
Abstract
Air pollution is an issue of increasing interest due to its globally relevant impacts on morbidity and mortality. Controlled human exposure (CHE) studies are often employed to investigate the impacts of pollution on human health, with diesel exhaust (DE) commonly used as a surrogate of traffic related air pollution (TRAP). This paper will review the results derived from 104 publications of CHE to DE (CHE-DE) with respect to health outcomes. CHE-DE studies have provided mechanistic evidence supporting TRAP’s detrimental effects on related to the cardiovascular system (e.g., vasomotor dysfunction, inhibition of fibrinolysis, and impaired cardiac function) and respiratory system (e.g., airway inflammation, increased airway responsiveness, and clinical symptoms of asthma). Oxidative stress is thought to be the primary mechanism of TRAP-induced effects and has been supported by several CHE-DE studies. A historical limitation of some air pollution research is consideration of TRAP (or its components) in isolation, limiting insight into the interactions between TRAP and other environmental factors often encountered in tandem. CHE-DE studies can help to shed light on complex conditions, and several have included co-exposure to common elements such as allergens, ozone, and activity level. The ability of filters to mitigate the adverse effects of DE, by limiting exposure to the particulate fraction of polluted aerosols, has also been examined. While various biomarkers of DE exposure have been evaluated in CHE-DE studies, a definitive such endpoint has yet to be identified. In spite of the above advantages, this paradigm for TRAP is constrained to acute exposures and can only be indirectly applied to chronic exposures, despite the critical real-world impact of living long-term with TRAP. Those with significant medical conditions are often excluded from CHE-DE studies and so results derived from healthy individuals may not apply to more susceptible populations whose further study is needed to avoid potentially misleading conclusions. In spite of limitations, the contributions of CHE-DE studies have greatly advanced current understanding of the health impacts associated with TRAP exposure, especially regarding mechanisms therein, with important implications for regulation and policy.
Similar content being viewed by others

Background
Air pollution, one of the leading causes of death worldwide, is linked to an estimated 7 million deaths per year [1]. Numerous adverse health outcomes are associated with air pollution affecting the cardiovascular, respiratory, and neurological systems [2, 3]. Air quality is a persistent and growing global issue as humans continue to generate pollution through industrial sources, vehicles, and household energy consumption. While agencies such as the World Health Organization (WHO) [4, 5] and the US Environmental Protection Agency [6] have set standards for air quality, in 2019 over 90% of the world’s population lived in regions with pollutant levels above 2005 WHO air quality standards [4]. Traffic related air pollution (TRAP) constitutes a substantial portion of air pollution globally, with a significant source of TRAP being diesel engines, used in vehicles such as trucks, buses, boats, cars, vans, and trains.
The health effects of diesel exhaust (DE) are commonly investigated in controlled human exposure (CHE) studies. In this experimental design, participants are exposed to a known quantity of pollutant under controlled conditions, typically inside a specialized exposure chamber. For CHE to DE (CHE-DE) studies, DE exposure is typically quantified by the concentration of particulate matter (PM) with diameter less than 2.5 μm (PM2.5) or, less frequently, 10 μm (PM10) as these are common PM metrics linked in voluminous epidemiological studies to date [7]. In addition to PM, DE is composed of gases, notably nitrogen dioxide (NO2), carbon monoxide (CO), and gaseous hydrocarbons [8] which are often measured but not titrated as the primary exposure metric in these studies. CHE-DE are valuable for studying the acute effects of DE exposure due to their ability to control exposure duration, PM concentration, and other exposure characteristics such as humidity and temperature. Furthermore, the use of individual participants as their own control, given the crossover design, is a powerful factor in effectively eliminating confounding factors that may vex other study designs. This crossover design also facilitates clear statistical analysis of such variables (sex, age, baseline health-related phenotypic characteristics, host genotype, etc.) as potential modifiers of the primary effect of DE across a range of endpoints. The design of CHE-DE experiments is conducive to analysis of wide variety of endpoints derived from blood, urine, or airway samples, or in terms of a range of physiologic parameters derived from respiratory or cardiovascular outputs, or from repeated administration of questionnaires.
These CHE-DE studies have investigated a broad range of health outcomes, from cardiovascular effects such as vasomotor function and thrombosis, to pulmonary effects including inflammation and lung function. Mechanisms of DE-associated health effects on the genetic, epigenetic, and biochemical levels have also been studied in CHE-DE papers. As the set-up of CHE-DE studies makes it possible to control concurrent exposures, many papers have investigated the influence of co-exposures such as ozone (O3) and allergens on the impact of DE inhalation. While findings from CHE-DE studies have been previously reviewed [9,10,11] the literature with respect to CHE-DE has grown profoundly in recent years and continues to do so. Thus, the focus of this paper is to provide a broad, updated review of the health-related results gleaned from this body of literature.
Methods
A literature search was conducted using the PubMed and Web of Science databases. The keyword “diesel exhaust” or “was included in all search queries, in combination with “controlled human exposure”, “human exposure”, or “exposure” (e.g., “diesel exhaust” AND “human exposure”). Eligible studies must have exposed participants to a controlled quantity of DE via inhalation and been published in December 2020 or earlier. We excluded letters, abstracts, and academic theses. The reference lists of included articles as well as the Clinicaltrials.gov registration page for publications that reported a clinical trial number were also searched for eligible studies. Through this process, we identified 104 publications eligible for review. The methodology of each paper, such as the DE concentration, gaseous composition, exposure conditions, and exposure durations is reviewed in detail in a separate companion paper currently under review. Main findings with respect to health outcomes were extracted and summarized in a table together with brief details of study methodology (PM concentration(s) of DE exposures, concurrent exposures, and participant characteristics). Based on their main findings, eligible publications were then categorized by a primary topic: oxidative stress and antioxidants, systemic inflammation, respiratory, cardiovascular, neurological, exercise, co-exposures, filtered DE, markers and quantification of DE exposure, and other. Detailed evaluation of each paper was then conducted to synthesize this review.
Results
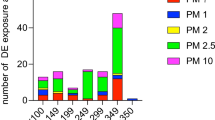
Main health outcomes of the 104 reviewed publications along with an abbreviated description of study methodology are outlined in Table 1. 24 studies included co-exposure to DE and additional agents such as allergens (6 studies), antioxidants (5 studies), ozone (3 studies), and various other agents. 46 studies included healthy participants only, 19 studies included participants with asthma or atopy, 7 studies included participants with metabolic syndrome, and 7 studies included participants with other morbidities such as heart failure [94, 95], COPD [26, 35], and coronary artery disease [51, 59, 69]. Further details of study methodology can be found in a separate companion paper which is currently under review. With respect to the primary category of health outcomes reported, 9 publications reported oxidative stress outcomes, 3 publications reported systemic inflammation outcomes, 25 publications reported respiratory outcomes, 23 publications reported cardiovascular outcomes, 2 publications reported neurological outcomes, 8 publications reported exercise outcomes, 12 publications reported co-exposure outcomes, 7 publications reported filtered DE outcomes, and 15 publications reported outcomes that did not fall under any of the above categories. Fig. 1 provides a brief summary of all health outcomes in the reviewed articles.

Summary of health outcome findings from controlled human exposure to diesel exhaust studies. DE diesel exhaust
Oxidative stress outcomes
Diesel exhaust exposure induces oxidative stress
Robust evidence from CHE-DE studies supports oxidative stress as a mechanism of DE-associated effects. DE exposure at 300 μg/m3 PM10 has been shown to induce activation and nuclear translocation of factors involved in response to oxidative stress, such as nuclear factor (NF)-κB (p = 0.02), as well as the mitogen-activated protein kinases p38 (p = 0.01) and JNK (p = 0.04), in the bronchial epithelium of healthy participants [18]. Other studies have demonstrated changes in gene expression and DNA methylation linked to oxidative stress pathways in peripheral blood mononuclear cells after DE exposure [15, 21].
Studies that demonstrate enhanced effects amongst those with variants of genes of the glutathione-s-transferase (GST) family, and others related to oxidative stress metabolism, further support the role of oxidative stress herein. Some CHE-DE studies estimate genetic susceptibility to oxidative stress by stratifying participants based on GSTM1 status, a gene coding one of several glutathione S-transferases involved in response to oxidative stress [116]. The GSTM1 null phenotype is common and is linked to decreased tolerance to oxidative stress as well as increased lung inflammation [13, 83, 86, 116], though this effect is not always observed [30]. Accordingly, there is evidence from CHE-DE studies that individuals with variant GST-family genotypes are more susceptibile to adverse effects of DE exposure [30], though this area of the literature is not entirely consistent and further investigation is necessary.
Potential benefit of supplemental and endogenous antioxidants
The potential of exogenous antioxidants, as supplements, in reducing the harmful effects of DE inhalation has been evaluated in CHE-DE studies as well. As noted above, in participants with baseline airway hyperresponsiveness, exposure to PM2.5 at 300 μg/m3 (subsequently within this review, this will be abbreviated simply to the mass concentration, e.g. DE300 unless otherwise indicated) increased this hyperresponsiveness, and N-acetylcysteine supplementation for 6 days prior to DE exposure eliminated this effect [13]. Another analysis from this study showed that antioxidant supplementation attenuated DE-induced changes in blood miRNA and associated oxidative stress genes, further implicating such stress as a mechanism for the effects of DE [14]. However, other studies have failed to demonstrate a protective effect of antioxidant supplementation. Another study found that supplementation with N-acetylcysteine and ascorbate prior to DE300 exposure did not abrogate DE-induced changes in markers of oxidative stress [12] and pre-treatment with antioxidant in a separate study was associated with enhancement of DE-induced vasoconstriction [50]. Therefore, while antioxidant supplementation is a relatively convenient and low-cost intervention, its ability to mitigate the adverse effects of DE has has not been consistently validated in CHE-DE studies [117]. It is likely that the uncertainty therein relates to the specific anti-oxidants, their administration (timing and dose), the host phenotype, and the pollution exposure context, amongst other factors.
The ability of endogenous antioxidants to moderate oxidative stress has also been investigated in a CHE-DE study. In healthy participants exposed to DE at 100 μg/m3 PM10, the endogenous antioxidants ascorbate, urate, and reduced glutathione in the bronchial and nasal airways were not depleted; rather airway reduced glutathione levels were instead elevated (nasal lavage p < 0.05, bronchial wash p = 0.004) while markers of inflammation were not, suggesting endogenous antioxidant systems in healthy populations may have been mobilized to sufficiently combat DE-associated oxidative stress [19]. Interestingly, a study of participants with metabolic syndrome did not find evidence of oxidative stress or systemic antioxidant responses after exposure to DE200, perhaps as a result of adaptations to chronic oxidative challenge [16]. This heterogeneity prompts further investigation of the effects of anti-oxidants, whether endogenous or exogenously supplied, including potential modification of effect by relevant gene variants. Furthermore, inference from these studies requires careful attention to the overall methods and results for any given investigation.
Diesel exhaust induces systemic inflammation
CHE-DE studies have reported robust links between acute DE exposure and systemic inflammation. DE exposure at 276 μg/m3 PM1 was shown to increase peripheral blood leukocyte count (increase of 0.16 ± 1.01 × 109 cells/L from pre-exposure count) and monocyte count (increase of 0.01 ± 0.10 × 109 cells/L from pre-exposure count) compared to FA (p = 0.007 and p = 0.017 respectively) [22]. The same study also reported a trend towards increased IL-6 after DE exposure (increase of 0.12 ± 0.78 pg/mL compared to pre-exposure, p = 0.066 compared to FA) [22]. A 1999 study of healthy volunteers reported significant increases in neutrophil (p = 0.04) and platelets (p = 0.02) counts in peripheral blood with DE exposure compared to FA, though HLA-DR + lymphocyte count was decreased (p = 0.02) [47]. In a 2012 study, plasma obtained from healthy volunteers exposed to DE at 100 μg/m3 was incubated with human endothelial cells, and this demonstrated increased endothelial cell expression of the inflammatory marker vascular cell adhesion molecule-1 by 20% (p < 0.05) [23]. Notably, one CHE-DE study suggests that COPD patients may be more susceptible to the pro-inflammatory effects of DE exposure [26]. In this study, markers of neutrophil activation such as CD16, CXCR2, as well as percentage of activated neutrophils (+ 12.2% change) in peripheral blood were increased in COPD patients compared to healthy subjects after DE exposure (p = 0.006, p = 0.002, p = 0.046 respectively) [26]. CHE-DE studies have also described DE-induced changes in DNA methylation [21] and gene expression [17] within inflammatory pathways. In sum, CHE-DE studies provide compelling evidence of systemic pro-inflammatory effects from acute DE exposure.
