
Abstract
The ACTA2 gene encodes for smooth muscle specific α-actin, a critical component of the contractile apparatus of the vascular smooth muscle cell. Pathogenic variants in the ACTA2 gene are the most frequently encountered genetic cause of non-syndromic hereditary thoracic aortic disease (HTAD). Although thoracic aortic aneurysm and/or dissection is the main clinical manifestation, a variety of occlusive vascular disease and extravascular manifestations occur in ACTA2-related vasculopathy. Current data suggest possible mutation-specific manifestations of vascular and extra-aortic traits.
Despite its relatively high prevalence, comprehensive recommendations on the care of patients and families with pathogenic variants in ACTA2 have not yet been established. We aimed to develop a consensus document to provide medical guidance for health care professionals involved in the diagnosis and treatment of patients and relatives with pathogenic variants in ACTA2.
The HTAD Working Group of the European Reference Network for Rare Vascular Diseases (VASCERN) convened to review current literature and discuss expert opinions on clinical management of ACTA2 related vasculopathy. This consensus statement summarizes our recommendations on diagnosis, monitoring, treatment, pregnancy, genetic counselling and testing in patients with ACTA2-related vasculopathy. However, there is a clear need for additional prospective multicenter studies to further define proper guidelines.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Background
Thoracic aortic aneurysm/dissection (TAAD) represents a vascular condition with life-threatening complications, including aortic dissection or rupture. Depending on the presence or absence of manifestations in other organ systems, TAAD can be subdivided into syndromic and non-syndromic. Heritable thoracic aortic disease (HTAD) refers to thoracic aortic disease caused by a pathogenic variant in a gene that confers a high risk for TAAD.
The ACTA2 gene (OMIM #102620, ORPHA 91387, 2573, 404,463) encodes for smooth muscle cell specific isoform of alpha-actin. Pathogenic variants in the gene are the most frequently encountered cause of non-syndromic HTAD with detection rates varying between 1,5–21%, probably depending on the inclusion criteria of the screened population [1,2,3,4,5,6,7,8,9,10,11].
ACTA2-related vasculopathy is an autosomal dominant condition with aortic aneurysm and risk of dissection as main vascular phenotype, and occasional extravascular traits such as skin (i.e. livedo reticularis) and ocular abnormalities (i.e. iris flocculi). The penetrance of the vascular phenotype is incomplete and age-related: according to the limited data available thus far, children usually do not manifest aortic dilatation/aneurysm. Thus far, some case reports and one comprehensive series comprising 277 ACTA2 mutation carriers have been reported; the main findings are listed in Table 1 [1, 3, 6, 12,13,14,15].
Multisystem smooth muscle dysfunction syndrome (MSMD syndrome, OMIM #613834, ORPHA 404463) is a severe phenotype caused by de novo pathogenic variants affecting the Arg179 residue of the ACTA2 gene and is characterized by aortic and cerebrovascular disease, fixed mydriatic pupils, hypotonic bladder, intestinal hypoperistalsis, pulmonary hypertension and white matter brain anomalies [16,17,18,19,20,21,22,23]. Specific management recommendations have been suggested for MSMD syndrome patients and therefore MSMD is not included in these recommendations [24].
This consensus statement focuses on the diagnosis, monitoring and treatment of patient with ACTA2-related vasculopathy including genetic counselling. This document represents a disease-specific, preliminary recommendation that certainly will require adjustment and expansion of contents as scientific and clinical insight evolves.
Methods
The international ACTA2 consensus group comprised 21 participants from 15 institutions in 9 countries with a shared interest in improving the care for patients with ACTA2 mutations. All participants (apart from two cooperating guests) are members of the Heritable Thoracic Aortic Disease (HTAD) working group of the European Reference Network on rare vascular diseases (VASCERN), and includes cardiologists, cardiac surgeons and clinical geneticists and all are experts of the disease for having diagnosed, monitored and treated more than 65 families together. The aim of the working group was to develop consensus recommendations in order to standardize and optimize the care for patients and families with pathogenic variants in the ACTA2 gene.
Our group evaluated the relevant literature to formulate recommendations. An inventory of nine questions on the most debatable ACTA2-related topics was sent to all members of the working group. (Additional file 1: Table S1) The inventory and its recommendations were then discussed at two plenary sessions and one video conference call held between September 2017 and May 2018 and were used to formulate the following European recommendations on ACTA2-related vasculopathy. The consensus document was approved by all members of the working group.
