
Abstract
Ichthyosis with confetti (IWC) is an autosomal dominant congenital ichthyosis also known as ichthyosis variegata or congenital reticular ichthyosiform erythroderma. It manifests at birth with generalized ichthyosiform erythroderma or with a collodion baby picture. The erythrodermic and ichthyotic phenotype persists during life and its severity may modify. However, the hallmark of the disease is the appearance, in childhood or later in life, of healthy skin confetti-like spots, which increase in number and size with time. IWC is a very rare genodermatosis, with a prevalence <1/1,000,000 and only 40 cases reported worldwide. The most important associated clinical features include ear deformities, mammillae hypoplasia, palmoplantar keratoderma, hypertrichosis and ectropion. IWC is due to dominant negative mutations in the KRT10 and KRT1 genes, encoding for keratins 10 and keratin 1, respectively. In this context, healthy skin confetti-like spots represent “repaired” skin due to independent events of reversion of keratin gene mutations via mitotic recombination. In most cases, IWC clinical suspicion is delayed until the detection of white skin spots. Clinical features, which may represent hint to the diagnosis of IWC even before appearance of confetti-like spots, include ear and mammillae hypoplasia, the progressive development of hypertrichosis and, in some patients, of adherent verrucous plaques of hyperkeratosis. Altogether the histopathological finding of keratinocyte vacuolization and the nuclear staining for keratin 10 and keratin 1 by immunofluorescence are pathognomonic. Nevertheless, mutational analysis of KRT10 or KRT1 genes is at present the gold standard to confirm the diagnosis. IWC has to be differentiated mainly from congenital ichthyosiform erythroderma. Differential diagnosis also includes syndromic ichthyoses, in particular Netherton syndrome, and the keratinopathic ichthyoses. Most of reported IWC cases are sporadic, but familial cases with autosomal dominant mode of inheritance have been also described. Therefore, knowledge of the mutation is the only way to properly counsel the couples. No specific and satisfactory therapy is currently available for IWC. Like for other congenital ichthyoses, topical treatments (mainly emollients and keratolytics) are symptomatic and offer only temporary relief. Among systemic treatments, retinoids, in particular acitretin, improve disease symptoms in most patients. Although at present there is no curative therapy for ichthyoses, treatments have improved considerably over the years and the best therapy for each patient is always the result of both physician and patient efforts.
Similar content being viewed by others


Review
Disease name/synonyms
Ichthyosis with confetti
Congenital reticular ichthyosiform erythroderma
Ichthyosis variegata
Orphanumber: ORPHA281190
OMIM: 609165
Definition
Ichthyosis with confetti (IWC) [1] is an autosomal dominant congenital ichthyosis also known as ichthyosis variegata [2] or congenital reticular ichthyosiform erythroderma (CRIE) [3], the latter being the disease name recommended in the clinical-genetic classification of inherited ichthyoses, developed by the First Ichthyosis Consensus Conference in 2009 [4]. According to the current classification, IWC belongs to non-syndromic ichthyoses [4]. IWC manifests at birth with generalized ichthyosiform erythroderma [1, 3, 5–16] or with a collodion baby picture [17–19]. The hallmark of this form of ichthyosis is the appearance, in childhood or later in life, of confetti-like spots of pale and normal-appearing skin, which increase in number and size with time [1, 3, 5–19].
Methods
A literature search was performed on PubMed from 1984, when IWC was first described, to April 30, 2015. In addition, the Orphanet database [20] was looked at. The following search terms were used: “ichthyosis with confetti”, “congenital reticular ichthyosiform erythroderma”, “ichthyosis variegata”, “MAUIE syndrome”. We then checked the bibliography of each article to identify additional references. Altogether, 17 reports containing a total of 40 IWC cases were identified.
Epidemiology
IWC is a very rare genodermatosis, with a prevalence <1/1,000,000 [20]. To our knowledge, only 40 IWC cases have been reported (Table 1) [1, 3, 5–19]. The disease may be underdiagnosed as 9 cases have been described between 1984 and 2010 [1, 3, 5, 7, 8, 10–12], while ten cases have been collected by Choate et al. who deciphered the causative gene in 2010 [13] and 21 additional cases have been reported since then [15–17, 19].
Clinical description
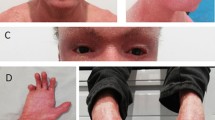
IWC manifests at birth as a non-bullous generalized ichthyosiform erythroderma [1, 3, 10–12, 16, 19] or as a collodion baby [17–19]. The collodion membrane is typically shed within the first days of life [17–19]. The erythrodermic and ichthyotic phenotype (Fig. 1a–d) persists during life, even if its severity may modify [19]. In addition, the extent and type of scaling and hyperkeratosis vary significantly, ranging from white fine scales [11, 12], highly reminiscent of congenital ichthyosiform erythroderma, to verrucous hyperkeratotic adherent plaques more evident on the limbs [17] (Fig. 1a–c). Of note, with time patients develop confetti-like spots of healthy skin (Fig. 1d–e) on the erythrodermic background. These are the hallmark of the disease and frequently suggest the correct diagnosis. Spots start appearing during childhood, gradually increase in number to hundreds [13] and enlarge from 2 to 10 mm up to 4 cm in diameter [1, 11, 13, 14, 17]. Sometimes, they are recognized only after hyperkeratosis shedding due to retinoid therapy [1, 3, 5, 11, 12, 17]. The spots are regularly present on the trunk [6, 17, 18] and may spread to limbs and the rest of the body. Healthy spots seem to follow a distribution gradient across the skin, being largest and more numerous on the neck, décolleté and scapula regions, fewer and smaller on arms and legs, and rare on the face [19]. Moreover, they have been described either as depressed [1, 11] or slightly elevated [6] with respect to the surrounding skin. Due to the presence of many, at instance confluent, confetti-like spots, the adjacent ichthyotic skin may assume a reticulate pattern [3, 6, 12]. Of note, a later age at first appearance of confetti-like spots (22 years), a smaller size (maximum 4 mm) and a predominant involvement of flexures characterize the recently described IWC-II subtype [16].