Few controlled human exposure studies to diesel exhaust examine genotoxicity
The genotoxic effects of DE and its components have been extensively investigated in animal models, in vitro experiments, and observational studies [118,119,120,121,122,123,124]. A 2015 CHE-DE study by Hemmingsen and colleagues examined genotoxicity-related outcomes; while there was no statistically appreciable effect of DE exposure on markers of DNA damage, they did report a significant increase (p < 0.05) in the level of hOGG1-sensitive sites with exposure to 75 dB of traffic noise, suggesting a genotoxic effect of traffic noise [91]. Currently, there is paucity of CHE-DE studies pertaining to genotoxicity of DE exposure (Table 1). Future CHE-DE studies investigating this topic would greatly strengthen the pre-existing literature, and serve to establish a stronger link between DE exposure and genotoxicity as a mechanism of DE-associated health effects.
Respiratory outcomes
Diesel exhaust increases pulmonary inflammation in healthy subjects
TRAP exposure has been associated with increased asthma exacerbations [125, 126], development of chronic obstructive pulmonary disease (COPD) [127,128,129,130], and symptoms of respiratory irritation [131,132,133]. CHE-DE studies published in the early 2000’s have consistently demonstrated elevations in biomarkers of pulmonary inflammation after DE exposure, providing an explanation for these observations. DE exposure at concentrations of 200 to 300 μg/m3 PM10 have been shown to acutely increase inflammatory markers such as interleukin (IL)-6, IL-8, IL-13, methylhistamine, neutrophils, and myeloperoxidase (MPO) in the respiratory tract of healthy subjects [41, 44,45,46]. Interestingly, pulmonary inflammation induced by DE in asthmatics is less clear. A 2011 study found that exposure to DE at 100 μg/m3 PM10 increased neutrophils (p = 0.01), IL-6 (p = 0.03), and MPO (p = 0.04) in bronchial wash samples from healthy participants while these markers were not elevated in asthmatic subjects [36]. Another study comparing healthy versus asthmatic volunteers reported similar results, with significantly increased airway neutrophils (p < 0.05), lymphocytes (p < 0.05), and IL-8 (p < 0.05) after DE exposure at 100 μg/m3 PM10 in only the healthy group [42]. While the asthmatic subjects in both studies had significantly elevated levels of eosinophils (p < 0.001 [42]; p = 0.013 [36]) and mast cells (p < 0.05 [42]; p < 0.001 [36]) compared to their healthy controls at baseline or after FA exposure, DE exposure did not significantly increase these markers of allergic inflammation in the asthmatic groups [36, 42].
Diesel exhaust worsens asthmatic airway function
Though inflammatory markers are not consistently increased by controlled DE in asthmatic subjects, CHE-DE does transiently worsen relevant asthma physiology. Exposure to DE300 has been shown to increase airway hyperreactivity (methacholine PC20 = 14.9 mg/mL after DE compared to 19.7 mg/mL after FA, p = 0.012) and obstruction (3.3% decrease in FEV1% after DE compared to 3.1% increase after FA at 24 h post exposure, p = 0.043) in subjects with asthma [34]. Similarly, in a different study of asthmatics, inhalation of DE at 300 μg/m3 PM10 increased both airway hyperreactivity (methacholine PC20 = 1.77 ± 1.35 mg/mL (DE) compared to 3.47 ± 1.36 mg/mL (FA), p < 0.001) and resistance (p = 0.004) [43]. Increased airway resistance has also been reported with a lower level of DE exposure, at 100 μg/m3 PM10 (4.1% increase in healthy subjects (p < 0.01 DE compared to FA), 6.5% increase in asthmatics (p < 0.01 DE compared to FA)) [42]. One proposed mechanism as to how DE exposure impacts the lungs of asthmatics is through oxidative stress. In one study of 16 participants with mild to moderate asthma, nitrite in exhaled breath condensate was increased immediately after DE exposure (p = 0.052) [34]. A separate study found that subjects with baseline airway hyperreactivity had a 42% increase in airway responsiveness after DE exposure compared to FA exposure (p = 0.03), with this increase abated by prior anti-oxidant supplementation [13]. Interestingly, this same study also showed that anti-oxidant supplementation in individuals with airway hyper-reactivity reduced baseline airway responsiveness by 20% (p = 0.001) [13], further implicating the role of oxidative stress. Other mechanistic insight has come through focus on underemphasized pathways [29, 134] and by interrogating the role of epigenetics on lung function [24]. However, the exact mechanisms underlying the impact of DE on asthmatic lungs is still poorly understood and elucidating the differential pulmonary effects of DE is an important focus for further study.
Cardiovascular outcomes
Diesel exhaust exposure impairs vasomotor function
Exposure to air pollution has been consistently associated with cardiovascular morbidity and mortality [3, 135,136,137]. One pathophysiological mechanism thought to underlie this association is the impairment of vasomotor function. Indeed, multiple CHE-DE studies have demonstrated a link between DE exposure and vascular dysfunction. A study of 30 healthy male participants showed that 1 h exposure to DE, titrated to a nominal concentration of DE300 significantly impaired vasodilation in response to bradykinin (p < 0.05), acetylcholine (p < 0.05), and sodium nitroprusside (p < 0.001) [71], with similar results observed at DE250 in a different study [60]. Two studies that assessed brachial artery diameter reported acute vasoconstriction, a corollary of impaired dilation, after DE200 (decrease in diameter with DE versus FA = 0.11 mm, p = 0.01 [67] and 0.09 mm, p = 0.03 [50]) [50, 67]. Another study demonstrated impairment of vasodilation at 24 h after DE exposure, implying this effect may persist even into the day following inhalation [70]. Dysfunction of the endothelial nitric oxide (NO) pathway has been implicated in DE-associated vascular dysfunction. In a study with 12 healthy volunteers, DE exposure reduced vasodilation in response to acetylcholine (p < 0.01) but not sodium nitroprusside, suggesting DE inhalation only impairs endothelium-dependent vasodilation [55]. The study further demonstrated a correlation between the impairment of vasomotor function and production of reactive oxygen species (ROS), indicating these effects of DE may be mediated by oxidative stress [55]. In another study, DE inhalation increased plasma nitrite concentration (68 ± 48 nmol/L after DE versus 41 ± 32 nmol/L after FA, p = 0.006) in healthy volunteers, suggesting increased NO generation as a potential physiological mechanism to the vasoconstrictive effect of DE [54]. The same study also demonstrated a greater increase in blood pressure (p = 0.048) and central arterial stiffness (p = 0.007) with DE inhalation compared to filtered air (FA) in the presence of systemic NO synthase inhibition, implying that while DE exposure increases NO generation, this is offset by a greater increase in NO consumption leading to an overall reduction in NO bioavailability [54]. The mechanism of reduced NO bioavailability has been further implicated in animal models [138] as well as other CHE-DE studies [61]. While there is strong evidence that DE exerts its vasomotor effects via the NO pathway, the exact mechanism and the potential involvement of other mediators remains uncertain.
Diesel exhaust exposure increases likelihood of thrombosis and decreases fibrinolytic capacity
Another mechanism through which DE increases susceptibility to cardiovascular disease is through its effects on fibrinolysis and thrombosis. DE has been shown to suppress bradykinin-induced release of plasma tissue plasminogen activator in healthy volunteers at DE250 and DE300 [60, 71], suggesting DE inhalation impairs endogenous fibrinolytic function. DE exposure has been linked to increased propensity of thrombus formation, as reported in an ex vivo study conducted with blood drawn from healthy volunteers exposed to DE at 350 μg/m3 [65]. However, a different study that also involved healthy participants found no difference in thrombotic markers such as D-dimer, von Willebrand factor (vWF), platelets, and plasminogen activator inhibitor-1 (PAI-1) after exposure to DE100 or DE200 [68]. This discrepancy may be due to sample size, as the authors noted their results trended in the expected direction but failed to meet statistical significance [68]. Another reason could be the DE concentration was insufficient to induce a significant prothrombotic effect, as the Lucking study [65] used a concentration of DE at 350 μg/m3. Interestingly, in a similar study conducted in volunteers with metabolic syndrome, hypothesized to be more susceptible to cardiovascular risk, again no significant increase in D-dimer, vWF, and PAI-1 was observed after DE100 and DE200 exposure [64]. In these studies from Seattle, it may be that the biomarker approach was insufficiently sensitive, relative to the Badimon chamber technique used routinely in Umea, to thrombotic phenomena. Though there seems to be a general consensus that DE inhalation is prothrombotic, this effect has most observable at DE concentrations at or above 250 μg/m3 and further mechanistic detail remains to be elucidated.
Diesel exhaust adversely affects heart rate variability and blood pressure
CHE-DE studies have also investigated the effects of DE exposure on measures of cardiac function. A paper by Langrish et al. [51] using data pooled from multiple studies, including a number of CHE-DE studies [58, 60, 69, 71, 73], reported no significant increase in the short term risk of arrhythmia after acute DE exposure in healthy individuals or those with coronary artery disease [51]. CHE-DE studies have also examined other cardiac parameters such as heart rate and blood pressure. A 2014 study found that 2 h exposure to DE at 300 μg/m3 increased diastolic blood pressure (DBP) by an average of 5 mmHg (p = 0.04), a change that was less pronounced at DE at 100 μg/m3 and 200 μg/m3 [52]. DE inhalation at 300 μg/m3 also decreased indices of the frequency domain of heart rate variability (HRV), a marker of cardiac autonomic function [52]. Of note, the 6 healthy volunteers who participated in this study were null for the GSTM1 gene, a deletion that is associated with increased susceptibility to oxidative stress [52]. Another study involving both healthy and participants with metabolic syndrome found no consistent effect of DE exposure on heart rate variability at DE100 and DE200 [66], perhaps due to the lower concentrations used. A different study found that exposure to DE200 did not significantly affect DBP but did increase systolic blood pressure (SBP) by an average of 4.4 mmHg (95% CI: 1.1, 7.7, p = 0.0009) post-exposure [56]. This study, involving both healthy and participants with metabolic syndrome, did not find a significant effect on heart rate [56]. Observational study designs have reported associations between TRAP exposure and both increased blood pressure [139,140,141] and changes in heart rate variability [142,143,144]. Though there is a lack of concordance in the specific findings, CHE-DE studies have overall demonstrated an adverse impact of acute DE exposure on heart rate variability and blood pressure.
Limited evidence from controlled human exposures to diesel exhaust exposure suggest adverse neurological effects
The neurological effects of acute DE exposure have also been explored in a 2008 CHE-DE study. 10 healthy volunteers were exposed to DE at 300 μg/m3, with brain activity monitored via electroencephalography (EEG) during and one hour after the exposure [73]. The results of the study demonstrated a significant increase in median power frequency in the frontal cortex during DE exposure compared to FA (p < 0.05), with this elevation attributed predominantly to increased fast wave activity (β2) [73]. Elevated β2 is associated with increased cortical stress, and is often seen in patients with neurological and neuropsychological disorders such as post-traumatic stress disorder, traumatic brain injury, and headache [145, 146]. However, the implications of this increased cortical activity on clinical outcomes such as cognition or neurological disease are unclear. Epidemiological studies, animal models, and in vitro experiments have linked air pollution exposure to the development of neurodegenerative diseases such as Alzheimer’s disease [147] and Parkinson’s disease [148, 149]. Long-term exposure to air pollution has been also shown to adversely impact cognitive performance on verbal and math tests [150]. Children are believed to be particularly susceptible to the cognitive consequences of air pollution. Exposure to TRAP has been shown to negatively impact neurobehavioural function in adolescents [151] as well as cognitive development in children [152]. Pathways that have been proposed to mediate the neurological impacts of DE exposure include neuroinflammation and oxidative stress [148, 153], though the exact mechanisms remain unclear. Cruts et al. [73] is the only CHE-DE study to date that has clearly documented concerning neurological impacts of DE exposure; another performed has revealed some preliminarily reassuring results [72, 111] but with major endpoints not yet resulted in detail, precluding complete conclusions. CHE-DE as a paradigm of TRAP exposure is a tool that promises to elucidate not only the clinical effects of DE on neurological function, but also the pathways that mediate these outcomes such that observational data would be given more credence. Given the paucity of research in this area, and the massive public health implications, future CHE-DE studies focus on neurological effects of DE exposure is warranted.
