
Abstract
Background
The Amaranthus genus contains at least 20 weedy and invasive species, including Amaranthus palmeri (palmer’s amaranth) and Amaranthus tuberculatus (tall waterhemp), two species of regulatory concern in North America, impacting production and yield in crops like corn, soybean and cotton. Amaranthus tuberculatus is regulated in Canada with limited establishment, while current climate models predict a range expansion of A. palmeri impacting crop growing areas in Ontario, Quebec and Manitoba. Since many Amaranthus species are similar in their morphology, especially at the seed stage, this demands the development of additional methods that can efficiently aid in the detection and identification of these species. Protein biotyping using Matrix-Assisted Laser Desorption Ionization Time of Flight Mass Spectrometry (MALDI-TOF-MS) has been traditionally used to identify microorganism species, races and pathotypes. Major protein fractions extracted from an organism, ionized and run through a biotyper using mass spectrometry, result in protein spectra that represent a fingerprint at the species or lower taxonomic rank, providing an efficient molecular diagnostics method. Here we use a modified protein biotyping protocol to extract major protein fractions from seeds of the family Brassicaceae to test our protocol, and then implemented the standardized approach in seeds from Amaranthus species. We then created a database of Amaranthus protein spectra that can be used to test blind samples for a quick identification of species of concern.
Results
We generated a protein spectra database with 16 Amaranthus species and several accessions per species, spanning target species of regulatory concern and species which are phylogenetically related or easily confused at the seed stage due to phenotypic plasticity. Testing of two Amaranthus blind sample seed sets against this database showed accuracies of 100% and 87%, respectively.
Conclusions
Our method is highly efficient in identifying Amaranthus species of regulatory concern. The mismatches between our protein biotyping approach and phenotypic identification of seeds are due to absence of the species in the database or close phylogenetic relationship between the species. While A. palmeri cannot be distinguished from A. watsonii, there is evidence these two species have the same native range and are closely related.
Similar content being viewed by others

Background
Identification of plant species is an important part of biodiversity studies and the regulatory framework in many countries. Federally regulated species and invasive species pose a risk to the agricultural production and natural habitats or ecosystems, can affect human health and cause significant economic loss in trade. In Canada regulation of invasive plant species or noxious weeds is done under the authority of the Plant Protection Act [1], Seeds Act [2] and Feeds Act [3]. Identification of seeds or plants can be performed using morphological characterization by expert botanists. However, while expert analysts can identify species from seeds or plants, the plasticity and similarity of weedy species could make the identification very challenging. In these cases, molecular tools can aid in classifying the specimens.
The Amaranthus genus contains over 70 species, including species cultivated for their grain or edible leaves, but also highly weedy and invasive species [4] that impact yield and production of major crops in North America including soybean, corn and cotton [5,6,7,8]. Losses associated with A. palmeri for 2015 were estimated to be 250 million USD for cotton, 1.3 billion USD for maize and 2.5 billion USD for soybean [8]. A 91% yield reduction was calculated for maize in Kansas when the density of A. palmeri was 10.5 plants per square meter [9]. And yield losses for soybean reached 78.7%, 56.2% and 38% at a density of 8 plants per square meter for palmer amaranth, tall waterhemp and redroot pigweed (A. retroflexus), respectively [10].
A major concern of weedy species like A. tuberculatus and A. palmeri is their increased resistance to multiple herbicide modes of action [5, 8, 11, 12], resulting in limiting the number of strategies for control. Additionally, trade can be jeopardized when contaminating seed is found in commodity shipments. For example, A. palmeri glyphosate-resistant populations have been established in less than 10 years in Japan from contaminated grain seed imported from the United States [13].
While not federally regulated in the US, A. palmeri is regulated as a noxious weed in Delaware, Iowa, Minnesota, North Dakota, Ohio, and Pennsylvania [14], and it is present in 32 states [8]. Canada regulates A. tuberculatus, but not A. palmeri, for which risk assessment is ongoing. However, while no established populations have been yet found in Canada, A. palmeri has been reported in Ontario and Manitoba. Viable seeds have been reported as part of import shipments [15]. Furthermore, an import ban from China that started in 2019, on two of the largest Canadian-based canola exporters (Viterra and Richardson) was based on seed contamination with quarantine seeds and pathogens, among which palmer amaranth was allegedly present [8]. Since China buys 40% of the Canadian canola exports, this has resulted in losses of over 2 billion dollars for the Canadian economy.
Climate change modeling and the strong phenotypic plasticity of A. palmeri [4], shows that the most likely scenario in the next few years is a constant expanding range into Canada for this species, affecting growing regions of corn and soybean in Ontario, Quebec and Manitoba [4, 6, 16, 17]. These factors demand effective diagnostic tools to distinguish species of concern from related species to avoid negative impacts on the environment and the economy.
Molecular identification of plant species has traditionally been done using DNA barcodes, which entail short DNA regions (e.g., 300 – 800 bp) that work as a fingerprint bearing specific polymorphisms that are unique to the species in question. Many barcodes have been tested for plants as potential regions for identification of most taxa, including several chloroplast barcodes (trnH-psbA, rbcL, matK) and the internal transcribed spacers from ribosomal DNA (ITS1 and ITS2) [18,19,20,21,22,23,24]. However, these regions can fail to provide resolution for specific taxa as when distinguishing regulated species from their non-regulated counterparts. One solution to this issue it to continue exploring taxa-specific genomic regions with strong polymorphic signals. Usually this can be done by sequencing full chloroplast genomes or full nuclear rDNA regions that constitute reservoirs of diversity where new DNA barcodes can be found. The exploration of these larger regions for new sources of polymorphism has been facilitated by approaches like genome skimming [25,26,27,28,29], a technique that uses low-pass sequencing of a full genome to achieve a high read number covering the highly repeated fractions of the genome (e.g., chloroplast and nrDNA). Alternatively, specific nuclear genes/regions used in phylogenetic studies [30,31,32,33,34,35], can also be explored as sources of polymorphic markers when nrDNA or chloroplast DNA do not suffice to provide polymorphic signal among the studied species. Finally, the increased use of long-read sequencing technology (Nanopore and PacBio) has open new doors not only to mine genomes for new polymorphic regions, but to generate DNA barcodes in the kilobase range [36, 37], thus allowing to compile multiple barcodes in a single read and test thousands of samples concurrently.
In Amaranthus the study of traditional barcoding regions and other genomic regions, has resulted in the development of species-specific barcodes and PCR-based assays, allowing identification of some species in the genus [12, 38, 39]. While these methodologies are effective in detecting some of the species, the process of finding sequences for DNA barcoding can be long, and PCR based analysis require multiple primers and sometimes specific conditions to detect specific species.