Major clinical features of ichthyosis with confetti. Severe ichthyosiform erythroderma: massive verrucous hyperkeratosis on the buttocks (a), knees (b) and feet (c) in the absence of retinoid therapy. Confetti-like spots of healthy skin are visible on the trunk and cheek (d, e: black rings) of the same patient. Note the presence of mammillae hypoplasia (d, arrowheads). Ear malformations: hypoplasia of the ear helix (e, f), and lobule (e). Clinical pictures are from a 8-year-old male (a-e) and a 1-year-old female (f)
Table 1 summarizes all the clinical features, when described, in previously reported IWC patients. Ectodermal malformations, specifically ear deformities (Fig. 1e–f) and mammillae hypoplasia (Figs. 1d and 2a) are typical of IWC patients [17–19]. Ear deformities, comprising micropinna (small ears and external auditory canal) and, in a more general way, ear hypoplasia, are already evident at birth [18]. Mammillae hypoplasia has been only reported by Spoerri et al. [19]. However, it can be underestimated as Spoerri noticed that it seems visible on previous published pictures of some IWC patients [3, 12] and a reevaluation of our two patients [17] confirmed its presence (Figs. 1d and 2a).

Major and minor clinical features of ichthyosis with confetti. Mammillae hypoplasia (a, arrowheads), palmoplantar hyperkeratosis (b), hypertrichosis of acral dorsal areas (arrowheads) (c) and back (d), scaling and hyperkeratosis of the scalp (e), large lunulae (f), long nail plates and unguis inflexus (g), severe eyelid ectropion (h). Clinical pictures are taken from a female at the age of 1 year (b, e, f), 2.5 (c) and 2.7 years (a, d) and a male at the age of 5 (g) and 8 (h) years. Note the healthy skin confetti-like spots (a, d: black rings) which became evident after hyperkeratosis shedding due to retinoid therapy
Palmoplantar keratoderma (PPK) is associated with most IWC cases (Fig. 2b) [3, 5, 6, 10–12, 14, 17–19]. It may be characterized by an orange-red color [6, 12], increased palmar skin marks [6, 12, 18] and absent finger dermatoglyphs [14]. Disproportionately severe PPK compared with body hyperkeratosis has been reported in the IWC-II subtype [16].
Hyperpigmented, irregular-shaped macules on the erythrokeratotic skin have been reported in a few cases [3, 9]. More frequently, hyperpigmentation within healthy skin spots has been described [19].
Hypohidrosis with temperature intolerance [18] and pruritus [6, 12, 17, 19] are likely due to the ichthyosiform skin condition.
Hypertrichosis is a frequent and characteristic IWC sign [10, 11, 14, 17, 19]. It can be apparent already in early childhood, and is preferentially localized to dorsal acral areas (Fig. 2c) [17, 19], with long hair even on the back of hands and fingers [14], but can be generalized (Fig. 2d) [17]. Hypertrichosis is confined to areas of ichthyotic skin [19] and never present in confetti-like spots [14]. Scaling usually also involves the scalp (Fig. 2e) [17, 19], sometimes causing alopecia [19]. IWC patients may show reduced eyebrows and eyelashes [19] and diffuse hair loss [11]. Scalp hair heterochromia has been reported [14].
Nails may appear thickened [18], with elongated nail plates and enlarged lunulae (Fig. 2f) [19]. Subungual hyperkeratosis may induce nail curving (Fig. 2g) [3, 18, 19], the so called “unguis inflexus” [19].
Other frequent manifestations of IWC are eclabion (outward lip eversion) and ectropion (out turning of the eyelids) (Fig. 2h) [6, 11, 14, 17–19]. Additional but rarely reported signs of eye involvement are strabismus and nystagmus [19].
Finger length relative to palms may be decreased [19]. Joint contractions of the fingers, caused by marked hyperkeratosis, worsen mobility of the hands [19]. Tautness of the skin may also involve elbows, shoulders, hips and knees, thus resulting in a forced limb flexion and motor impairment with gait abnormality [16–19].
Almost all IWC individuals have small height and weight for age [1, 17, 19], but usually normal neural development. However, peripheral hyperreflexia has been detected by neurologic examination in four patients [15] and psychomotor retardation has been reported in one case [17]. Multiple non-melanoma skin cancers (NMSCs) have been described in three adult IWC patients [15].
The majority of IWC manifestations are present in the two previously reported cases of MAUIE (micropinnae, alopecia universalis, congenital ichthyosis and ectropion) syndrome (Table 1, cases 3 and 4) [7, 8]. These patients showed congenital ichthyosiform erythroderma at birth, normal-appearing skin spots, ear deformity (micropinna or external ear hypoplasia), ectropion, eclabion and nail thickening [7, 8]. At the time of examination, the two adult men also presented a complete absence of hair all over the body [7, 8]. NMSCs were additional findings in both cases [7, 8]. Although not further characterized, these patients may be considered suffering from IWC [2, 15].
Spoerri et al. [19] analysed a cohort of 6 unrelated, genetically characterized, IWC patients and suggested that clinical features of the disease may be grouped into major and minor criteria for diagnosis. Major criteria include all symptoms that the Authors identified as constantly associated with IWC in their cohort, i.e. erythroderma since birth, confetti-like spot appearance, scaling with changing severity, dorsal acral hypertrichosis, hypoplasia of mammillae and malformation of ears. Clinical features which were not always present in every patient were considered as minor criteria. Based on literature data (Table 1), palmoplantar keratoderma and ectropion appear as frequent as the major clinical findings identified by Spoerri et al. [19].
Aetiology
IWC is a disorder of keratins. Keratins are major structural proteins of epithelial cells. They are obligate heterodimers of an acidic type I and neutral-basic type II polypeptide [21, 22]. Keratins have a characteristic expression patterns in normal human epithelial tissues according to the function and body site of each cell type. Some of them can have a very restricted tissue-specificity [23, 24]. For example, differentiating keratinocytes of the epidermis express a particular pair of type I and type II cytokeratins, keratin 10 (K10) and keratin 1 (K1) [25]. All keratins share a common structure composed of a central rod domain, through which they interact to form dimers, flanked by a N- (head) and C-terminal (tail) domain, which are important for elongation and lateral alignment of K1/K10 heterodimers [22, 25, 26]. The C-terminal “tail domain” of K1 and K10 is unusually glycine rich. In addition, the K10 tail shows extensive size polymorphism due to variable numbers and sizes of glycine loops [27]. K1/K10 heterodimers assemble to form the intermediate filaments (IF) cytoskeleton of differentiating epidermal keratinocytes [21].