Exercising during diesel exhaust exposure is generally not harmful in healthy populations
Those living in areas with sub-optimal air quality are often concerned about outdoor exercise due to the risk of breathing in larger quantities of pollutants. The majority of CHE-DE studies have participants alternate between rest and exercise during exposure, in order to simulate real world variations in activity. Exercise is typically done on a stationary bicycle with a modest target ventilation rate. A smaller number of CHE-DE studies have specifically evaluated the effect of exercise during DE exposure on health endpoints. In three papers from the same group, 18 healthy male participants cycled for 30 min at low or high intensity after inhalation of DE300 [76, 77]. While exposure to DE increased plasma NOx (nitrite and nitrate) levels (p < 0.05), this effect was not significantly different between different exercise intensities [76]. Exercise during DE exposure did not increase levels of adhesion molecules or systemic inflammatory markers, nor did it affect blood pressure or flow mediated dilation, a measure of endothelial function [75, 76]. Furthermore, the effects of exercise intensity on heart rate variation and norepinephrine were not modulated by DE exposure [77]. A separate study reported no significant difference in parameters of micro- and macrovascular vasodilation after exercise during DE300 exposure compared to FA, suggesting that DE does not impair vascular effects of exercise [74]. Data from observational literature suggests the benefits of exercise typically outweigh the risks of air pollution and that it may not be necessary for most general populations to avoid exercising during periods of increased air pollution except perhaps when extreme [155,156,157,158,159]. However, some epidemiological studies have reported benefits of physical activity are negated in polluted regions, or even an overall adverse effect of exercise in polluted environments [160,161,162,163]. Taken together, results from CHE-DE and observational studies imply exercise in areas of air pollution likely imparts beneficial effects in general, however the detrimental impact of air pollution may attenuate the health benefits of activity.
Co-exposures
The ability of CHE studies to precisely control exposure conditions is a double-edged sword. While this facilitates casual relationships being inferred between focused parameters and health endpoints, the tight elimination of other environmental variables is scarcely encountered in ambient settings. It is common that people are subject to TRAP while going about their daily activities, simultaneously experiencing many other types of exposures, both airborne and not. One way CHE-DE studies have attempted to remedy this issue is by introducing various co-exposures alongside DE. Examples of these co-exposures include aeroallergens, ozone, noise and viruses. These co-exposure studies help delineate interactions between DE with other environmental factors while also enhancing the real-world relevance of CHE-DE exposures, although the full range of potential co-exposure combinations can never be fully captured in a laboratory-based algorithm [154].
Diesel exhaust magnifies allergenic effects
The relationship between allergens and TRAP exposure is one of great interest, as both have the potential to exacerbate atopic airway disease and they occur frequently together in many settings. Co-exposures to DE and allergen have been investigated in several more recent CHE-DE studies. In these experiments, participants are first tested for sensitization to common environmental allergens such as house dust mite, grasses, or birch. Participants then undergo a DE or FA exposure, followed by exposure to an allergen to which they demonstrate being already sensitized (either by inhalation or, alternatively, by instillation of allergen or saline into different lung segments via bronchoscopy, a technique known as segmental allergen challenge). Exposure to allergen alone in atopic subjects has been shown to increase airway markers of allergic inflammation, including surfactant protein D (SPD), MPO, and eosinophils, with an additive effect from DE on some but not all of these markers [30, 84]. DE and allergen co-exposure has also been shown to increase non-allergic inflammatory markers such as CD4 (p = 0.035), IL-4 (p = 0.034), and neutrophil elastase (p = 0.031) in submucosal tissue of atopic participants [31]. Inhalation of DE may also inhibit protective responses to allergen-triggered phenomena in the lung. In a 2020 study, atopic participants were exposed to allergen alone, combination of DE and allergen, combination of particle-depleted DE (PDDE) and allergen, and filtered air control [25]. SPD, a protein that modulates pulmonary immune responses, was increased in BAL samples compared to FA-saline control following exposure to allergen only (p = 0.02) but not after exposure to combination DE and allergen (p = 0.19) [25]. Exposure to the combination of PDDE and allergen restored the protective increase in SPD (p = 0.007), suggesting the PM fraction of DE was responsible for suppressed SPD levels [25]. Interestingly, there may be a complex gene-environment interaction that mediates the effects of allergen and DE exposure. A 2016 study found that individuals null for GSTT1, encoding a glutathione S-transferase involved in mitigating oxidative stress, experienced a significantly greater decrease in FEV1 after co-exposure to DE and allergen than those with normal GSTT1 (24.5 ± 19.6% decrease (GSTT1 null) compared to 9.2 ± 7.3% decrease (normal GSTT1), p = 0.001) [86]. CHE-DE studies have also produced evidence that DE and allergen co-exposure often uniquely affects lung gene expression, DNA methylation, and secreted proteins [27, 82, 85], while also showing how some pathways are less effected by the combination [28, 82, 164]. Given that TRAP and aeroallergens are commonly encountered together in urban and suburban environments, delineating the impacts of co-exposure is a relevant and meaningful area of future research. This is particularly important as evidence mounts for greenness as a health-enhancing exposure, as a major caveat therein is the risk for worsening allergenic phenomena when greenness including allergen-rich species in close proximity to TRAP [165]. Species with lower allergenic potential, such as flowering trees [166], are alternatives that can be considered in green space planning, though further investigation is needed to establish if there is indeed a meaningful difference in health outcomes compared with allergen-rich species.
Ozone and diesel exhaust have compounding effects
O3 is a prevalent component of ambient air pollution and has been shown to negatively impact the cardiovascular and pulmonary systems [167, 168]. Given the near ubiquity of both DE and O3 in the air we breathe, several CHE-DE studies have investigated the complex interaction between these two pollutants. DE and O3 appear to have compounding effects on airway inflammation. A 2008 paper reported increased bronchial wash neutrophil (5.4 × 104 cells/L (DE and O3) versus 3.6 × 104 cells/L (FA and O3), p = 0.006) and macrophage (8.2 × 104 cells/L (DE and O3) versus 7.1 × 104 cells/L (FA and O3), p = 0.046) numbers in healthy volunteers exposed to DE at 300 μg/m3 PM10 followed by 0.2 ppm O3, compared to FA followed by O3 [90]. An earlier paper from this group found increased neutrophils and MPO in sputum samples with combination DE and O3 versus combination DE and FA (p < 0.05 and < 0.05 respectively) ([39]. A potentially enhanced effect of DE and O3 co-exposure on inflammatory cytokines and white blood cell counts was noted by a different study group as well [87]. With respect to lung function, co-exposure to DE and O3 has been shown to magnify decreases in FEV1 induced by either exposure alone (p = 0.057 when comparing change in FEV1 post DE and O3 co-exposure to sum of FEV1 changes post DE mono-exposure and post O3 mono-exposure) [88]. Mechanistically, the effects of DE and O3 may be mediated through different pathways. Interestingly, exposure to DE at 300 μg/m3 PM10 but not 0.3 ppm O3 has been shown to increase fraction of exhaled nitric oxide (FeNO) (p = 0.01 for DE compared to FA), an indicator of airway inflammation [89]. This may be due to FeNO’s reflection of more eosinophilic inflammation (perhaps induced by DE’s complex mixture including organic elements) while ozone-induced inflammation is primarily neutrophilic. Given the potent ability of CHE studies in investigating contributions of co-exposures, this study design has contributed significantly to our understanding of the interplay between DE and O3, whilst recognizing that the laboratory setting imperfectly simulates related ambient mixtures.
Limited evidence of interactions between diesel exhaust and traffic noise
One CHE-DE study involved co-exposure to traffic noise, with results reported in four of the presently reviewed publications [22, 91, 103, 104]. Transportation-related noise has been associated with cardiovascular disease risk, though the mechanisms are poorly understood [169]. In this CHE-DE study, healthy participants were exposed to DE titrated to 300 μg/m3 PM1, alongside traffic noise at 48 or 75 dB [91]. Exposure to the higher level of traffic noise, but not DE, was associated with significantly increased signs of DNA damage in peripheral blood mononuclear cells (p < 0.05 effect of 75 dB traffic noise) [91]. However, traffic noise did not modulate DE-induced effects on peak expiratory flow and inflammatory markers [22]. As such, evidence on the interaction between DE and traffic noise remains limited.
Diesel exhaust may exacerbate allergic inflammation induced by influenza virus
Two CHE-DE studies involved co-exposure to live attenuated influenza virus (LAIV) [92, 93]. Compared to FA, acute exposure to DE at 100 μg/m3 prior to intranasal administration of LAIV was shown to increase markers of eosinophil activation (eotaxin-1, p = 0.01; eosinophil cationic protein, p < 0.01) in subjects with allergic rhinitis, indicating DE may exacerbate LAIV-induced allergic inflammation [93]. A follow up study produced similar results with respect to eosinophil activation and also presented evidence that this effect is mediated by natural killer (NK) cells [92]. DE was shown to significantly decrease IP-10, a marker of NK cell activation (p < 0.05 compared to baseline), suggesting DE reduces eosinophil clearance by NK cells [92]. These studies provide novel insight into the interplay between DE, allergic inflammation, and viral infection, demonstrating how increasing complexity integrated into CHE studies can contribute markedly to our understanding of the interaction of TRAP with various environmental factors.
Use of particle filters may mitigate adverse effects of diesel exhaust
Given the association of fine particulate matter with negative health outcomes, the ability of particulate filters to mitigate the consequences of air pollution has been widely studied. Several CHE-DE papers have found promising data with regards to the beneficial impact of such filters. For brevity, we will use PDDE (particle-depleted DE) to refer to DE which has undergone particle filtration. The FILTER-HF trial investigated the impact of a particle filter on various endpoints in 26 patients with heart failure [94, 95]. Use of the filter reduced DE concentration from DE325 to DE25 but did not affect levels of gaseous DE components [94, 95]. Exposure to unfiltered DE increased levels of B-type natriuretic peptide (BNP) (47.0 pg/mL (FA) versus 66.5 pg/mL (unfiltered DE), p = 0.004) and impaired endothelial function (21% decrease in reactive hyperemia index (RHI) during unfiltered DE exposure compared to FA, p = 0.002), but these effects were reduced with PDDE (BNP = 66.5 pg/mL (unfiltered DE) versus 44.0 pg/mL (PDDE), p = 0.015; 20% increase in RHI during PDDE compared to unfiltered DE, p = 0.019) [94]. During a modified version of the 6-min walking test, exposure to unfiltered DE adversely impacted markers of exercise tolerance, such as 6-min walking distance (p = 0.03), maximal oxygen uptake (VO2) (p < 0.001), and oxygen uptake per heartbeat (O2 pulse) (p < 0.001) [95]. Use of the particle filter was able to reverse some of these DE-induced changes (p < 0.001 (VO2) and p < 0.001 (O2 pulse) comparing PDDE to unfiltered DE) [95]. Another study conducted in healthy volunteers showed that filtration through a particle trap was able to reduce the harmful effects of DE inhalation on vasomotor function, thrombus formation, and fibrinolysis (p = 0.04, p = 0.02, and p = 0.03 respectively, for PDDE versus unfiltered DE) [97]. However, evidence from other CHE-DE studies serve as reminders that the particulate portion of DE is not the only mediator of adverse health effects. A 2019 study exposed healthy volunteers to allergen in combination with unfiltered DE or PDDE [83]. The combination of PDDE and allergen impaired lung function to a greater extent than did unfiltered DE with allergen (7.5% greater decrease in FEV1 with PDDE and allergen compared to unfiltered DE and allergen, p = 0.047) [83]. Notably, the level of NO2 in PDDE exposure was greater than that of unfiltered DE exposure (150 ppb compared to 53 ppb, p < 0.0001), implying the detrimental effect of particle filtration may have been mediated by NO2 enrichment known to occur with some PM-reducing technologies [83].