Among alternative methodologies to DNA barcoding and PCR-based assays for species identification is protein biotyping. The technique is based on a simple protocol for rapid extraction of the major acid soluble protein fractions of the biological samples (including ribosomal proteins), followed by immobilization on a solid matrix and ionization of the proteins followed by mass spectrometry using Matrix-Assisted Laser Desorption Ionization or MALDI [40]. The result of this process is a protein mass spectra per each tested sample, providing a fingerprint for the species. Protein biotyping has been used often in the identification of microorganisms, especially in pathogen identification [40,41,42,43,44], and also specifically in plant pathogen identification [45,46,47,48,49]. Recently, some research has been developed to test protein biotyping in plants. Importantly, work was performed to distinguish Impatiens glandulifera from other species in the genus, and also to distinguish its regional biotypes using leaves and seeds [50,51,52]. Using MALDI-TOF MS spectra from acid-soluble proteins, four different I. glandulifera biotypes differing in susceptibility to a rust biological control agent, were identified [52]. Extraction of proteins from seed material [51, 52], seemed to contain a more stable protein fraction compared to tissue undergoing development (e.g., young leaves) [50]. Further work studied protein biotyping of tomato varieties, showing good reproducibility for the technique, but low accuracy in distinguishing the test varieties [53].
In the current study we tested protein biotyping on seeds from the family Brassicaceae from different samples and years, to demonstrate consistency on the generation of protein spectra from single seeds. Then we applied the protocol to seeds from Amaranthus species, which comprise species of regulatory concern, including the two species of highest concern in Canada due to their weediness and invasiveness (A. palmeri and A. tuberculatus). Our results showed high accuracy in identifying two sets of 15 and 60 blind sample batches, using a database of 16 Amaranthus species. Protein biotyping using MALDI-TOF-MS is promising as a suitable, cheap and efficient technique to identify Amaranthus species of regulatory concern. The method has the potential to be easily transferred to other taxa to aid in identification of species that impact trade and weed management.
Results
Brassicaceae preliminary tests
To test our protein biotyping methodology we set out to complete both protein extractions and biotyping using seeds from four species of the family Brassicaceae. These seeds were used because they were readily available and in large amounts at the National Archive of Legal Reference Material at the CFIA Seed Science Unit. This allowed for multiple testing to standardize conditions before testing Amaranthus spp. where material was initially more scarce.
We performed a preliminary test with small modifications to previously established protocols (see materials and methods) on five seed batches corresponding to two varieties of Brassica napus (spring and winter), and one variety of each of other three species (B. rapa, B. juncea and B. carinata). Three seeds of each batch, with two different solvent dilutions (1:1 and 1:10 - see methods) and two technical replicates were obtained. Out of the 12 potential protein spectral profiles expected for each seed batch, we obtained 12 spectra for B. napus (spring), 11 for B. napus (winter), 12 for B. rapa, 7 for B. carinata and 12 for B. juncea. Both a composite Principal Component Analysis (PCA) (Fig. 1), and a dendrogram analysis (Additional file 1 A), showed that all seeds belonging to the same species had similar protein spectra and clustered together. There was no specific clustering linked to dilution or technical replicates.

Principal Component Analysis (PCA) of protein spectra corresponding to four Brassica species. B. c (Brassica carinata), B. r (Brassica rapa), B. j (Brassica juncea), B. n (Brassica napus)
To test potential variability among seed batches from the same species but from different years we conducted a second run where we compared seed batches from three different years in B. napus (spring) and B. rapa along with the available single-year batches of B. napus (winter), B. juncea and B. carinata. To increase the probability of obtaining successful protein spectra we improved the protocol by using a more reliable approach to grind the seeds (tissue lyzer), and modified the preliminary protocol so that spotting of the samples in the target plate would require less manipulation (compare preliminary test with final protocol in materials and methods). Out of 45 total seeds (5 per accession) we obtained protein spectra for 43, with only two seeds failing from B. carinata. Samples clustered correctly for each one of the species (Additional File 1B). While our pilot study did not show that the age of the seeds constitutes a factor that could introduce variability into the protein spectral profiles, conditions of storage of seeds and the access to moisture could trigger oxidative processes on different biomolecules including proteins [54]. Therefore, we recommend having this in mind when testing the technology
Amaranthus species protein biotyping
Protein biotyping is based on differences and similarities of protein spectra of major protein fractions of the different species. Figure 2 shows the difference and similarities between protein spectra generated from different accessions of A. palmeri and A. tuberculatus (two species of regulatory concern).

Protein spectra of two Amaranthus species. Three different A. palmeri accessions and three different A. tuberculatus accessions are shown. GRIN ID refers to their identification number from: https://npgsweb.ars-grin.gov/gringlobal/search
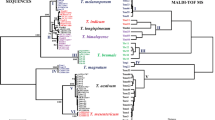
We performed protein biotyping with 15 Amaranthus species using five biological replicates (5 seeds) per accession. For some species like A. tuberculatus and A. palmeri we tested more than one accession due to regulatory concern of these two species in Canada or the United States. The protein spectra were clustered in three major groups with sub-clusters within the clusters (Fig. 3). Cluster 1 had two sub-clusters grouping the protein profiles corresponding to A. powellii - A.hybridus - A. retroflexus, and A.hypochondriacus - A. caudatus, respectively. However, the subgroups from this first cluster could not be clearly delimited by species. Cluster 2 had six species which could all be separated by their own sub-cluster. Importantly, a regulated species in Canada (A. tuberculatus) was separated from all other species on this cluster. In Cluster 3 we found A. palmeri, A. watsonii, A. spinosus and A. arenicola. While A. spinosus samples can be distinguished in a subgroup, the spectra from the two phylogenetically sister species (A. palmeri and A. watsonii) cannot be distinguished. A single spectra from A. palmeri was clustered with A. arenicola.

Clustering of protein spectra corresponding to 15 Amaranthus species. Species were clustered into three major clusters, with sub-clusters within each cluster
Database generation
While spectra clustering is a rapid way of visualizing relationships between spectra of individual samples, PCA and dendrograms are not based on structured phylogenetic algorithms. Protein biotyping is meant to be used to characterize samples for which a phenotypic identification is not possible, to confirm phenotypes and to classify blind samples, using a spectral database of known samples to which unknown samples can be compared to. Enrichment of this database with multiple accessions per species (e.g., coming from different geographical regions), provides a way of accounting for potential intra-species variability and increases accuracy of determination of test samples. Furthermore, even when a dendrogram or PCA analysis shows different species clustering together, a rich database comprising several accessions of each of the species to be identified, will increase accuracy in sample identification [55, 56].