Results
Existing international literature on patients with a pathogenic variant in the ACTA2 gene are often too general with regard to screening recommendations. The results are constituted of recommendations with regard to screening, monitoring, treatment, life-style advices, preconception and pregnancy management for patients with a pathogenic variant in ACTA2 (excluding MSDM patients). In addition, recommendations with regard to genetic counselling and testing of index patients and their families are composed.
Diagnosis and monitoring
At initial diagnosis, we recommend 2D-transthoracic echocardiography (2D-TTE) and complete vascular imaging of thorax and abdomen (neck to pelvis) from the age of 18 years. Imaging has to be performed according to established guidelines for each corresponding technique and obtained values have to be corrected for age, BSA and gender. 2D-TTE is the method of choice in the assessment and follow-up of the aortic root and proximal ascending aorta diameters [25, 26]. Measurements are obtained from the parasternal long axis view at end-diastole according to the leading-edge to leading-edge convention. CT and MRI provide information of the entire aorta and peripheral vessels [27]. CT has the best special resolution but low-dose electrocardiographically gated protocols should be used to decrease radiation exposure. MRI has significant advantages in the follow-up of patients with genetic disorders adding functional information to anatomical data without the burden of ionizing radiation. Aortic root measurement is performed by SSFP cine sequence images and the rest of the aorta by 3D-angiogram. Multiplanar reconstruction of the axial source data can create aortic images in a plane perpendicular to the aortic lumen direction (double-oblique or true short-axis images of the aorta) by CT and MRI. There is general consensus that in both imaging techniques the aortic diameter should be measured with the inner-to-inner convention at end-diastole.
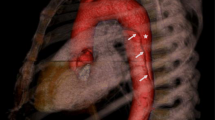
With an aneurysm of the ascending aorta we recommend 2D-TTE with a time schedule based on the individual characteristics of the patient (age, sex) and the TAA (diameter, growth, shape). With an aneurysm in another artery or in the distal part of the aorta, imaging is recommended yearly, the technique depending on the location of the aneurysm. If no aneurysm is found, enlargement of the ascending aorta is monitored by yearly 2D-TTE of the aortic root and the ascending aorta only. We recommend complete imaging of the vascular tree from neck to pelvis once every two to 5 years (Fig. 1). In children, we recommend yearly 2D-TTE from the age of diagnosis, although intervals can be extended depending on clinical judgement. With normal diameters on echocardiography, computerised tomography angiography (CTA)/magnetic resonance angiography (MRA) imaging of the thoracoabdominal aorta is recommended at the age of 18 years (Fig. 1). The choice for CTA or MRA depends on local expertise and availability, always aiming to keep radiation exposure as low as possible.

Summary of the screening and management of patients with pathogenic ACTA2 variants
No systematic imaging of the cerebral vessels is recommended since there is at present insufficient data to make recommendations for treatment. In patients with symptoms potentially related to cerebral vasculopathy, we recommend imaging according to local guidance and clinical review by Neurology and/or Neurosurgery services. Imaging of the coronary arteries should be limited to patients with symptoms suggesting ischemic heart disease and in patients scheduled for elective aortic surgery.
Ophthalmological examination for detection of iris flocculi is recommended at diagnosis and in patients with symptoms. In young children, the ophthalmological examination can be postponed until they can more easily comply.
Treatment of aortic aneurysm
Elective aortic root replacement should be considered in asymptomatic patients with a maximal aortic diameter between 45 and 50 mm. The American Heart Association (AHA) recommends a threshold of 45 mm [28] and there is currently no European Society of Cardiology (ESC) recommendation for this topic. Since no additional data have become available, there is no consensus on the threshold. The indications for preventive surgery should consider risk factors such as rate of aortic growth, aortic valve dysfunction, body surface area, family history of aortic dissection, and hypertension. Women planning a pregnancy should be considered for preventive surgery at aortic diameters ≥45 mm (see pregnancy section). For elective intervention, we recommend valve sparing aortic root replacement according to David reimplantation technique of valve sparing root replacement when feasible and in the absence of surgical contraindications such as leaflet fenestrations or relevant aortic valve regurgitation. External aortic support procedures, offered in a limited number of centres, has no established role in this population.
Elective replacement of the descending thoracic aorta should be performed according to the ESC, AHA and European Society for Thoraco Vascular Surgery (ESTVS) guidelines [28,29,30]. Operative decisions are often difficult and therefore the benefits and risks should be a matter of collective discussion of the patient with the vascular surgeons. There is, at present, no consensus on appropriateness or indications for endovascular procedures in ACTA2 aortopathy. The general rule is to avoid endoprostheses in patients with genetic aortopathies [29].