CHE-DE studies involving activated charcoal filters have attempted to address the additional benefit of controlling the gaseous fraction of DE. In a 2014 study, 30 healthy subjects were exposed to unfiltered DE and DE filtered through two different particle filters, including one filter with an active charcoal component [96]. Levels of PM10 were reduced by 47% using the particle filter alone (p < 0.001) and by 74% using the filter containing active charcoal (p < 0.001) [96]. The combination with charcoal filter, but not the particle filter alone, reduced the levels of NO2 by 85% (p < 0.001) and of hydrocarbon by 58% (p < 0.001) compared to unfiltered DE [96]. Participants reported fewer subjective symptoms after exposure to DE run through the particle filter without charcoal component compared to unfiltered DE; though this was not statistically significant, symptom reduction in the active charcoal filter condition was significant (p < 0.05) compared to unfiltered DE) [96]. An earlier CHE-DE study had also reported greater reduction in symptoms with combination particle and charcoal filter compared to particle filter alone [98]. Though particle filters alone seem to have some protective effects, the benefits of filtration may be improved by adding an activated charcoal component.
No reliable methods of quantifying personal diesel exhaust exposure in controlled human exposure studies
Chambers used in CHE-DE are typically connected to a host of instruments able to precisely measure airborne concentrations of particulates and gaseous co-pollutants. However, it is rather difficult to quantify exactly how much DE is truly inhaled as this depends on a multitude of participant-dependent factors such as variations in respiratory rate and tidal volume (even when design attempts to control and keep these consistent, due to individual anatomy and other factors). Development of methods to most accurately quantify personal exposure to DE are ongoing, but several CHE-DE studies have assessed the utility of specific biomarkers as surrogate indicators of DE exposure, while recognizing that variability in individual factors related to metabolism makes such markers imperfect proxies for proximal exposure. In a preliminary study, the airborne concentrations of DE constituents that can be found in biological fluids were measured during exposure to DE100 [110]. Naphthalene and phenanthrene, two polycyclic aromatic hydrocarbons (PAHs), were identified as potential markers of DE exposure and were further investigated in a follow up study. The urinary concentrations of these two compounds along with 12 other PAHs were measured prior and after DE exposure ranging from DE106–DE276, but none of these compounds were found to be a suitable biomarker [103]. Blood concentrations of plasma PAHs from the Sobus et al. 2008 [110] study were visualized using a heat map in a separate paper [115], demonstrating a novel approach to displaying data generated in CHE-DE studies. Another compound that has been assessed for this purpose is 1-nitropyrene, a marker of DE exposure that is excreted in urine as 1-aminopyrene [108]. Urinary concentration of 1-aminopyrene in healthy volunteers exposed to DE at 300 μg/m3 PM10 increased upon DE exposure [108]. However, the utility of this marker is limited by a high degree of variability between subjects [108], likely related to the aforementioned constraint (given inevitable co-exposures to other products of common combustion). While a consistent marker of DE exposure has yet to be found, exploration of this area should continue as identification of such a compound would greatly benefit air pollution research.
Controlled human studies to diesel exhaust: limitations and future directions
Though CHE-DE studies are important tools in research aiming to assess health effects of TRAP, these experiments carry an inherent set of limitations. As discussed earlier, CHE studies are, by necessity, constrained environments and thus unable to fully replicate the complex co-exposures encountered in the real world. This is remedied to some extent by the increasing sophistication of co-exposures in CHE-DE studies, but the addition of each additional exposure layer imposes further budgetary costs, participant burden, and analytical complexity. CHE studies using ambient pollution [170,171,172] as well as concentrated ambient particles [173,174,175,176,177] have also been employed to better reflect the intricacies of real world exposures. Another significant weakness of CHE-DE studies is the inherent uncertainty in terms of how the setting relates to the long-term effects of DE exposure. While chronic effects are inevitably the result of a series of acute exposures, the precise relationship between the short and long terms therein is a complex subject of ongoing investigation. Observational studies have provided robust evidence for the detrimental effects of chronic diesel exhaust exposure across multiple physiological systems [178]. One important avenue therein is to integrate or compare the results of CHE studies with data obtained via observational methods, and examples of this approach are emerging [179], albeit with methodological challenges that need further refinement.
Another noteworthy limitation is that the concentrations of DE used in CHE-DE studies are typically on the high end relative to real world exposures. This has typically been motivated by a desire to quantify significant changes in endpoints within a relatively short amount of time, by a need to induce an effect detectable beyond the background exposure levels that participants will routinely experience in daily life, and because a focus on mechanistic insight typically justifies some excursion from typical levels. Furthermore, it is logistically impractical and costly for CHE studies to bring participants in for multiple low-level exposures over an extended period. While the cohort and longitudinal designs often used in studies of chronic TRAP exposure come with their own set of advantages and disadvantages, the approach of collating insight from various study designs—each with their valuable angles of insight—remains the gold standard for ultimate decision-making.
Studies designed to allow assessment of concentration–response relationships can be informative as the whether or not concentrations typical of CHE are exceptional [180]. For example, one study exposed healthy participants to whole DE at 100 μg/m3, 200 μg/m3, 300 μg/m3 and found exposure at a concentration of 300 μg/m3, but not the lower concentrations induced cardiovascular responses [52]. While this study might be interpreted as 300 μg/m3 being a minimal concentration to induce measureable effects in this specific context, the concentration–response function likely varies according to participant phenotype, details of exposure protocol, and particular outcomes. Indeed, another study examined the effects of 2-h exposures of DE at a modest concentration of DE25 and demonstrated significant differences in measurements of endothelial function and fibrinolysis [49]. And yet, in contrast, Giles et al. [64] used the same measure of endothelial function as Tousoulis et al. [118] but did not find effect of DE exposure, despite using a concentration of DE300 and a shorter exposure duration of 30 min [76]. In epidemiological studies as well, the minimal concentration required to demonstrate discernible effects of chronic PM exposure is unclear. One large scale multicenter European study reported an increase in natural cause mortality in participants chronically exposed to PM2.5 concentrations under 20 μg/m3 [181]. Furthermore, even at the same chamber concentration of DE, inter-individual variability in the quantity of DE inhaled likely plays a role clouding the threshold concentration for observable effects. As discussed above, a reliable marker of DE exposure has yet to be identified and research in this area is still ongoing. A method of accurately quantifying personal DE exposure would, among its many other potential applications, greatly aid in elucidating the minimum concentrations needed for detection of various endpoints in both CHE-DE and observational studies. Thus, the minimum concentration needed to produce observable or significant effects likely depends not only on the health outcome being investigated, but factors related to study design and participants as well.
As noted above, there is also a lack of consistency among the findings of CHE-DE studies investigating heart rate and blood pressure, though in general CHE-DE studies have revealed a negative effect of acute DE exposure on cardiovascular function. Observational study designs have demonstrated associations between TRAP exposure and both increased blood pressure [139,140,141] and changes in heart rate variability [142,143,144]. A 2015 study utilizing CHE to ambient air pollution reported changes in heart rate variability as well [172]. Inter-study variability in the results of CHE-DE studies may indicate that exposures within such protocols are not sufficiently long and/or reflective of complex real-world ambient aerosols to produce a significant and detectable effects, or simply that effects of acute TRAP exposures are distinct from those observed in epidemiology over a longer time course.
Out of an abundance of caution in CHE-DE studies, those with significant medical comorbidities have often been excluded from participating, particularly in the earlier era of such studies. Indeed, results from the small number of CHE-DE studies involving subjects with notable medical conditions have suggested these individuals may respond differently to TRAP exposure relative to healthy populations. As noted previously, in a recent study, patients with mild to moderate COPD were shown to have more activated peripheral neutrophils after acute DE300 exposure compared to healthy never-smokers, suggesting this cohort is more susceptible to the inflammatory effects of DE inhalation [26]. Another CHE-DE study of COPD patients proposed differential deposition of DE particles in the respiratory tract as a cause of their apparent risk in the setting of TRAP [35]. The rate of deposited DE particles during spontaneous breathing was higher in the COPD group versus healthy controls and was also correlated with increasing disease severity [35]. Participants with significant cardiovascular disease have been included in a few studies, such as the FILTER-HF trial reviewed earlier [94, 95]. Another study exposed 20 males with prior myocardial infarction (MI) and stable coronary artery disease to DE at 300 μg/m3 PM10 [69]. DE inhalation decreased bradykinin-induced release of tissue plasminogen activator in these participants with cardiovascular disease [69]. However, vasodilation in response to acetylcholine was impaired in the MI group compared to healthy controls [69]. Taken together, these results suggest DE exposure in men with stable coronary artery disease may exacerbate myocardial ischemia and impair fibrinolysis [69].
Notably, while this limited set of studies confirms some effects of DE inhalation in these groups, consistent with their chronic cardio-pulmonary disease, these effects have been sub-clinical, anticipated by the study design, and recognized as tolerable by approving ethics boards. Furthermore, and most importantly, they were unassociated with adverse clinical events, across more than a thousand of participants of various phenotypes, and are instead broadly recognized as important advances in our understanding of the pathophysiology of these diseases in the context of a nearly ubiquitous exposure of global concern. Moreover, from a public health standpoint, illustrating the biological plausibility of previously observed consequences of pollution has supported and buttressed the establishment of stronger air quality regulations.
While distinct adverse clinical events from CHE-DE have not reported, studies have noted that undesirable symptoms were more frequent with DE exposure at a concentration of 300 μg/m3 PM1 [22] and at DE at 300 μg/m3 PM2.5 [114] compared to filtered air. However, another study, notable for having demonstrated effective blinding to exposure (importantly, as nominal blinding is much more common), suggested that symptoms may be more related to the perception of exposure rather than the actual exposure itself [182], an effect that is especially relevant for self-reported measures. Future studies should not only be designed in a rigourously blinded manner but also assess specifically for effectiveness of that blinding.
Finally, all systems for CHE-DE studies to date have been within a contained infrastructure that is not exposed to the typical ambient environment and associated photochemistry known to ‘age’ the aerosol, in a manner that generally increases oxidative potential. While the use of ambient air pollution in CHE studies does account for this aging effect to an extent, studies that incorporate a method of photochemical aging mimicking that of common urban airsheds would provide yet another incremental step towards optimizing the translational capacity of these already highly informative investigations.
Conclusion
CHE-DE studies have contributed greatly to our current understanding of health outcomes linked to TRAP exposure, particular in terms of elucidating mechanisms that substantiate—or in some cases put into question—observations from other study contexts. Research has focused on the cardiovascular and pulmonary impacts of DE inhalation, with oxidative stress thought to be the dominant mechanism of DE-induced effects, while other systems such as neuro-cognitive have been more recently explored. Co-exposure studies have delineated powerful interactions between DE and commonly encountered environmental factors, such as allergens and ozone. Particle filters, particularly in combination with activated charcoal filters, are a promising method of reducing the detrimental impacts of TRAP. To date, no robust biomarker of DE exposure has been identified. The main limitation of CHE-DE studies is their inability to directly examine chronic effects of DE inhalation, a niche better filled by other experimental designs [183,184,185,186]. While few CHE-DE experiments have included participants with significant medical co-morbidities, evidence from those that have suggests this can be done safely and can reveal aspects of pathophysiology particular to these populations. CHE-DE studies have proven to be an invaluable research tool and continue to advance relevant and applicable knowledge as we strive to further limit exposure to, and adverse effects of, air pollution.
Availability of data and materials
Not applicable.
