
Abstract
Background
Brain microvascular endothelial cells (BMECs) astrocytes, neurons, and pericytes form the neurovascular unit (NVU). Interactions with NVU cells endow BMECs with extremely tight barriers via the expression of tight junction proteins, a host of active efflux and nutrient transporters, and reduced transcellular transport. To recreate the BMEC-enhancing functions of NVU cells, we combined BMECs, astrocytes, neurons, and brain pericyte-like cells.
Methods
BMECs, neurons, astrocytes, and brain like pericytes were differentiated from human induced pluripotent stem cells (iPSCs) and placed in a Transwell-type NVU model. BMECs were placed in co-culture with neurons, astrocytes, and/or pericytes alone or in varying combinations and critical barrier properties were monitored.
Results
Co-culture with pericytes followed by a mixture of neurons and astrocytes (1:3) induced the greatest barrier tightening in BMECs, supported by a significant increase in junctional localization of occludin. BMECs also expressed active P-glycoprotein (PGP) efflux transporters under baseline BMEC monoculture conditions and continued to express baseline active PGP efflux transporters regardless of co-culture conditions. Finally, brain-like pericyte co-culture significantly reduced the rate of non-specific transcytosis across BMECs.
Conclusions
Importantly, each cell type in the NVU model was differentiated from the same donor iPSC source, yielding an isogenic model that could prove enabling for enhanced personalized modeling of the NVU in human health and disease.
Similar content being viewed by others


Introduction
The blood–brain barrier (BBB) is both a passive and active barrier between the CNS and its surrounding vasculature [1]. The BBB is critical in hindering the movement of pathogens and toxins from the blood to the brain, while simultaneously allowing the passage of essential nutrients [2]. Brain microvascular endothelial cells (BMECs) possess specialized features such as tight junctions, efflux transporter activity and reduced nonspecific transcytosis compared to non-brain endothelium. BMECs gain their specialized BBB functions through interactions with supporting cells such as pericytes, astrocytes and neurons that along with BMECs form the so-called neurovascular unit (NVU). The interaction and cross-talk between BMECs and the cells of the NVU is vital in the development, regulation, and maintenance of the BBB [2,3,4,5,6,7,8].
A number of in vitro models have been utilized to understand the role of the BBB in both physiological and pathological states [9,10,11,12]. The majority of BBB models have been based on freshly isolated BMECs from a number of different species, although interpretation of results need to be carefully weighted due to inter-species variations [13,14,15]. Use of human primary or immortalized BMECs addresses some of the limitations of animal-sourced BMECs, however issues with de-differentiation and suboptimal phenotypes can limit their utility [16,17,18,19,20]. Human induced pluripotent stem cell (iPSC)-derived BMECs have recently become available and exhibit multiple critical phenotypes: elevated TEER, reduced permeability, and the expression of tight junction proteins, nutrient transporters and efflux transporters [21,22,23,24,25,26]. Additionally, iPSC-derived BMECs and their supporting NVU cell types have been utilized in several human disease models, and could be especially powerful in modeling human genetic diseases [11, 27].
A number of approaches have been employed to further enhance iPSC-based in vitro BBB models, including small molecule addition, shear stress (flow), tissue engineering approaches, and the co-culture with other NVU cell types [23, 28,29,30]. Astrocyte, neuron and pericyte co-culture with BMECs individually have been shown to enhance several BBB properties, including elevated TEER, reduced para-cellular permeability and continuous tight junction formation [23, 31]. Enhancement of BMEC properties was observed when BMECs were placed in co-culture with a multicellular combination of primary pericytes, and neural progenitor-derived astrocytes and neurons [21, 25, 26, 31,32,33,34]. Moreover, the addition of retinoic acid to the iPSC-derived BMECs significantly elevated barrier tightness in conjunction with primary brain pericytes and neural progenitor-derived astrocytes and neurons, approaching in vivo TEER measurements [23]. Recently, we reported an NVU model consisting of iPSC-derived neurons and astrocytes in co-culture with iPSC-derived BMECs that were all differentiated from the same parental iPSC line [25]. Such an isogenic model could facilitate the study of BBB function in human health and disease when using patient derived iPSC lines. However, a limitation of the iPSC-derived isogenic NVU model was the lack of stem cell-derived brain pericyte influences [25, 35]. We recently described the differentiation of brain-like pericytes from human iPSCs via a neural crest lineage, and demonstrated their capacity to induce barrier properties and reduced nonspecific transcytosis in iPSC-derived BMECs [36]. Furthermore, iPSC-derived brain-like pericytes could be incorporated with neurons and astrocytes to further elevate BMEC TEER [36]. Here we investigate whether retinoic acid stimulation can synergize with this multicellular co-culture to further enhance the BBB properties of iPSC-derived BMECs. Such an isogenic human model would provide an important in vitro tool for helping to understand the human BBB in development, function, disease, and potentially identifying novel therapeutic approaches.
