
Abstract
Background
Mu heavy chain disease is a rare lymphoid neoplasm characterized by vacuolated bone marrow plasma cells and secretion of defective mu immunoglobulin heavy chains. The biological basis of mu heavy chain disease is poorly understood.
Case presentation
We report a case of mu heavy chain disease with MYD88 L265P mutation and deletion of 6q, genetic aberrations that are both strongly associated with lymphoplasmacytic lymphoma/Waldenström macroglobulinemia. Identification of the truncated mu immunoglobulin was facilitated by mass spectrometric analysis of the patient’s serum.
Conclusions
Mu heavy chain disease has been described as similar to chronic lymphocytic leukemia; however, the frequency of lymphocytosis in mu heavy chain disease has not been previously reported. We reviewed all previously published mu heavy chain disease reports and found that lymphocytosis is uncommon in the entity. This finding, along with the emerging genetic feature of recurrent MYD88 mutation in mu heavy chain disease, argues that at least a significant subset of cases are more similar to lymphoplasmacytic lymphoma than to chronic lymphocytic leukemia.
Similar content being viewed by others


Background
Mu heavy chain disease (mu-HCD) is an exceptionally rare entity described as resembling chronic lymphocytic leukemia (CLL), with vacuolated plasma cells and secretion of a defective mu heavy chain [1]. We report a case of mu-HCD with MYD88 L265P mutation along with deletion of 6q, supporting a biologic relationship between mu-HCD and lymphoplasmacytic lymphoma (LPL), a lymphoma that most often presents as Waldenström macroglobulinemia (WM).
Case presentation
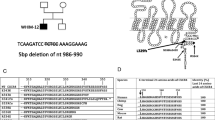
A 68-year-old female presented with fatigue, dyspnea, weight loss, and anemia (8.8 g/dL). The patient had an absolute lymphocyte count (ALC) of 2.2 × 109/L. A PET/CT scan did not demonstrate any lytic bone lesions or enlarged lymph nodes; the spleen size was reported as at the upper limit of normal. A bone marrow biopsy and aspirate were obtained. The bone marrow was markedly hypercellular due to a diffuse proliferation of small lymphocytes and plasma cells with prominent cytoplasmic vacuolization (Fig. 1 A and B). The B cells and plasma cells were positive for IgM by immunohistochemistry (Fig. 1C), and the plasmacytic component expressed kappa light chains (Fig. 1D). A CD5-negative, CD10-negative, kappa-restricted B-cell population was detected by flow cytometry. Serum and urine protein electrophoresis, serum free light chain testing, and serum immunoglobulin-enriched matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MASS-FIX) [2] were performed. The patient had markedly elevated free kappa light chains in the serum (13,285 mg/L, reference range 3.3–19.4 mg/L), along with kappa light chains in the urine, without a significant serologically detectable heavy chain (Fig. 2A). MASS-FIX identified an abnormally small mu heavy chain (26.3 kDa) in the serum that was further characterized using liquid chromatography coupled with electrospray ionization quadrupole time of flight mass spectrometry (LC-ESI-Q-TOF) [3] (Fig. 2B). Mutational studies performed on the peripheral blood demonstrated a MYD88 L265P mutation, and array comparative genomic hybridization performed on the bone marrow revealed a 6q deletion (Fig. 2C). The patient had normal renal function despite the Bence-Jones proteinuria. She was treated with 3 cycles of single-agent rituximab with normalization of hemoglobin and improvement of the white blood cell count.