IWC is due to dominant negative mutations in the K10 gene, KRT10, which maps to chromosome 17q21.2 and consists of 8 exons. The large majority of the 13 KRT10 mutations identified so far represent de novo events. All the mutations are single nucleotide substitutions or small insertions and deletions located in exon 6, intron 6 splice sites and exon 7 (Table 1) [13, 14, 17, 19]. Of note, each mutation results in a C-terminal frameshift that converts the glycine/serine-rich K10 tail to an arginine-rich carboxy-terminal sequence [13]. As arginine-rich motifs are positively charged and encode nuclear localization sequences leading to nuclear entry, mutant K10, and its natural partner K1, accumulates within the nucleus, specifically within nucleoli, which are site of active synthesis of ribosomal RNA [13, 15].
Choate et al. [16] have recently described a new IWC subtype, named IWC-II or IWC-K1, due to a heterozygous de novo single base-pair insertion in the last exon of K1 gene (KRT1) which maps to chromosome 12q13.13. Similarly to KRT10 gene defects, the KRT1 mutation introduces a C-terminal frameshift, which results in the replacement of the last 22 K1 aminoacids by a novel 30-aminoacid peptide. In both IWC subtypes the frameshift peptide is implicated in the dominant negative effect that disrupts the interactions of K1/K10 tails, leading to the collapse of IF network and mislocalization of the mutant proteins to the nucleus [13, 16].
Of note, the KRT10 gene mutations identified in the germline of IWC patients are not found in the DNA extracted from the “confetti-like” spots [13, 14]. Spots are not visible at birth, appear and expand over time, and have normal tissue architecture. Such observations led Choate et al. to investigate white spot-purified DNA using high-density single nucleotide polymorphism genotyping platforms [13]. In paired analysis with blood-purified genomic DNA, each DNA sample from 28 white spots taken from six independent individuals showed homozygous genotypes of a single region of chromosome 17q with copy number equals to two, indicating loss-of-heterozygosity (LOH). The delineated LOH intervals span from a proximal breakpoint to the telomere and varied among samples. However they overlapped and identified the proximal boundary of the KRT10 locus. Thus, white spots represent “repaired” skin due to independent events of reversion of KRT10 mutations via mitotic recombination (Fig. 3). Since white spots can be hundreds and expand over time, the recombination event is expected to occur at a high frequency and to confer selective growth advantage to revertant cell clones. A similar copy-neutral mechanism of genetic recombination was discovered in IWC-II. In this subtype the revertant tissue DNA from several revertant spots harbors overlapping LOH intervals on chromosome 12 with breakpoints proximal to the KRT1 locus, indicating frequent somatic reversion of the KRT1 mutation via mitotic recombination [16].

Schematic of the copy-neutral mechanism of mitotic recombination leading to revertant mosaicism in a patient affected with ichthyosis with confetti. The patient is heterozygous for a pathogenic mutation in the KRT10 locus (17q21.2, indicated by a horizontal bar within the long arm of the chromosome). During somatic cell division, the parental affected keratinocyte bears homologous chromosomes with wild-type (wt) and mutant (mut) genotypes (a). Following a crossover event proximal to the KRT10 locus both homologous chromosomes will have one chromatid carrying each genotype (b). Then, daughter cells receiving the same allele will be homozygous at that locus for either wild-type (revertant cell) or mutant (affected cell) genotype (c). A single revertant daughter cell will expand and give rise to the “confetti-like” skin spot
Diagnosis
IWC diagnosis is based on dermatologic evaluation, and personal and family history. Skin manifestations, in particular the presence of verrucous hyperkeratotic plaques and associated findings such as ear and mammillae hypoplasia and hypertrichosis, should be carefully evaluated. However, IWC clinical suspicion is usually delayed until the detection of white skin spots. In addition, family history is rarely informative, as most cases are sporadic. Laboratory analyses are mandatory to confirm the diagnosis and represent the only mean to assess it before the appearance of healthy skin macules.
At first, biopsy should be taken for histological examination from affected skin. Histopathologically, the ichthyotic skin in IWC shows the following alterations of the epidermis: hyperkeratosis with focal parakeratosis (retention of cell nuclei in the stratum corneum), a reduced granular layer and pronounced perinuclear vacuolization of scattered keratinocytes in the suprabasal epidermal layers (Fig. 4a, b) [6, 11, 13, 14, 17]. The presence of binucleated keratinocytes has also been reported [3, 6]. IWC-II skin shows a thickened stratum corneum without parakeratosis, milder perinuclear vacuolization with rare binucleated cells and prominent coarse keratohyalin granules, the latter being absent in the classical IWC form [16]. On the other hand, a skin biopsy taken within a spot of normal appearing skin always reveals normal architecture. Overall, the histopathological findings of IWC, and in particular the presence of keratinocyte vacuolization, are characteristic and should prompt further diagnostic investigations. Specifically, immunopathological analysis of K10 and K1 expression in the epidermis should be performed. This is carried out by immunofluorescence labelling on formalin-fixed, paraffin-embedded skin sections with commercially available monoclonal antibodies to K10 and K1.

Histology and immunofluorescence findings in ichthyosis with confetti caused by KRT10 mutation. The epidermis appears acanthotic and hyperkeratotic with parakeratosis, a reduced granular layer and cytoplasmic vacuolization in suprabasal keratinocytes (a). Higher magnification of the inset depicted in panel (a) highlighting the perinuclear vacuolization in suprabasal keratinocytes (b). Immunofluorescence labelling for keratin 10 (K10) shows reduced cytoplasmic staining in epidermal suprabasal cell layers, dot-like labelling of numerous nuclei in suprabasal epidermis and bright perinuclear rings (c). Higher magnification of the inset depicted in panel c showing that nuclear labeling is mainly localised to nucleoli (d). Haematoxylin-eosin staining (a and b), original magnification × 200 (a). Nuclear DAPI counterstaining (c and d), original magnification × 200 (c)
Immunofluorescence labelling for K10 in IWC shows a marked reduction of the cytoplasmic staining in epidermal suprabasal cell layers, collapsed filament networks visible as bright perinuclear rings in scattered keratinocytes and a dot-like labelling of numerous nuclei in suprabasal epidermis (Fig. 4c, d), while in the healthy control skin K10 is only localized to the keratinocyte cytoplasm [13, 14, 17]. Similar findings have also been described for the partner keratin, K1 [13]. Counterstaining with the nucleolar markers fibrillarin shows that K10 and also K1 localize to the nucleolus [13]. Immunofluorescence labelling for K1 in the IWC-II subtype reveals perinuclear rings in affected skin [16]. The nuclear staining for K10 has been described only in IWC epidermis and can be considered a diagnostic hallmark. In addition, K10 immunolabelling performed on archival skin biopsies can allow retrospective diagnosis [17].
