
Abstract
The Cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) serves as a key innate immune signaling axis involved in the regulation of various human diseases. It has been found that cGAS-STING pathway can recognize a variety of cytosolic double-stranded DNA (dsDNA), contributing to cause a robust type I interferon response thereby affecting the occurrence and progression of viral infection. Accumulating evidence indicates RNA virus-derived components play an important role in regulating cGAS-STING signaling, either as protective or pathogenic factors in the pathogenesis of diseases. Thus, a comprehensive understanding of the function of RNA virus-derived components in regulating cGAS-STING signaling will provide insights into developing novel therapies. Here, we review the existing literature on cGAS-STING pathway regulated by RNA virus-derived components to propose insights into pharmacologic strategies targeting the cGAS-STING pathway.
Similar content being viewed by others


Introduction
RNA viruses are responsible for many infectious diseases, including influenza virus, hepatitis C virus, polio, and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1]. The virus genome continues to mutate, which results in changes in viral infectivity and pathogenicity, creating an escape from antibody as well as vaccine protection thereby posing a greater risk [2]. The main symptoms of virus infection are characterized by fever, cough, fatigue and so on. In severe cases, acute respiratory distress, shock and multi-organ failure may even occur [3, 4]. The innate immune system, as the body’s first line of immune defense, can non-specifically recognize viral pathogen-associated molecular patterns (PAMPs) and launch a signaling cascade that produce proinflammatory cytokines and chemokines [5]. These inflammatory factors could further trigger the cytokine storm, posing a threat to the life of patients.
The Cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway serves as a critical mechanism for detecting both endogenous or exogenous DNA, thereby eliciting innate immune responses [6]. cGAS is capable of identifying cytoplasmic DNA anomalies and catalyzing the synthesis of cyclic GMP-AMP (cGAMP) from GTP and ATP. Subsequently, cGAMP binds to and activates the endoplasmic reticulum protein STING, then STING transduces signals to the nucleus through downstream molecules, ultimately leading to the transcription of type I interferons (IFN-I) and interferon-stimulator genes (ISGs), which play an important role in antiviral response [6, 7]. Previous studies have proposed the involvement of the cGAS-STING signaling pathway in RNA virus infections, thereby influencing disease progression [8,9,10]. In this paper, we primarily delve into the RNA virus-derived components regulate the cGAS-STING signaling pathway and explore the therapeutic potential of targeting this pathway in virus infections.
Overview of cGAS-STING signaling
cGAS, functioning as an innate immune sensor, possesses the capability to recognize a diverse array of cytosolic dsDNA, originating from viruses, bacteria, mitochondria, and micronuclei, which can be primarily categorized into pathogen-derived DNA and self-DNA [11, 12]. In addition to intracellular dsDNA and dsRNA, cytoplasmic RNA: DNA hybrids can directly activate cGAS, eliciting an effective antiviral immune response [13]. In instances such as simian virus 40 infection, slight DNA damage leakage fosters GAS recruitment and activation [14]. cGAS can also be involved in the detection of tissue damage/formation of DNA traps. After phagocytosis of neutrophil extracellular traps (NETs) by peripheral blood mononuclear cells (PBMCs), their DNA is transferred to the cytoplasm and cGAS is activated in the cytoplasm. Evidence of NETs activation of cGAS in vivo was also obtained in a model of autoimmune hepatitis induced by injection of the lectin concanavalin A [15, 16].
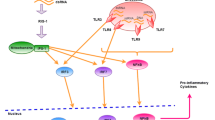
STING, discovered before cGAS, plays a critical role in DNA recognition and TLR9-independent IFN production [17,18,19]. In the cytoplasm, cGAS undergoes activation through interaction with dsDNA, a process independent of DNA sequence but reliant on DNA length [20, 21]. Structural analyses have unveiled a distinctive zinc thumb in cGAS responsible for recognizing B-form dsDNA [22]. Activated cGAS catalyzed the ATP and GTP into 2′3′-cyclic GMP-AMP (2′3′-cGAMP) [23]. Then cGAMP bound to and activated STING in the endoplasmic reticulum, promoting tetramer formation of STING through the oligomerization and translocated to the ER-Golgi intermediate compartments [24, 25]. Afterwards, TANK-binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3) are recruited by STING, followed by TBK1 autophosphorylation along with phosphorylation of STING and IRF3. The phosphorylated IRF3 dimerizes and localized to the nucleus to initiate IFN-I expression and subsequent induction of ISG expression, thereby instigating antiviral defense (Fig. 1) [6, 26]. Simultaneously, IRF7 and nuclear factor (NF-κB) are activated by TBK1, leading to the expression of other inflammatory cytokines [27, 28]. Notably, STING is also capable of directly detecting viral particles independent of cGAS [29].