Materials and methods
iPSC differentiation to BMECs, Astrocytes, Neurons and Brain-like Pericytes
IMR90C4 (WiCell, Madison, WI) iPSCs were utilized for differentiations of all cell types. iPSCs were cultured between passages 30–50 on Matrigel (BD Biosciences, San Jose, CA) with daily mTESR1 (WiCell) medium changes. BMECs were differentiated as previously described [24]. In brief, iPSCs were singularized with Accutase (Life Technologies, Carlsbad, CA) and expanded to 30,000 cells/cm2 prior to the initiation of the differentiation. Unconditioned medium (UM; 100 mL Knock-out serum replacement, 5 mL non-essential amino acids (NEAA), 2.5 mL of gluta-max, 392.5 mL of Dulbecco’s modified Eagle’s medium (DMEM)/F12 (1:1) (Life Technologies) and 3.5 μL of β-mercapto-ethanol (Sigma, St Louis, MO) was exchanged daily for 6 days. UM was exchanged with EC +/+ medium (200 mL human Endothelial Serum-Free medium (hESFM; Life Technologies), 20 ng/mL basic fibroblast growth factor (bFGF; WiCell) and 1% platelet-derived bovine serum (PDS; Biomedical Technologies, Tewksbury, MA)) for 48 h supplemented with (treated) or without (non-treated) 10 μM retinoic acid (RA; Sigma). Neurons and astrocytes were derived from intermediary EZ-sphere and astro-sphere populations (respectively) as previously described [25]. EZ- and astro-spheres were singularized with Accutase and seeded at 25,000 cells/cm2 onto Matrigel coated plates. Neurons were differentiated and cultured in neuron medium [DMEM/F12 (70:30) supplemented with 1% penicillin–streptomycin, 2% B27 minus Vitamin A (Life Technologies) and 2 μg/mL heparin (Sigma)] with every-other day medium exchanges for 2 weeks. Astrocytes were differentiated and cultured in astrocyte medium (DMEM/F12 (1:1) supplemented with 1% NEAA, 1% N2 supplement; Life Technologies, and 2 μg/mL heparin) with every-other day medium exchanges for 2 weeks. Pericytes were differentiated from iPSCs as previously described [36]. Pericytes were derived in a two-step process. Initially iPSCs were differentiated in E6 medium [DMEM/F12, l-ascorbic acid-2-phosphate magnesium (Sigma), sodium selenium (Sigma), insulin (Sigma), NaHCO3 (Sigma), transferrin (Sigma)] supplemented with dorsomorphin (Tocris, Bristol, United Kingdom), SB431542 (Tocris) CHIR99021 (Tocris), bFGF (Waisman Biomanufacturing, Madison, WI), and heparin (Sigma)) to a neural-crest stem cell population. CD271 positive cells were enriched with neural crest stem cell microbeads (Miltenyi, Bergisch Gladbach, Germany) and seeded at 10,000 cells/cm2 onto non-coated plates (Corning). Following 6 days of expansion in E6 medium supplemented with 10% FBS (Thermofisher), pericytes were seeded at 10,000 cells/cm2 for co-culture studies with BMECs.
Culture of 3T3 fibroblasts
3T3 mouse fibroblasts (ATCC, Manassas, VA) were cultured on Matrigel coated-plates with daily medium changes of DMEM (Life Technologies) supplemented with 10% FBS (Life Technologies). Mouse 3T3 fibroblasts served as a non-neural co-culture control.
Initiation of co-culture experiments
BMECs were differentiated from iPSCs as previously discussed. Following differentiation, BMECs were singularized with Accutase and seeded onto Transwells (Corning Transwell polyester filters, 0.4 μm pore diameter; 1.12 cm2, Corning, NY) coated with collagen IV/fibronectin (Sigma) at a density of 1 × 106 cells/cm2. Immediately following seeding, BMEC-coated Transwells were then placed into plates with either no cell types (monoculture), neuron: astrocyte (1:3) mixture, 3T3 fibroblasts, pericytes, or pericytes for the initial 24 h followed by a mixture of neurons: astrocytes (1:3) for the duration of the experiment. All experimental groups were in EC +/+ medium (hESFM supplemented with 1% PDS and 20 ng/mL bFGF) with or without retinoic acid for the initial 24 h and switched to EC ± medium (hESFM supplemented with 1% PDS) for the remainder of the experiment. All experiments occurred immediately following 48 h in co-culture unless otherwise stated.
Transcytosis/accumulation measurements
Dextran (Alexa-Fluor® 488, 10 kDa, 10 μM; Sigma) was used to measure the amount of transcytotic activity in the BMECs. Transwells were removed from co-culture and replaced in EC ± medium in the basolateral chamber. Dextran, diluted in EC ± was added to the apical side of the transwells on a rotating platform at 37 °C or 4 °C for 2 h. To determine the rate of transcytosis, media was collected from the basolateral chamber and read on a fluorescent plate reader. BMECs were then rinsed twice in phosphate buffer solution (PBS; Sigma) and lysed with radioimmunoprecipitation assay (RIPA; Sigma). After trituration the lysate was collected and quantified using a fluorescent plate reader, this value is the Dextran accumulated at this time point. Both transcytosis and accumulation values were normalized to protein content as determined by a bicinchoninic acid assay (BCA; Sigma).
Trans-endothelial electrical resistance (TEER) measurements
Barrier tightness was measured by the voltage difference from the movement of ions across the Transwell membrane. Epithelial Volt/Ohm Meter (EVOM) with STX2 electrodes (World Precision Instruments) was used to measure the TEER value in ohms starting at day 9 of differentiation and continuing up to a week after purification to create an extended barrier tightness profile. The values are normalized by subtracting the background (TEER from a blank well) and then multiplied by the surface area (1.12 cm2) of the transwell filter and reported as ohms × cm2.
Immunocytochemistry and analysis of tight junctions
Cells were fixed with cold methanol (100%; Sigma) or 2% paraformaldehyde (PFA; Sigma) in PBS for 15 min at room temperature. Following three washes in PBS the cells were blocked in 10% goat serum (Sigma) for 30 min. Cells were incubated with primary antibodies at 4 °C overnight on a rotating platform (Table 1). Following three PBS washes, cells were incubated with secondary antibodies for 1 h on a rotating platform at room temperature. Cells were incubated with a nuclear counterstain 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI; Sigma) for 15 min then washed in PBS and viewed on the Olympus epifluorescence microscope (Center Valley, PA). Images were analyzed using Image J, tight junction proteins (claudin-5, occludin and zo-1) were analyzed using threshold analysis and perimeter counting tools to determine the area of each image that exhibited occludin, claudin-5, or zo-1 immunoreactivity to determine area fraction index as previously discussed [25].
P-glycoprotein efflux transporter assay
BMECs were removed from co-culture groups and placed in EC ± . Cyclosporin A (CsA, 10 μM; Sigma) was used as an inhibitor to P-glycoprotein efflux transporter (PGP), whereas the small molecule Rhodamine123 (Rh123, 10 μM; Sigma) is a PGP substrate. Half the wells of each experimental group were treated with CsA for 30 min in both apical and basolateral chambers of the transwell. A solution of diluted Rh123 in EC ± with and without CsA, was added to the apical side of all transwells for 1 h at 37 °C on a rotating platform. Media collected from the basolateral chamber is quantified on the fluorescent plate reader. To ensure that barrier integrity was maintained for the duration of the experiment, TEER values were monitored throughout the experimental timeline. NVU co-culture did not affect baseline Rh123 transport therefore the data is presented as a percentage change from no CsA treatment with each respective co-culture condition.