At the ultrastructural level, the most significant findings in IWC are the presence of binuclear keratinocytes and cup- or bowl-like perinuclear masses of granular material in the vacuolized superficial keratinocytes [3, 6, 9, 12]. A closer examination demonstrates that this material is composed by thin interlacing filaments [6, 9, 12]. Additional electron microscopy abnormalities include marked reduction in the total number of keratin tonofilaments in cytoplasm, poorly formed desmosomes and sparse and tapered bundles of keratin filaments attached to desmosomes [13].
Altogether, histopathological and immunofluorescence findings of IWC are pathognomonic. Electron microscopy may further support the diagnosis. Finally, mutational analysis of KRT10 and KRT1 genes is at present the gold standard to confirm IWC diagnosis and provides a firm basis for genetic counseling of affected individuals and families. Figure 5 summarizes the proposed diagnostic algorithm for IWC.

Proposed diagnostic algorithm for ichthyosis with confetti. Medical history and physical examination raise diagnosis suspicion. Histological examination and keratin immunolocalization show specific findings, such as suprabasal keratinocyte vacuolization and nuclear keratin staining, respectively. Electron microscopy may further support the diagnosis. Finally, mutational analysis of KRT10/KRT1 genes represents the gold standard to confirm the diagnosis. Findings relevant to differential diagnosis are in bold
Differential diagnosis
IWC at its onset is usually confused with the erythrodermic form of autosomal recessive congenital ichthyosis (ARCI), congenital ichthyosiform erythroderma (CIE) [4, 28]. The following clinical criteria should be used for the differential diagnosis of IWC: i. the evolution over time of skin manifestations, in particular a progressive worsening of the hyperkeratosis may be suggestive of IWC; ii. the type of scaling and hyperkeratosis, as severe, adherent hyperkeratotic plaques are not usually seen in CIE; iii. the presence of additional dermatologic features, in particular ear and mammillae hypoplasia, and hypertrichosis; iv. the family history which may point toward a particular mode of inheritance and, above all, v. the appearance of confetti-like spots which drives the correct diagnosis. Histopathological and immunopathological features also allow to differentiate IWC from CIE. Indeed, keratinocyte vacuolization and K10 reduced cytoplasmic labeling and nuclear staining are not found in CIE. Table 2 summarizes differences between CIE and IWC [4, 29, 30]. IWC can also be differentiated from syndromic ichthyoses, in particular Netherton syndrome [31] which manifests at birth with ichthyosiform erythroderma, but also presents specific hair alterations (i.e. trichorrhexis invaginata). The absence of overt skin blistering in infancy allows to distinguish IWC from other keratinopathic ichthyoses [4].
Genetic counselling
Genetic counselling in IWC is usually required by the dermatologist who has in charge the affected patient/family. It is aimed to: i. confirm the diagnosis, ii. ascertain disease recurrence risk in future pregnancies, and iii. plan prenatal diagnosis. Histopathological and immunopathological analyses should be considered as first steps in the diagnostic pathway as they permit to put in evidence the peculiar tissue architecture and keratin nuclear staining of affected skin, thus allowing early diagnosis before revertant skin spots become evident [17]. Then, molecular analysis of KRT10 and, if IWC-II subtype is suspected, of KRT1 gene is aimed at identifying the heterozygous frameshift mutation in the hotspot genomic region coding for the C-terminal tail of keratins 10 and 1. Mutation identification provides confirmation of the disease subtype and allows to support the autosomal dominant mode of inheritance, which has a 50 % recurrence risk for subsequent pregnancies of affected individuals. Knowledge of the mutation also provides a tool to monitor recurrence risk by prenatal testing. However, IWC often manifests as a sporadic case, making difficult the diagnosis in infancy when “confetti like” spots are not yet evident and, thus, CIE is usually suspected. The latter is classically inherited as an autosomal recessive trait, which bears a 25 % recurrence risk for subsequent pregnancies, while sporadic IWC has a lower risk of recurrence as it results from de novo event(s) during gametogenesis or at conception. Therefore, molecular analysis is the only way to properly counsel the couples in sporadic IWC. Molecular confirmation after diagnosis by immunofluorescence also provides a prenatal diagnostic tool in selected pregnancies to exclude gonadal mosaicism.
Management including treatment
Due to the rarity of the disease, there are no controlled studies on IWC management. In general, care of the disease follows the rules for treatment of ARCI patients.
Like for other congenital ichthyosis forms, the management of IWC in the neonate, presenting either with ichthyosiform erythroderma or colloidon baby, requires admission to a neonatal intensive care unit and a multidisciplinary approach, with nursing staff, neonatologists, dermatologists and other specialists [32]. In particular, the skin barrier function of the newborn is greatly compromised and ongoing water loss can lead to dehydration and electrolyte imbalance [33]. Moreover defective barrier allows bacteria and yeast colonization, increasing risks of infection and sepsis [17]. Thus, neonate care includes providing a temperature controlled environment via a humidified incubator, and frequent application of lubricants such as petrolatum-based products under sterile conditions [32, 33]. Urinary output, electrolytes and weight should be monitored and managed with intravenous hydration, electrolyte repletion and additional nutritional input when necessary. Close monitoring and prompt systemic treatment with antibiotics for infection is essential.
Therapy of infants, children and adults affected with ichthyosis, including IWC, is not curative but rather aimed at symptom relieving. Daily bathing with water and sodium bicarbonate alkalinizes the epidermis and is helpful for many patients, especially for mechanical scale removal. If sodium bicarbonate is not tolerated, rice starch can be used and is preferred in the first year of life. During bathing, gentle mechanical keratolysis can be obtained using sponges or microfiber cloths. Bland emollients such as petrolatum-based products should be applied several times a day, especially after bathing, to prevent drying. Starting from the second year of life, keratolysis may be obtained through alpha-hydroxy (e.g. lactic and glycolic) acids and urea or combination of these ingredients. Alpha-hydroxy acids-based lotions reduce corneocyte adhesion and skin thickness and relieve itching. Urea creams decrease dryness and scaling and improve skin permeability barrier function by regulating epidermal gene activity [34]. Of note, topical urea and alpha-hydroxy acids can also cause skin irritation and should be applied with caution and only on stubborn areas in the first three years of life. Moreover, alpha-hydroxy acids and urea creams may cause systemic absorption with metabolic acidosis or elevating plasma urea levels, respectively, when applied over large body surfaces in infants and toddlers [35, 36]. The use of a topical retinoid, tazarotene cream, has been reported in a single IWC patient, specifically on skin areas presenting marked hyperkeratosis and around eyes to prevent ectropion [18]. Although topical tazarotene is increasingly used in ichthyosis patients [37, 38], it can be irritating [37, 39] and thus not tolerated in erythrodermic forms of the disease. Its use in IWC should be therefore cautiously considered for selected skin areas, such as hyperkeratotic ones or eyelids, and tolerance evaluated in each patient. Despite low systemic absorption [40], rules for pregnancy avoidance during systemic retinoid treatments should also be applied to tazarotene topical therapy.