Schematic diagram of cGAS-STING signaling pathway. (cGAS, as an innate immune sensor, is able to recognize various cytoplasmic dsDNA from pathogens, apoptotic/dead cells, mitochondria, and others. The interaction between cGAS and dsDNA leads to enzymatic activation of cGAS and catalyzing the formation of 2’,3’-cyclic GMP-AMP (cGAMP) from ATP and GTP. cGAMP binds to the dimer of interferon gene stimulatory factor (STING) located on the endoplasmic reticulum (ER) membrane. Then, STING moves from the ER to Golgi body via the ER–Golgi intermediate compartment, and then serves as a signaling platform for TBK1 phosphorylation. TBK1 phosphorylates the C-terminal domains of STING, and then IRF3 is recruited and phosphorylated. Finally, dimerized IRF3 can act as a transcription factor to initiate the transcription of type-I IFN and subsequent induction of ISG expression, eliciting antiviral defense)
Regulation of cGAS-STING signaling by virus-derived components
Though most studies focused on its role in DNA virus infection, increasing evidence has confirmed that cGAS-STING signaling also participated in RNA virus infection [30, 31]. As a self-protective mechanism, multiple RNA virus-derived components have been reported to be involved in the regulation of cGAS-STING signaling, such as SARS-CoV-2-derived open reading frames, and non-structural proteins, HIV-1-derived Vif, and HCoV-NL63-derived Papin-like protease (PLP) [32,33,34,35]. Thus, we will discuss the role of viral components in regulating cGAS-STING signaling in the following sections (Table 1).
cGAS-STING signaling in SARS-CoV-2 infection
SARS-CoV-2 inhibits the expression of IFN-I at the early stage of infection to counteract the IFN-I-mediated innate immune responses through a variety of mechanisms [36,37,38]. In the early stages of infection, patients with severe COVID-19 exhibited a weak IFN-I response but with a hyper-inflammatory profile, revealing a distinctive and inappropriate inflammatory response [39,40,41]. However, large but delayed IFN-I responses have also been reported in COVID-19 patients and animal models [42,43,44]. cGAS-STING signaling, regulated by different components of SARS-CoV-2, is a pivotal pathway involved in the IFN-I production.
SARS-CoV-2 activates cGAS-STING signaling
Upon SARS-CoV-2 infection, the production of 2′3′-cGAMP and phosphorylation of STING at Ser366 were hallmarks of cGAS-STING signaling pathway activation [31]. Consistent with the above, SARS-CoV-2 spike protein has been found to induce the expression of IFN-I and ISGs with the cooperation of host proteases [8, 45]. During viral infection-induced syncytium formation in angiotensin-converting enzyme 2-expressing cells, spike proteins damage the nucleus through DNA damage responses, contributing to the formation of micronuclei. Then the micronuclei were sensed by cGAS which localized to the fused cells, subsequently leading to the activation of STING-IRF3 signaling and production of IFN-I [8, 31, 46].
SARS-CoV-2 suppresses cGAS-STING signaling
In addition to spike protein, other components of SARS-CoV-2 have been found to inhibit the activation of cGAS-STING signaling [9, 34, 47, 48]. Open reading frame 10 (ORF10), an accessory protein of SARS-CoV-2, has been reported to interfere with STING-TBK1 interaction and inhibit STING ER-Golgi translocation to suppress IFN activation [9]. In addition, ORF10 antagonized the anti-viral response in a STING-mediated autophagy way. Further exploration also found that ORF9b co-localized and associated with STING and TBK-1, reducing the phosphorylation of TBK-1 and IRF3 as well as the nuclear translocation of IRF3, thereby antagonizing IFN-I production [47]. ORF3a, another accessory protein, has been revealed to block cGAS-STING-mediated IFN-β promoter activity in a NF-κB-dependent way [34, 48]. Post-translational modifications play a key role in regulating cGAS-STING signaling [49]. As a protease of SARS-CoV-2, 3CL could inhibit cGAS-STING-mediated NF-κB signaling via suppressing K63-linked ubiquitination of STING [48]. Another component of SARS-CoV-2, PLP, removed K63-linked polyubiquitin chains of STING, thereby disrupting the STING-IKKε-IRF3 complex for the production of IFN-β and ISGs, consequently inhibiting the IFN-I-mediated anti-viral responses [50]. The balance of dual roles in both promoting and suppressing cGAS-STING signaling in SARS-CoV-2 infection still remains to be investigated.