Statistical analysis
Data for Figs. 2, 3, 4 are presented as mean ± SD of three independent differentiations. Due to variable TEER baseline recordings, TEER values presented in Fig. 1 are mean ± SD of three replicates from a single differentiation. Two additional independent differentiations were repeated for verification of reported statistical trends. SigmaStat 3.0 software (Systat Software, San Jose, CA) was used for statistical analysis. Student’s t-test and one-way analysis of variance (ANOVA) with Holm-Sidak correction for all comparisons were utilized when appropriate and described within the figure legends for each particular data set.

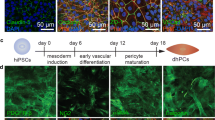
Induction of barrier properties brain microvascular endothelial cells (BMECs) following co-culture with pericytes, neurons, and/or astrocytes. a A completely isogenic blood brain barrier model was obtained by differentiating BMECs, neurons, astrocytes and pericytes from IMR90C4 induced pluripotent stem cells (iPSCs). BMECs cultured in monoculture conditions (MC) were directly compared to BMECs in co-culture up to 96 h past initial seeding onto Transwell filters. BMECs were co-cultured with mouse 3T3 fibroblasts (F), a mixture of iPSC-derived neurons and astrocytes (1:3; NA) or Pericytes (P). BMECs were initially co-cultured with pericytes for 24 h and then transferred to co-culture with a mixture of neurons and astrocytes (1:3) for the remainder of the experiment (PNA). b Trans-endothelial electrical resistance (TEER) profile of the experimental groups defined in panel (a) over a 96 h period. Statistical significance calculated using ANOVA. *p < 0.05 versus MC, F, #p < 0.05 versus P, †p < 0.05 versus NA. Values are mean ± SD of three replicates from a single differentiation and experiments were repeated for two additional differentiations for verification of reported statistical trends. c TEER profile of retinoic acid- treated BMECs over a 96 h period. *p < 0.05 versus MC, F, #p < 0.05 versus P, †p < 0.05 versus NA. Values are mean ± SD of three replicates from a single differentiation and experiments were repeated for two additional differentiations for verification of reported statistical trends. d Immunostaining of iPSC-derived BMECs following co-culture for 48 h in the PNA experimental condition probing for tight junction proteins, claudin-5 and ZO-1, endothelial cell markers, PECAM and VE-Cadherin, and glucose transporter, GLUT1. Scale bar = 100 μm. e Immunostaining of iPSC-derived neurons, astrocytes, and pericytes following co-culture with RA-treated BMECs for 48 h probing for β-tubulin III, GFAP, PDGFR-β, and NG2. Scale Bar = 100 μm
Results
Co-culture of BMECs with Pericytes, Astrocytes and Neurons
Established protocols were used to differentiate BMECs [24], astrocytes (from EZ spheres) [25], neurons (from EZ-spheres) [25, 37], and brain-like pericytes (from neural crest progenitors) [36] from the IMR90C4 iPSC line. Following differentiation, iPSC-derived BMECs were seeded onto Transwells with different combinations of fully differentiated astrocytes, pericytes and neurons. The co-cultured cells were differentiated and maintained in their respective media prior to co-culture with BMECs. Immediately following seeding onto Transwells, BMECs were co-cultured with either iPSC-derived pericytes alone (P), a neuron and astrocyte mixture (1:3 NA) or a sequential pericyte co-culture followed by a neuron and astrocyte co-culture (PNA) (Fig. 1a). Monocultured BMECs (MC) and 3T3 fibroblast co-cultures (F) were used as comparative controls (Fig. 1a). All co-culture conditions were conducted initially in EC +/+ medium (hESFM supplemented with bFGF and PDS) for 24 h and then switched to an EC ± medium (hESFM supplemented with PDS) for the duration of the time course. To determine the effects that co-cultured cell types had on BMEC barrier properties, transendothelial electrical resistance (TEER) was monitored from 24 h after initiation of co-culture to 96 h (Fig. 1b). TEER values reached a maximum value at 48–72 h in all experimental conditions. Monocultured BMECs exhibited a maximum TEER value of 310 ± 19 Ω × cm2, whereas BMECs co-cultured with pericytes reached a maximum TEER value of 564 ± 21 Ω × cm2 (p < 0.05) and remained significantly elevated above monoculture and fibroblast co-culture for the duration of the experiment (p < 0.05). As previously demonstrated, EZ-sphere derived neurons and astrocytes (1:3) significantly elevated barrier tightening in BMECs with a maximum TEER of 720 ± 38 Ω × cm2 (p < 0.05), and remained significantly elevated (p < 0.05). Sequential BMEC co-culture with pericytes followed by a mixture of neurons and astrocytes resulted in the greatest induction of TEER above all other experimental groups at 48 h (1155 ± 64 Ω × cm2; p < 0.05) and remained significantly elevated above all co-culture groups (p < 0.05). All NVU co-culture groups elevated BMEC barrier tightness above monoculture and 3T3 fibroblast co-culture at all the time points of the experiment (p < 0.05). Finally, mouse 3T3 fibroblasts were utilized as a non-inductive co-culture cell type and did not elevate TEER above monoculture TEER levels at any time point (Fig. 1b, p > 0.05).
