
Abstract
Background
Porcine cytomegalovirus (PCMV) is a porcine roseolovirus (PCMV/PRV) which is widely distributed in pigs. Transmission of PCMV/PRV in preclinical xenotransplantations was shown to significantly reduce the survival time of the pig transplants in non-human primates. PCMV/PRV was also transmitted in the first transplantation of a pig heart into a human patient. To analyze how PCMV/PRV could be introduced into pig breeds, especially considering cloned transgenic pigs, and subsequently spread in breeding facilities, we screened ovaries and derived materials which are used to perform somatic cell nuclear transfer (SCNT).
Methods
DNA was isolated from ovarian tissues, follicular fluids, oocytes with cumulus cells, denuded oocytes and parthenotes. A real-time PCR with PCMV/PRV-specific primers and a probe was performed to detect PCMV/PRV. Furthermore, a Western blot assay using a recombinant fragment of the gB protein of PCMV/PRV was performed to screen for virus-specific antibodies in the follicular fluids.
Results
PCMV/PRV was found by real-time PCR in ovarian tissues, in the follicular fluid and in oocytes. In parthenotes the virus could not be detected, most-likely due to the low amount of DNA used. By Western blot assay specific antibodies against PCMV/PRV were found in 19 of 20 analyzed follicular fluids.
Conclusion
PCMV/PRV was found in ovarian tissues, in the follicular fluids and also in denuded oocytes, indicating that the virus is present in the animals of which the oocytes were taken from. Despite several washing steps of the denuded oocytes, which are subsequently used for microinjection or SCNT, the virus could still be detected. Therefore, the virus could infect oocytes during genetic modifications or stay attached to the surface of the oocytes, potentially infecting SCNT recipient animals.
Similar content being viewed by others
Background
Porcine cytomegalovirus (PCMV) is a porcine roseolovirus (PCMV/PRV) [1], which poses a considerable risk for xenotransplantation using pig cells, tissues and organs. Xenotransplantation is one solution to alleviate the shortage of human transplants and it has achieved great success in the last years with long survival times of pig hearts, kidneys and other organs in non-human primates [2, 3]. However, PCMV/PRV transmission to the recipients drastically reduced the survival time of the xenotransplant, in the case of orthotopic heart transplantation into baboons from 195 days to less than 30 days [4], in the case of kidney transplantation into baboons from to 53 to 14 days [5] and in the case of kidney transplantation into cynomolgus monkeys from to 28 to 9 days [6]. For review and schematic presentation of the results see [7, 8]. In the baboons which received a PCMV/PRV-positive heart, an increase of certain cytokines such as interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-alpha) was observed [4]. Furthermore, high levels of tissue plasminogen activator–plasminogen activator inhibitor type 1 (tPA–PAI‑1) complexes were found in the transplanted baboons, suggesting a complete loss of the pro‑fibrinolytic properties of the endothelium [4]. The presence of PCMV/PRV was usually associated with consumptive coagulopathy (CC) [9], but when PCMV/PRV-free transplants were used, CC was significantly reduced [10]. PCMV was also transmitted by the first transplantation of a pig heart into a human patient, who survived for remarkable 2 months [11]. The clinical features observed in the patient were similar to the features seen in the baboons which received a PCMV/PRV-positive pig heart [4], suggesting that the virus may have contributed among other factors to the death of the patient.
The donor animal of the heart for the patient had 10 genetic modifications (10-GE pigs), including targeted insertion of two human complement inhibitor genes (human decay-accelerating factor, hDAF or hCD55; and human membrane cofactor protein, hCD46), two human anticoagulant genes (human thrombomodulin, hTM, and human endothelial protein C receptor, hEPCR), and two immunomodulatory genes (human signal regulatory protein alpha, hCD47, and human heme oxygenase 1, hHO1), as well as knockouts (KO) of 3 enzymes creating the porcine carbohydrate antigens: α1,3-galactosyltransferase (GGTA1), cytidine monophosphate-N-acetylneuraminic acid hydroxylase (CMAH), and β-1,4-N-acetyl-galactosaminyl transferase 2 (B4GALNT2) as well as the porcine growth hormone receptor (GHR). These 10-GE pigs do not express red blood cell antigens and are therefore universal donors with respect to blood type [11]. For the genetic modification and the cloning of the pigs, oocytes had to be used for SCNT. As each SCNT requires hundreds of oocytes, almost all laboratories use oocytes from slaughterhouse pigs. For maturation and modifications, these oocytes are cultivated in vitro, based on follicular fluid culture media in the past, which meanwhile has been replaced by fully synthetic maturation media. After having performed the desired modifications, e.g., by microinjection or SCNT, these oocytes are transferred to a surrogate mother to give birth to gene-modified piglets. We recently summarized the literature clearly showing the risk of viral transmission, including herpesviruses, by oocytes or follicular fluid in numerous cases [12]. In order to analyze, whether this may happen in the case of PCMV/PRV, ovarian tissues, oocytes with and without cumulus cells, follicular fluid and other tissues were screened for the presence of PCMV/PRV.