Morphologic features of mu heavy chain disease. The bone marrow was markedly hypercellular due to a diffuse proliferation of small lymphocytes and plasma cells with prominent cytoplasmic vacuolization (A: bone marrow aspirate, Wright-Giemsa stain, 1000 × and B: bone marrow core biopsy, hematoxylin and eosin stain, 1000 ×). The immunohistochemical studies performed on the bone marrow biopsy demonstrated that the B-cells and plasma cells were strongly positive for IgM (C: 1000 ×) and the plasmacytic component strongly expressed kappa light chains (D: 1000 ×). Scale bars indicate 20 µm

Serum protein and genetic characterization of the case and lymphocyte counts in reported cases of mu heavy chain disease. A Serum immunoelectrophoresis identified a monoclonal protein composed of kappa light chains with a possible faint band at IgM. B The serum immunoglobulin-enriched matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MASS-FIX) and monoclonal immunoglobulin rapid accurate mass measurement IgM spectra for the patient demonstrating a 26,320 Da fragment of IgM heavy chain. Upper left panel: MASS-FIX IgM spectra demonstrating 3 M-proteins (An IgM kappa plus free kappa plus a suspected IgM heavy chain). Upper right panel: MASS-FIX IgM spectra for a healthy donor. Lower left panel: An LC-ESI-Q-TOF of the suspected IgM fragment for the + 11 charge state. Lower right panel: Fragmentation spectra of the suspected IgM heavy chain, demonstrating a pattern consistent with an IgM heavy chain. C Array comparative genomic hybridization revealed a 6q deletion. D Absolute lymphocyte count of mu heavy chain disease cases previously reported in the literature and including this case, plotted on a log scale. Dashed line indicates 5 × 109/L threshold for lymphocytosis. Abbreviations: LC-ESI-Q-TOF: liquid chromatography coupled with electrospray ionization quadrupole time of flight mass spectrometry
Discussion and conclusions
This case of mu-HCD with classic morphologic, phenotypic, and serologic features demonstrates genetic similarity to LPL with a MYD88 L265P mutation and deletion of 6q, both common abnormalities in LPL [4, 5]. Vergneault and colleagues recently reported two cases of mu-HCD with MYD88 L265P mutations, although vacuolated plasma cells were described in only one case and none were illustrated [6]. Detailed analysis of the mu heavy chain was also not provided. These cases, however, in addition to ours, establish that MYD88 mutations are recurrent in mu-HCD, suggesting a relationship with LPL. The generalizability of this finding is difficult to assess as mu-HCD is extraordinarily rare, with only 30–40 cases previously reported; however, additional indirect evidence suggests that a relationship with LPL is plausible in many mu-HCD cases.
The first patient described with mu-HCD had a high ALC of approximately 50 × 109/L, and the patient was clinically diagnosed with CLL [7, 8]. In 1992, Wahner-Roedler and Kyle published a summary of the then-extant literature on mu-HCD, describing the clinicopathologic features of 28 affected patients in what remains the most comprehensive description of the disease [9]. In their review, 9 of the 28 patients were reported as having been diagnosed with CLL. The 2017 WHO Classification states that mu-HCD is “a B-cell neoplasm resembling CLL” [1]; however, mu heavy chain secretion and plasmacytic differentiation are not typical features of CLL. Lymphocytosis is the sine qua non of CLL, but the frequency of lymphocytosis in mu-HCD is not established, as it has not been summarized in previous reviews of the entity. We attempted to review all prior reports of mu-HCD, collecting peripheral blood white blood cell and differential count information from 29 previously reported patients with mu-HCD [6, 8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29]. Including our case, ~ 73% (22 of 30) of patients with mu-HCD for whom information was available had no evidence of lymphocytosis (defined as ALC > 5 × 109/L), and only 5 reported patients have had ALC greater than 10 × 109/L (Fig. 2D), a much lower frequency of lymphocytosis than would be expected for a cohort of CLL patients.
Nearly all cases of CLL are positive for CD5, but the immunophenotypic characteristics of mu-HCD are not well established, as many of the reports of mu-HCD predate comprehensive immunophenotyping of lymphoid neoplasia and the recognition of the CD5 antigen on CLL cells [30]. However, at least one previously reported patient with mu-HCD with lymphocytosis (ALC ~ 29 × 109/L) was negative for CD5 [14], as was our case and one of the two cases reported by Vergneault and colleagues [6]. CD5 may be expressed on LPL [31], and so reports of CD5 expression in some mu-HCD cases do not preclude classification as LPL [6].
These features suggest that most previously reported cases of mu-HCD are unlike CLL as currently understood. Our case and those of Vergneault and colleagues [6] establish a biologic relationship with LPL in at least some mu-HCD cases, which is also congruent with the presence of plasmacytic differentiation and production of IgM heavy chains in both entities. Some clinical features such as hepatosplenomegaly appear to be more common in mu-HCD than in LPL. It may be that conventional LPL associated with WM tends to manifest earlier in the course of disease than does mu-HCD due to increased pathogenicity of intact IgM paraproteins versus the truncated IgM proteins produced by mu-HCD. It remains possible that mu-HCD is a biologically heterogeneous entity, and some cases may have CLL-like genetics.