Abbreviations
- BNP:
-
B-type natriuretic peptide
- CHE:
-
Controlled human exposure
- CHE-DE:
-
Controlled human exposure to diesel exhaust
- CO:
-
Carbon monoxide
- COPD:
-
Chronic obstructive pulmonary disease
- DBP:
-
Diastolic blood pressure
- DE:
-
Diesel exhaust
- EEG:
-
Electroencephalography
- FA:
-
Filtered air
- FeNO:
-
Fraction of exhaled nitric oxide
- HRV:
-
Heart rate variability
- IL:
-
Interleukin
- MI:
-
Myocardial infarction
- MPO:
-
Myeloperoxidase
- NF:
-
Nuclear factor
- NK:
-
Natural killer cells
- NO:
-
Nitric oxide
- NO2 :
-
Nitrogen dioxide
- NOx :
-
Nitrite and nitrate
- O3 :
-
Ozone
- PAH:
-
Polycyclic aromatic hydrocarbons
- PAI-1:
-
Plasminogen activator inhibitor-1
- PDDE:
-
Particle depleted diesel exhaust
- PM:
-
Particulate matter
- PM1 :
-
PM with aerodynamic diameter under 1 μm
- PM2.5 :
-
PM with aerodynamic diameter under 2.5 μm
- PM10 :
-
PM with aerodynamic diameter under 10 μm
- RHI:
-
Reactive hyperemia index
- SBP:
-
Systolic blood pressure
- SPD:
-
Surfactant protein D
- TRAP:
-
Traffic related air pollution
- VO2 :
-
Maximal oxygen uptake
- vWF:
-
Von Willebrand factor
- WHO:
-
World Health Organization
References
Murray CJL, Aravkin AY, Zheng P, Abbafati C, Abbas KM, Abbasi-Kangevari M, et al. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. The Lancet. 2020;396:1223–49.
Sun Z, Zhu D. Exposure to outdoor air pollution and its human health outcomes: a scoping review. PLoS ONE. 2019;14:e0216550. https://doi.org/10.1371/journal.pone.0216550.
Al-Kindi SG, Brook RD, Biswal S, Rajagopalan S. Environmental determinants of cardiovascular disease: lessons learned from air pollution. Nat Rev Cardiol. 2020;17:656–72.
World Health Organization. WHO global air quality guidelines: particulate matter (PM2.5 and PM10), ozone, nitrogen dioxide, sulfur dioxide and carbon monoxide. Geneva: World Health Organization; 2021.
World Health Organization. Air quality guidelines. Global update 2005. Geneva: World Health Organization; 2006.
EPA. Criteria Air Pollutants. United States Environmental Protection Agency. [cited 2020 Dec 28]
Kim KH, Kabir E, Kabir S. A review on the human health impact of airborne particulate matter. Environ Int. 2015;74:136–43.
Reşitoğlu İA, Altinişik K, Keskin A. The pollutant emissions from diesel-engine vehicles and exhaust aftertreatment systems. Clean Technol Environ Policy. 2015;17:15–27. https://doi.org/10.1007/s10098-014-0793-9.
Alexis NE, Carlsten C. Interplay of air pollution and asthma immunopathogenesis: a focused review of diesel exhaust and ozone. Int Immunopharmacol. 2014;23:347–55.
Ghio A, Sobus J, Pleil J, Madden M. Controlled human exposures to diesel exhaust. Swiss Med Wkl. 2012;142:w13597.
Weitekamp CA, Kerr LB, Dishaw L, Nichols J, Lein M, Stewart MJ. A systematic review of the health effects associated with the inhalation of particle-filtered and whole diesel exhaust. Inhal Toxicol. 2020;32:1–13.
Cosselman KE, Allen J, Jansen KL, Stapleton P, Trenga CA, Larson TV, et al. Acute exposure to traffic-related air pollution alters antioxidant status in healthy adults. Environ Res. 2020;191:110027.
Carlsten C, MacNutt MJ, Zhang Z, Sava F, Pui MM. Anti-oxidant N-acetylcysteine diminishes diesel exhaust-induced increased airway responsiveness in person with airway hyper-reactivity. Toxicol Sci. 2014;139:479–87.
Yamamoto M, Singh A, Sava F, Pui M, Tebbutt SJ, Carlsten C. MicroRNA expression in response to controlled exposure to diesel exhaust: attenuation by the antioxidant N-acetylcysteine in a randomized crossover study. Environ Health Perspect. 2013;121:670–5.
Pettit AP, Brooks A, Laumbach R, Fiedler N, Wang Q, Strickland PO, et al. Alteration of peripheral blood monocyte gene expression in humans following diesel exhaust inhalation. Inhal Toxicol. 2012;24:172–81.
Allen J, Trenga CA, Peretz A, Sullivan JH, Carlsten CC, Kaufman JD. Effect of diesel exhaust inhalation on antioxidant and oxidative stress responses in adults with metabolic syndrome. Inhal Toxicol. 2009;21:1061–7.
Peretz A, Peck EC, Bammler TK, Beyer RP, Sullivan JH, Trenga CA, et al. Diesel exhaust inhalation and assessment of peripheral blood mononuclear cell gene transcription effects: an exploratory study of healthy human volunteers. Inhal Toxicol. 2007;19:1107–19.
Pourazar J, Mudway IS, Samet JM, Helleday R, Blomberg A, Wilson SJ, et al. Diesel exhaust activates redox-sensitive transcription factors and kinases in human airways. Am J Physiol Lung Cell Mol Physiol. 2005;289:L724–30.
Mudway IS, Stenfors N, Duggan ST, Roxborough H, Zielinski H, Marklund SL, et al. An in vitro and in vivo investigation of the effects of diesel exhaust on human airway lining fluid antioxidants. Arch Biochem Biophys. 2004;423:200–12.
Blomberg A, Sainsbury C, Rudell B, Frew AJ, Holgate ST, Sandströw T, et al. Nasal cavity lining fluid ascorbic acid concentration increases in healthy human volunteers following short term exposure to diesel exhaust. Free Radic Res. 1998;28:59–67.
Jiang R, Jones MJ, Sava F, Kobor MS, Carlsten C. Short-term diesel exhaust inhalation in a controlled human crossover study is associated with changes in DNA methylation of circulating mononuclear cells in asthmatics. Part Fibre Toxicol. 2014;11:71.
Xu Y, Barregard L, Nielsen J, Gudmundsson A, Wierzbicka A, Axmon A, et al. Effects of diesel exposure on lung function and inflammation biomarkers from airway and peripheral blood of healthy volunteers in a chamber study. Part Fibre Toxicol. 2013;10:60.
Channell MM, Paffett ML, Devlin RB, Madden MC, Campen MJ. Circulating factors induce coronary endothelial cell activation following exposure to inhaled diesel exhaust and nitrogen dioxide in humans: evidence from a novel translational in vitro model. Toxicol Sci. 2012;127:179–86.
Rabinovitch N, Jones MJ, Gladish N, Faino AV, Strand M, Morin AM, et al. Methylation of cysteinyl leukotriene receptor 1 genes associates with lung function in asthmatics exposed to traffic-related air pollution. Epigenetics. 2021;16:177–85.
Ryu MH, Lau KS-K, Wooding DJ, Fan S, Sin DD, Carlsten C. Particle depletion of diesel exhaust restores allergen-induced lung-protective surfactant protein D in human lungs. Thorax. 2020;75:640–7.
Wooding DJ, Ryu MH, Li H, Alexis NE, Pena O, Carlsten C. Acute air pollution exposure alters neutrophils in never-smokers and at-risk humans. Eur Respir J. 2020;55:1901495.
Mookherjee N, Piyadasa H, Ryu MH, Rider CF, Ezzati P, Spicer V, et al. Inhaled diesel exhaust alters the allergen-induced bronchial secretome in humans. Eur Respir J. 2018;51:1701385.
Clifford RL, Jones MJ, MacIsaac JL, McEwen LM, Goodman SJ, Mostafavi S, et al. Inhalation of diesel exhaust and allergen alters human bronchial epithelium DNA methylation. J Allergy Clin Immunol. 2017;139:112–21.
Kramer MM, Hirota JA, Sood A, Teschke K, Carlsten C. Airway and serum adipokines after allergen and diesel exposure in a controlled human crossover study of atopic adults. Transl Res. 2017;182:49–60.
Carlsten C, Blomberg A, Pui M, Sandstrom T, Wong SW, Alexis N, et al. Diesel exhaust augments allergen-induced lower airway inflammation in allergic individuals: a controlled human exposure study. Thorax. 2016;71:35–44.
Hosseini A, Hirota JA, Hackett TL, McNagny KM, Wilson SJ, Carlsten C. Morphometric analysis of inflammation in bronchial biopsies following exposure to inhaled diesel exhaust and allergen challenge in atopic subjects. Part Fibre Toxicol. 2016;13:2.
Behndig AF, Shanmuganathan K, Whitmarsh L, Stenfors N, Brown JL, Frew AJ, et al. Effects of controlled diesel exhaust exposure on apoptosis and proliferation markers in bronchial epithelium—an in vivo bronchoscopy study on asthmatics, rhinitics and healthy subjects. BMC Pulm Med. 2015;15:99.
Larsson N, Brown J, Stenfors N, Wilson S, Mudway IS, Pourazar J, et al. Airway inflammatory responses to diesel exhaust in allergic rhinitics. Inhal Toxicol. 2013;25:160–7.
Hussain S, Laumbach R, Coleman J, Youssef H, Kelly-McNeil K, Ohman-Strickland P, et al. Controlled exposure to diesel exhaust causes increased nitrite in exhaled breath condensate among subjects with asthma. J Occup Environ Med. 2012;54:1186–91.
Löndahl J, Swietlicki E, Rissler J, Bengtsson A, Boman C, Blomberg A, et al. Experimental determination of the respiratory tract deposition of diesel combustion particles in patients with chronic obstructive pulmonary disease. Part Fibre Toxicol. 2012;9:30.
Behndig AF, Larsson N, Brown JL, Stenfors N, Helleday R, Duggan ST, et al. Proinflammatory doses of diesel exhaust in healthy subjects fail to elicit equivalent or augmented airway inflammation in subjects with asthma. Thorax. 2011;66:12–9.
Sehlstedt M, Behndig AF, Boman C, Blomberg A, Sandström T, Pourazar J. Airway inflammatory response to diesel exhaust generated at urban cycle running conditions. Inhal Toxicol. 2010;22:1144–50.
Sawant AA, Cocker DR III, Miller JW, Taliaferro T, Diaz-Sanchez D, Linn WS, et al. Generation and characterization of diesel exhaust in a facility for controlled human exposures. J Air Waste Manag Assoc. 2008;58:829–37.
Bosson J, Pourazar J, Forsberg B, Ädelroth E, Sandström T, Blomberg A. Ozone enhances the airway inflammation initiated by diesel exhaust. Respir Med. 2007;101:1140–6.
Behndig AF, Mudway IS, Brown JL, Stenfors N, Helleday R, Duggan ST, Wilson SJ, Boman C, Cassee FR, Frew AJ, Kelly FJ, T. Sandström AB,. Airway antioxidant and inflammatory responses to diesel exhaust exposure in healthy humans. Eur Respiratory J. 2006;27:359–65. https://doi.org/10.1183/09031936.06.00136904.
Pourazar J, Frew AJ, Blomberg A, Helleday R, Kelly FJ, Wilson S, et al. Diesel exhaust exposure enhances the expression of IL-13 in the bronchial epithelium of healthy subjects. Respir Med. 2004;98:821–5.
Stenfors N, Nordenhall C, Salvi SS, Mudway I, Soderberg M, Blomberg A, et al. Different airway inflammatory responses in asthmatic and healthy humans exposed to diesel. Eur Respir J. 2004;23:82–6.
Nordenhäll C, Pourazar J, Ledin M-C, Levin J-O, Sandström T, Ädelroth E. Diesel exhaust enhances airway responsiveness in asthmatic subjects. Eur Respir J. 2001;17:909–15.
Nightingale JA, Maggs R, Cullinan P, Donnelly LE, Rogers DF, Kinnersley R, et al. Airway inflammation after controlled exposure to diesel exhaust particulates. Am J Respir Crit Care Med. 2000;162:161–6.
Nordenhäll C, Pourazar J, Blomberg A, Levin J-O, Sandström T, Ädelroth E. Airway inflammation following exposure to diesel exhaust: a study of time kinetics using induced sputum. Eur Respir J. 2000;15:1046.
Salvi SS, Nordenhall C, Blomberg A, Rudell B, Pourazar J, Kelly FJ, et al. Acute exposure to diesel exhaust increases IL-8 and GRO- α production in healthy human airways. Am J Respir Crit Care Med. 2000;161:550–7.