Due to disease severity, oral retinoids, at first etretinate [1, 3, 6] and then acitretin [11, 17], have been used in most IWC patients with positive results, including extension of the areas of normal confetti-like skin [1, 6, 17]. Systemic retinoids decrease cell proliferation, thus thinning the stratum corneum, normalize keratinocyte differentiation, facilitate desquamation through downregulation of desmosomal proteins, and have anti-inflammatory properties [41, 42]. Of note, they have been also shown to downregulate K10 expression [41, 43]. However, the lowest possible dose of a systemic retinoid producing desired clinical outcomes needs to be titrated. Few IWC patients require more than 0.5 mg/kg of acitretin once a day [11, 17]. Importantly, in women of childbearing age pregnancy must be excluded by negative pregnancy test (serum levels of human chorionic gonadotropin) within 2 weeks prior to therapy and effective contraception has to be initiated 4 weeks before, during and for 3 years after retinoid therapy [44].
Acute adverse effects of systemic retinoids include skin irritation, fragility and tenderness, mucocutanous toxicities (in particular cheilitis, epistaxis, eye irritation), hair loss, and laboratory abnormalities in blood cell counts, transaminases and serum lipids [44]. Complete blood count, liver function, cholesterol and lipid levels must be monitored.
Possible long-term retinoid complications include premature epiphyseal closure, hyperostosis and tendon calcifications [42, 44]. Although not reported to date in IWC, they represent an indication to limit the duration of retinoid therapy or to periodically interrupt their administration, e.g. during summertime. Baseline radiographs should also be obtained before programming long-term retinoid therapy. Frequency of X-ray follow-ups, complemented if required with bone density scan, will depend on baseline findings and on clinical manifestations and symptoms. In addition, it has been hypothesized that retinoids might interfere with vitamin D metabolism, as their vitamin A-like biologic activity may hinder vitamin D action [45]. Of note, vitamin D deficiency has been described in most types of ichthyoses, in particular severe forms, and attributed to different factors, such as poor sunlight penetration due to hyperkeratotic skin and limited sun exposure for cosmetic reasons or sun-induced pain and pruritus [46]. Therefore, it is currently recommended to screen all congenital ichthyoses for calcium, phosphorus and vitamin D3 levels and to provide supplementation if required, in order to treat vitamin D3 deficiency and prevent possible complications, such as osteoporosis and rickets. This recommendation should apply also to IWC, although no data on ions and vitamin D3 levels in these patients have been reported to date.
For infection control in IWC, topical antiseptics and antimicrobials must be combined with appropriate systemic therapy. Among other IWC complications, severe ectropion should be managed by an ophthalmologist to avoid desiccation, e.g. through regular application of liquid tears and eye lubricants, and plastic surgery may be required. Routine otolaryngologist visits are required to remove desquamated skin from ear canals. Contractures and gait abnormalities should be evaluated by a combined team of orthopedists, neurologists and plastic surgeons and physical therapy should be instituted for patients with these findings. Psychological support may be of benefit to both patients and their families. Regular examination for skin cancer risk is recommended in adulthood.
Prognosis
IWC, as most other heritable ichthyoses, is a condition that requires significant attention in the neonatal period. Although at present there is no curative therapy for all forms of ichthyoses, including IWC, treatments have improved considerably over the years. Topical medications can reduce scaling and, thus, patient quality of life. In addition, retinoid therapies decrease scaling and hyperkeratosis, and may help in preventing and treating ectropion. Nevertheless, treatments remain symptomatic and topical ones are time-consuming, challenging patient and caregiver compliance. Therefore, personalized multidisciplinary care plans should be set up and regularly up-dated by combined dermatologist and patient efforts. In this context, the support of patient organizations (see Additional file 1: Table S1) will help affected individuals and their families to better cope with the disease.
Unresolved questions
There have been recently major advances in our understanding of the genetic basis of IWC, with the identification of KRT10 and KRT1 mutations. However, several questions remain unanswered. First, although IWC is considered a nonsyndromic ichthyosis, it is also true that a spectrum of ectodermal malformations and neurological findings leading to different degrees of disability have been described in affected individuals without any apparent genotype-phenotype correlation. Indeed, among 40 previously reported IWC patients, 7 showed both ear deformities and mammillae hypoplasia [17, 19], whereas 6 presented ear deformities [7, 8, 18] or mammillae hypoplasia [3, 12, 19] alone (Table 1). Moreover, 4 out of 40 IWC cases were diagnosed with peripheral hyperreflexia [15] (Table 1). This led some Authors to suggest a reclassification of IWC as a syndromic ichthyosis [19].
Mutations affecting K10/K1 heterodimers trigger keratinocyte hyperproliferation [24], which explains acanthosis and hyperkeratosis in IWC patients. Although K10/K1 are not expressed in nail bed, they are detectable in all other regions of the nail unit, including eponychium, hyponychium and the apical matrix [47, 48]. Therefore KRT10/KRT1 mutations may also cause nail dystrophies in IWC patients. In contrast, K10/K1 are absent from the hair follicle, except for the infundibulum [47], thus raising the question of pathogenetic mechanisms underlying hypertrichosis in IWC. Hypertrichosis is limited to areas of ichthyotic skin and Spoerri et al. hypothesized that it may be due to inflammation and hyperemia, as it happens in postcast hypertrichosis [19].
Another open issue is the timing of mitotic recombination leading to the generation of revertant cells. Spots become evident as early as 2.5 years of age [17] and can reach a diameter of 4 cm in adulthood [13]. Since a white spot represents the expansion of a single homozygotic revertant daughter cell in the absence of phenotypic evidence of homozygotic mutant daughter cell [13], the recombination event should involve the epidermal stem cell unit, the specification of which is thought to occur during embryogenesis [49]. This leads to think that generation of revertant cells might begin already in fetal skin. Moreover, as mutant K10 is thought to be involved in triggering the recombination event, KRT10 should be transcriptionally active in the epidermal stem cell. However, evidence of KRT10 expression in epidermal stem cells is still quite limited [50].