We have previously demonstrated that retinoic acid (RA) can significantly elevate the barrier properties of iPSC-derived BMECs [23]. To determine the potential synergistic effects of co-culture on a BMEC population that was exposed to RA during differentiation, we monitored TEER of RA-treated BMECs in all of the previously mentioned NVU co-culture groups (Fig. 1c). Similar to the untreated BMECs, we observed peak TEER values 48 h following seeding onto Transwells and the initiation of co-culture. Monocultured, RA-treated BMECs (M) had a maximum TEER value of 2525 ± 149 Ω × cm2 and were undistinguishable from BMECs in co-culture with 3T3 fibroblasts (F) 2321 ± 92 Ω × cm2 (p > 0.05). Sequential pericyte co-culture followed by neuron and astrocyte co-culture (PNA) significantly elevated TEER (5486 ± 396 Ω × cm2; p < 0.05) compared with pericyte co-culture (P) (4015 ± 173 Ω × cm2; p < 0.05) or neuron and astrocyte co-culture (NA) (4726 ± 55 Ω × cm2; p < 0.05). Similar to untreated BMECs, NVU co-culture elevated BMEC barrier tightness at all time points throughout the experiment (p < 0.05). To confirm that BMECs, neurons, astrocytes, and pericytes still expressed key markers after co-culture, we verified that RA-treated BMECs expressed the endothelial cell markers PECAM-1 and VE-cadherin, the BBB glucose transporter Glut-1, and tight junction proteins: claudin-5 and ZO-1 (Fig. 1d). Neurons expressed β-tubulin III, astrocytes expressed glial fibrillary acidic protein (GFAP), and pericytes expressed platelet derived growth factor receptor-β (PDGFR-β) and neuron glial antigen-2 (NG2) (Fig. 1e). Taken together, the PNA configuration provided the most significant induction of barrier properties and increased the TEER in RA-treated and untreated BMECs.
PNA co-culture increases occludin localization in BMECs
Immunocytochemistry was utilized to investigate changes in tight junction protein localization during BMEC barrier tightening induced by NVU cell co-culture. Monoculture, RA-treated BMECs expressed junctionally localized occludin, claudin-5 and ZO-1 (Fig. 2a). Compared with monocultured RA-treated BMECs, co-culture with pericytes, a neuron: astrocyte mixture or pericytes followed by a neuron: astrocyte mixture, qualitatively increased occludin localization to the tight junctions (Fig. 2a). Quantification of area fraction index revealed that after 48 h of pericyte co-culture (P), BMECs had a slight, but statistically insignificant elevation in junctional occludin immuno-reactivity (19 ± 11%; p > 0.05) (Fig. 2b). However, BMECs in co-culture with pericytes for 24 h followed by a neuron: astrocyte (1:3) mixture for 24 h (PNA), exhibited significantly elevated junctional occludin levels (54 ± 17%; p < 0.05); however, the most significant boost in junctional occludin occurred after BMECs in co-culture for 48 h with a neuron: astrocyte (1:3) mixture (69 ± 17%; p < 0.05). However, the area fraction index of claudin-5 and ZO-1 were unchanged in BMECs following co-culture with pericytes, neurons, and astrocytes (Fig. 2b). These results suggest that enhanced barrier tightness following co-culture, at least in part, were the result of improved occludin localization and maintenance.

Analysis of tight junction continuity following co-culture. Localization of tight junction proteins, occludin, claudin-5, and ZO-1 were investigated in RA treated iPSC-derived BMECs following 48 h of co-culture with IMR90-iPSC-derived pericytes (P), neurons and astrocytes (1:3; NA) or pericytes (initial 24 h) followed by 24 h of neurons and astrocytes (1:3; PNA). a Immunocytochemistry of occludin revealed numerous discontinuous junctions (white arrows) in BMECs in monoculture compared to co-culture. Scale bar = 100 μm. b Quantification of the tight junction localization in BMECs in monoculture and co-culture conditions were calculated by the area of each image having immune-reactive occludin, claudin-5, or ZO1, resulting in area fraction index. Statistical significance calculated using ANOVA. Values are mean ± SD of three independent differentiations. *p < 0.05 versus MC, #p < 0.05 versus P, and †p < 0.05 versus the NA group
P-glycoprotein efflux transporter activity is not affected by co-culture
To determine the effects of co-culture on P-glycoprotein (PGP) efflux transporter activity in RA-treated BMECs, the transport of rhodamine 123 across the BMEC monolayer was measured. Rhodamine 123 transport was compared between BMECs in monoculture and BMEC in co-culture with either pericytes (P), a neuron: astrocyte (1:3) mixture (NA), or pericytes followed by a neuron: astrocyte mixture (PNA). Immunocytochemistry revealed that, qualitatively, BMECs either in monoculture or in co-culture expressed similar levels of PGP (Fig. 3a). Correlating with the immunocytochemistry images, PGP function was also unchanged by any co-culture configuration. BMECs in monoculture exhibited an increase in rhodamine 123 following CsA inhibition (45 ± 7%; p < 0.05), benchmarking the baseline PGP activity in RA-treated BMECs (Fig. 3b). A similar increase in rhodamine 123 following CsA inhibition was observed in BMECs following co-culture (P: 45 ± 19%; PNA: 42 ± 7%; NA: 41 ± 5%; p < 0.05); however, these data were indistinguishable between BMECs in monoculture and co-culture (p > 0.05 n.s.). These data reveal that PGP is present and active in RA-treated BMECs as previously demonstrated [23] and co-culture with pericytes, neurons and astrocytes alone or in combination does not affect this activity.