Treatment of aortic dissection
Type A aortic dissection always requires immediate surgical intervention whether or not a pathogenic ACTA2 variant is present. Valve sparing aortic root replacement should also be considered in this and the risk of coronary artery dissection should be taken into account.
Type B aortic dissections require initial medical management and surgery in case of complications as in patients without ACTA2 mutations [12]. Endovascular therapy should be avoided in the presence of genetic aortopathies [29].
Medical treatment
Scientific proof for medical benefit in patients treated with β-blockers is not available. Medications that reduce hemodynamic stress, such as β-blockers, should be considered in ACTA2 patients, with aortic dilatation (aortic diameter at z-score ≥ 2) or hypertension or progression of aortic dilatation (> 0,3 cm/year) and is reasonable to propose in ACTA2 patients without dilatation. The use of other antihypertensive drugs can be discussed with the treating clinician.
Hypertension should be treated according to the ESC hypertension guidelines which state that all hypertensive patients with aortic dilatation, should have their blood pressure controlled ≤130/80 mmHg [31]. Patients with a pathogenic variant in ACTA2 should be managed according to standard strategies for other cardiovascular risk factors, including dyslipidemia [28, 29].
Lifestyle
Patients should be advised to avoid smoking and substance abuse as for all patients. Endurance sports can be of great value to control body weight, blood pressure and fitness. In patients with aortic dilatation/aneurysm, we recommend low static, dynamic sports such as swimming, walking, running and cycling, without competitive spirit. The physical activity level, both in children and adults, should be adjusted by the cardiologist based on the evaluation of aortic dimensions and valvular function. However, we recommend the avoidance of sports with abrupt isometric or static manoeuvers or any other activities with significant increase in arterial blood pressure, such as weightlifting, competitive football, basketball, handball, and tennis.
Preconception
For preconception care we recommend counselling of all carriers by a pregnancy heart team with cardiologist (specialized in preconception care), obstetrician, and clinical geneticist for evaluation of the aortic risk, counselling of the risks for the future mother, father and children, including discussion of invasive prenatal diagnostic testing and pre-implantation genetic diagnostics [32]. Imaging of the entire aorta is recommended prior to pregnancy. Women who have a medical history of aortic dissection should be advised not to become pregnant. Women with aortic root diameters ≥45 mm should be advised to undergo prophylactic aortic root replacement prior to pregnancy. Teratogenic drugs (e.g. angiotensin receptor blockers such as losartan and irbesartan) should be discontinued – β-blockers should be considered throughout pregnancy (preferably metoprolol or labetalol).
Pregnancy management
Foetal surveillance
If the genetic status of the foetus is unknown or the foetus carries an ACTA2 mutation, standardized cardiac ultrasound of the foetus around a gestational age of 20 weeks should be offered due to the slightly increased risk of congenital heart malformations. Many congenital heart malformations associated with ACTA2 mutations might however be difficult to detect prenatally on ultrasound (e.g. patent ductus, bicuspid valve) [3, 12]. Regular foetal growth monitoring should also be performed in pregnant women who are taking β-blockers because of the associated small risk of foetal growth retardation.
Maternal surveillance
Since little data on pregnancies in ACTA2 mutation carriers are available, we recommend management in pregnant women as for pregnant women with Marfan syndrome [32].
Monitoring (2D-TTE) of the aortic diameter of the pregnant woman with ascending aortic pathology every 4–12 weeks (depending on the severity of dilatation) throughout the pregnancy and 6 months post-partum is recommended.
Delivery
In low risk pregnancies (aortic diameter < 40 mm or Z-score < 2 without growth, no aortic dissections in family) the indications and decision for vaginal delivery in hospital should be discussed with the patient who should be aware that, although rare, the risk of dissection during or after delivery may also occur at an aortic diameter lower or around 40 mm [13, 14].
Moderate and high risk pregnancies should be managed by a cardiac and obstetric team with experience in managing patients with aortopathies in an expert center with the option of acute cardiothoracic surgery support. In patients with moderate risk (ascending aortic diameter 40-45 mm), vaginal delivery should be carefully considered on an individual basis. Vaginal delivery should be assisted with expedited second-stage and regional anaesthesia. In high-risk pregnancies (ascending aortic diameter > 45 mm) caesarean delivery should be considered [32].