cGAS-STING signaling in HIV infection
HIV infection leads to progressive CD4+ T-cell loss and immune dysfunction, resulting in an increased risk of infections and tumor [30, 51]. Type I interferons are well-characterized innate antiviral proteins that contribute to resistance to HIV-1 infection [52]. Studies have shown that HIV-1 infection induces the cGAS-STING-TBK1-IRF3 signaling pathway which activates innate immunity to produce IFN-I [30, 53]. In recent years, several lines of evidence revealed that cGAS-STING signaling could be regulated by the components of HIV [32, 54]. Src homology 2 (SH2) domain-containing protein tyrosine phosphatase 1 (SHP-1), a protein tyrosine phosphatase, is comprised of two SH2 domains (N-SH2 and C-SH2) and a catalytic domain [55]. A recent study revealed that SHP-1 (residues 243–595) bound to STING (residues 1–137) and thereby inhibited the K63-linked ubiquitination of STING at Lys337 by dephosphorylating STING at Tyr162, thereby reducing the production of IFN-I [32]. This study also pointed out that HIV infection enhanced the inhibitory effect of SHP-1 on STING activation. Mechanistically, HIV-1-derived viral infectivity factor (Vif) promoted the recruitment of SHP-1 to STING and enhanced their interaction, which enhanced SHP-1-mediated inhibition of STING phosphorylation and K63-linked ubiquitination, accompanied by the inhibition of STING oligomerization and the interaction between STING and TBK1, consequently downregulating the IFN-I expression [32]. Another study reported that HIV-2 Vpx (a naturally immunogenic virion-associated protein) suppressed cGAS-STING-mediated NF-κB signaling to promote viral infection [54]. At the same time, Vpx markedly inhibited cGAS-STING-triggered DC maturation, which may have contributed to immune silencing [56]. Vpr and Vpu interact with STING to selectively inhibit NF-ĸB signaling by interfering with the degradation of IκBα and the recruitment of IκBβ [57, 58]. As HIV is cunning enough to evade cGAS-STING-mediated antiviral immune responses in disguise, limiting the inhibitory ingredient of the virus to ‘defenders’ has become a promising therapeutic strategy [32, 54].
cGAS-STING signaling in HCV infection
Hepatitis C virus (HCV) is a significant pathogen that causes chronic hepatitis liver cirrhosis, and hepatocellular carcinoma worldwide [59]. Ding Q et al. found that activation of the STING-mediated innate immune response to produce IFNs and cytokines inhibited replication of the HCV genotype 1b/Con1 replicon in Huh7.5 cells. STING was crucial for HCV PAMP-induced interferon activation [60]. However, HCV-derived NS4B was found to resist cGAMP stimulation and inhibit STING accumulation, thereby blocking the production of IFN-I and pro-inflammatory cytokines [60, 61]. NS4B silences interferon signaling by disrupting the collaboration between STING and TBK1, and NS4B and NS3/4A may synergistically inhibit different steps of IFN signaling during HCV infection [60].Another study found that NS4B targeted STING and abrogated RIG-I-mediated IFN-I response [62]. To sum up, disruption of these interactions mentioned above may restore IFN-I-mediated antiviral responses and may shed some light on the emergence of novel therapeutic strategies for HCV infection.
cGAS-STING signaling in ZIKV infection
Zika virus (ZIKV) is a flavivirus transmitted by mosquitoes that can cause significant neurological diseases [63]. According to published data, ZIKV has host tropism and can impair agonist-induced cGAS-STING signaling activation after infecting human cells but not in rodents [64,65,66,67]. Specifically, ZIKV blocks the anti-viral function of human STING (hSTING) not only through protease-dependent non-structural protein 2B3 (NS2B3) cleavage, but also potentially through NS2B3 protease cleavage-dependent mechanisms (increased permissiveness) [64]. Recent studies in Drosophila have revealed that insect STING homologues exert anti-viral activity against ZIKV infection by inducing autophagy in the brain [65, 67]. Liu et al. demonstrated that ZIKV infection led to the activation of NF-κB signaling, which in turn induces the expression of Drosophila STING (dSTING) in the Drosophila brain [67]. Mechanistically, they claimed that NF-κB-dependent dSTING-dependent autophagy controls ZIKV infection [67]. Overall, the cGAS-STING signaling pathway in restricted ZIKV infection has been well summarized in several papers [67,68,69].
cGAS-STING signaling in DENV infection
Dengue fever is a vector-borne viral disease caused by dengue virus (DENV), which evades host “pursuit” by expressing proteins that antagonize cellular innate immunity [70, 71]. DENV has been shown to manipulate cGAS-STING-mediated innate immunity through protein hydrolysis of STING and activation or degradation of cGAS [72,73,74,75]. Aguirre et al. demonstrated, for the first time, a clear mechanism of cGAS-STING activation in RNA virus infection [75]. Their study found that the DENV NS2B3 protease complex targeted cGAS for lysosomal degradation to avoid mtDNA sensing, which inhibited IFN-I expression and weakened the antiviral response [75]. A recent study showed that the DENV protease NS2B3 cleaves cGAS in the N-terminal region without destroying the C-terminal catalytic center, resulting in an N-terminal cleavage product (CP-N) and a C-terminal cleavage product (CP-C, including the catalytic center) [72]. Interestingly, the authors found that the DNA-binding affinity of CP-C was lower than that of cGAS, which was also associated with reduced CP-C enzyme activity. In contrast, the DNA binding affinity of CP-N was comparable to that of cGAS. Thus, CP-N competitively inhibited cGAMP production by both cGAS and CP-C [72]. Besides, this study revealed the physical interaction of NS2B3 with cGAS and CP-C, setting the stage for their degradation [72]. Another study revealed the dual role of STING in response to DENV infection [76]. On the one hand, replication of DENV2, a DENV mutant, destroys host DNA, which induces cGAS-STING signaling and the IFN-I response, inhibiting the spread of infection [76]. On the other hand, STING activation also supports DENV2 replication in infected cells through STING-induced autophagy [76].