Salvi S, Blomberg A, Rudell B, Kelly F, Sandström T, Holgate ST, et al. Acute inflammatory responses in the airways and peripheral blood after short-term exposure to diesel exhaust in healthy human volunteers. Am J Respir Crit Care Med. 1999;159:702–9.
Rudell B, Sandström T, Hammarström U, Ledin ML, Hörstedt P, Stjernberg N. Evaluation of an exposure setup for studying effects of diesel exhaust in humans. Int Arch Occup Environ Health. 1994;66:77–83.
Tousoulis D, Fountoulakis P, Oikonomou E, Antoniades C, Siasos G, Tsalamandris S, et al. Acute exposure to diesel affects inflammation and vascular function. Eur J Prevent Cardiol. 2020;2047487319898020.
Sack CS, Jansen KL, Cosselman KE, Trenga CA, Stapleton PL, Allen J, et al. Pretreatment with antioxidants augments the acute arterial vasoconstriction caused by diesel exhaust inhalation. Am J Respir Crit Care Med. 2016;193:1000–7.
Langrish JP, Watts SJ, Hunter AJ, Shah ASV, Bosson JA, Unosson J, et al. Controlled exposures to air pollutants and risk of cardiac arrhythmia. Environ Health Perspect. 2014;122:747–53.
Tong H, Rappold AG, Caughey M, Hinderliter AL, Graff DW, Berntsen JH, et al. Cardiovascular effects caused by increasing concentrations of diesel exhaust in middle-aged healthy GSTM1 null human volunteers. Inhal Toxicol. 2014;26:319–26.
Krishnan RM, Sullivan JH, Carlsten C, Wilkerson H-W, Beyer RP, Bammler T, et al. A randomized cross-over study of inhalation of diesel exhaust, hematological indices, and endothelial markers in humans. Part Fibre Toxicol. 2013;10:7.
Langrish JP, Unosson J, Bosson J, Barath S, Muala A, Blackwell S, et al. Altered nitric oxide bioavailability contributes to diesel exhaust inhalation-induced cardiovascular dysfunction in man. J Am Heart Assoc. 2013;2:e004309.
Wauters A, Dreyfuss C, Pochet S, Hendrick P, Berkenboom G, van de Borne P, et al. Acute exposure to diesel exhaust impairs nitric oxide-mediated endothelial vasomotor function by increasing endothelial oxidative stress. Hypertension. 2013;62:352–8.
Cosselman KE, Krishnan RM, Oron AP, Jansen K, Peretz A, Sullivan JH, et al. Blood pressure response to controlled diesel exhaust exposure in human subjects. Hypertension. 2012;59:943–8.
Lund AK, Lucero J, Harman M, Madden MC, McDonald JD, Seagrave JC, et al. The oxidized low-density lipoprotein receptor mediates vascular effects of inhaled vehicle emissions. Am J Respir Crit Care Med. 2011;184:82–91.
Mills NL, Miller MR, Lucking AJ, Beveridge J, Flint L, Boere AJF, et al. Combustion-derived nanoparticulate induces the adverse vascular effects of diesel exhaust inhalation. Eur Heart J. 2011;32:2660–71.
Mills NL, Finlayson AE, Gonzalez MC, Tornqvist H, Barath S, Vink E, et al. Diesel exhaust inhalation does not affect heart rhythm or heart rate variability. Heart. 2011;97:544–50.
Barath S, Mills NL, Lundbäck M, Törnqvist H, Lucking AJ, Langrish JP, et al. Impaired vascular function after exposure to diesel exhaust generated at urban transient running conditions. Part Fibre Toxicol. 2010;7:19.
Langrish JP, Lundbäck M, Mills NL, Johnston NR, Webb DJ, Sandström T, et al. Contribution of endothelin 1 to the vascular effects of diesel exhaust inhalation in humans. Hypertension. 2009;54:910–5.
Lund AK, Lucero J, Lucas S, Madden MC, McDonald JD, Seagrave J-C, et al. Vehicular emissions induce vascular MMP-9 expression and activity associated with endothelin-1-mediated pathways. Arterioscler Thromb Vasc Biol. 2009;29:511–7.
Lundbäck M, Mills NL, Lucking A, Barath S, Donaldson K, Newby DE, et al. Experimental exposure to diesel exhaust increases arterial stiffness in man. Part Fibre Toxicol. 2009;6:7.
Carlsten C, Kaufman JD, Trenga CA, Allen J, Peretz A, Sullivan JH. Thrombotic markers in metabolic syndrome subjects exposed to diesel exhaust. Inhal Toxicol. 2008;20:917–21.
Lucking AJ, Lundback M, Mills NL, Faratian D, Barath SL, Pourazar J, et al. Diesel exhaust inhalation increases thrombus formation in man. Eur Heart J. 2008;29:3043–51.
Peretz A, Kaufman JD, Trenga CA, Allen J, Carlsten C, Aulet MR, et al. Effects of diesel exhaust inhalation on heart rate variability in human volunteers. Environ Res. 2008;107:178–84.
Peretz A, Sullivan JH, Leotta DF, Trenga CA, Sands FN, Allen J, et al. Diesel exhaust inhalation elicits acute vasoconstriction in vivo. Environ Health Perspect. 2008;116:937–42.
Carlsten C, Kaufman JD, Peretz A, Trenga CA, Sheppard L, Sullivan JH. Coagulation markers in healthy human subjects exposed to diesel exhaust. Thromb Res. 2007;120:849–55.
Mills NL, Törnqvist H, Gonzalez MC, Vink E, Robinson SD, Söderberg S, et al. Ischemic and thrombotic effects of dilute diesel-exhaust inhalation in men with coronary heart disease. N Engl J Med. 2007;357:1075–82.
Törnqvist H, Mills NL, Gonzalez M, Miller MR, Robinson SD, Megson IL, et al. Persistent endothelial dysfunction in humans after diesel exhaust inhalation. Am J Respir Crit Care Med. 2007;176:395–400.
Mills NL, Törnqvist H, Robinson SD, Gonzalez M, Darnley K, MacNee W, et al. Diesel exhaust inhalation causes vascular dysfunction and impaired endogenous fibrinolysis. Circulation. 2005;112:3930–6.
Cliff R, Curran J, Hirota JA, Brauer M, Feldman H, Carlsten C. Effect of diesel exhaust inhalation on blood markers of inflammation and neurotoxicity: a controlled, blinded crossover study. Inhal Toxicol. 2016;28:145–53.
Cruts B, van Etten L, Tornqvist H, Blomberg A, Sandstrom T, Mills NL, et al. Exposure to diesel exhaust induces changes in EEG in human volunteers. Part Fibre Toxicol. 2008;5:4.
Koch S, Zelembaba A, Tran R, Laeremans M, Hives B, Carlsten C, et al. Vascular effects of physical activity are not modified by short-term inhaled diesel exhaust: Results of a controlled human exposure study. Environ Res. 2020;183:109270.
Giles LV, Tebbutt SJ, Carlsten C, Koehle MS. Effects of low-intensity and high-intensity cycling with diesel exhaust exposure on soluble P-selectin, E-selectin, I-CAM-1, VCAM-1 and complete blood count. BMJ Open Sport Exerc Med. 2019;5:e000625.
Giles LV, Tebbutt SJ, Carlsten C, Koehle MS. The effect of low and high-intensity cycling in diesel exhaust on flow-mediated dilation, circulating NOx, endothelin-1 and blood pressure. PLoS ONE. 2018;13:419.
Giles LV, Carlsten C, Koehle MS. The pulmonary and autonomic effects of high-intensity and low-intensity exercise in diesel exhaust. Environ Health. 2018;17:87.
Wauters A, Esmaeilzadeh F, Bladt S, Beukinga I, Wijns W, van de Borne P, et al. Pro-thrombotic effect of exercise in a polluted environment: a P-selectin- and CD63-related platelet activation effect. Thromb Haemost. 2015;113:118–24.
Wauters A, Vicenzi M, de Becker B, Riga J-P, Esmaeilzadeh F, Faoro V, et al. At high cardiac output, diesel exhaust exposure increases pulmonary vascular resistance and decreases distensibility of pulmonary resistive vessels. Am J Physiol Heart Circ Physiol. 2015;309:H2137–44.
Giles LV, Brandenburg JP, Carlsten C, Koehle MS. Physiological responses to diesel exhaust exposure are modified by cycling intensity. Med Sci Sports Exerc. 2014;46:1999–2006.
Giles LV, Carlsten C, Koehle MS. The effect of pre-exercise diesel exhaust exposure on cycling performance and cardio-respiratory variables. Inhal Toxicol. 2012;24:783–9.
Li H, Ryu MH, Rider CF, Tse W, Clifford RL, Aristizabal MJ, et al. Predominant DNMT and TET mediate effects of allergen on the human bronchial epithelium in a controlled air pollution exposure study. J Allergy Clin Immunol. 2021;147:1671–82.
Wooding DJ, Ryu MH, Hüls A, Lee AD, Lin DTS, Rider CF, et al. Particle depletion does not remediate acute effects of traffic-related air pollution and allergen. A randomized, double-blind crossover study. Am J Respir Crit Care Med. 2019;200:565–74.
Biagioni BJ, Tam S, Chen Y-WR, Sin DD, Carlsten C. Effect of controlled human exposure to diesel exhaust and allergen on airway surfactant protein D, myeloperoxidase and club (Clara) cell secretory protein 16. Clin Exp Allergy. 2016;46:1206–13.
Rider CF, Yamamoto M, Günther OP, Hirota JA, Singh A, Tebbutt SJ, et al. Controlled diesel exhaust and allergen coexposure modulates microRNA and gene expression in humans: Effects on inflammatory lung markers. J Allergy Clin Immunol. 2016;138:1690–700.
Zhang X, Hirota JA, Yang C, Carlsten C. Effect of GST variants on lung function following diesel exhaust and allergen co-exposure in a controlled human crossover study. Free Radical Biol Med. 2016;96:385–91.
Stiegel MA, Pleil JD, Sobus JR, Madden MC. Inflammatory cytokines and white blood cell counts response to environmental levels of diesel exhaust and ozone inhalation exposures. PLoS ONE. 2016;11:e0152458.
Madden MC, Stevens T, Case M, Schmitt M, Diaz-Sanchez D, Bassett M, et al. Diesel exhaust modulates ozone-induced lung function decrements in healthy human volunteers. Part Fibre Toxicol. 2014;11:37.
Barath S, Mills NL, Ädelroth E, Olin A-C, Blomberg A. Diesel exhaust but not ozone increases fraction of exhaled nitric oxide in a randomized controlled experimental exposure study of healthy human subjects. Environ Health. 2013;12:36.
Bosson J, Barath S, Pourazar J, Behndig AF, Sandstrom T, Blomberg A, et al. Diesel exhaust exposure enhances the ozone-induced airway inflammation in healthy humans. Eur Respir J. 2008;31:1234–40.
Hemmingsen JG, Møller P, Jantzen K, Jönsson BAG, Albin M, Wierzbicka A, et al. Controlled exposure to diesel exhaust and traffic noise—effects on oxidative stress and activation in mononuclear blood cells. Mutat Res Fund Mol Mech Mutagen. 2015;775:66–71.
Pawlak EA, Noah TL, Zhou H, Chehrazi C, Robinette C, Diaz-Sanchez D, et al. Diesel exposure suppresses natural killer cell function and resolution of eosinophil inflammation: a randomized controlled trial of exposure in allergic rhinitics. Part Fibre Toxicol. 2015;13:24.
Noah TL, Zhou H, Zhang H, Horvath K, Robinette C, Kesic M, et al. Diesel exhaust exposure and nasal response to attenuated influenza in normal and allergic volunteers. Am J Respir Crit Care Med. 2012;185:179–85.
Vieira JL, Guimaraes GV, de Andre PA, Cruz FD, Saldiva PHN, Bocchi EA. Respiratory filter reduces the cardiovascular effects associated with diesel exhaust exposure. JACC Heart Fail. 2016;4:55–64.