This report additionally underscores the value of novel techniques such as MASS-FIX in the detection of abnormal serum proteins. mu-HCD may be underrecognized, and patients with increased free serum light chains or Bence-Jones proteinuria without a corresponding serum heavy chain should be considered for further assessment using methods such as MASS-FIX to better evaluate for abnormal proteins that could be missed using conventional serologic techniques. Discrimination between a B-cell neoplasm with plasmacytic differentiation and a plasma cell neoplasm is highly clinically relevant, and accurate M-protein characterization is a critical component of the diagnosis. Our findings suggest that mu-HCD cases should be screened for MYD88 mutation and establish a rationale for potential LPL-directed management of this rare entity.
Change history
20 October 2022
A Correction to this paper has been published: https://doi.org/10.1186/s13000-022-01265-w
Abbreviations
- mu-HCD:
-
Mu heavy chain disease
- CLL:
-
Chronic lymphocytic leukemia
- LPL:
-
Lymphoplasmacytic lymphoma
- WM:
-
Waldenström macroglobulinemia
- ALC:
-
Absolute lymphocyte count
- MASS-FIX:
-
Serum immunoglobulin-enriched matrix-assisted laser desorption ionization time-of-flight mass spectrometry
- LC-ESI-Q-TOF:
-
Liquid chromatography coupled with electrospray ionization quadrupole time of flight mass spectrometry
References:
Swerdlow SHCE, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised. 4th ed. Lyon: IARC; 2017.
Murray DL, Puig N, Kristinsson S, Usmani SZ, Dispenzieri A, Bianchi G, et al. Mass spectrometry for the evaluation of monoclonal proteins in multiple myeloma and related disorders: an International Myeloma Working Group Mass Spectrometry Committee Report. Blood Cancer J. 2021;11(2):24.
Barnidge DR, Dasari S, Ramirez-Alvarado M, Fontan A, Willrich MAV, Tschumper RC, et al. Phenotyping Polyclonal Kappa and Lambda Light Chain Molecular Mass Distributions in Patient Serum Using Mass Spectrometry. J Proteome Res. 2014;13(11):5198–205.
Mansoor A, Medeiros LJ, Weber DM, Alexanian R, Hayes K, Jones D, et al. Cytogenetic Findings in Lymphoplasmacytic Lymphoma /Waldenström Macroglobulinemia: Chromosomal Abnormalities Are Associated With the Polymorphous Subtype and an Aggressive Clinical Course. Am J Clin Pathol. 2001;116(4):543–9.
Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P Somatic Mutation in Waldenström’s Macroglobulinemia. N Engl J Med. 2012;367(9):826–33.
Vergneault H, Bengoufa D, Frazier-Mironer A, Brocheriou I, Bitoun S, Villesuzanne C, et al. Light chain proteinuria revealing mu-heavy chain disease: an atypical presentation of Waldenström macroglobulinemia in two cases. Haematologica. 2021;106(7):2034–6.
Ballard HS, Hamilton LM, Marcus AJ, Illes CH. A new variant of heavy-chain disease (mu-chain disease). N Engl J Med. 1970;282(19):1060–2.
Forte FA, Prelli F, Yount WJ, Jerry LM, Kochwa S, Franklin EC, et al. Heavy chain disease of the gamma (gamma M) type: report of the first case. Blood. 1970;36(2):137–44.
Wahner-Roedler DL, Kyle RA. μ-heavy chain disease: Presentation as a benign monoclonal gammopathy. Am J Hematol. 1992;40(1):56–60.
Andreeva NE, Antipova LG, Chernokhvostova EV, Batalova TN. Disease of heavy chains mu (new forms of paraproteinemic hemoblastoses). Ter Arkh. 1976;48(8):10–5.
Bakhshi A, Guglielmi P, Coligan JE, Gamza F, Waldmann TA, Korsmeyer SJ. A pre-translational defect in a case of human mu heavy chain disease. Mol Immunol. 1986;23(7):725–32.
Bonhomme J, Seligmann M, Mihaesco C, Clauvel JP, Danon F, Brouet JC, et al. MU-chain disease in an African patient. Blood. 1974;43(4):485–92.
Brouet J-C, Seligmann M, Danon F, Belpomme D, Fine J-M. µ-Chain Disease: Report of Two New Cases. Arch Intern Med. 1979;139(6):672–4.
Cogné M, Aucouturier P, Brizard A, Dreyfus B, Duarte F, Preud’homme J-L. Complete variable region deletion in Aα heavy chain disease protein (roul). Correlation with light chain secretion. Leuk Res. 1993;17(6):527–32.
Dammacco F, Bonomo L, Franklin EC. A new case of mu heavy chain disease: clinical and immunochemical studies. Blood. 1974;43(5):713–9.