Genetic counselling and genetic testing
Genetic counselling should be systematically offered to all patients carrying (or at risk for carrying) a pathogenic variant in ACTA2. The counselling aims to collect information and clinical reports of family members, and to inform the consultants of the possible genetic origin of their disease. The genetic work-up should include review with a clinical geneticist who will assess for traits that can potentially recur in patients with ACTA2-related vasculopathy as well as other, previously unreported traits. Clinical annotation is fundamental for expanding and collecting observations that may complete the list of the traits of the disease (either markers or abnormalities deserving care).
Cascade genetic testing should be offered in families with probands harbouring pathogenic variants in ACTA2. In the case of variants of unknown significance (VUS), in particular novel missense variants, clinical family screening is recommended. Segregation studies in families are essential especially when functional studies are not available or are not feasible.
Each family member that accepts counselling and testing should give his/her informed consent. The counselling process provides the opportunity for informing and discussing current knowledge and data on potential disease-specific risks, and the benefits of genetic testing in order to facilitate autonomous decision-making. Studies of psychological implication of genetic testing for other cardiovascular diseases have shown that the genetic risk is perceived to be manageable {Oliveri, [33] #47}.
Predictive genetic testing of minors is generally accepted for childhood-onset conditions, if preventative or therapeutic measures are available to reduce morbidity or mortality. Although there is no scientific proof for medical benefit in children treated with β-blockers, we advise to propose treatment from the age of 4 years as stated above. Therefore the inclusion of children in the family genetic testing program appears beneficial. The genetic test to evaluate for the ACTA2 mutation identified in the proband of the family can be performed in the paediatric age using a saliva sample, to avoid the discomfort of blood sampling. In particular, when genetic testing is performed in children under the age of 16 we strongly recommend proper genetic counselling involving psychosocial support.
Conclusions
Patients affected by ACTA2-related vasculopathy will usually be diagnosed due to an incidental detection of aortic dilatation, or after an acute dissection. Coronary or cerebrovascular disease should be assessed in symptomatic patients. Monitoring programs should be tailored to each single patient. Medications such as β- blockers can be considered with the major aim to reduce haemodynamic stress in the aorta. Patients should be carefully screened and monitored for arterial hypertension and treated in order to fully control blood pressure. Preventive aortic surgery should be considered based on general indications, individual needs and risk factors. Since ACTA2-related vasculopathy is a recently described condition, the phenotypic spectrum and precise risk factors are probably not fully understood. It is therefore essential to follow these patients at tertiary reference centres, and subsequently report recurring features of the disease. This would enrich the clinical characterisation and facilitate the suspect, thus addressing patients and families to referral centres.
Availability of data and materials
All data generated or analysed during this study are included in this manuscript.
Abbreviations
- 2D-TTE:
-
2D-transthoracic echocardiography
- AHA:
-
American heart association
- CTA:
-
Computerised tomography angiography
- ESC:
-
European society of cardiology
- ESTVS:
-
European society for thoraco vascular surgery
- HTAD:
-
Hereditary thoracic aortic disease
- MRA:
-
Magnetic resonance angiography
- MSMD:
-
Multisystem smooth muscle dysfunction
- TAAD:
-
Thoracic aortic aneurysm/dissection
- VASCERN:
-
European reference network for rare vascular diseases
- VUS:
-
Variants of unknown significance
References
Disabella E, Grasso M, Gambarin FI, Narula N, Dore R, Favalli V, et al. Risk of dissection in thoracic aneurysms associated with mutations of smooth muscle alpha-actin 2 (ACTA2). Heart. 2011;97(4):321–6.
Renard M, Callewaert B, Baetens M, Campens L, Macdermot K, Fryns JP, et al. Novel MYH11 and ACTA2 mutations reveal a role for enhanced TGFbeta signaling in FTAAD. Int J Cardiol. 2011;165(2):314–21.
Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39(12):1488–93.
Ke T, Han M, Zhao M, Wang QK, Zhang H, Zhao Y, et al. Alpha-actin-2 mutations in Chinese patients with a non-syndromatic thoracic aortic aneurysm. BMC Med Genet. 2016;17(1):45.
Morisaki H, Akutsu K, Ogino H, Kondo N, Yamanaka I, Tsutsumi Y, et al. Mutation of ACTA2 gene as an important cause of familial and nonfamilial nonsyndromatic thoracic aortic aneurysm and/or dissection (TAAD). Hum Mutat. 2009;30(10):1406–11.
Guo DC, Papke CL, Tran-Fadulu V, Regalado ES, Avidan N, Johnson RJ, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84(5):617–27.