Regulation of cGAS-STING signaling by other RNA virus components
It is worth noting that the cGAS-STING signaling can also be regulated by other RNA virus components, such as influenza virus (IAV), encephalomyocarditis virus (EMCV), and lymphocytic choriomeningitis virus (LCMV) [73, 77,78,79]. In IAV infection, researchers have demonstrated that STING-dependent IFN-β gene expression was indispensable for limiting viral replication. The influenza virus M2 or EMCV 2B protein triggered mtDNA release to initiate cGAS-STING-dependent anti-viral signaling to restrict disease, whereas the influenza virus NS1 binds to mtDNA to attenuate innate immunity [80]. Papain-like proteases (PLP), an important component of human coronavirus (HCoV) NL63 and SARS-CoV, have been reported to antagonize the STING signaling [33, 81]. PLP inhibited STING-mediated IRF-3 nuclear translocation and induction of IRF-3 dependent promoters [33]. Another study found that PLP could regulate STING-mediated innate immune response in an autophagy-dependent manner [81]. PLP2-TM interacted with the key autophagy regulators, LC3 and Beclin1, and promoted Beclin1 interaction with STING. Additionally, knockdown of Beclin1 partially reversed the inhibitory effect of PLP2-TM on innate immune responses [81]. This process was dependent on the interaction between PLP and STING, which blocked dimerization of STING and inhibited the assembly of STING-MAVS-TBK1/IKKe complexes required for activation of IRF-3 [33]. With the deepening of research, cGAS-STING signaling pathway has become a non-negligible presence in the treatment of RNA virus infections.
Potential therapy based on cGAS-STING signaling
Ongoing clinical trials of cGAS-STING signaling pathway primarily focused on antitumor immunity, whereas, due to growing evidence tapping into the potential of anti-viral immunity, making it a new strategy for the treatment of infections. Considering the pivotal roles of STING in viral infection, several studies have explored the function of STING agonists in therapy [31, 82,83,84]. Diaminobenzimidazole (diABZI), a small-molecule STING agonist that induces rapid short-term activation of STING, has been reported to inhibit viral replication in infected cells (∼ 1000-fold inhibition) in an IFN-dependent manner [82]. Similar phenomena have been revealed in studies of other STING agonists. Given the inhibitory effect of SARS-CoV-2-dereived 3CL on STING, 3CL inhibitors (such as flavonoids and PF-00835231) have been widely used in the treatment of COVID-19 [85, 86]. Also, it might be promising in the treatment of other 3CL-containing virus-induced infection [87]. However, not all cases of cGAS-STING activation lead to ameliorating the symptoms. Researchers have found that excessive activation of cGAS-STING can exacerbate the virus-induced inflammatory factor storm, which may correlate with the poor prognosis of patients [88]. It has been shown that in different RNA virus-infected cells, immunological differences between viruses can result in different regulatory mechanisms of cGAS-STING. In a study by Christopher J Neufeldt et al., it was found that SARS-CoV-2 infection leads to a storm of inflammatory factors through selective activation of the cGAS-STING signaling axis and thus NF-κB. Inflammatory gene activation was reduced by 60–75% with STING inhibitors [88]. Similar phenomena have been observed in other plus-stranded RNA viruses such as flaviviruses, SARS-CoV and NL63 coronavirus [89]. Application of H-151, a STING inhibitor, attenuates SARS-CoV-2-induced severe lung inflammation and improves disease prognosis [10]. An alternative inhibitor of TBK1/IKKε signaling that disrupts TBK1/IKKε signaling and prevents phosphorylation of S172, thereby blocking IRF3 and STING-mediated NF-κB-mediated transcriptional programs, shows strength in limiting excessive inflammation in SARS-CoV-2 [90]. Thus, the exploration and improvement of STING agonists and protease inhibitors and how to avoid pathological activation of STING during treatment may be the future orientation of treatment. In addition, studies have shown that the cGAS-STING signaling pathway is closely related to a variety of diseases such as tumors, autoimmune diseases, cardiovascular diseases, metabolic diseases, and neurodegenerative diseases, and has great potential to enhance tumor immunity and improve diseases. Therefore, guiding the development of novel targeted drugs for cGAS-STING signaling pathway and taking it into account the safety and efficacy of diseases are also imminent.