We generated a database with 16 Amaranthus species (seeds obtained from the Germplasm Resource Information Centre, U.S. National Plant Germplasm System – see methods), including species of regulatory concern, weedy species, and species that show phenotypic plasticity at the seed level that may be easily confused when performing phenotypic characterization (Additional file 2). To increase accuracy and resolution power of our database we included at least three different accessions per species when available, and used three seeds per accession (each one with 30 spectral readings – see methods), to account for biological and technical variation. We also had a larger number of accessions for species which pose the largest regulatory concern in Canada (A. tuberculatus and A. palmeri).
We generated spectral information for each seed, and produced a consensus Main Spectra (MSP) from at least 20 spectra per sample (Additional file 2). Each newly produced MSP was compared to the full database of MSPs to test if each MSP matched itself as the highest hit, and if the secondary hit (second highest similarity) also corresponded to another accession of the same species. This analysis showed that when the generated MSPs are used as unknown samples, they match themselves as top hits and match other accessions of the same species as secondary hits (Additional File 3). There were only four exceptions where the second best hit was not the expected species: A. watsonii (PI633593-1) second hit was A. spinosus, A. palmeri (PI667167-2) second hit was A. spinosus, A. watsonii (PI633593-RE2) second hit was A. palmeri, and A. caudatus (PI553073-1) second hit = was A. hybridus. This phenomena could be attributed to similarity in the protein spectra among species which are closely related phylogenetically. For these four exceptions, the second hits are phylogenetically related [7] to the first hits in all cases.
Blind sample testing
We received blind samples from three different labs doing work in Amaranthus spp. Six blind samples were received from AAFC Saint-Jean-sur-Richelieu, 9 blind samples from AAFC Harrow and 60 blind samples from CFIA’s Seed Science and Technology Sections in Saskatoon. From the 6 samples received from Saint-Jean-sur-Richelieu, using 3 seeds per accession, we obtained 100% correct identification with matching average scores > 2 in most cases (Table 1, samples MIRL22-A.unknown-07 to 12). In one case a batch that was identified as A. tuberculatus (MIRL22-A.unknown-09) had one out of three seeds identified as A. arenicola. However, since two out of the three seeds for the respective accession were correctly identified, the final assigned identification matched the original identification uncovered by the providers after the protein biotyping analysis was completed. A similar situation happened with sample MIRL22.A.unknown-11, which had one of three seeds identified as A. rudis instead of A. tuberculatus. While historically these two were at times identified as different species, the latest consensus is that both are the same species, or varieties of the same species (Waterhemp | CALS (cornell.edu), Amaranthus rudis J.D.Sauer — The Plant List), which supports our identification. In this sense we classified all samples that were A. rudis or A. tuberculatus as A. tuberculatus. Two samples that were originally sent by the provider as A. powellii and A. viridis (see superscript information 3–4 from Table 1) were identified by us as A. retroflexus. After the provider of the seeds grew plants from the seeds of these two accessions, their phenotypic characterization confirmed our identification by protein biotyping, showing that our method was effective in correcting initial misidentifications from the seed batches from the provider.
In the case of the samples obtained from AAFC Harrow (Table 1, samples MIRL22-A.unknown-14 to 23), all samples were correctly identified according to the identification uncovered by the seed providers after our analyses were complete. We learned that a sample which was originally part of the blind samples (MIRL22-A.unknown-13 – Additional file 4), and initially classified as A. caudatus, corresponds to seeds whose correct taxonomic identification could not be confirmed by the provider (the sample was provided to them by an external collaborator years ago without verification). Therefore, this sample was excluded from our analysis as there was no morphological confirmation. Finally, MIRL22-A.unknown-17, identified as A. hypochondriacus, had technical replicates which diverged from the consensus identification (Additional file 4), but was nevertheless identified correctly by our majority rule.
Overall the application of our methodology to identify blind samples obtained from AAFC centres in Quebec and Ontario was 100% accurate when using our majority rule (at least 2 of the 3 tested seeds matched the expected species). When accounting for all biological replicates (3 seeds per accession) our success rate was 96%, due to a single seed from 3 replicates resulting in incorrect identification in two cases (Table 1 - MIRL22-A.unknown-09 and MIRL22-A.unknown-11).
We then examined 60 blind samples sent from the CFIA SSST (Saskatoon Seed Science and Technology) unit. Out of 60 individual seeds, only two failed to produce a protein spectra in the three technical replicates (MIRL22-A.unknown 68 and 70 in Table 2 and Additional file 5); which can be most likely attributed to a technical error during processing of the samples. This means that our method was 97% effective in producing protein spectral profiles in this second set of blind samples. Out of 60 seeds where a phenotypic identification was performed by seed analysts, our protein biotyping assay was able to correctly predict the species for 52 samples, which means an accuracy of 87%. Three samples provided by the SSST but not identified by seed analysts, were tested by our protein biotyping assay but excluded from the validation analysis (MIRL22-A.unknown-37, 49 and 71 – Additional file 5). Out of the 8 samples where we could not correctly identify the source sample species, two were the samples where we did not obtain protein spectra. On the 6 samples where our biotyping identification did not match the phenotypic identification, there was no apparent relationship to the Brukker score, with some mismatches scoring below 2 and some scoring above 2 (Table 2). Three of the mismatches corresponded to seeds phenotypically identified as A. palmeri, where our identification matched A. watsonii (A. palmeri’s sister species) in two of those cases. In two cases a seed classified as A. cruentus matched A. hypochondriacus protein spectra, but our database did not have A. cruentus protein profiles, so this misidentification is expected. And in one case, a seed classified as A. caudatus matched a A. hypochondriacus MSP from our database.
Discussion
Protein biotyping using MALDI-TOF MS has commonly been used to identify pathogens [47, 49, 57,58,59], due to its high efficiency, throughput and manageable cost. In pathogens, protein profiles generated by species and strains [47, 49, 60] show a reliable identification methodology which can be efficiently used to classify samples and cluster related organisms. In plants, protein biotyping was tested to distinguish Impatiens species and regional biotypes of the invasive weed Impatiens glandulifera [52], yielding accuracy in identification of two sets of 12 blind samples of 100% and 92% respectively. Our methodology, which was derived from the method reported on [52], resulted in species identification accuracies of 100% and 87% for the two sets of 15 and 60 blind samples from the genus Amaranthus.