Hoffjan S, Waldmuller S, Blankenfeldt W, Kotting J, Gehle P, Binner P, et al. Three novel mutations in the ACTA2 gene in German patients with thoracic aortic aneurysms and dissections. Eur J Hum Genet. 2011;19(5):520–4.
Campens L, Callewaert B, Muino Mosquera L, Renard M, Symoens S, De Paepe A, et al. Gene panel sequencing in heritable thoracic aortic disorders and related entities - results of comprehensive testing in a cohort of 264 patients. Orphanet J Rare Dis. 2015;10:9.
Lerner-Ellis JP, Aldubayan SH, Hernandez AL, Kelly MA, Stuenkel AJ, Walsh J, et al. The spectrum of FBN1, TGFbetaR1, TGFbetaR2 and ACTA2 variants in 594 individuals with suspected Marfan syndrome, Loeys-Dietz syndrome or thoracic aortic aneurysms and dissections (TAAD). Mol Genet Metab. 2014;112(2):171–6.
Bee KJ, Wilkes DC, Devereux RB, Basson CT, Hatcher CJ. TGFbetaRIIb mutations trigger aortic aneurysm pathogenesis by altering transforming growth factor beta2 signal transduction. Circ Cardiovasc Genet. 2012;5(6):621–9.
Akutsu K, Morisaki H, Okajima T, Yoshimuta T, Tsutsumi Y, Takeshita S, et al. Genetic analysis of young adult patients with aortic disease not fulfilling the diagnostic criteria for Marfan syndrome. Circ J. 2010;74(5):990–7.
Regalado ES, Guo DC, Prakash S, Bensend TA, Flynn K, Estrera A, et al. Aortic disease presentation and outcome associated with ACTA2 mutations. Circ Cardiovasc Genet. 2015;8(3):457–64.
Regalado ES, Guo DC, Estrera AL, Buja LM, Milewicz DM. Acute aortic dissections with pregnancy in women with ACTA2 mutations. Am J Med Genet A. 2014;164A(1):106–12.
Yoo EH, Choi SH, Jang SY, Suh YL, Lee I, Song JK, et al. Clinical, pathological, and genetic analysis of a Korean family with thoracic aortic aneurysms and dissections carrying a novel Asp26Tyr mutation. Ann Clin Lab Sci. 2010;40(3):278–84.
Chamney S, McGimpsey S, McConnell V, Willoughby CE. Iris Flocculi as an ocular marker of ACTA2 mutation in familial thoracic aortic aneurysms and dissections. Ophthalmic Genet. 2015;36(1):86–8.
Milewicz DM, Ostergaard JR, Ala-Kokko LM, Khan N, Grange DK, Mendoza-Londono R, et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am J Med Genet A. 2010;152A(10):2437–43.
Meuwissen ME, Lequin MH, Bindels-de Heus K, Bruggenwirth HT, Knapen MF, Dalinghaus M, et al. ACTA2 mutation with childhood cardiovascular, autonomic and brain anomalies and severe outcome. Am J Med Genet A. 2013;161A(6):1376–80.
Moosa AN, Traboulsi EI, Reid J, Prieto L, Moran R, Friedman NR. Neonatal stroke and progressive leukoencephalopathy in a child with an ACTA2 mutation. J Child Neurol. 2013;28(4):531–4.
Richer J, Milewicz DM, Gow R, de Nanassy J, Maharajh G, Miller E, et al. R179H mutation in ACTA2 expanding the phenotype to include prune-belly sequence and skin manifestations. Am J Med Genet A. 2012;158A(3):664–8.
Munot P, Saunders DE, Milewicz DM, Regalado ES, Ostergaard JR, Braun KP, et al. A novel distinctive cerebrovascular phenotype is associated with heterozygous Arg179 ACTA2 mutations. Brain. 2012;135(Pt 8):2506–14.
Al-Mohaissen M, Allanson JE, O'Connor MD, Veinot JP, Brandys TM, Maharajh G, et al. Brachial artery occlusion in a young adult with an ACTA2 thoracic aortic aneurysm. Vasc Med. 2012;17(5):326–9.
Yetman AT, Starr LJ, Bleyl SB, Meyers L, Delaney JW. Progressive aortic dilation associated with ACTA2 mutations presenting in infancy. Pediatrics. 2015;136(1):e262–6.
Amans MR, Stout C, Fox C, Narvid J, Hetts SW, Cooke DL, et al. Cerebral arteriopathy associated with Arg179His ACTA2 mutation. J Neurointerv Surg. 2014;6(9):e46.