Conclusion
Extensive researches have shed light on the scope and significance of cGAS-STING in antiviral immunity. With its dual role in inhibiting infection dissemination induced by the IFN-I response and promoting viral replication via STING-induced autophagy, STING plays a dual role in response to virus infection. In addition to stimulating IRF3-IFN-I signaling, STING strengthened the transcriptional activity of NF-κB to coordinate innate and adaptive immunity. This review aims to outline the role of components from different RNA viruses in the cGAS-STING signaling pathway, aiming to provide a sound theoretical basis for further studies on viral camouflage to evade cGAS-STING-mediated antiviral immune responses. Studies revealed that activation of STING signaling during RNA virus infection is regulated by a variety of components, such as spike-induced activation and ORF10, ORF3a, ORF9b, 3CL, Vif, and PLP, among others. These components affected cGAS-STING signaling pathway-mediated antiviral responses in IRF-3-dependent, NF-κB-dependent, and autophagy-dependent manner. With the emergence of STING antagonists, therapeutic means to block viral immune evasion have become possible. Furthermore, the interference of accessoryprotein and non-structural protein (NSP, NSP13/14/15) with the production of IFN-I presents new therapeutic avenues for infection. As an important target in antiviral immunity and tumor immunotherapy, it is also imperative to guide the development of novel targeted drugs against the cGAS-STING signaling pathway.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- cGAS:
-
Cyclic GMP-AMP synthase
- STING:
-
Stimulator of interferon genes
- dsDNA:
-
Double-stranded DNA
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- PAMPs:
-
Pathogen-associated molecular patterns
- cGAMP:
-
Cyclic GMP-AMP
- ISGs:
-
Interferon-stimulator genes
- TLR9:
-
Toll-like receptor 9
- TBK1:
-
TANK-binding kinase 1
- IRF3:
-
Interferon regulatory factor 3
- PLP:
-
Papin-like protease
- ORF:
-
Open reading frame
- Vif:
-
Viral infectivity factor
- SHP-1:
-
Src homology 2 domain-containing protein tyrosine phosphatase 1
- HCV:
-
Hepatitis C virus
- ZIKV:
-
Zika virus
- DENV:
-
Dengue virus
- IAV:
-
Influenza virus
- EMCV:
-
Encephalomyocarditis virus
- LCMV:
-
Lymphocytic choriomeningitis virus
- HCoV:
-
Human coronavirus
- diABZI:
-
Diaminobenzimidazole
- NSP:
-
Non-structural protein
References
Fan YM, Zhang YL, Luo H, Mohamud Y. Crosstalk between RNA viruses and DNA sensors: role of the cGAS-STING signalling pathway. Rev Med Virol. 2022;32(5):e2343.
Tao K, Tzou PL, Nouhin J, Gupta RK, de Oliveira T, Kosakovsky Pond SL, et al. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat Rev Genet. 2021;22(12):757–73.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506.
Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of Coronavirus Disease 2019 in China. New Engl J Med. 2020;382(18):1708–20.
Fischer S, Deindl E. State of the art of Innate Immunity-An overview. Cells. 2022;11(17).
Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol. 2020;21(9):501–21.
Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786–91.
Liu X, Wei L, Xu F, Zhao F, Huang Y, Fan Z, et al. SARS-CoV-2 spike protein-induced cell fusion activates the cGAS-STING pathway and the interferon response. Sci Signal. 2022;15(729):eabg8744.
Han L, Zheng Y, Deng J, Nan ML, Xiao Y, Zhuang MW, et al. SARS-CoV-2 ORF10 antagonizes STING-dependent interferon activation and autophagy. J Med Virol. 2022;94(11):5174–88.
Domizio JD, Gulen MF, Saidoune F, Thacker VV, Yatim A, Sharma K, et al. The cGAS-STING pathway drives type I IFN immunopathology in COVID-19. Nature. 2022;603(7899):145–51.
Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17(10):1142–9.
Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell. 2014;54(2):289–96.
Mankan AK, Schmidt T, Chauhan D, Goldeck M, Höning K, Gaidt M, et al. Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. Embo j. 2014;33(24):2937–46.
Pépin G, Nejad C, Ferrand J, Thomas BJ, Stunden HJ, Sanij E et al. Topoisomerase 1 inhibition promotes cyclic GMP-AMP synthase-dependent antiviral responses. mBio. 2017;8(5).
Aubé FA, Bidias A, Pépin G. Who and how, DNA sensors in NETs-driven inflammation. Front Immunol. 2023;14:1190177.
Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. 2014;505(7485):691–5.
Sun W, Li Y, Chen L, Chen H, You F, Zhou X, et al. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci U S A. 2009;106(21):8653–8.
Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455(7213):674–8.
Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29(4):538–50.
Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339(6121):826–30.
Luecke S, Holleufer A, Christensen MH, Jønsson KL, Boni GA, Sørensen LK, et al. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 2017;18(10):1707–15.
Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013;498(7454):332–7.
Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, et al. cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498(7454):380–4.