Vieira JL, Guimaraes GV, de Andre PA, Saldiva PHN, Bocchi EA. Effects of reducing exposure to air pollution on submaximal cardiopulmonary test in patients with heart failure: analysis of the randomized, double-blind and controlled FILTER-HF trial. Int J Cardiol. 2016;215:92–7.
Muala A, Sehlstedt M, Bion A, Österlund C, Bosson JA, Behndig AF, et al. Assessment of the capacity of vehicle cabin air inlet filters to reduce diesel exhaust-induced symptoms in human volunteers. Environ Health. 2014;13:16.
Lucking AJ, Lundbäck M, Barath SL, Mills NL, Sidhu MK, Langrish JP, et al. Particle traps prevent adverse vascular and prothrombotic effects of diesel engine exhaust inhalation in men. Circulation. 2011;123:1721–8.
Rudell B, Wass U, Horstedt P, Levin JO, Lindahl R, Rannug U, et al. Efficiency of automotive cabin air filters to reduce acute health effects of diesel exhaust in human subjects. Occup Environ Med. 1999;56:222–31.
Rudell B, Blomberg A, Helleday R, Ledin MC, Lundbäck B, Stjernberg N, et al. Bronchoalveolar inflammation after exposure to diesel exhaust: comparison between unfiltered and particle trap filtered exhaust. Occup Environ Med. 1999;56:527–34.
Rudell B, Ledin MC, Hammarstrom U, Stjernberg N, Lundback B, Sandstrom T. Effects on symptoms and lung function in humans experimentally exposed to diesel exhaust. Occup Environ Med. 1996;53:658–62.
Gouveia-Figueira S, Karimpour M, Bosson JA, Blomberg A, Unosson J, Sehlstedt M, et al. Mass spectrometry profiling reveals altered plasma levels of monohydroxy fatty acids and related lipids in healthy humans after controlled exposure to biodiesel exhaust. Anal Chim Acta. 2018;1018:62–9.
Gouveia-Figueira S, Karimpour M, Bosson JA, Blomberg A, Unosson J, Pourazar J, et al. Mass spectrometry profiling of oxylipins, endocannabinoids, and N-acylethanolamines in human lung lavage fluids reveals responsiveness of prostaglandin E2 and associated lipid metabolites to biodiesel exhaust exposure. Anal Bioanal Chem. 2017;409:2967–80.
Lu SS, Sobus JR, Sallsten G, Albin M, Pleil JD, Gudmundsson A, et al. Are urinary PAHs biomarkers of controlled exposure to diesel exhaust? Biomarkers. 2014;19:332–9.
Wierzbicka A, Nilsson PT, Rissler J, Sallsten G, Xu Y, Pagels JH, et al. Detailed diesel exhaust characteristics including particle surface area and lung deposited dose for better understanding of health effects in human chamber exposure studies. Atmos Environ. 2014;86:212–9.
Rissler J, Swietlicki E, Bengtsson A, Boman C, Pagels J, Sandström T, et al. Experimental determination of deposition of diesel exhaust particles in the human respiratory tract. J Aerosol Sci. 2012;48:18–33.
Huyck S, Ohman-Strickland P, Zhang L, Tong J, Xu XU, Zhang J. Determining times to maximum urine excretion of 1-aminopyrene after diesel exhaust exposure. J Eposure Sci Environ Epidemiol. 2010;20:650–5.
Hubbard HF, Sobus JR, Pleil JD, Madden MC, Tabucchi S. Application of novel method to measure endogenous VOCs in exhaled breath condensate before and after exposure to diesel exhaust. J Chromatogr B. 2009;877:3652–8.
Laumbach R, Tong J, Zhang L, Ohman-Strickland P, Stern A, Fiedler N, et al. Quantification of 1-aminopyrene in human urine after a controlled exposure to diesel exhaust. J Environ Monit JEM. 2009;11:153–9.
Sawyer K, Samet JM, Ghio AJ, Pleil JD, Madden MC. Responses measured in the exhaled breath of human volunteers acutely exposed to ozone and diesel exhaust. J Breath Res. 2008;2:037019.
Sobus JR, Pleil JD, Madden MC, Funk WE, Hubbard HF, Rappaport SM. Identification of surrogate measures of diesel exhaust exposure in a controlled chamber study. Environ Sci Technol. 2008;42:8822–8.
Curran J, Cliff R, Sinnen N, Koehle M, Carlsten C. Acute diesel exhaust exposure and postural stability: a controlled crossover experiment. J Occup Med Toxicol. 2018;13:2.
Carlsten C, Oron AP, Curtiss H, Jarvis S, Daniell W, Kaufman JD. Symptoms in response to controlled diesel exhaust more closely reflect exposure perception than true exposure. PLoS ONE. 2013;8:e83573.
Kipen HM, Gandhi S, Rich DQ, Ohman-Strickland P, Laumbach R, Fan Z-H, et al. Acute decreases in proteasome pathway activity after inhalation of fresh diesel exhaust or secondary organic aerosol. Environ Health Perspect. 2011;119:658–63.
Laumbach RJ, Kipen HM, Kelly-McNeil K, Zhang J, Zhang L, Lioy PJ, et al. Sickness response symptoms among healthy volunteers after controlled exposures to diesel exhaust and psychological stress. Environ Health Perspect. 2011;119:945–50.
Pleil JD, Stiegel MA, Madden MC, Sobus JR. Heat map visualization of complex environmental and biomarker measurements. Chemosphere. 2011;84:716–23.
Wu W, Peden D, Diaz-Sanchez D. Role of GSTM1 in resistance to lung inflammation. Free Radic Biol Med. 2012;53:721–9.
Tashakkor AY, Chow KS, Carlsten C. Modification by antioxidant supplementation of changes in human lung function associated with air pollutant exposure: a systematic review. BMC Public Health. 2011;11:532. https://doi.org/10.1186/1471-2458-11-532.
Douki T, Corbière C, Preterre D, Martin PJ, Lecureur V, André V, et al. Comparative study of diesel and biodiesel exhausts on lung oxidative stress and genotoxicity in rats. Environ Pollut. 2018;235:514–24.
Cadrazco M, Agudelo JR, Orozco LY, Estrada V. Genotoxicity of diesel particulate matter emitted by port-injection of hydrous ethanol and n-butanol. J Energy Resour Technol. 2017. https://doi.org/10.1115/1.4036253/373377/Genotoxicity-of-Diesel-Particulate-Matter-Emitted.
Topinka J, Milcova A, Schmuczerova J, Mazac M, Pechout M, Vojtisek-Lom M. Genotoxic potential of organic extracts from particle emissions of diesel and rapeseed oil powered engines. Toxicol Lett. 2012;212:11–7.
León-Mejía G, Luna-Rodríguez I, Trindade C, Oliveros-Ortíz L, Anaya-Romero M, Luna-Carrascal J, et al. Cytotoxic and genotoxic effects in mechanics occupationally exposed to diesel engine exhaust. Ecotoxicol Environ Saf. 2019;171:264–73.
Shen M, Bin P, Li H, Zhang X, Sun X, Duan H, et al. Increased levels of etheno-DNA adducts and genotoxicity biomarkers of long-term exposure to pure diesel engine exhaust. Sci Total Environ. 2016;543:267–73.
Duan H, Jia X, Zhai Q, Ma L, Wang S, Huang C, et al. Long-term exposure to diesel engine exhaust induces primary DNA damage: a population-based study. Occup Environ Med. 2016;73:83–90. https://doi.org/10.1136/oemed-2015-102919.
Knudsen LE, Gaskell M, Martin EA, Poole J, Scheepers PTJ, Jensen A, et al. Genotoxic damage in mine workers exposed to diesel exhaust, and the effects of glutathione transferase genotypes. Mutat Res Genet Toxicol Environ Mutagen. 2005;583:120–32.
Spira-Cohen A, Chen LC, Kendall M, Lall R, Thurston GD. Personal exposures to traffic-related air pollution and acute respiratory health among bronx schoolchildren with asthma. Environ Health Perspect. 2011;119:559–65. https://doi.org/10.1289/ehp.1002653.
Perez L, Declercq C, Iñiguez C, Aguilera I, Badaloni C, Ballester F, et al. Chronic burden of near-roadway traffic pollution in 10 European cities (APHEKOM network). Eur Respir J. 2013;42:594–605. https://doi.org/10.1183/09031936.00031112.
Schikowski T, Sugiri D, Ranft U, Gehring U, Heinrich J, Wichmann H-E, et al. Long-term air pollution exposure and living close to busy roads are associated with COPD in women. Respir Res. 2005;6:152. https://doi.org/10.1186/1465-9921-6-152.
Andersen ZJ, Hvidberg M, Jensen SS, Ketzel M, Loft S, Sørensen M, et al. Chronic obstructive pulmonary disease and long-term exposure to traffic-related air pollution. Am J Respir Crit Care Med. 2011;183:455–61. https://doi.org/10.1164/rccm.201006-0937OC.
To T, Zhu J, Larsen K, Simatovic J, Feldman L, Ryckman K, et al. Progression from asthma to chronic obstructive pulmonary disease. Is air pollution a risk factor? Am J Respir Crit Care Med. 2016;194:429–38. https://doi.org/10.1164/rccm.201510-1932OC.
Gan WQ, FitzGerald JM, Carlsten C, Sadatsafavi M, Brauer M. Associations of ambient air pollution with chronic obstructive pulmonary disease hospitalization and mortality. Am J Respir Crit Care Med. 2013;187:721–7. https://doi.org/10.1164/rccm.201211-2004OC.
Escamilla-Nuñez M-C, Barraza-Villarreal A, Hernandez-Cadena L, Moreno-Macias H, Ramirez-Aguilar M, Sienra-Monge J-J, et al. Traffic-related air pollution and respiratory symptoms among asthmatic children, resident in Mexico City: the EVA cohort study. Respir Res. 2008;9:74. https://doi.org/10.1186/1465-9921-9-74.
Oosterlee A, Drijver M, Lebret E, Brunekreef B. Chronic respiratory symptoms in children and adults living along streets with high traffic density. Occup Environ Med. 1996;53:241–7.
Bayer-Oglesby L, Schindler C, Hazenkamp-von Arx ME, Braun-Fahrländer C, Keidel D, Rapp R, et al. Living near main streets and respiratory symptoms in adults. Am J Epidemiol. 2006;164:1190–8.
Sava F, MacNutt MJ, Carlsten CR. Nasal neurogenic inflammation markers increase after diesel exhaust inhalation in individuals with asthma. Am J Respir Crit Care Med. 2013;188:759–60. https://doi.org/10.1164/rccm.201302-0330LE.
Pope CA, Burnett RT, Thurston GD, Thun MJ, Calle EE, Krewski D, et al. Cardiovascular mortality and long-term exposure to particulate air pollution. Circulation. 2004;109:71–7. https://doi.org/10.1161/01.CIR.0000108927.80044.7F.
Miller KA, Siscovick DS, Sheppard L, Shepherd K, Sullivan JH, Anderson GL, et al. Long-term exposure to air pollution and incidence of cardiovascular events in women. N Engl J Med. 2007;356:447–58. https://doi.org/10.1056/NEJMoa054409.
Dominici F, Peng RD, Bell ML, Pham L, McDermott A, Zeger SL, et al. Fine Particulate air pollution and hospital admission for cardiovascular and respiratory diseases. JAMA. 2006;295:1127. https://doi.org/10.1001/jama.295.10.1127.
Miller MR, Borthwick SJ, Shaw CA, McLean SG, McClure D, Mills NL, et al. Direct impairment of vascular function by diesel exhaust particulate through reduced bioavailability of endothelium-derived nitric oxide induced by superoxide free radicals. Environ Health Perspect. 2009;117:611–6. https://doi.org/10.1289/ehp.0800235.
Coogan PF, White LF, Jerrett M, Brook RD, Su JG, Seto E, et al. Air pollution and incidence of hypertension and diabetes mellitus in black women living in Los Angeles. Circulation. 2012;125:767–72. https://doi.org/10.1161/CIRCULATIONAHA.111.052753.
Schwartz J, Alexeeff SE, Mordukhovich I, Gryparis A, Vokonas P, Suh H, et al. Association between long-term exposure to traffic particles and blood pressure in the Veterans Administration Normative Aging Study. Occup Environ Med. 2012;69:422–7.