One of our main objectives in testing biotyping to identify Amaranthus seeds is to find a rapid diagnostics test to support seed identification during trade under the Canadian regulatory framework for international export. At times, phenotypic plasticity and similarity from seeds will result in difficulties to accurately identify an Amaranthus seed using morphological methods. While DNA sequencing has uncovered polymorphisms that can be utilized to differentiate species in Amaranthus species, diagnostic assays are usually targeted towards one species [39, 61, 62], and do not examine related species. Comparatively, the identification of Amaranthus samples using protein biotyping is more efficient than developing and using DNA barcodes. Protocols tested in our lab show that two 96-well plates can be processed in a day, covering seed grinding, protein extraction, dilution, plate spotting and MALDI biotyping. In the meantime, obtaining a barcode comprises performing a PCR, gel electrophoresis, PCR purification, sequencing, sequence purification, and a sequencing run, which can take at least 2–3 days for the same number of samples. While both methodologies would require the generation of a database to identify blind samples or validate known samples, the generation of the protein spectra itself does not require any previous knowledge to obtain the protein profile. DNA barcoding requires some previous knowledge of the target region to design primers that can amplify the barcode. While traditional chloroplast and Internal Transcribed Spacer (ITS) regions can use conserved primers, these regions do not always provide the best resolution in all plant genera. This has also been seen in studies outside the plant realm, where the resolution of traditional 16 S rDNA markers at the species level was below the power of MALDI-TOF biotyping [63]. In our case, our lab has used genome skimming [29] to assemble full chloroplast genomes to find new Amaranthus spp. barcodes. However, the generation of the protein spectra database does require some time investment since it is necessary to have several accessions per species that represent the variability of a species, so that blind samples can be accurately identified. While we did not test infra-species resolution, previous studies show that genotype and variety discrimination is possible [52, 53]. Additionally, we standardized our methodology for seeds since previous research has shown that other tissue under development (like young leaves) might not have a stable protein composition [50]. However, we expect to test other plant tissues in the future. A comparison of protein biotyping and DNA barcoding for molecular identification is shown below (Table 3).
Our preliminary dendrograms did not show distinctive clustering for samples of some of the species in cluster 1 (Fig. 3), that traditionally belong to the Hybridus clade [7]. While the protein spectra dendrograms are not based on a phylogenetics algorithm, many studies of rapidly evolving markers have produced polytomies of the Hybridus clade, and have concluded a sister-lineage relationship of A. powellii and A. retroflexus to the A. hybridus group [7, 12]. In fact, a previous study using Genotyping By Sequencing (GBS) in the Amaranthus genus showed that there was no strong separation by species in the Hybridus complex, similarly to what was found in our results [64], reflecting strong gene flow and potential hybridization in this clade. Nevertheless, several species from the Hybridus clade were part of the blind samples provided to us and were correctly classified when compared to our database, which was enriched with multiple accessions per species. This shows that dendrograms can be used as a preliminary step for comparison of the protein profiles, but an enriched database with multiple accessions per species, is necessary to represent inherent variability between populations of a species, therefore increasing the accuracy of identification.
We then performed identification of blind samples using our protein spectra database (Additional file 2). On six blind samples where we did not achieve correct identification according to the phenotypic classification of samples provided, there was no apparent relationship to the Bruker matching score, meaning both high (> 2) and low (< 2) scores were present in mismatches. Bruker scores are calculated based on comparison of the number of matching peaks between test and database spectra and on the concordance of peak height and symmetry [52]. In our case, only one score (1.30) indicated low relatedness, on a mismatch between A. spinosus and A. palmeri (MIRL22-A.unknown-24 – Table 2). These two species, are nevertheless closely related [7, 64], as is the case for two other mismatches where our biotyping indicating A. watsonii had A. palmeri as phenotypic identification. In this latter case, previous research has shown that A. watsonii and A. palmeri are sister species according to morphological and molecular characterization [7, 65]. This is also suggested by comparison of full chloroplast genomes done in our lab (unpublished), and by a recently published plastome comparison analysis [66]. Additionally, comparative genomics has shown that A. palmeri and A. watsonii have similar genome sizes, transposon content, higher heterozigosity and a common origin of dioecy among dioecious species [65]. Work comparing their plastomes shows these two species have the closest relationship among all dioecious species studied [66], and even suggests that due to their morphological similarity and very small genetic distance, they could be a single polymorphic species instead of two different species. This would be in agreement with the inability of the protein biotyping to distinguish between the two alleged species.
Three samples that were identified as A. hypochondriacus by protein biotyping (MIRL-A.unknown 43, 50 and 60 – Table 2) did not match the seed phenotypic identification of A. cruentus, for the two first samples, and A. caudatus from the third sample. Our inability to identify A. cruentus seeds stems from the absence of this species in our database. However, A. hypochondriacus, A. cruentus and A. caudatus are all part of the hybridus clade from Amaranthus species, and closely related according to previous studies [7, 64,65,66,67,68]. The three species are grain domesticated amaranths originated from A. hybridus [7, 69]. Close evolutionary relationship potentially plays a role in common proteins showing up between these species in our biotyping analyses, but we also expect that continuing to enrich our Amaranthus protein spectra library with more accessions in each species, will result in better resolution.
Conclusions
Protein biotyping is an efficient methodology to identify species in the genus Amaranthus. While some of the main problems that are present with DNA barcoding can arise when studying this complex genus, we determined that enrichment with a larger number of diverse populations in each species can increase accuracy in calling the correct species. As we gather more species and populations for our Amaranthus library we expect our accuracy and power to identify these species will increase.
While DNA barcoding and PCR tests derived from sequence analysis are still at the core of species identification, new alternate methods can provide complementary tools where difficulties exist with these more traditional methods and for specific situations. In our case protein biotyping has provided a quick method to identify species from seed protein, aiding seed analysts when seed plasticity makes it difficult to do a phenotypic characterization. This tool becomes important in the regulatory context of trade as a last step of classification when other tools or analysis are not sufficient. We also expect that the methodology can be easily transferred to other plants that are regulated at the federal level, but outside the regulatory context the protocols shown here can be applied to different plant genera for identification purposes.
Methods
Plant material
Amaranthus spp. seeds for our protein database were obtained from collaborators at Agriculture and Agri-Food Canada, Harrow, ON, and from the Germplasm Resource Information Centre (GRIN-Global) which is part of the U.S. National Plant Germplasm System. Blind Amaranthus spp. seed samples were provided by Dr. Marie-Josée Simard (Agriculture Agri-Food Canada, Saint-Jean-sur-Richelieu, QC), Dr. Robert Nurse (Agriculture and Agri-Food Canada, Harrow, ON), and Dr. Ruojing Wang (Canadian Food Inspection Agency, Saskatoon, SK).