Shang G, Zhang C, Chen ZJ, Bai XC, Zhang X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature. 2019;567(7748):389–93.
Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, et al. Structure-function analysis of STING activation by c[G(2’,5’)pA(3’,5’)p] and targeting by antiviral DMXAA. Cell. 2013;154(4):748–62.
Tao J, Zhou X, Jiang Z. cGAS-cGAMP-STING: the three musketeers of cytosolic DNA sensing and signaling. IUBMB Life. 2016;68(11):858–70.
Yum S, Li M, Fang Y, Chen ZJ. TBK1 recruitment to STING activates both IRF3 and NF-κB that mediate immune defense against tumors and viral infections. Proc Natl Acad Sci U S A. 2021;118(14).
Ruangkiattikul N, Nerlich A, Abdissa K, Lienenklaus S, Suwandi A, Janze N, et al. cGAS-STING-TBK1-IRF3/7 induced interferon-beta contributes to the clearing of non tuberculous mycobacterial infection in mice. Virulence. 2017;8(7):1303–15.
Holm CK, Rahbek SH, Gad HH, Bak RO, Jakobsen MR, Jiang Z, et al. Influenza a virus targets a cGAS-independent STING pathway that controls enveloped RNA viruses. Nat Commun. 2016;7:10680.
Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341(6148):903–6.
Zhou Z, Zhang X, Lei X, Xiao X, Jiao T, Ma R, et al. Sensing of cytoplasmic chromatin by cGAS activates innate immune response in SARS-CoV-2 infection. Signal Transduct Target Ther. 2021;6(1):382.
Wang Y, Qian G, Zhu L, Zhao Z, Liu Y, Han W, et al. HIV-1 Vif suppresses antiviral immunity by targeting STING. Cell Mol Immunol. 2022;19(1):108–21.
Sun L, Xing Y, Chen X, Zheng Y, Yang Y, Nichols DB, et al. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS ONE. 2012;7(2):e30802.
Su J, Shen S, Hu Y, Chen S, Cheng L, Cai Y et al. SARS-CoV-2 ORF3a inhibits cGAS-STING-mediated autophagy flux and antiviral function. J Med Virol. 2022.
Yuen CK, Lam JY, Wong WM, Mak LF, Wang X, Chu H, et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg Microbes Infect. 2020;9(1):1418–28.
Li JY, Liao CH, Wang Q, Tan YJ, Luo R, Qiu Y, et al. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020;286:198074.
Park A, Iwasaki A, Type I, Type III. Interferons - induction, signaling, evasion, and application to Combat COVID-19. Cell Host Microbe. 2020;27(6):870–8.
Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, et al. SARS-CoV-2 ORF3b is a potent Interferon Antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. 2020;32(12):108185.
Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci Immunol. 2020;5(49).
Ronit A, Berg RMG, Bay JT, Haugaard AK, Ahlstrom MG, Burgdorf KS, et al. Compartmental immunophenotyping in COVID-19 ARDS: a case series. J Allergy Clin Immunol. 2021;147(1):81–91.
Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al. Imbalanced host response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020;181(5):1036–e459.
Aid M, Busman-Sahay K, Vidal SJ, Maliga Z, Bondoc S, Starke C, et al. Vascular disease and thrombosis in SARS-CoV-2-Infected Rhesus macaques. Cell. 2020;183(5):1354–e6613.
Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z, et al. Heightened Innate Immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe. 2020;27(6):883–e902.
Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun. 2020;11(1):3810.
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–e808.
Ren H, Ma C, Peng H, Zhang B, Zhou L, Su Y, et al. Micronucleus production, activation of DNA damage response and cGAS-STING signaling in syncytia induced by SARS-CoV-2 infection. Biol Direct. 2021;16(1):20.
Han L, Zhuang MW, Deng J, Zheng Y, Zhang J, Nan ML, et al. SARS-CoV-2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG-I/MDA-5-MAVS, TLR3-TRIF, and cGAS-STING signaling pathways. J Med Virol. 2021;93(9):5376–89.
Rui Y, Su J, Shen S, Hu Y, Huang D, Zheng W, et al. Unique and complementary suppression of cGAS-STING and RNA sensing- triggered innate immune responses by SARS-CoV-2 proteins. Signal Transduct Target Ther. 2021;6(1):123.
Liu J, Rui K, Peng N, Luo H, Zhu B, Zuo X, et al. The cGAS-STING pathway: post-translational modifications and functional implications in diseases. Cytokine & growth factor reviews; 2022.
Cao D, Duan L, Huang B, Xiong Y, Zhang G, Huang H. The SARS-CoV-2 papain-like protease suppresses type I interferon responses by deubiquitinating STING. Sci Signal. 2023;16(783):eadd0082.
Herzner AM, Hagmann CA, Goldeck M, Wolter S, Kübler K, Wittmann S, et al. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat Immunol. 2015;16(10):1025–33.