Regalado ES, Mellor-Crummey L, De Backer J, Braverman AC, Ades L, Benedict S, et al. Clinical history and management recommendations of the smooth muscle dysfunction syndrome due to ACTA2 arginine 179 alterations. Genet Med. 2018;20(10):1206–15.
Rodríguez-Palomares JF, Teixidó-Tura G, Galuppo V, Cuéllar H, Laynez A, Gutiérrez L, González-Alujas MT, García-Dorado D, Evangelista A. Multimodality Assessment of Ascending Aortic Diameters: Comparison of Different Measurement Methods. J Am Soc Echocardiogr. 2016;29(9):819–826.
Goldstein SA, Evangelista A, Abbara S, Arai A, Asch FM, Badano LP, Bolen MA, Connolly H, Cuéllar-Calàbria H, Czerny M, Devereux RB, Erbel RA, Fattori R, Isselbacher EM, Lindsay JM, McCulloch M, Michelena H, Nienaber CA, Oh JK, Pepi M, Taylor AJ, Weinsaft JW, Zamorano JL, Dietz H, Eagle K, Elefteriades J, Jondeau G, Rousseau H, Schepens M. Multimodality imaging of diseases of the thoracic aorta in adults: from the American Society of Echocardiography and the European Association of Cardiovascular Imaging: endorsed by the Society of Cardiovascular Computed Tomography and Society for Cardiovascular Magnetic Resonance. J Am Soc Echocardiogr. 2015;28(2):119–82.
Evangelista A, Flachskampf FA, Erbel R, Antonini-Canterin F, Vlachopoulos C, Rocchi G, Sicari R, Nihoyannopoulos P, Zamorano J; European Association of Echocardiography; Document Reviewers: Pepi M, Breithardt OA, Plonska-Gosciniak E. Echocardiography in aortic diseases: EAE recommendations for clinical practice. Eur J Echocardiogr. 2010;11(8):645–58.
Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, Casey DE Jr, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation. 2010;121(13):e266–369.
Erbel R, Aboyans V, Boileau C, Bossone E, Bartolomeo RD, Eggebrecht H, et al. 2014 ESC guidelines on the diagnosis and treatment of aortic diseases: document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The task force for the diagnosis and treatment of aortic diseases of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(41):2873–926.
Riambau V, Bockler D, Brunkwall J, Cao P, Chiesa R, Coppi G, et al. Editor’s choice - Management of Descending Thoracic Aorta Diseases: clinical practice guidelines of the European Society for Vascular Surgery (ESVS). Eur J Vasc Endovasc Surg. 2017;53(1):4–52.
Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, et al. 2018 ESC/ESH guidelines for the management of arterial hypertension. Eur Heart J. 2018;39(33):3021–104.
Regitz-Zagrosek V, Roos-Hesselink JW, Bauersachs J, Blomstrom-Lundqvist C, Cifkova R, De Bonis M, et al. 2018 ESC guidelines for the management of cardiovascular diseases during pregnancy. Eur Heart J. 2018;39(34):3165–241.
Oliveri S, Ferrari F, Manfrinati A, Pravettoni G. A Systematic Review of the Psychological Implications of Genetic Testing: A Comparative Analysis Among Cardiovascular, Neurodegenerative and Cancer Diseases. Front Genet. 2018;9:624.
Acknowledgements
We would like to thank Liesbeth Wildero- Van Wouwe for setting up the surveys and coordinating the meetings and Marine Hurard, the VASCERN Project Manager, for management of the HTAD conference calls and meetings.
This work is generated within the European Reference Network for Rare Multisystemic Vascular Diseases (VASCERN) - Project ID: 769036.
Funding
This work was supported by the Dutch Heart Foundation (2014 T007) and by an Erasmus University Rotterdam Fellowship (I.M.B.H. van de Laar).
Author information
Authors and Affiliations
Contributions
All authors (IvdL, EA, BL, EB, LM, MG, MK, JT, JR, KB, GP, BM, ZS, GT, LR, YE, AE, AP, YvK, GJ, JDB) participated in the inventory, and contributed to the discussions. IvdL, EA, GJ, and JDB wrote the manuscript with input from all authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
ACTA2 clinical features.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
van de Laar, I.M.B.H., Arbustini, E., Loeys, B. et al. European reference network for rare vascular diseases (VASCERN) consensus statement for the screening and management of patients with pathogenic ACTA2 variants. Orphanet J Rare Dis 14, 264 (2019). https://doi.org/10.1186/s13023-019-1186-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-019-1186-2