MALDI-TOF-MS sample preparation
Preliminary test
Individual seeds from four Brassicaceae species were manually ground in 2 mL rounded bottom microfuge tubes. Seed protein extraction was modified from the original protocol [52] to place the ground seeds in 50 µL of a 1:1 Acetonitrile (for UHPCL, Sigma-Aldrich, Oakville, Canada)-Formic Acid (ACS reagent, ≥ 96%, Sigma-Aldrich, Oakville, Canada). This was followed by a centrifugation step at maximum speed (20,000 rcf) to separate the supernatant carrying the proteins from the seed debris. Since the expected amount of protein needed to create a clear spectra was initially unknown, we generated two testing dilutions (1:1 and 1:10), by mixing either 5 µL of supernatant with 5 µL of a 1:1 Acetonitrile-Formic Acid solution, or 1µL of supernatant with 9 µL of the Acetonitrile-Formic Acid solution. Then 0.5 µL of each dilution per sample was added to the 96-well MBT biotarget plate (Bruker, Billerica, MA, US) followed by addition of 0.5 µL of HCCA dissolved matrix solution (Bruker, Billerica, MA, US). This mix was left to dry at room temperature (without exceeding one hour), and then overlaid with one additional 1 µL of HCCA matrix solution which was left to dry. One microliter of BTS (Bruker Bacterial Test Standard - Bruker, Billerica, MA, US) was spotted in duplicate on the MALDI target plate and allowed to dry at room temperature. The entire spot was overlaid with 1 µL of HCCA solution and allowed to dry at room temperature.
Final protocol
Single seeds were placed in 2 mL rounded bottom microfuge tubes and a 5 mm steel bead was added per tube. Samples were loaded into a tissue lyzer (Tissue Lyser II, Qiagen, Germantown, MD, US) and fully ground for 1 min at 25 Hz. Tubes were centrifuged at 12,000 rcf for 1 min and then 50 µL of a 1:1 solution of Formic Acid (ACS reagent, ≥ 96%, Sigma-Aldrich, Oakville, Canada) and Acetonitrile (for UHPCL, Sigma-Aldrich, Oakville, Canada) was added, and the tube was briefly vortexed. Samples were then centrifuged at 12,000 rcf for 1 min. Using a new tube 2 µL of the supernatant were mixed with 4 µL of HCCA dissolved matrix solution (Bruker, Billerica, MA, US) and 2 µL of 1:1 Formic acid – Acetonitrile solution. One microliter of this solution was pipetted onto the 96-well MBT biotarget plate (Bruker, Billerica, MA, US) and dried at room temperature (without exceeding one hour). The sample was then overlaid with 1 µL of HCCA matrix solution and allowed to dry at room temperature. One microliter of BTS was spotted in duplicate on the MALDI target plate and allowed to dry at room temperature. The entire spot was overlaid with 1 µL of HCCA solution and allowed to dry at room temperature.
MALDI-TOF-MS
Mass spectrometry was carried out over a range of 2 kDa to 20 kDa using a Bruker MALDI Biotyper Sirius instrument (Bruker, Billerica, MA, US), together with the MBT software suite including FlexControl Sirius (version 3.4) and MBT Compass (version 4.1) for the acquisition of spectra. The Smartbeam MBT laser was used with a frequency of 200 Hz, initial output power of 30% and maximum output of 40%, with a 2000 μm spot size and 240 laser shots per sample. The laser’s settings were Global Attenuator Range (0%), Attenuator Offset (48%) and Attenuator Range (20%). The ion source voltage was 19.77 kV and 18.01 kV for each source. The samples were spotted onto Bruker MBT Biotarget 96 target plates. The calibration followed manufacturer’s instructions using Bruker’s BTS control, comprised of E. coli ribonuclease A and myoglobin. Calibration peaks had masses at 3637.8; 5096.8; 5381.4; 6255.4; 7274.5; 10,300.2; 13,683.2, and 16,952.3 Da. Spectra were acquired using FlexControl Sirius (version 3.4) and MBT Compass (version 4.1) software under the manufacturer’s default settings. The generated spectra were used for principal-component analysis (PCA) and to create PCA dendrograms using the 70 main peaks of each spectra.
Database entries
To generate a database of protein spectra that can be used to test blind samples, database entries require 10 technical replicates per sample and 3 spectral readings per technical replicate, so a total of 30 spectra are compiled at the end per sample. This is necessary in order to obtain the minimum of 20 high quality spectra to be included in the database as a Main Spectra (MSP), which is calculated as an average of the 20–30 spectral readings. We performed this process for three seeds for each one of the accessions that were included in the generated database.
Database samples were plated on the MBT target plate (including at least 1 BTS spot and 1 Blank spot for quality control), and the target plate was placed into the equipment to acquire data using the FlexControl Software (version 3.4). BTS Calibration was performed through detection of the major eight spectral peaks in the expected range of the protein profile (Da range of +/-300ppm). The resulting BTS spectra was used as input for analysis with the FlexAnalysis software (version 3.4), where BTS spectra were smoothed, baseline was subtracted and BTS masses were identified. Sample spectra to create the database were also smoothed, baseline subtracted and outliers eliminated. A minimum of 20 spectra were selected to generate each one of the MSPs for the database.
The MSPs to be added to the database were generated in Compass Explorer software (version 4.1). The newly created MSPs were matched against all created database spectra to validate identity of the species. Spectra validated in this way are expected to match the same species with a Bruker score of 2.00 or higher. Each of the major 70 peaks used to create the MSP is expected to be at least 25% of the times in the 20–30 spectral sample reading compiled. MSPs are then added to a library of choice using the “Start Taxonomy Tree Editor”. We generated a library with a total of 16 species and 56 accessions from the Amaranthus genus. Our database was created based on preliminary information of target species with North American distribution and that can be confused with species of regulatory concern when performing morphological classification.