A further intriguing question concerns the extremely high rate of mitotic recombination. As different types of keratin mutations lead to keratinization diseases without revertant mosaicism, this implicates the C-terminal frameshift peptide of mutant K10 and K1, and its nuclear localization in the elevated rate of reversion in IWC [15]. Interestingly, revertant spots associated with IWC-II are lesser in number and smaller in size compared to classic IWC and harbor clinically apparent affected skin islands [16]. Moreover, spot-derived keratinocytes of IWC-II give rise to mixed cultures of revertant and mutant cells, thus revertant cells seem to have a lesser growth selective advantage [16]. Finally, nuclear K1 staining is clearly evident in epithelial cells transfected with mutant K1, but not in IWC-II patient epidermis [16]. The explanation for these observations might be dual: i. the sequence of the mutant C-terminal frame-shifted K1 peptide harbors very few positive-charged residues compared to mutant K10 and ii. only the K10 protein has been linked to cell cycle control [24, 51, 52], suggesting that mutant K10 might gain and/or loss an activity that interferes with this process. However, the relationship between IWC KRT10/KRT1 mutations and the frequency of mitotic recombination events leading to revertant cells remains to be determined.
Adult individuals with IWC may also have a higher risk to develop NMSCs [7, 8, 15]. Although such risk should be carefully re-evaluated by long term patient follow-up, tumour susceptibility in IWC may have a genetic rationale related to the mechanism of somatic mosaicism. Indeed, LOH occurring on chromosome 17q not only involves the KRT10 locus but can also reduce to homozygosity mutations and/or variants in epithelial cancer susceptibility loci distal to the KRT10 allele. For example, within chromosomal band 17q21.31 several of these loci have been mapped, including the BRCA1 tumor suppressor [53, 54]. Homozygotic revertant cells might have a selective growth advantage also as a consequence of LOH events in tumor suppressor loci. However, available data on BRCA1 mutations and non-melanoma skin cancer show non-significant association [54]. In addition, it is not known whether squamous cell carcinomas reported in IWC individuals developed from revertant or erythrokeratotic skin. Finally, NMSCs have been reported also in other ichthyoses, in particular erythrodermic ones, which do not present revertant mosaicism [55, 56].
Future perspectives
Revertant skin spots could be exploited in future as a cell source for a “natural gene therapy” approach in IWC. The feasibility of ex-vivo gene therapy for genetic skin disorders has been demonstrated by the successful engraftment of LAMB3 cDNA retrovirally-corrected epidermal sheets and maintenance of a functional epidermis in a 36-year old male with non-Herlitz junctional EB [57, 58]. However, concerns remain regarding the safety of this approach as there is an oncogenic potential related to random insertion of retroviral vectors [59]. By contrast, the natural occurrence of revertant mosaicism creates a unique opportunity for therapy in patients, because the presence of reverted cells circumvents the need for viral vectors. Persistent ulcers in a patient with non-Herlitz junctional EB, caused by mutations in the LAMB3 gene, have been recently treated by transplantation of punch biopsy specimens taken from one of his revertant patches with complete re-epithelialization and restoration of a healthy phenotype and genotype in the grafted areas [60]. Stable reversion of the EB phenotype by transplantation of revertant skin in this patient may be a pioneering work for the development of the “revertant cell therapy” for other genetic diseases with somatic revertant mosaicism, such as IWC. To date, the only attempt to utilize revertant cell therapy in a clinical setting was performed in an individual with non-Herlitz junctional EB associated with mutations in COL17A1 who presented revertant skin patches [61]. In this case, revertant keratinocytes were isolated and expanded into epidermal sheets that were subsequently grafted back onto the patient. However fewer than 3 % of the cells remained corrected in the graft and there was no clinically relevant outcome.
The persistence of revertant clones in patients with revertant mosaicism indicates that reversion events occur in epidermal stem cells and that naturally-corrected stem cell clones may be under strong positive selection [13, 62]. Therefore, efficacy and long-term persistence of the regenerated epidermis after autotransplantation requires epidermal stem cell maintenance in cultured sheets and their grafting onto the patients [63]. We hypothesize that, with optimized culture conditions, the “revertant cell therapy” might be applied in future to IWC. Nevertheless, in this scenario, the possibility of homozygosity for mutations in tumor suppressors should be carefully considered together with the potential therapeutic advantage of reversion.
Conclusions
IWC is a very rare genodermatosis and represents the most impressive example of revertant somatic mosaicism. The IWC phenotype is characterized by a wide spectrum of clinical features, in addition to erythrodermic ichthyosis. The recent discoveries in IWC genetics have led to a better understanding of disease aetiology and made available molecular diagnostic tools. Disease care requires a multidisciplinary approach, but remains symptomatic. However, based on current scientific and technological progresses, new therapeutic strategies potentially able to cure skin manifestations might be developed in future.
Consent
Written informed consent for patient reexamination for the present review and for patient data and image publication was obtained from patients’ parents.
Abbreviations
- IWC:
-
Ichthyosis with confetti
- CRIE:
-
Congenital reticular ichthyosiform erythroderma
- CIE:
-
Congenital ichthyosiform erythroderma
- MAUIE:
-
Micropinnae, alopecia universalis, congenital ichthyosis and ectropion
- PPK:
-
Palmoplantar keratoderma
- NMSC:
-
Non-melanoma skin cancer
- K/KRT :
-
Keratin (protein/gene)
- IF:
-
Intermediate filaments
- LOH:
-
Loss of heterozygosity
- ARCI:
-
Autosomal recessive congenital ichthyosis
References
Camenzind M, Harms M, Chavaz P, Saurat JH. Confetti ichthyosis. Ann Dermatol Venereol. 1984;111:675–6.
Happle R, Küster W. Ichthyosis variegata: a new name for a neglected disease. J Am Acad Dermatol. 1997;36:500.
Marghescu S, Anton-Lamprecht I, Rudolph PO, Kaste R. Congenital reticular ichthyosiform erythroderma. Hautarzt. 1984;35:522–9.
Oji V, Tadini G, Akiyama M, Blanchet Bardon C, Bodemer C, Bourrat E, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol. 2010;63:607–41.
Rufli T, Schneider BV, Schnyder UW. Non-bullous erythroderma (congenital) ichthyosiforme with perinuclear shells. Hautarzt. 1990;41:442–7.