Evaluation of P-glycoprotein (PGP) efflux transporter expression and activity in RA-treated (BMECs). The expression and activity of PGP efflux transporters in BMECs in co-culture with iPSC-derived pericytes (P), neurons and astrocytes (NA), or pericytes, neurons, and astrocytes (PNA) were evaluated. a Immunolabeling of PGP efflux transporter following 48 h co-culture. b Activity of PGP efflux transporters in BMECs following 48 h of co-culture was assessed by the transport of the PGP substrate Rhodamine 123 with and without the PGP inhibitor cyclosporine A (CsA) from the apical to the basolateral chamber in the two-compartment co-culture model. Data is reported as percentage change from no-CsA treatment within each respective co-culture condition. Statistical significance was determined using a Student’s t-test. *p < 0.05 versus no CsA inhibition. One-way ANOVA determined there were no significant changes between each experimental group (co-culture conditions). p > 0.05 n.s.) Values are mean ± SD of three independent differentiations
Non-specific transcytosis in RA-treated BMECs is decreased in pericyte co-cultures
In previous studies, pericytes have been demonstrated to reduce the non-specific transcytosis of tracers and large molecules through BMECs [6, 38, 39]. To determine if co-culture with brain-like pericytes could also elicit similar effects in iPSC-derived RA-treated BMECs, the effects of co-culture on uptake and transcytosis of 10 kDa fluorescently tagged dextran was employed. BMECs in monoculture or 48 h following co-culture with pericytes (P), a neuron: astrocyte (1:3) mixture (NA), or pericytes followed by a neuron: astrocyte mixture (PNA) were incubated with fluorescently-tagged dextran in a Transwell setting. After addition of dextran to the top chamber, transport was measured both as cellular accumulation in BMECs and by accumulation in the lower chamber (transcytosis) then normalized to accumulation and transcytosis using monocultured BMECs (100%) (Fig. 4a, b). Following pericyte co-culture, BMECs exhibited significantly lower accumulation and transcytosis of dextran (65 ± 22% and 45 ± 5%; respectively; p < 0.05). While pericyte co-culture followed by neuron: astrocyte co-culture (PNA) did not yield a statistically significant decrease in dextran accumulation (77 ± 15%; p > 0.05), the amount of dextran that transcytosed across BMECs was significantly reduced (45 ± 8%; p < 0.05). Neuron: astrocyte co-culture (NA) lacking pericyte influences did not significantly affect dextran accumulation or transcytosis across BMECs (103 ± 14%, 83 ± 5%; respectively; p > 0.05). Non-specific transcytosis is minimal in BMECs compared to peripheral endothelial cells, similarly less than 0.05% of the dextran added to the apical chamber accumulated within the cell and less than 0.04% crossed the BMEC monolayer. To rule out the changes in transcytosis being a result of the increased passive barrier properties upon co-culture, dextran accumulation and transcytosis was also conducted at 4 °C conditions where vesicular transport processes are significantly inhibited (< 0.003% of the total dextran), but passive diffusion is comparatively unaffected (Fig. 4a, b). Under these conditions, the dextran transport was unaffected following pericyte co-culture compared to monoculture conditions, indicating that the increases in TEER do not explain the change in dextran transport. Taken together, these data indicate that brain-like pericyte co-culture alone or in conjunction with neurons and astrocytes can decrease the uptake and non-specific transcytosis of large molecules across RA-treated BMECs.

Determination of a fluorescently labeled dextran ability to cross RA-treated induced pluripotent stem cell (iPSC)-derived brain microvascular endothelial cells (BMECs) from the apical to the basolateral chamber in the two-compartment co-culture model. BMECs in monoculture (MC) or in co-culture for 48 h with either pericytes (P), neurons and astrocytes (NA), or pericytes, neurons, and astrocytes (PNA) were presented with a fluorescently tagged dextran for 2 h. Fluorescently-tagged dextran was measured within the BMEC population (a; accumulation) and from the bottom chamber (b; transcytosis) at both 37 °C and 4 °C. Raw fluorescence units are normalized to monoculture BMECs. Statistical significance was determined with ANOVA. Values are mean ± SD of three independent differentiations. *p < 0.05 versus MC
Discussion
This study demonstrates that human iPSCs can be utilized to derive BMECs, neurons, astrocytes, and brain-like pericytes that can be combined to generate an enhanced in vitro NVU model with elevated barrier and reduced nonspecific transcytosis properties. The unique advantage of the model presented here is that each of the cell types is derived from the same human iPSC source, enabling a host of applications from modeling human disease to personalized medicine. A major limitation of other NVU models is that one or more of the cell types is from a different source, either neonatal human or animal, impeding translational aspects [14, 32, 40,41,42]. However, iPSC technology allows for each cell type of the model to be generated independently under ideal conditions prior to co-culture [24, 36, 37, 43, 44]. Several studies have shown that BMECs can be successfully differentiated from iPSCs and they express a wide array of critical barrier properties including active nutrient and efflux transporters [11, 21, 24, 27, 33]. Previously, it has been demonstrated that astrocytes and neurons derived from iPSCs via an intermediary EZ-sphere/astro-sphere population, can enhance BMEC properties such as increased TEER, reduced para-cellular permeability, and enhanced junctional localization of occludin and claudin-5 [25, 33]. The greatest barrier tightening in BMECs was observed following coculture with a mixture of neurons and astrocytes (1:3), closely mimicking the composition of neurons and astrocytes in the human brain [45, 46]. Formerly, the limiting component of the stem cell-derived BBB model was the unavailability of iPSC-derived pericytes having brain-specific attributes [35, 47, 48]. The recent ability to derive brain-like pericytes from iPSCs is invaluable to modeling the NVU because pericytes enhance several critical BBB properties. While we have reported the elevation of TEER in the baseline PNA model [36], the contribution of brain-like pericytes to the isogenic BBB model in terms of non-specific transctytosis and efflux transporter function had not yet been evaluated. Moreover, the impact of combining enhanced RA-treated BMECs, with iPSC-derived brain-like pericytes, neurons, and astrocytes was unknown.
We observed the greatest barrier induction when BMECs were first co-cultured with pericytes for 24 h then co-cultured with a mixture of neurons: astrocytes (1:3). Our laboratory previously demonstrated that primary human brain pericytes and primary human neural progenitor cell-derived neurons and astrocytes also enhanced barrier properties in iPSC-derived BMECs [23]. In BBB development, BMECs are first subject to cues from pericytes and then later are influenced by astrocytes, motivating the sequential co-culture utilized here [5,6,7]. To further enhance the barrier tightening capabilities of BMECs, we manipulated retinoic acid signaling, a vital developmental pathway that has been implicated in the induction of brain-specific properties during development while also influencing brain and anterior spinal cord development [49,50,51,52]. Previously, it has been demonstrated that RA increased barrier function and junctional protein localization in iPSC-derived BMECs [23, 53]. Here we show that RA-treated BMECs further respond to co-culture with iPSC-derived brain-like pericytes, neurons, and astrocytes to elevate TEER to near in vivo levels (~ 2000–6000 Ω × cm2) [54]. Elevations in TEER are often associated with changes in tight junction protein levels or localization [41, 54,55,56,57]. Even in BMEC models where sufficient tight junctions are present, and their protein levels remain unchanged by a particular stimulus, the junctional continuity can be enhanced and correlate with increased TEER [23, 25]. Similar to these studies, we observed an increase in the junctional continuity of occludin following co-culture suggesting a potential role of occludin organization in increasing TEER levels. Interestingly, the greatest increase in occludin junctional continuity was observed following neuron: astrocyte (1:3) co-culture and did not correspond to the greatest increase in TEER amongst experimental groups. Pericyte co-culture alone did not yield a statistically significant change in junctional localization of occludin despite a qualitative improvement in junctional localization, suggesting that the corresponding increase in TEER observed in pericyte co-culture may be associated with mechanisms other than occludin localization.