Blind sample testing
The first set of blind seed samples provided by Dr. Robert Nurse (AAFC, Harrow, ON) and Dr. Marie-Josée Simard (AAFC, Saint-Jean-sur-Richelieu, QC) had multiple seeds per accession and therefore three biological replicates (individual seeds in each accession) and three technical replicates per blind sample (coming from each seed) were tested. A second set of blind samples was obtained from Dr. Ruojing Wang from the CFIA Saskatoon Seed Science and Technology (SSST) Lab. These latter samples were chosen from regulated Amaranthus species, and visually similar seeds from species that occur in Canada and the United States. For this second set, mature intact seeds were selected from a single source, and samples were shuffled and numbered using a random number generator. Individual seeds were then sent in vials to our lab, with each species/sample having 2–4 replicates throughout the total number of randomized vials. Three technical replicates per seed were run in the Bruker protein biotyper. All samples from the two blind sets received were compared against the generated Amaranthus spp. MSPs database built in-house. Spectra from blind samples were compared against database entries, and were scored using Bruker Identification scores that establish relatedness between spectra where a score > 2.00 indicates high relatedness and a score below 1.70 indicates low relatedness. Bruker Scores are computed as described in Reeve & Pollard [52]. However, we considered matches and mismatches based on the top hits when comparing a blind sample to the database, regardless of the score. Validation of our identification was done by sending our results back to the three seed providers so they could match their phenotypic characterization with our biotyping identification. AAFC providers usually grew plants for characterization while the CFIA SSST performed expert analyst seed characterization.
Data Availability
Data generated or analysed during this study are included in this published article [and its supplementary information files]. Protein spectra would be available upon request.
References
Plant Protection Act [https://laws-lois.justice.gc.ca/eng/acts/p-14.8/page-1html accessed 26 September 2023].
Seeds Act [https://laws-lois.justice.gc.ca/eng/acts/S-8/page-1.html accessed 26 September 2023].
Feeds Act [https://laws-lois.justice.gc.ca/eng/acts/f-9/page-1.html accessed 26 september 2023].
Oliveira MC, Jhala AJ, Bernards ML, Proctor CA, Stepanovic S, Werle R. Palmer Amaranth (Amaranthus palmeri) adaptation to US midwest agroecosystems. 2022, 4:887629.
Beckie HJ. Herbicide-resistant weed management: focus on glyphosate. Pest Manage Sci Vol. 2011;67:1037–48.
Ward SM, Webster TM, Steckel LE. Palmer Amaranth (Amaranthus palmeri): a review. Weed Technol. 2013;27(1):12–27.
Waselkov KE, Boleda AS, Olsen KM. A phylogeny of the genus Amaranthus (Amaranthaceae) based on several low-copy nuclear loci and chloroplast regions. Syst Bot. 2018;43(2):439–58.
Amaranthus palmeri S.Watson. In. Edited by EPPO, vol. 50. EPPO Bulletin; 2020: 535–542.
Massinga R, Currie R, Horak M, Boyer J. Interference of Palmer Amaranth in corn. Weed Sci. 2001;49(2):202–8.
Bensch CN, Horak MJ, Peterson D. Interference of redroot pigweed (Amaranthus retroflexus), Palmer Amaranth (A. Palmeri), and common waterhemp (A. Rudis) in soybean. Weed Sci. 2003;51(1):37–43.
Tranel PJ. Herbicide resistance in Amaranthus tuberculatus †. Pest Manag Sci. 2021;77(1):43–54.
Xu H, Pan X, Wang C, Chen Y, Chen K, Zhu S, van Klinken RD. Species identification, phylogenetic analysis and detection of herbicide-resistant biotypes of Amaranthus based on ALS and ITS. Sci Rep. 2020;10(1):1–9.
Shimono A, Kanbe H, Nakamura S, Ueno S, Yamashita J, Asai M. Initial invasion of glyphosate-resistant Amaranthus palmeri around grain-import ports in Japan. Plants People Planet. 2020;2(6):640–8.
Weed Risk Assessment for. Amaranthus palmeri (Amaranthaceae) – Palmer’s amaranth. In. United States Department of Agriculture; 2020.
Page ER, Nurse RE, Meloche S, Bosveld K, Grainger C, Obeid K, Filotas M, Simard MJ, Laforest M. Import of palmer amaranth (Amaranthus palmeri s. wats.) Seed with sweet potato (Ipomoea batatas (L.) lam) slips. Can J Plant Sci. 2021;101(5):774–8.
Kistner EJ, Hatfield JL. Potential geographic distribution of Palmer amaranth under current and future climates. Agricultural & Environmental Letters. 2018;3(1):170044–4.
Briscoe Runquist RD, Lake T, Tiffin P, Moeller DA. Species distribution models throughout the invasion history of Palmer Amaranth predict regions at risk of future invasion and reveal challenges with modeling rapidly shifting geographic ranges. Sci Rep. 2019;9(1):2426.
Kress WJ, Erickson DL. A two-locusglobal DNA barcode for land plants: the coding rbcL gene complements the non-coding trnh-psba spacer region. PLoS ONE. 2007;2(6):e508–8.
Fazekas AJ, Burgess KS, Kesanakurti PR, Graham SW, Newmaster SG, Husband BC, Percy DM, Hajibabaei M, Barrett SCH. Multiple multilocus DNA barcodes from the plastid genome discriminate plant species equally well. PLoS ONE. 2008;3:e2802–2.
Hollingsworth PM, Forrest LL, Spouge JL, Hajibabaei M, Ratnasingham S, van der Bank M, Chase MW, Cowan RS, Erickson DL, Fazekas AJ, et al. A DNA barcode for land plants. Proc Natl Acad Sci USA. 2009;106(31):12794–7.
Yao H, Song J, Liu C, Luo K, Han J, Li Y, Pang X, Xu H, Zhu Y, Xiao P, et al. Use of ITS2 region as the yniversal DNA barcode for pants and animals. PLoS ONE. 2010;5(10):e13102–2.
Yu J, Xue J-H, Zhou S-L. New universal matK primers for DNA barcoding angiosperms. J Syst Evol. 2011;49(3):176–81.
Hollingsworth PM, Graham SW, Little DP. Choosing and using a plant DNA barcode. PLoS ONE. 2011;6(5):e19254.
Braukmann TWA, Kuzmina ML, Sills J, Zakharov EV, Hebert PDN. Testing the efficacy of DNA barcodes for identifying the vascular plants of Canada. PLoS ONE. 2017;12(1):e0169515.
Alsos IG, Lavergne S, Merkel MKF, Boleda M, Lammers Y, Alberti A, Pouchon C, Denoeud F, Pitelkova I, Pușcaș M, et al. The treasure vault can be opened: large-scale genome skimming works well using herbarium and silica gel dried material. Plants. 2020;9(4):432–2.
Nevill PG, Zhong X, Tonti-Filippini J, Byrne M, Hislop M, Thiele K, Van Leeuwen S, Boykin LM, Small I. Large scale genome skimming from herbarium material for accurate plant identification and phylogenomics. Plant Methods. 2020;16(1):1–1.
Song F, Song F, Li T, Li T, Burgess KS, Feng Y, Feng Y, Ge XJ. Complete plastome sequencing resolves taxonomic relationships among species of Calligonum L. (Polygonaceae) in China. BMC Plant Biol. 2020;20(1):261–1.
