Durtschi J, Mallempati K, Kumanovics A, Katz BE, Voelkerding KV, Opitz JM. A novel germline PIGA mutation in Ferro-Cerebro-cutaneous syndrome: a neurodegenerative X-linked epileptic encephalopathy with systemic iron-overload. Am J Med Genet A. 2014;164A:17–28.
Kinoshita T, Maeda Y, Fujita M. Transport of glycosylphosphatidylinositol-anchored proteins from the endoplasmic reticulum. Biochim Biophys Acta. 1833;2013:2473–8.
Fan J, Li Y, Jia R, Fan X. An inherited FGFR2 mutation increased osteogenesis gene expression and result in Crouzon syndrome. BMC Med Genet. 2018;19:91.
Bryant MR, Marta CB, Kim FS, Bansal R. Phosphorylation and lipid raft association of fibroblast growth factor receptor-2 in oligodendrocytes. Glia. 2009;57:935–46.
Dufour C, Guenou H, Kaabeche K, Bouvard D, Sanjay A, Marie PJ. FGFR2-Cbl interaction in lipid rafts triggers attenuation of PI3K/Akt signaling and osteoblast survival. Bone. 2008;42:1032–9.
Hekman M, Hamm H, Villar AV, Bader B, Kuhlmann J, Nickel J, Rapp UR. Associations of B- and C-Raf with cholesterol, phosphatidylserine, and lipid second messengers: preferential binding of Raf to artificial lipid rafts. J Biol Chem. 2002;277:24090–102.
Gutiérrez J, Brandan E. A novel mechanism of sequestering fibroblast growth factor 2 by glypican in lipid rafts, allowing skeletal muscle differentiation. Mol Cell Biol. 2010;30:1634–49.
Ng A, Wong M, Viviano B, Erlich JM, Alba G, Pflederer C, Jay PY, Saunders S. Loss of glypican-3 function causes growth factor-dependent defects in cardiac and coronary vascular development. Dev Biol. 2009;335:208–15.
Zhao JJ, Halvardson J, Knaus A, Georgii-Hemming P, Baeck P, Krawitz PM, Thuresson A-C, Feuk L. Reduced cell surface levels of GPI-linked markers in a new case with PIGG loss of function. Hum Mutat. 2017;38:1394–401.
Chen R, Walter EI, Parker G, Lapurga JP, Millan JL, Ikehara Y, Udenfriend S, Medof ME. Mammalian glycophosphatidylinositol anchor transfer to proteins and posttransfer deacylation. Proc Natl Acad Sci U S A. 1998;95:9512–7.
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin-Salomon R, Rubinstein W, Maglott DR. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–8.
Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: online Mendelian inheritance in man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789–98.
Freeman PJ, Hart RK, Gretton LJ, Brookes AJ, Dalgleish R. VariantValidator: accurate validation, mapping, and formatting of sequence variation descriptions. Hum Mutat. 2018;39:61–8.
Acknowledgments
The authors would like to thank Taroh Kinoshita, Ph.D., for his critical comments regarding this paper. We would also like to thank Jane W. Cha and Jackson Laboratories Creative team for the design of figures used in this manuscript.
Funding
National Institutes of Health (NIH), Monarch Initiative [OD #5R24OD011883].
Author information
Authors and Affiliations
Contributions
LCC curated data, interpreted the resulting data analysis and wrote the manuscript. HB developed the analysis software and contributed to writing this paper. DD developed tools used for data curation collection. MDT helped write background information, and NV generated figures used in the paper. PNR developed the concepts for this paper, curated data, aided in the development of curation and analysis tools, interpretation, and writing of this manuscript. NV, MDT, HB, XAZ, JPG, and PK critically reviewed the manuscript and figures. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable. All data was previously published.
Competing interests
The authors declare they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Venn diagram representing overlaps of genes associated with the indicated phenotypic abnormalities in the Synthesis group. Figure S2. Venn diagram representing genes found in at diseases that overlap by at least 14 phenotypic abnormalities. Table S1. Case reports included in the present work. Table S2. GPI-anchored proteins. Table S3. Phenotypic comparison of Synthesis+Transamidase (S/T) vs. Remodeling groups. Table S4. Publications curated for this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Carmody, L.C., Blau, H., Danis, D. et al. Significantly different clinical phenotypes associated with mutations in synthesis and transamidase+remodeling glycosylphosphatidylinositol (GPI)-anchor biosynthesis genes. Orphanet J Rare Dis 15, 40 (2020). https://doi.org/10.1186/s13023-020-1313-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-020-1313-0