Another vital property of the BBB is the presence and activity of efflux transporters. Previous studies have indicated that co-culture conditions, primarily with astrocytes, can enhance PGP protein expression and activity levels in primary and immortalized BMECs [8, 58, 59]. iPSC-derived BMECs under baseline conditions express functional PGP [22,23,24,25, 43]. In this study, we demonstrated that regardless of co-culture conditions, BMECs expressed active efflux PGP transporters that were unaffected by co-culture. Similar to these observations, previous studies have demonstrated that co-culture did not alter PGP expression or activity [25, 60,61,62]. The final critical BBB component that was investigated is that of a significantly reduced rate of non-specific transcytosis in BMECs compared to endothelial cells of the periphery [6]. Following iPSC-derived brain-like pericyte co-culture, we observed a significant reduction in uptake and transport of a large dextran molecule in iPSC-derived BMECs, reprising a similar role for pericytes in BBB development and regulation [6, 38].We utilized a transwell co-culture system to evaluate the effects that astrocytes, neurons, and pericytes have on BMEC barrier phenotypes. A number of effectors secreted by NVU cell types have been previously identified to enhance barrier properties in non-stem cell BBB models. Specifically, pericytes induced claudin-5 expression and enhanced tight junction localization via platelet-derived growth factor and transforming growth factor beta-1 signaling [6, 8]. Astrocyte-induced effectors: sonic hedgehog, angiopoietin 1/2, and apolipoprotein E upregulate, alter the subcellular distribution, and modify tight junction proteins [63,64,65]. The effectors involved in the human BBB are relatively unknown, and further studies will be needed to examine whether previously identified factors or possibly new human-specific NVU secreted factors are acting in the model. The Trans-well system enabled several barrier properties to be monitored simultaneously while maintaining flexibility in altering co-culture schemes. Additional co-culture architectures such as engineered vessels [28, 29], microfluidic channels [66], and spheroid models can be utilized to further mimic the in vivo barrier [67].
Conclusions
Here we demonstrate a human isogenic BBB model comprised of BMECs, neurons, astrocytes and brain-like pericytes all derived from the same iPSC line. The model performed similarly to other non-isogenic BBB models with expression of several key BMEC proteins and enhanced tight junction continuity while also displaying near in vivo TEER levels and significantly reduced transcytosis rates. Importantly, an entirely isogenic human BBB model will likely enable personalized medicine approaches with potential applications in tissue engineering, disease modeling and pharmaceutical screening.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Change history
10 September 2019
Following publication of the original article [1], the author has reported that in Figure 1 (b and c) the y-axis TEER (© x cm2) should be replaced with TEER (Ω x cm2).
Abbreviations
- BBB:
-
blood–brain barrier
- BMECs:
-
brain microvascular endothelial cells
- iPSCs:
-
induced pluripotent stem cells
- NVU:
-
neurovascular unit
- RA:
-
retinoic acid
- PGP:
-
p-glycoprotein
- TEER:
-
trans-endothelial electrical resistance
Bibliography
Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and dysfunction of the blood–brain barrier. Cell. 2015;163(5):1064–78.
Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood–brain barrier. Neurobiol Dis. 2010;37(1):13–25.
Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006;7(1):41–53.
Ballabh P, Braun A, Nedergaard M. The blood–brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16(1):1–13.
Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci USA. 2009;106(2):641–6.
Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature. 2010;468(7323):562–6.
Daneman R, Zhou L, Agalliu D, Cahoy JD, Kaushal A, Barres BA. The mouse blood–brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PLoS ONE. 2010;5(10):e13741.
Dohgu S, Takata F, Yamauchi A, et al. Brain pericytes contribute to the induction and up-regulation of blood–brain barrier functions through transforming growth factor-beta production. Brain Res. 2005;1038(2):208–15.
Weiss N, Miller F, Cazaubon S, Couraud PO. The blood–brain barrier in brain homeostasis and neurological diseases. Biochim Biophys Acta. 2009;1788(4):842–57.
Sweeney MD, Sagare AP, Zlokovic BV. blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14(3):133–50.
Vatine GD, Al-Ahmad A, Barriga BK, et al. Modeling psychomotor retardation using iPSCs from MCT8-deficient patients indicates a prominent role for the blood–brain barrier. Cell Stem Cell. 2017;20(6):831–43.
Zlokovic BV. The blood–brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57(2):178–201.
Deli MA, Abrahám CS, Kataoka Y, Niwa M. Permeability studies on in vitro blood–brain barrier models: physiology, pathology, and pharmacology. Cell Mol Neurobiol. 2005;25(1):59–127.
Syvänen S, Lindhe O, Palner M, et al. Species differences in blood–brain barrier transport of three positron emission tomography radioligands with emphasis on P-glycoprotein transport. Drug Metab Dispos. 2009;37(3):635–43.
Warren MS, Zerangue N, Woodford K, et al. Comparative gene expression profiles of ABC transporters in brain microvessel endothelial cells and brain in five species including human. Pharmacol Res. 2009;59(6):404–13.
Weksler BB, Subileau EA, Perrière N, et al. blood–brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19(13):1872–4.