Acchioni C, Marsili G, Perrotti E, Remoli AL, Sgarbanti M, Battistini A. Type I IFN–a blunt spear in fighting HIV-1 infection. Cytokine Growth Factor Rev. 2015;26(2):143–58.
Lahaye X, Satoh T, Gentili M, Cerboni S, Conrad C, Hurbain I, et al. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity. 2013;39(6):1132–42.
Su J, Rui Y, Lou M, Yin L, Xiong H, Zhou Z, et al. HIV-2/SIV vpx targets a novel functional domain of STING to selectively inhibit cGAS-STING-mediated NF-kappaB signalling. Nat Microbiol. 2019;4(12):2552–64.
Lim S, Lee KW, Kim JY, Kim KD. Consideration of SHP-1 as a Molecular Target for Tumor Therapy. Int J Mol Sci. 2023;25(1).
Hilligan KL, Ronchese F. Antigen presentation by dendritic cells and their instruction of CD4 + T helper cell responses. Cell Mol Immunol. 2020;17(6):587–99.
Kogan M, Deshmane S, Sawaya BE, Gracely EJ, Khalili K, Rappaport J. Inhibition of NF-κB activity by HIV-1 vpr is dependent on vpr binding protein. J Cell Physiol. 2013;228(4):781–90.
Langer S, Hammer C, Hopfensperger K, Klein L, Hotter D, De Jesus PD et al. HIV-1 Vpu is a potent transcriptional suppressor of NF-κB-elicited antiviral immune responses. Elife. 2019;8.
Zeisel MB, Felmlee DJ, Baumert TF. Hepatitis C virus entry. Curr Top Microbiol Immunol. 2013;369:87–112.
Ding Q, Cao X, Lu J, Huang B, Liu YJ, Kato N, et al. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J Hepatol. 2013;59(1):52–8.
Yi G, Wen Y, Shu C, Han Q, Konan KV, Li P, et al. Hepatitis C Virus NS4B can suppress STING Accumulation to evade Innate Immune responses. J Virol. 2016;90(1):254–65.
Nitta S, Sakamoto N, Nakagawa M, Kakinuma S, Mishima K, Kusano-Kitazume A, et al. Hepatitis C virus NS4B protein targets STING and abrogates RIG-I-mediated type I interferon-dependent innate immunity. Hepatology (Baltimore MD). 2013;57(1):46–58.
Ferraris P, Yssel H, Missé D. Zika virus infection: an update. Microbes Infect. 2019;21(8–9):353–60.
Ding Q, Gaska JM, Douam F, Wei L, Kim D, Balev M, et al. Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proc Natl Acad Sci U S A. 2018;115(27):E6310–8.
Delorme-Axford E, Klionsky DJ. Inflammatory-dependent Sting activation induces antiviral autophagy to limit Zika virus in the Drosophila brain. Autophagy. 2019;15(1):1–3.
Coyne CB. STING’ing Zika virus in neurons. Nat Microbiol. 2018;3(9):975–6.
Liu Y, Gordesky-Gold B, Leney-Greene M, Weinbren NL, Tudor M, Cherry S. Inflammation-Induced, STING-Dependent Autophagy restricts Zika Virus infection in the Drosophila brain. Cell Host Microbe. 2018;24(1):57–e683.
Eaglesham JB, McCarty KL, Kranzusch PJ. Structures of diverse poxin cGAMP nucleases reveal a widespread role for cGAS-STING evasion in host-pathogen conflict. Elife. 2020;9.
Souza MPM, Freitas BCG, Holanda GM, Diniz Junior JAP, Cruz ACR. Correlation of cGAS, STING, INF-α and INF-β gene expression with Zika virus kinetics in primary culture of microglia and neurons from BALB/c mice. Acad Bras Cienc. 2022;94(suppl 3):e20211189.
Roy SK, Bhattacharjee S. Dengue virus: epidemiology, biology, and disease aetiology. Can J Microbiol. 2021;67(10):687–702.
Wrighton K. The STING behind dengue virus infection. Nat Rev Microbiol. 2018;16(6):330.
Bhattacharya M, Bhowmik D, Tian Y, He H, Zhu F, Yin Q. The Dengue virus protease NS2B3 cleaves cyclic GMP-AMP synthase to suppress cGAS activation. J Biol Chem. 2023:102986.
Sun B, Sundström KB, Chew JJ, Bist P, Gan ES, Tan HC, et al. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci Rep. 2017;7(1):3594.
Stabell AC, Meyerson NR, Gullberg RC, Gilchrist AR, Webb KJ, Old WM et al. Dengue viruses cleave STING in humans but not in nonhuman primates, their presumed natural reservoir. Elife. 2018;7.
Aguirre S, Luthra P, Sanchez-Aparicio MT, Maestre AM, Patel J, Lamothe F, et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat Microbiol. 2017;2:17037.