Brusasco A, Tadini G, Cambiaghi S, Ermacora E, Grimalt R, Caputo R. A case of congenital reticular ichthyosiform erythroderma-ichthyosis ‘en confettis’. Dermatology. 1994;188:40–5.
Elbaum DJ, Kurz G, MacDuff M. Increased incidence of cutaneous carcinomas in patients with congenital ichthyosis. J Am Acad Dermatol. 1995;33:884–6.
Hendrix Jr JD, Patterson JW, Greer KE. Skin cancer associated with ichthyosis: the MAUIE syndrome. J Am Acad Dermatol. 1997;37:1000–2.
Brusasco A, Cambiaghi S, Tadini G, Berti E, Caputo R. Unusual hyperpigmentation developing in congenital reticular ichthyosiform erythroderma (ichthyosis variegata). Br J Dermatol. 1998;139:893–6.
Metze D, Raghunath M, Traupe H. Congenital reticular ichthyosiform erythroderma: a histologic, immunohistochemical and ultrastructural study of a rare keratinization disorder. J Invest Dermatol. 2000;115:923.
Torrelo A, Marrero MD, Mediero IG, Martínez MA, Zambrano A. Progressive macular leucoderma in a patient with congenital ichthyosiform erythroderma. Br J Dermatol. 2001;144:1280–2.
Krunic AL, Palcesky D, Busbey S, Medenica M. Congenital reticular ichthyosiform erythroderma-ichthyosis variegata: a case report and review of the literature. Acta Derm Venereol. 2003;83:36–9.
Choate KA, Lu Y, Zhou J, Choi M, Elias PM, Farhi A, et al. Mitotic recombination in patients with ichthyosis causes reversion of dominant mutations in KRT10. Science. 2010;330:94–7.
Burger B, Spoerri I, Schubert M, Has C, Itin PH. Description of the natural course and clinical manifestations of ichthyosis with confetti caused by a novel KRT10 mutation. Br J Dermatol. 2012;166:434–9.
Choate KA, Milstone LM. Phenotypic expansion in ichthyosis with confetti. JAMA Dermatol. 2015;151:15–6.
Choate KA, Lu Y, Zhou J, Elias PM, Zaidi S, Paller AS, et al. Frequent somatic reversion of KRT1 mutations in ichthyosis with confetti. J Clin Invest. 2015;125:1703–7.
Diociaiuti A, Fortugno P, El Hachem M, Angelo C, Proto V, De Luca N, et al. Early immunopathological diagnosis of ichthyosis with confetti in two sporadic cases with new mutations in keratin 10. Acta Derm Venereol. 2014;94:579–82.
Long MC. Ichthyosis with confetti: a rare diagnosis and treatment plan. BMJ Case Rep. 2014. doi:10.1136/bcr-2014-204509.
Spoerri I, Brena M, De Mesmaeker J, Schlipf N, Fischer J, Tadini G, et al. The phenotypic and genotypic spectra of ichthyosis with confetti plus novel genetic variation in the 3' end of KRT10: from disease to a syndrome. JAMA Dermatol. 2015;151:64–9.
Orphanet. The portal for rare diseases and orphan drugs. http://www.orpha.net/consor/cgi-bin/index.php. Accessed on April 4, 2015.
Moll R, Divo M, Langbein L. The human keratins: biology and pathology. Histochem Cell Biol. 2008;129:705–33.
Chamcheu JC, Siddiqui IA, Syed DN, Adhami VM, Liovic M, Mukhtar H. Keratin gene mutations in disorders of human skin and its appendages. Arch Biochem Biophys. 2011;508:123–37.
Haines RL, Lane EB. Keratins and disease at a glance. J Cell Sci. 2012;125:3923–8.
Pan X, Hobbs RP, Coulombe PA. The expanding significance of keratin intermediate filaments in normal and diseased epithelia. Curr Opin Cell Biol. 2013;25:47–56.
McLean WH, Irvine AD. Disorders of keratinisation: from rare to common genetic diseases of skin and other epithelial tissues. Ulster Med J. 2007;76:72–82.
Toivola DM, Boor P, Alam C, Strnad P. Keratins in health and disease. Curr Opin Cell Biol. 2015;32:73–81.
Korge BP, Gan SQ, McBride OW, Mischke D, Steinert PM. Extensive size polymorphism of the human keratin 10 chain resides in the C-terminal V2 subdomain due to variable numbers and sizes of glycine loops. Proc Natl Acad Sci U S A. 1992;89:910–4.
Traupe H, Fischer J, Oji V. Nonsyndromic types of ichthyoses - an update. J Dtsch Dermatol Ges. 2014;12:109–21.
Grall A, Guaguere E, Planchais S, Grond S, Bourrat E, Hausser I, et al. PNPLA1 mutations cause autosomal recessive congenital ichthyosis in golden retriever dogs and humans. Nat Genet. 2012;44:140–7.
Radner FP, Marrakchi S, Kirchmeier P, Kim GJ, Ribierre F, Kamoun B, et al. Mutations in CERS3 cause autosomal recessive congenital ichthyosis in humans. PLoS Genet. 2013;9:e1003536.
Hovnanian A. Netherton syndrome: skin inflammation and allergy by loss of protease inhibition. Cell Tissue Res. 2013;351:289–300.
Craiglow BG. Ichthyosis in the newborn. Semin Perinatol. 2013;37:26–31.
Dyer JA, Spraker M, Williams M. Care of the newborn with ichthyosis. Dermatol Ther. 2013;26:1–15.
Grether-Beck S, Felsner I, Brenden H, Kohne Z, Majora M, Marini A, et al. Urea uptake enhances barrier function and antimicrobial defense in humans by regulating epidermal gene expression. J Invest Dermatol. 2012;132:1561–72.
Oji V, Traupe H. Ichthyosis: clinical manifestations and practical treatment options. Am J Clin Dermatol. 2009;10:351–64.
Rodríguez-Pazos L, Ginarte M, Vega A, Toribio J. Autosomal recessive congenital ichthyosis. Actas Dermosifiliogr. 2013;104:270–84.
Hofmann B, Stege H, Ruzicka T, Lehmann P. Effect of topical tazarotene in the treatment of congenital ichthyoses. Br J Dermatol. 1999;141:642–6.
Craiglow BG, Choate KA, Milstone LM. Topical tazarotene for the treatment of ectropion in ichthyosis. JAMA Dermatol. 2013;149:598–600.