Cecchelli R, Berezowski V, Lundquist S, et al. Modelling of the blood–brain barrier in drug discovery and development. Nat Rev Drug Discov. 2007;6(8):650–61.
Calabria AR, Shusta EV. A genomic comparison of in vivo and in vitro brain microvascular endothelial cells. J Cereb Blood Flow Metab. 2008;28(1):135–48.
Förster C, Burek M, Romero IA, Weksler B, Couraud PO, Drenckhahn D. Differential effects of hydrocortisone and TNFalpha on tight junction proteins in an in vitro model of the human blood–brain barrier. J Physiol. 2008;586(Pt 7):1937–49.
Man S, Ubogu EE, Williams KA, Tucky B, Callahan MK, Ransohoff RM. Human brain microvascular endothelial cells and umbilical vein endothelial cells differentially facilitate leukocyte recruitment and utilize chemokines for T cell migration. Clin Dev Immunol. 2008;2008:384982.
Hollmann EK, Bailey AK, Potharazu AV, Neely MD, Bowman AB, Lippmann ES. Accelerated differentiation of human induced pluripotent stem cells to blood–brain barrier endothelial cells. Fluids Barriers CNS. 2017;14(1):9.
Lippmann ES, Azarin SM, Kay JE, et al. Derivation of blood–brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol. 2012;30(8):783–91.
Lippmann ES, Al-Ahmad A, Azarin SM, Palecek SP, Shusta EV. A retinoic acid-enhanced, multicellular human blood–brain barrier model derived from stem cell sources. Sci Rep. 2014;4:4160.
Stebbins MJ, Wilson HK, Canfield SG, Qian T, Palecek SP, Shusta EV. Differentiation and characterization of human pluripotent stem cell-derived brain microvascular endothelial cells. Methods. 2016;101:93–102. https://doi.org/10.1016/j.ymeth.2015.10.016.
Canfield SG, Stebbins MJ, Morales BS, et al. An isogenic blood–brain barrier model comprising brain endothelial cells, astrocytes, and neurons derived from human induced pluripotent stem cells. J Neurochem. 2017;140(6):874–88.
Ribecco-Lutkiewicz M, Sodja C, Haukenfrers J, et al. A novel human induced pluripotent stem cell blood–brain barrier model: applicability to study antibody-triggered receptor-mediated transcytosis. Sci Rep. 2018;8(1):1873.
Lim RG, Quan C, Reyes-Ortiz AM, et al. Huntington’s disease iPSC-derived brain microvascular endothelial cells reveal WNT-mediated angiogenic and blood–brain barrier deficits. Cell Rep. 2017;19(7):1365–77.
DeStefano JG, Xu ZS, Williams AJ, Yimam N, Searson PC. Effect of shear stress on iPSC-derived human brain microvascular endothelial cells (dhBMECs). Fluids Barriers CNS. 2017;14(1):20.
Katt ME, Linville RM, Mayo LN, Xu ZS, Searson PC. Functional brain-specific microvessels from iPSC-derived human brain microvascular endothelial cells: the role of matrix composition on monolayer formation. Fluids Barriers CNS. 2018;15(1):7.
Helms HC, Abbott NJ, Burek M, et al. In vitro models of the blood–brain barrier: an overview of commonly used brain endothelial cell culture models and guidelines for their use. J Cereb Blood Flow Metab. 2016;36(5):862–90.
Brown JA, Pensabene V, Markov DA, et al. Recreating blood–brain barrier physiology and structure on chip: a novel neurovascular microfluidic bioreactor. Biomicrofluidics. 2015;9(5):054124.
Lippmann ES, Weidenfeller C, Svendsen CN, Shusta EV. Blood–brain barrier modeling with co-cultured neural progenitor cell-derived astrocytes and neurons. J Neurochem. 2011;119(3):507–20.
Qian T, Maguire SE, Canfield SG, et al. Directed differentiation of human pluripotent stem cells to blood–brain barrier endothelial cells. Sci Adv. 2017;3(11):e1701679.
Appelt-Menzel A, Cubukova A, Günther K, et al. Establishment of a human blood–brain Barrier co-culture model mimicking the neurovascular unit using induced pluri-and multipotent stem cells. Stem Cell Reports. 2017;8(4):894–906.
Xiang J, Andjelkovic AV, Wang MM, Keep RF. Blood–brain barrier models derived from individual patients: a new frontier: an Editorial Highlight on ‘An isogenic blood–brain barrier model comprising brain endothelial cells, astrocytes, and neurons derived from human induced pluripotent stem cells’. J Neurochem. 2017;140(6):843–4.
Stebbins MJ, Gastfriend BD, Canfield SG, et al. Human pluripotent stem cell-derived brain pericyte-like cells induce blood–brain barrier properties. Sci Adv. 2019;5(3):eaau7375.
Ebert AD, Shelley BC, Hurley AM, et al. EZ spheres: a stable and expandable culture system for the generation of pre-rosette multipotent stem cells from human ESCs and iPSCs. Stem Cell Res. 2013;10(3):417–27.
Armulik A, Genové G, Mäe M, et al. Pericytes regulate the blood–brain barrier. Nature. 2010;468(7323):557–61.
Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21(2):193–215.
Schiera G, Sala S, Gallo A, et al. Permeability properties of a three-cell type in vitro model of blood–brain barrier. J Cell Mol Med. 2005;9(2):373–9.
Nakagawa S, Deli MA, Kawaguchi H, et al. A new blood–brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem Int. 2009;54(3–4):253–63.
Lippmann ES, Al-Ahmad A, Palecek SP, Shusta EV. Modeling the blood–brain barrier using stem cell sources. Fluids Barriers CNS. 2013;10(1):2.
Wilson HK, Canfield SG, Hjortness MK, Palecek SP, Shusta EV. Exploring the effects of cell seeding density on the differentiation of human pluripotent stem cells to brain microvascular endothelial cells. Fluids Barriers CNS. 2015;12:13.
Sareen D, Gowing G, Sahabian A, et al. Human induced pluripotent stem cells are a novel source of neural progenitor cells (iNPCs) that migrate and integrate in the rodent spinal cord. J Comp Neurol. 2014;522(12):2707–28.