Ng WC, Kwek SS, Sun B, Yousefi M, Ong EZ, Tan HC, et al. A fast-growing dengue virus mutant reveals a dual role of STING in response to infection. Open Biol. 2022;12(12):220227.
Dixon CR, Malik P, de Las Heras JI, Saiz-Ros N, de Lima Alves F, Tingey M, et al. STING nuclear partners contribute to innate immune signaling responses. iScience. 2021;24(9):103055.
Hervas-Stubbs S, Riezu-Boj JI, Mancheño U, Rueda P, Lopez L, Alignani D, et al. Conventional but not plasmacytoid dendritic cells foster the systemic virus-induced type I IFN response needed for efficient CD8 T cell priming. J Immunol. 2014;193(3):1151–61.
Liu H, Zhu Z, Xue Q, Yang F, Li Z, Xue Z, et al. Innate sensing of picornavirus infection involves cGAS-STING-mediated antiviral responses triggered by mitochondrial DNA release. PLoS Pathog. 2023;19(2):e1011132.
Moriyama M, Koshiba T, Ichinohe T. Influenza a virus M2 protein triggers mitochondrial DNA-mediated antiviral immune responses. Nat Commun. 2019;10(1):4624.
Chen X, Wang K, Xing Y, Tu J, Yang X, Zhao Q, et al. Coronavirus membrane-associated papain-like proteases induce autophagy through interacting with Beclin1 to negatively regulate antiviral innate immunity. Protein Cell. 2014;5(12):912–27.
Li M, Ferretti M, Ying B, Descamps H, Lee E, Dittmar M, et al. Pharmacological activation of STING blocks SARS-CoV-2 infection. Sci Immunol. 2021;6:59.
Humphries F, Shmuel-Galia L, Jiang Z, Wilson R, Landis P, Ng SL et al. A diamidobenzimidazole STING agonist protects against SARS-CoV-2 infection. Sci Immunol. 2021;6(59).
Wu J-J, Chen F-Y, Han B-B, Zhang H-Q, Zhao L, Zhang Z-R, et al. CASTING: a potent Supramolecular Strategy to cytosolically deliver STING agonist for Cancer Immunotherapy and SARS-CoV-2 vaccination. CCS Chem. 2022;0(0):1–17.
Jo S, Kim S, Shin DH, Kim MS. Inhibition of SARS-CoV 3CL protease by flavonoids. J Enzyme Inhib Med Chem. 2020;35(1):145–51.
Boras B, Jones RM, Anson BJ, Arenson D, Aschenbrenner L, Bakowski MA, et al. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat Commun. 2021;12(1):6055.
Xiong B, Gui CS, Xu XY, Luo C, Chen J, Luo HB, et al. A 3D model of SARS_CoV 3CL proteinase and its inhibitors design by virtual screening. Acta Pharmacol Sin. 2003;24(6):497–504.
Neufeldt CJ, Cerikan B, Cortese M, Frankish J, Lee JY, Plociennikowska A, et al. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-κB. Commun Biology. 2022;5(1):45.
Ma Z, Damania B. The cGAS-STING defense pathway and its counteraction by viruses. Cell Host Microbe. 2016;19(2):150–8.
Ullah TR, Johansen MD, Balka KR, Ambrose RL, Gearing LJ, Roest J, et al. Pharmacological inhibition of TBK1/IKKε blunts immunopathology in a murine model of SARS-CoV-2 infection. Nat Commun. 2023;14(1):5666.
Neufeldt CJ, Cerikan B, Cortese M, Frankish J, Lee JY, Plociennikowska A, et al. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-kappaB. Commun Biology. 2022;5(1):45.
Su J, Shen S, Hu Y, Chen S, Cheng L, Cai Y, et al. SARS-CoV-2 ORF3a inhibits cGAS-STING-mediated autophagy flux and antiviral function. J Med Virol. 2023;95(1):e28175.
Yan S, Wu G. Spatial and temporal roles of SARS-CoV PL(pro) -A snapshot. Faseb j. 2021;35(1):e21197.
Su J, Rui Y, Lou M, Yin L, Xiong H, Zhou Z, et al. HIV-2/SIV vpx targets a novel functional domain of STING to selectively inhibit cGAS-STING-mediated NF-κB signalling. Nat Microbiol. 2019;4(12):2552–64.
Bhattacharya M, Bhowmik D, Tian Y, He H, Zhu F, Yin Q. The Dengue virus protease NS2B3 cleaves cyclic GMP-AMP synthase to suppress cGAS activation. J Biol Chem. 2023;299(3):102986.
Acknowledgements
The authors thank all researchers who contributed to the advancement of science.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
FX conceived the topic, drafted the manuscript, and revised the figure. QZ revised the manuscript and prepared the figure. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xie, F., Zhu, Q. The regulation of cGAS-STING signaling by RNA virus-derived components. Virol J 21, 101 (2024). https://doi.org/10.1186/s12985-024-02359-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-024-02359-1