Stege H, Hofmann B, Ruzicka T, Lehmann P. Topical application of tazarotene in the treatment of nonerythrodermic lamellar ichthyosis. Arch Dermatol. 1998;134:640.
Nguyen V, Cunningham BB, Eichenfield LF, Alió AB, Buka RL. Treatment of ichthyosiform diseases with topically applied tazarotene: risk of systemic absorption. J Am Acad Dermatol. 2007;57(5 Suppl):S123–5.
Torma H. Regulation of keratin expression by retinoids. Dermatoendocrinol. 2011;3:136–40.
Digiovanna JJ, Mauro T, Milstone LM, Schmuth M, Toro JR. Systemic retinoids in the management of ichthyoses and related skin types. Dermatol Ther. 2013;26:26–38.
Li H, Torma H. Retinoids reduce formation of keratin aggregates in heat-stressed immortalized keratinocytes from an epidermolytic ichthyosis patient with a KRT10 mutation. Acta Derm Venereol. 2013;93:44–9.
Ormerod AD, Campalani E, Goodfield MJ. BAD Clinical Standards Unit. British Association of Dermatologists guidelines on the efficacy and use of acitretin in dermatology. Br J Dermatol. 2010;162:952–63.
Neema S, Mukherjee S, Vasudevan B, Verma R, Moorchung N, Chatterjee M. Vitamin D deficiency after oral retinoid therapy for ichthyosis. Pediatr Dermatol. 2015. doi: 10.1111/pde.12614.
Frascari F, Dreyfus I, Rodriguez L, Gennero I, Ezzedine K, Salles JP, et al. Prevalence and risk factors of vitamin D deficiency in inherited ichthyosis: a French prospective observational study performed in a reference center. Orphanet J Rare Dis. 2014;9:127.
Perrin C, Langbein L, Schweizer J. Expression of hair keratins in the adult nail unit: an immunohistochemical analysis of the onychogenesis in the proximal nail fold, matrix and nail bed. Br J Dermatol. 2004;151:362–71.
Fleckman P, Jaeger K, Silva KA, Sundberg JP. Comparative anatomy of mouse and human nail units. Anat Rec (Hoboken). 2013;296:521–32.
Strachan LR, Ghadially R. Tiers of clonal organization in the epidermis: the epidermal proliferation unit revisited. Stem Cell Rev. 2008;4:149–57.
Jensen UB, Lowell S, Watt FM. The spatial relationship between stem cells and their progeny in the basal layer of human epidermis: a new view based on whole-mount labelling and lineage analysis. Development. 1999;126:2409–18.
Paramio JM, Segrelles C, Ruiz S, Jorcano JL. Inhibition of protein kinase B (PKB) and PKC zeta mediates keratin K10-induced cell cycle arrest. Mol Cell Biol. 2001;21:7449–59.
Chen J, Cheng X, Merched-Sauvage M, Caulin C, Roop DR, Koch PJ. An unexpected role for keratin 10 end domains in susceptibility to skin cancer. J Cell Sci. 2006;119:5067–76.
Permuth-Wey J, Lawrenson K, Shen HC, Velkova A, Tyrer JP, Chen Z, et al. Identification and molecular characterization of a new ovarian cancer susceptibility locus at 17q21.31. Nat Commun. 2013;4:1627.
Gumaste PV, Penn LA, Cymerman RM, Kirchhoff T, Polsky D, McLellan B. Skin cancer risk in BRCA1/2 mutation carriers. Br J Dermatol. 2015;172:1498–506.
Natsuga K, Akiyama M, Shimizu H. Malignant skin tumours in patients with inherited ichthyosis. Br J Dermatol. 2011;165:263–8.
Guerra L, Fortugno P, Sinistro A, Proto V, Zambruno G, Didona B, et al. Betapapillomavirus in multiple non-melanoma skin cancers of Netherton syndrome: Case report and published work review. J Dermatol. 2015;42:786–94.
Mavilio F, Pellegrini G, Ferrari S, Di Nunzio F, Di Iorio E, Recchia A, et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat Med. 2006;12:1397–402.
De Rosa L, Carulli S, Cocchiarella F, Quaglino D, Enzo E, Franchini E, et al. Long-term stability and safety of transgenic cultured epidermal stem cells in gene therapy of junctional epidermolysis bullosa. Stem Cell Reports. 2013;2:1–8.
Lai-Cheong JE, McGrath JA, Uitto J. Revertant mosaicism in skin: natural gene therapy. Trends Mol Med. 2011;17:140–8.
Gostynski A, Pasmooij AM, Jonkman MF. Successful therapeutic transplantation of revertant skin in epidermolysis bullosa. J Am Acad Dermatol. 2014;70:98–101.
Gostynski A, Deviaene FC, Pasmooij AM, Pas HH, Jonkman MF. Adhesive stripping to remove epidermis in junctional epidermolysis bullosa for revertant cell therapy. Br J Dermatol. 2009;161:444–7.
Kiritsi D, Garcia M, Brander R, Has C, Meijer R, Escámez MJ, et al. Mechanisms of natural gene therapy in dystrophic epidermolysis bullosa. J Invest Dermatol. 2014;134:2097–104.
Guerra L, Dellambra E, Panacchia L, Paionni E. Tissue engineering for damaged surface and lining epithelia: stem cells, current clinical applications and available engineered tissues. Tissue Eng Part B Rev. 2009;15:91–112.
Acknowledgements
We thank patient families for contributing to and supporting our study. Authors also acknowledge the Italian Ministry of Health (Ricerca Corrente Program) and Association Athina Ichtyose Monaco for the support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have competing interests.
Authors’ contributions
LG performed the literature search and wrote the manuscript. DC participated in writing and reviewing the “aetiology”, “genetic counseling” and “unresolved questions” sections and provided molecular expertise. AD and MEH contributed to the clinical, diagnostic and therapeutic sections. GZ provided clinical and immunopathological advice, and supervised and critically revised the entire manuscript. The figures are from Istituto Dermopatico dell’Immacolata and Bambino Gesù Children's Hospital. All authors read and approved the final manuscript.
Additional file
Additional file 1: Table S1.
Worldwide ichthyosis foundations and patient organizations. (DOC 31 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Guerra, L., Diociaiuti, A., El Hachem, M. et al. Ichthyosis with confetti: clinics, molecular genetics and management. Orphanet J Rare Dis 10, 115 (2015). https://doi.org/10.1186/s13023-015-0336-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-015-0336-4