Azevedo FA, Carvalho LR, Grinberg LT, et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol. 2009;513(5):532–41.
Herculano-Houzel S, Lent R. Isotropic fractionator: a simple, rapid method for the quantification of total cell and neuron numbers in the brain. J Neurosci. 2005;25(10):2518–21.
van der Meer AD, Orlova VV, ten Dijke P, van den Berg A, Mummery CL. Three-dimensional co-cultures of human endothelial cells and embryonic stem cell-derived pericytes inside a microfluidic device. Lab Chip. 2013;13(18):3562–8.
Kusuma S, Facklam A, Gerecht S. Characterizing human pluripotent-stem-cell-derived vascular cells for tissue engineering applications. Stem Cells Dev. 2015;24(4):451–8.
Nordström U, Maier E, Jessell TM, Edlund T. An early role for WNT signaling in specifying neural patterns of Cdx and Hox gene expression and motor neuron subtype identity. PLoS Biol. 2006;4(8):e252.
Maden M, Gale E, Kostetskii I, Zile M. Vitamin A-deficient quail embryos have half a hindbrain and other neural defects. Curr Biol. 1996;6(4):417–26.
Liu JP, Laufer E, Jessell TM. Assigning the positional identity of spinal motor neurons: rostrocaudal patterning of Hox-c expression by FGFs, Gdf11, and retinoids. Neuron. 2001;32(6):997–1012.
Mizee MR, Wooldrik D, Lakeman KA, et al. Retinoic acid induces blood–brain barrier development. J Neurosci. 2013;33(4):1660–71.
Stebbins MJ, Lippmann ES, Faubion MG, Daneman R, Palecek SP, Shusta EV. Activation of RARα, RARγ, or RXRα increases barrier tightness in human induced pluripotent stem cell-derived brain endothelial cells. Biotechnol J. 2018;13(2):1700093.
Butt AM, Jones HC, Abbott NJ. Electrical resistance across the blood–brain barrier in anaesthetized rats: a developmental study. J Physiol. 1990;429:47–62.
Weidenfeller C, Svendsen CN, Shusta EV. Differentiating embryonic neural progenitor cells induce blood–brain barrier properties. J Neurochem. 2007;101(2):555–65.
Weidenfeller C, Schrot S, Zozulya A, Galla HJ. Murine brain capillary endothelial cells exhibit improved barrier properties under the influence of hydrocortisone. Brain Res. 2005;1053(1–2):162–74.
Calabria AR, Weidenfeller C, Jones AR, de Vries HE, Shusta EV. Puromycin-purified rat brain microvascular endothelial cell cultures exhibit improved barrier properties in response to glucocorticoid induction. J Neurochem. 2006;97(4):922–33.
Berezowski V, Landry C, Dehouck MP, Cecchelli R, Fenart L. Contribution of glial cells and pericytes to the mRNA profiles of P-glycoprotein and multidrug resistance-associated proteins in an in vitro model of the blood–brain barrier. Brain Res. 2004;1018(1):1–9.
Perrière N, Yousif S, Cazaubon S, et al. A functional in vitro model of rat blood–brain barrier for molecular analysis of efflux transporters. Brain Res. 2007;1150:1–13.
Lim JC, Wolpaw AJ, Caldwell MA, Hladky SB, Barrand MA. Neural precursor cell influences on blood–brain barrier characteristics in rat brain endothelial cells. Brain Res. 2007;1159:67–76.
Freese C, Reinhardt S, Hefner G, Unger RE, Kirkpatrick CJ, Endres K. A novel blood–brain barrier co-culture system for drug targeting of Alzheimer’s disease: establishment by using acitretin as a model drug. PLoS ONE. 2014;9(3):e91003.
Appelt-Menzel A, Cubukova A, Metzger M. Establishment of a human blood–brain barrier co-culture model mimicking the neurovascular unit using induced pluripotent stem cells. Curr Protoc Stem Cell Biol. 2018;47(1):e62.
Alvarez JI, Dodelet-Devillers A, Kebir H, et al. The Hedgehog pathway promotes blood–brain barrier integrity and CNS immune quiescence. Science. 2011;334(6063):1727–31.
Wosik K, Cayrol R, Dodelet-Devillers A, et al. Angiotensin II controls occludin function and is required for blood brain barrier maintenance: relevance to multiple sclerosis. J Neurosci. 2007;27(34):9032–42.
Nishitsuji K, Hosono T, Nakamura T, Bu G, Michikawa M. Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood–brain barrier model. J Biol Chem. 2011;286(20):17536–42.
Wang YI, Abaci HE, Shuler ML. Microfluidic blood–brain barrier model provides in vivo-like barrier properties for drug permeability screening. Biotechnol Bioeng. 2017;114(1):184–94.
Gastfriend BD, Palecek SP, Shusta EV. Modeling the blood–brain barrier: beyond the endothelial cells. Curr Opin Biomed Eng. 2018;5:6–12.
Acknowledgements
Not applicable.
Funding
This work was supported by National Institutes of Health Grants NS083688 and NS103844 to EVS and SPP, The Hartwell Foundation to SGC, and the National Institutes of Health Biotechnology Training Program grant T32GM008349 and the National Science Foundation Graduate Research Fellowship Program 1747503 to BDG.
Author information
Authors and Affiliations
Contributions
SGC, MJS, MGF, BDG, SPP, EVS conceived and designed the experiments. SGC, MJS, BDG, and MGF performed the experiments. SGC analyzed the data. SGC, SPP, and EVS wrote and corrected the manuscript. SGC, SPP and EVS supervised the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: the author has reported that in Figure 1 (b and c) the y-axis TEER (© x cm2) should be replaced with TEER (Ω x cm2)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Canfield, S.G., Stebbins, M.J., Faubion, M.G. et al. An isogenic neurovascular unit model comprised of human induced pluripotent stem cell-derived brain microvascular endothelial cells, pericytes, astrocytes, and neurons. Fluids Barriers CNS 16, 25 (2019). https://doi.org/10.1186/s12987-019-0145-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12987-019-0145-6