Zeng CX, Hollingsworth PM, Yang J, He ZS, Zhang ZR, Li DZ, Yang JB. Genome skimming herbarium specimens for DNA barcoding and phylogenomics. Plant Methods. 2018;14(1):43–3.
Straub SCK, Parks M, Weitemier K, Fishbein M, Cronn RC, Liston A. Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. Am J Bot. 2012;99(2):349–64.
Zimmer EA, Wen J. Using nuclear gene data for plant phylogenetics: progress and prospects. Mol Phylogenet Evol. 2012;65(2):774–85.
Zhao L, Li X, Zhang N, Zhang SD, Yi TS, Ma H, Guo ZH, Li DZ. Phylogenomic analyses of large-scale nuclear genes provide new insights into the evolutionary relationships within the rosids. Mol Phylogenet Evol. 2016;105:166–76.
Cai L, Ma H. Using nuclear genes to reconstruct angiosperm phylogeny at the species level: a case study with Brassicaceae species. J Syst Evol. 2016;54(4):438–52.
Huang CH, Sun R, Hu Y, Zeng L, Zhang N, Cai L, Zhang Q, Koch MA, Al-Shehbaz I, Edger PP, et al. Resolution of Brassicaceae phylogeny using nuclear genes uncovers nested radiations and supports convergent morphological evolution. Mol Biol Evol. 2016;33(2):394–4.
Huang J, Xu W, Zhai J, Hu Y, Guo J, Zhang C, Zhao Y, Zhang L, Martine C, Ma H, et al. Nuclear phylogeny and insights into whole genome duplications and reproductive development of Solanaceae plants. Plant Commun. 2023;4(4):100595.
Ogutcen E, Christe C, Nishii K, Salamin N, Möller M, Perret M. Phylogenomics of Gesneriaceae using targeted capture of nuclear genes. Mol Phylogenet Evol. 2021;157:107068–8.
Srivathsan A, Lee L, Katoh K, Hartop E, Kutty SN, Wong J, Yeo D, Meier R. ONTbarcoder and MinION barcodes aid biodiversity discovery and identification by everyone, for everyone. BMC Biol. 2021;19(1):1–21.
Wang Y, Zhao Y, Bollas A, Wang Y, Au KF. Nanopore sequencing technology, bioinformatics and applications. Nat Biotechnol. 2021;39(11):1348–65.
Wright AA, Molin WT, Nandula VK. Distinguishing between weedy Amaranthus species based on intron 1 sequences from the 5-enolpyruvylshikimate-3-phosphate synthase gene. Pest Manag Sci. 2016;72(12):2347–54.
Brusa A, Patterson EL, Gaines TA, Dorn K, Westra P, Sparks CD, Wyse D. A needle in a seedstack: an improved method for detection of rare alleles in bulk seed testing through KASP. Pest Manag Sci. 2021;77(5):2477–84.
Singhal N, Kumar M, Kanaujia PK, Virdi JS. MALDI-TOF mass spectrometry: an emerging technology for microbial identification and diagnosis. Front Microbiol. 2015;6:791.
Murugaiyan J, Lewin A, Kamal E, Bakula Z, van Ingen J, Ulmann V, Unzaga Barañano MJ, Humiecka J, Safianowska A, Roesler UH, et al. MALDI spectra database for rapid discrimination and subtyping of Mycobacterium kansasii. Front Microbiol. 2018;9:587–7.
Luethy PM, Zelazny AM. Rapid one-step extraction method for the identification of molds using MALDI-TOF MS. Diagn Microbiol Infect Dis. 2018;91(2):130–0.
Oros D, Ceprnja M, Zucko J, Cindric M, Hozic A, Skrlin J, Barisic K, Melvan E, Uroic K, Kos B, et al. Identification of pathogens from native urine samples by MALDI-TOF/TOF tandem mass spectrometry. Clin Proteomics. 2020;17(1):25.
Huguenin A, Kincaid-Smith J, Rô Me Depaquit J, Rô Me Boissier J, Ferté H. MALDI-TOF: a new tool for the identification of Schistosoma cercariae and detection of hybrids. PLoS Negl Trop Dis. 2023;17(3):e0010577.
Ferreira L, Sánchez-Juanes F, García-Fraile P, Rivas R, Mateos PF, Martínez-Molina E, González-Buitrago JM, Velázquez E. MALDI-TOF mass spectrometry is a fast and reliable platform for identification and ecological studies of species from family Rhizobiaceae. PLoS ONE. 2011;6(5):e20223.
Zaluga J, Heylen K, Van Hoorde K, Hoste B, Van Vaerenbergh J, Maes M, De Vos P. GyrB sequence analysis and MALDI-TOF MS as identification tools for plant pathogenic Clavibacter. Syst Appl Microbiol. 2011;34(6):400–7.
Ahmad F, Babalola OO, Tak HI. Potential of MALDI-TOF mass spectrometry as a rapid detection technique in plant pathology: identification of plant-associated microorganisms. Anal Bioanal Chem. 2012;404:1247–55.
Ribeiro DG, Carmo LST, Santos IR, Almeida RF, Silva LP, Oliveira-Neto OB, Scherwinski-Pereira JE, Mehta A. MALDI TOF MS-profiling: applications for bacterial and plant sample differentiation and biological variability assessment. J Proteom. 2020;213:103619.
Santos C, Ventura JA, Costa H, Fernandes PMB, Lima N. MALDI-TOF MS to identify the pineapple pathogen Fusarium guttiforme and its antagonist Trichoderma Asperellum on decayed pineapple. Trop Plant Pathol. 2015;40(4):227–32.
Reeve MA, Pollard KM, Kurose D. Differentiation between closely-related Impatiens spp. And regional biotypes of Impatiens glandulifera using a highly-simplified and inexpensive method for MALDI-TOF MS. Plant Methods. 2018;14(1):1–13.
Reeve MA, Pollard KM. MALDI-TOF MS-based analysis of dried seed proteins immobilized on filter paper. Biology Methods & Protocols. 2019;4(1):1–12.
Reeve MA, Pollard KM. Discrimination between regional biotypes of Impatiens glandulifera using a simple MALDI-TOF MS-based method for use with seeds. Plant Methods. 2019;15(1):1–12.
Reeve M. MALDI-TOF MS-based analysis of seed proteins from catalogue varieties of Solanum lycopersicum/Lycopersicon esculentum. Horticulturae. 2019;5(3):48.
Ranganathan U, Groot SPC. Seed Longevity and Deterioration. In: Seed Science and Technology: Biology, Production, Quality Edited by Dadlani M, Yadava DK. Singapore: Springer Nature Singapore; 2023: 91–108.