Fuks KB, Weinmayr G, Foraster M, Dratva J, Hampel R, Houthuijs D, et al. Arterial blood pressure and long-term exposure to traffic-related air pollution: an analysis in the European Study of Cohorts for Air Pollution Effects (ESCAPE). Environ Health Perspect. 2014;122:896–905. https://doi.org/10.1289/ehp.1307725.
Zanobetti A, Gold DR, Stone PH, Suh HH, Schwartz J, Coull BA, et al. Reduction in heart rate variability with traffic and air pollution in patients with coronary artery disease. Environ Health Perspect. 2010;118:324–30. https://doi.org/10.1289/ehp.0901003.
Shields KN, Cavallari JM, Hunt MJO, Lazo M, Molina M, Molina L, et al. Traffic-related air pollution exposures and changes in heart rate variability in Mexico City: a panel study. Environ Health. 2013;12:7. https://doi.org/10.1186/1476-069X-12-7.
Schwartz J, Litonjua A, Suh H, Verrier M, Zanobetti A, Syring M, et al. Traffic related pollution and heart rate variability in a panel of elderly subjects. Thorax. 2005;60:455–61.
de Carlo L, Cavaliere B, Arnaldi C, Faggioli R, Soriani S, Scarpa P. EEG evaluation in children and adolescents with chronic headaches. Eur J Pediatr. 1999;158:247–8. https://doi.org/10.1007/s004310051060.
Thornton K. The electrophysiological effects of a brain injury on auditory memory functioning The QEEG correlates of impaired memory. Arch Clin Neuropsychol. 2003;18:363–78.
Kilian J, Kitazawa M. The emerging risk of exposure to air pollution on cognitive decline and Alzheimer’s disease—evidence from epidemiological and animal studies. Biomedical Journal. 2018;41:141–62.
Block ML, Calderón-Garcidueñas L. Air pollution: mechanisms of neuroinflammation and CNS disease. Trends Neurosci. 2009;32:506–16.
Kasdagli M-I, Katsouyanni K, Dimakopoulou K, Samoli E. Air pollution and Parkinson’s disease: a systematic review and meta-analysis up to 2018. Int J Hyg Environ Health. 2019;222:402–9.
Zhang X, Chen X, Zhang X. The impact of exposure to air pollution on cognitive performance. Proc Natl Acad Sci. 2018;115:9193–7. https://doi.org/10.1073/pnas.1809474115.
Kicinski M, Vermeir G, van Larebeke N, den Hond E, Schoeters G, Bruckers L, et al. Neurobehavioral performance in adolescents is inversely associated with traffic exposure. Environ Int. 2015;75:136–43.
Sunyer J, Esnaola M, Alvarez-Pedrerol M, Forns J, Rivas I, López-Vicente M, et al. Association between traffic-related air pollution in schools and cognitive development in primary school children: a prospective cohort study. PLoS Med. 2015;12:e1001792. https://doi.org/10.1371/journal.pmed.1001792.
Durga M, Devasena T, Rajasekar A. Determination of LC50 and sub-chronic neurotoxicity of diesel exhaust nanoparticles. Environ Toxicol Pharmacol. 2015;40:615–25.
Carlsten C. Synergistic environmental exposures and the airways capturing complexity in humans. Chest. 2018;154:918–24.
de Hartog JJ, Boogaard H, Nijland H, Hoek G. Do the health benefits of cycling outweigh the risks? Environ Health Perspect. 2010;118:1109–16. https://doi.org/10.1289/ehp.0901747.
Rojas-Rueda D, de Nazelle A, Tainio M, Nieuwenhuijsen MJ. The health risks and benefits of cycling in urban environments compared with car use: health impact assessment study. BMJ. 2011;343:d4521–d4521. https://doi.org/10.1136/bmj.d4521.
Wong C-M, Ou C-Q, Thach T-Q, Chau Y-K, Chan K-P, Ho S-Y, et al. Does regular exercise protect against air pollution-associated mortality? Prev Med. 2007;44:386–92.
Vieira RDP, Toledo AC, Silva LB, Almeida FM, Damaceno-Rodrigues NR, Caldini EG, et al. Anti-inflammatory effects of aerobic exercise in mice exposed to air pollution. Med Sci Sports Exerc. 2012;44:1227–34.
Giles LV, Koehle MS. The health effects of exercising in air pollution. Sports Med. 2014;44:223–49. https://doi.org/10.1007/s40279-013-0108-z.
Yu ITS, Wong TW, Liu HJ. Impact of air pollution on cardiopulmonary fitness in schoolchildren. J Occup Environ Med. 2004;46:946–52.
Hankey S, Marshall JD, Brauer M. Health impacts of the built environment: within-urban variability in physical inactivity, air pollution, and ischemic heart disease mortality. Environ Health Perspect. 2012;120:247–53. https://doi.org/10.1289/ehp.1103806.
McConnell R, Berhane K, Gilliland F, London SJ, Islam T, Gauderman WJ, et al. Asthma in exercising children exposed to ozone: a cohort study. The Lancet. 2002;359:386–91.
Lovinsky-Desir S, Jung KH, Rundle AG, Hoepner LA, Bautista JB, Perera FP, et al. Physical activity, black carbon exposure and airway inflammation in an urban adolescent cohort. Environ Res. 2016;151:756–62.
Piyadasa H, Hemshekhar M, Carlsten C, Mookherjee N. Inhaled diesel exhaust decreases the antimicrobial peptides a-Defensin and S100A7 in human bronchial secretions. Am J Respir Crit Care Med. 2018;197:1358–61.
Fuertes E, Markevych I, Bowatte G, Gruzieva O, Gehring U, Becker A, et al. Residential greenness is differentially associated with childhood allergic rhinitis and aeroallergen sensitization in seven birth cohorts. Allergy. 2016;71:1461–71. https://doi.org/10.1111/all.12915.
Green BJ, Levetin E, Horner WE, Codina R, Barnes CS, Filley WV. landscape plant selection criteria for the allergic patient. J Allergy Clin Immunol Pract. 2018;6:1869–76.
Devlin RB, Duncan KE, Jardim M, Schmitt MT, Rappold AG, Diaz-Sanchez D. Controlled exposure of healthy young volunteers to ozone causes cardiovascular effects. Circulation. 2012;126:104–11. https://doi.org/10.1161/CIRCULATIONAHA.112.094359.
Kim CS, Alexis NE, Rappold AG, Kehrl H, Hazucha MJ, Lay JC, et al. Lung function and inflammatory responses in healthy young adults exposed to 0.06 ppm ozone for 6.6 hours. Am J Respir Crit Care Med. 2011;183:1215–21. https://doi.org/10.1164/rccm.201011-1813OC.
Babisch W. Transportation noise and cardiovascular risk: Updated Review and synthesis of epidemiological studies indicate that the evidence has increased. Noise Health. 2006;8:1–29.
Hemmingsen JG, Jantzen K, Møller P, Loft S. No oxidative stress or DNA damage in peripheral blood mononuclear cells after exposure to particles from urban street air in overweight elderly. Mutagenesis. 2015;30:635–42. https://doi.org/10.1093/mutage/gev027.
Bräuner EV, Møller P, Barregard L, Dragsted LO, Glasius M, Wåhlin P, et al. Exposure to ambient concentrations of particulate air pollution does not influence vascular function or inflammatory pathways in young healthy individuals. Partic Fibre Toxicol. 2008;5:13. https://doi.org/10.1186/1743-8977-5-13.
Hemmingsen JG, Rissler J, Lykkesfeldt J, Sallsten G, Kristiansen J, et al. Controlled exposure to particulate matter from urban street air is associated with decreased vasodilation and heart rate variability in overweight and older adults. Particle Fibre Toxicol. 2015;12:6. https://doi.org/10.1186/s12989-015-0081-9.
Mills NL, Robinson SD, Fokkens PHB, Leseman DLAC, Miller MR, Anderson D, et al. Exposure to concentrated ambient particles does not affect vascular function in patients with coronary heart disease. Environ Health Perspect. 2008;116:709–15. https://doi.org/10.1289/ehp.11016.
Tong H, Rappold AG, Diaz-Sanchez D, Steck SE, Berntsen J, Cascio WE, et al. Omega-3 fatty acid supplementation appears to attenuate particulate air pollution-induced cardiac effects and lipid changes in healthy middle-aged adults. Environ Health Perspect. 2012;120:952–7. https://doi.org/10.1289/ehp.1104472.
Gong H Jr, Linn WS, Sioutas C, Terrell SL, Clark KW, Anderson KR, et al. Controlled exposures of healthy and asthmatic volunteers to concentrated ambient fine particles in Los Angeles. Inhal Toxicol. 2003;15:305–25. https://doi.org/10.1080/08958370304455.
Samet JM, Rappold A, Graff D, Cascio WE, Berntsen JH, Huang Y-CT, et al. Concentrated ambient ultrafine particle exposure induces cardiac changes in young healthy volunteers. Am J Respir Crit Care Med. 2009;179:1034–42. https://doi.org/10.1164/rccm.200807-1043OC.
Ghio AJ, Hall A, Bassett MA, Cascio WE, Devlin RB. Exposure to concentrated ambient air particles alters hematologic indices in humans. Inhal Toxicol. 2003;15:1465–78. https://doi.org/10.1080/08958370390249111.
Reis H, Reis C, Sharip A, Reis W, Zhao Y, Sinclair R, et al. Diesel exhaust exposure, its multi-system effects, and the effect of new technology diesel exhaust. Environ Int. 2018;114:252–65.
Gref A, Merid SK, Gruzieva O, Ballereau S, Becker A, Bellander T, et al. Genome-wide interaction analysis of air pollution exposure and childhood asthma with functional follow-up. Am J Respir Crit Care Med. 2017;195:1373–83. https://doi.org/10.1164/rccm.201605-1026OC.
Orach J, Rider CF, Carlsten C. Concentration-dependent health effects of air pollution in controlled human exposures. Environ Int. 2021;150:106424.
Beelen R, Raaschou-Nielsen O, Stafoggia M, Andersen ZJ, Weinmayr G, Hoffmann B, et al. Effects of long-term exposure to air pollution on natural-cause mortality: an analysis of 22 European cohorts within the multicentre ESCAPE project. The Lancet. 2014;383:785–95.
Carlsten C, Oron AP, Curtiss H, Jarvis S, Daniell W, Kaufman JD. Symptoms in response to controlled diesel exhaust more closely reflect exposure perception than true exposure. PLoS ONE. 2013;8:e83573.
Hansel NN, Putcha N, Woo H, Peng R, Diette GB, Fawzy A, et al. Randomized clinical trial of air cleaners to improve indoor air quality and COPD Health: results of the CLEAN AIR STUDY. Am J Respir Crit Care Med. 2021. https://doi.org/10.1164/rccm.202103-0604OC.
Dong W, Liu S, Chu M, Zhao B, Yang D, Chen C, et al. Different cardiorespiratory effects of indoor air pollution intervention with ionization air purifier: findings from a randomized, double-blind crossover study among school children in Beijing. Environ Pollut. 2019;254:113054.
Yoda Y, Tamura K, Adachi S, Otani N, Nakayama SF, Shima M. Effects of the use of air purifier on indoor environment and respiratory system among healthy adults. Int J Environ Res Public Health. 2020;17:3687.
Chuang H-C, Ho K-F, Lin L-Y, Chang T-Y, Hong G-B, Ma C-M, et al. Long-term indoor air conditioner filtration and cardiovascular health: a randomized crossover intervention study. Environ Int. 2017;106:91–6.
Acknowledgements
We are grateful to the members of the Air Pollution Exposure Laboratory (Vancouver, Canada) for their valuable input on improving this manuscript.
Funding
This project did not receive funding from agencies in the public, commercial, or not-for-profit sectors. CC is supported by the Canada Research Chairs program.
Author information
Authors and Affiliations
Contributions
EL conducted the literature search and wrote the manuscript with input from CC. CC conceived the project and provided supervision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Long, E., Carlsten, C. Controlled human exposure to diesel exhaust: results illuminate health effects of traffic-related air pollution and inform future directions. Part Fibre Toxicol 19, 11 (2022). https://doi.org/10.1186/s12989-022-00450-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12989-022-00450-5