Danon F, Mihaesco C, Bouvry M, Clerc M, Seligmann M. A new case of heavy mu-chain disease. Scand J Haematol. 1975;15(1):5–9.
Fujii H, Shimizu T, Seki S, Isemura T, Yamamoto K, Kanoh T, et al. Combined features of mu-heavy chain disease and primary macroglobulinemia in a single patient: clinical and immunological studies. Nihon Ketsueki Gakkai Zasshi. 1982;45(3):622–32.
Germann HJ, Westerhausen M, Kickhöfen B. μ-Kettenkrankheit*. Dtsch Med Wochenschr. 1972;97(49):1902–5.
Jønsson V, Videbæk A, Axelsen NH, Harboe M. μ-Chain Disease in a Case of Chronic Lymphocytic Leukaemia and Malignant Histiocytoma. Scandinavian Journal of Haematology. 1976;16(3):209–17.
Josephson AS, Nicastri A, Price E, Biro L. H chain fragment and monoclonal IgA in a lymphoproliferative disorder. Am J Med. 1973;54(1):127–35.
Kinoshita K, Yamagata T, Nozaki Y, Sugiyama M, Ikoma S, Funauchi M, et al. μ-Heavy Chain Disease Associated with Systemic Amyloidosis. Hematology. 2004;9(2):135–7.
Leach IH, Jenkins JS, Murray-Leslie CF, Powell RJ. Mu-heavy chain and monoclonal IgG K paraproteinaemia in systemic lupus erythematosus. Br J Rheumatol. 1987;26(6):460–2.
Lee SL, Rosner F, Ruberman W, Glasberg S. Mu-chain disease. Ann Intern Med. 1971;75(3):407–14.
O’Reilly DS, Adjukiewicz A, Whicher JT. Biochemical findings in a case of mu-chain disease. Clin Chem. 1981;27(2):331–3.
Pruzanski W, Hasselback R, Katz A, Parr DM. Multiple myeloma (light chain disease) with rheumatoid-like amyloid arthropathy and mu-heavy chain fragment in the serum. Am J Med. 1978;65(2):334–41.
Silva-Moreno M, Ruiz-Arguelles GJ, Lopez-Karpovitch X, Labardini-Mendez J. Heavy chain disease. Report of four cases. Sangre (Barc). 1983;28(1):89–98.
Tamura A, Yamashiro A, Mizutani F, Oita T, Maeda A, Takahashi T. Immunochemical properties of free mu-chain protein in a patient with mu-heavy chain disease. Rinsho Byori. 2003;51(9):847–51.
Wetter O, Schmidt CG, Linder KH, Leene W. H-Kettenkrankheit: Humorale und celluläre Befunde bei sechs Fällen vom μ-Kettentyp. J Cancer Res Clin Oncol. 1979;94(2):207–23.
Witzens M, Egerer G, Stahl D, Werle E, Goldschmidt H, Haas R. A case of μ heavy-chain disease associated with hyperglobulinemia, anemia, and a positive Coombs test. Ann Hematol. 1998;77(5):231–4.
Schroff RW, Foon KA, Billing RJ, Fahey JL. Immunologic Classification of Lymphocytic Leukemias Based on Monoclonal Antibody-Defined Cell Surface Antigens. Blood. 1982;59(2):207–15.
Hunter ZR, Branagan AR, Manning R, Patterson CJ, Ditzel Santos D, Tournilhac O, et al. CD5, CD10, and CD23 Expression in Waldenström’s Macroglobulinemia. Clin Lymphoma. 2005;5(4):246–9.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
VB and NGB wrote the manuscript and analyzed data. SHS edited the manuscript. SEW performed immunologic studies. DLM and MCK performed mass spectrometric studies. YAS performed genetic studies. MEA provided clinical care to the patient. JAV, MD, SHS, and NGB interpreted the bone marrow specimen and flow cytometric data. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Consent for publication
Not applicable.
Competing interests
David L Murray has intellectual property rights to the MASS-FIX assay and patents. The other authors have no conflicts related to this work.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: "Following publication of the original article [1], the authors found a typo in one of the author names in the author group section. Instead of the accurate author name "Jeffrey A. Vos," it displays the wrong author name "Jeffrey A. VosUPMC" in the web article that has been published. The original article has been updated.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Baloda, V., Wheeler, S.E., Murray, D.L. et al. Mu heavy chain disease with MYD88 L265P mutation: an unusual manifestation of lymphoplasmacytic lymphoma. Diagn Pathol 17, 63 (2022). https://doi.org/10.1186/s13000-022-01244-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13000-022-01244-1