Crossay T, Antheaume C, Redecker D, Bon L, Chedri N, Richert C, Guentas L, Cavaloc Y, Amir H. New method for the identification of arbuscular mycorrhizal fungi by proteomic-based biotyping of spores using MALDI-TOF-MS. Sci Rep. 2017;7(1):14306.
Mather CA, Rivera SF, Butler-Wu SM. Comparison of the Bruker Biotyper and Vitek MS matrix-assisted laser desorption ionization-time of flight mass spectrometry systems for identification of mycobacteria using simplified protein extraction protocols. J Clin Microbiol. 2014;52(1):130–8.
Ashfaq MY, Da’na DA, Al-Ghouti MA. Application of MALDI-TOF MS for identification of environmental bacteria: a review. J Environ Manage. 2022;305:114359.
Chun S, Gopal J, Muthu M. A consolidative synopsis of the MALDI-TOF MS accomplishments for the rapid diagnosis of microbial plant Disease pathogens. TrAC - Trends in Analytical Chemistry. 2022;156:116713.
Nishnitha K, Chaitrha M. MALDI-TOF MS: a novel tool for rapid detection of plant pathogens. Food and Scientific Reports. 2022;3:27–9.
Tarfeen N, Nisa KU, Nisa Q. MALDI-TOF MS: application in diagnosis, dereplication, biomolecule profiling and microbial ecology. Proceedings of the Indian National Science Academy 2022, 88(3):277–291.
Murphy BP, Tranel PJ. Identification and validation of Amaranthus species-specific snps within the ITS region: applications in quantitative species identification. Crop Sci. 2018;58(1):304–11.
Murphy BP, Plewa DE, Phillippi E, Bissonnette SM, Tranel PJ. A quantitative assay for Amaranthus palmeri identification. Pest Manag Sci. 2017;73(11):2221–4.
Brauge T, Trigueros S, Briet A, Debuiche S, Leleu G, Gassilloud B, Wilhelm A, Py J-S, Midelet G. MALDI-TOF mass spectrometry fingerprinting performance versus 16S rDNA sequencing to identify bacterial microflora from seafood products and sea water samples. 2021, 8:650116.
Stetter MG, Schmid KJ. Analysis of phylogenetic relationships and genome size evolution of the Amaranthus genus using GBS indicates the ancestors of an ancient crop. Mol Phylogenet Evol. 2017;109:80–92.
Raiyemo DA, Bobadilla LK, Tranel PJ. Genomic profiling of dioecious Amaranthus species provides novel insights into species relatedness and sex genes. BMC Biol. 2023;21(1):37.
Raiyemo DA, Tranel PJ. Comparative analysis of dioecious Amaranthus plastomes and phylogenomic implications within Amaranthaceae s.s. BMC Ecol Evol 2023, 23(1).
Ma X, Vaistij FE, Li Y, van Jansen WS, Harvey S, Bairu MW, Venter SL, Mavengahama S, Ning Z, Graham IA, et al. A chromosome-level Amaranthus cruentus genome assembly highlights gene family evolution and biosynthetic gene clusters that may underpin the nutritional value of this traditional crop. Plant J. 2021;107(2):613–28.
Thapa R, Edwards M, Blair MW. Relationship of cultivated grain amaranth species and wild relative accessions. Genes. 2021;12(12):1849.
Kietlinski KD, Jimenez F, Jellen EN, Maughan PJ, Smith SM, Pratt DB. Relationships between the weedy Amaranthus hybridus (Amaranthaceae) and the grain amaranths. Crop Sci. 2014;54(1):220–8.
Acknowledgements
We would to thank Laurena Matthies (Seed Science Unit - CFIA, Fallowfield, Ottawa) for providing the Brassicaceae seeds for the preliminary tests. We would also like to thank Daniel Klein and Stephan Briere (Plant Pathology lab - CFIA, Fallowfield, Ottawa) for their support in access to protein biotyping equipment and training. We also acknowledge Jennifer Neudorf (Seed Science and Technology Section – CFIA, Saskatoon) for preparation of the blind samples for protein biotyping. We want to thank Dr. Marie-Josée Simard, Dr. Robert Nurse and Kerry Bosveld from AAFC for providing the blind samples for our evaluation process. Finally, we would like to thank the GRIN-global US National Plant Germplasm system for providing Amaranthus seeds for our experiments.
Funding
This research was funded by CFIA internal projects N-000239 (Expansion of the CFIA weed DNA barcode collection and development of complementary genomic tools for the identification of regulated plant and seed species), N-000365 (Protein biotyping for seed species identification), N-000352 (Method development and testing decision tree for Identifying Amaranthus Seeds), and N-000451(Amaranth single seed blind sample validation).
Author information
Authors and Affiliations
Contributions
LGG conceived and led the project, supervised the experiments and wrote the article. MM generated the MSP library and ran all blind samples. JH performed the preliminary tests with Brassicaceae and standardized the base methodology. RW provided insight on project targets, shared data and blind samples. MM, JH and RW reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
13007_2023_1116_MOESM1_ESM.pptx
Additional file 1 (.Pptx) (A) Clustering of protein spectra corresponding to four Brassica species. (B) c (Brassica carinata), B. r (Brassica rapa), B. j (Brassica juncea), B. n (Brassica napus), alt = Winter variety. B. Clustering of protein spectra corresponding to four Brassica species including two species with accessions from different years. Cluster 1: Brassica rapa, 2: Brassica juncea, 3: Brassica carinata, 4: Brassica napus. Winter B. napus (w). Color legends show the year the accession was collected.
13007_2023_1116_MOESM3_ESM.pdf
Supplementary Material 3: Additional file 3 (.Pdf) MSP Validation Check 2023-05-10 Library. Bruker Daltonik MALDI Biotyper Classification Results.
13007_2023_1116_MOESM4_ESM.xlsx
Supplementary Material 4: Additional file 4. (.Xlsx) Blind sample testing for samples from AAFC in Table 1. For each sample, three seeds were tested, and for each seed three technical replicates were ran.
13007_2023_1116_MOESM5_ESM.xlsx
Supplementary Material 5: Additional file 5. (.Xlsx) Blind sample testing for samples from CFIA SSST in Table 2. Three technical replicates were run per seed.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Murphy, M., Hubert, J., Wang, R. et al. Seed protein biotyping in Amaranthus species: a tool for rapid identification of weedy amaranths of concern. Plant Methods 19, 143 (2023). https://doi.org/10.1186/s13007-023-01116-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13007-023-01116-9