Methods
Pigs and porcine tissues
Porcine oocytes and tissues were collected at a local slaughterhouse. As this slaughterhouse receives pigs from Germany, Austria and Czech Republic, animals had several genetic backgrounds including among others Deutsche Landrasse, two-breed crossing out of Edelschwein and Pietrain, and three-breed crossing of Edelschwein, Deutscher Landrasse and Pietrain. Ovaries from this slaughterhouse have been regularly used by several groups to isolate oocytes and to perform genetic modifications. The age of the pigs was about 6 months with a weight between 100 and 120 kg.
Isolation of oocytes
Ovaries from prepubertal gilts were isolated and transported to the laboratory at 38 °C in phosphate buffered saline (PBS) supplemented with antibiotics and antimycotics. Ovaries were rinsed several times with warm PBS, supplemented with 1% cetyl trimethyl ammonium bromide (CTAB), an antiseptic solution with various antibacterial, antifungal and antiviral properties. Subsequent washing steps were performed with warm PBS solution. Ovaries were placed in warm PBS and kept at 38 °C. Follicles with a diameter of 3–6 mm were punctured using a 10 ml syringe and a 18G needle. Porcine follicular fluid was extracted and stored at 38 °C. Oocytes with cumulus cells complexes settled at the bottom of the tube were subsequently isolated. 6–8 ml of working medium (WM), which consists of medium 199 and 10% FCS supplemented with 1% amphotericin B and 1% penicillin–streptomycin, were mixed with the cells and transferred to a petri dish for collection. High quality oocytes with dark, evenly granulated cytoplasm and several compact layers of cumulus cells were identified under a stereomicroscope. Oocytes were rinsed twice in WM to remove cell debris. Oocytes were transferred with a mouth pipette and self-made glass capillaries of about 300 µm diameter, also to make washing steps as efficient as possible.
In vitro maturation
For in vitro maturation, oocytes were transferred to a triple gas incubator (5% O2, 5% CO2, 90% N2, set to 38.5 °C humidified atmosphere). Oocytes were rinsed with maturation medium and transferred to a separate maturation well. After 45 h, successful maturation was confirmed by visual assessment of polar body extrusion from a sample group of ovaries. Maturation was carried out in a chemically defined maturation medium which consists of 500 ml medium 199, 27.5 mg glucose, 5 mg sodium pyruvate, 0.5 mL penicillin–streptomycin and 1 mL 3% polyvinyl alcohol.
Denuding and parthenogenesis
Mature oocytes were denuded in WM supplemented with 1 mg/ml hyaluronidase and rinsed twice in working medium. They were controlled for granulated cytoplasm and extrusion of the first polar body. Chemical activation was conducted in WM supplemented with 25 µm ionomycin (calcium ionophore) for 10 min. Oocytes were washed twice in WM and once in PZM5, a defined medium for embryos, and subsequently placed in the incubator in 500 µl of PZM5 supplemented with 5 µg/ml of cytochalasin for 3 h. Afterwards they were rinsed twice in working medium and once in PZM5. Parthenotes were used for our assays to increase the extractable DNA amount from single oocytes and to avoid possible PCMV/PRV viral transfer by sperm.
Sample storage
Samples of porcine ovaries and follicular fluids were frozen and stored at − 20 °C. Oocytes with cumulus cells, denuded oocytes and parthenotes were collected in PBS and stored at − 20 °C.
DNA extraction
DNA was isolated from ovarian tissues (20 mg), follicular fluid (200–400 µl), maturation medium (200 µl), oocytes with or without cumulus cells (5–250 oocytes per sample) and parthenotes (5–15 cells per sample) using the innuPREP Virus DNA/RNA Kit (Analytik Jena, Jena, Germany). The DNA/RNA was eluated in 30 or 60 µl nuclease-free water. Samples were stored at − 20 °C until further processing.
Real-time polymerase chain reaction (PCR)
The detection of PCMV/PRV was performed using a real-time PCR assay with specific primers and a probe developed by Mueller et al. [13] (Table 1). All assays were performed as duplex real-time PCR using as reference gene porcine glyceraldehyde-3-phosphate-dehydrogenase (pGAPDH) with a specific primer–probe mixture (Table 1) [14] running 45 cycles as described previously [15,16,17]. All experiments were performed with the SensiFAST Probe No-ROX kit (Meridian Bioscience, Cincinnati, OH, USA) and the qTOWER3 G qPCR cycler (Analytik Jena, Jena, Germany). A reaction volume of 20 µl was prepared containing 1.8 µl of PCMV/PRV-FAM mix with 1.8 µl of pGAPDH-HEX mix as internal control and 4.0 µl of extracted DNA. The reaction for the PCMV/PRV real-time PCR was carried out for 2 min at 50 °C for activation, 10 min at 95 °C followed by 45 cycles comprising 15 s at 95 °C for denaturation and 60 s at 60 °C for annealing and elongation. As positive control a PCMV/PRV-specific gene block, as negative control water or medium were used as described [15].
Western blot analysis
Western blot analysis was performed as previously described [16, 18]. Briefly, for the detection of antibodies against PCMV/PRV, the Western Blot assay designed by Plotzki et al. [18] was re-established, but only the C-terminal fragment R2 of the gB protein of PCRV/PRV was used as antigen because the R2 protein was shown to be immunodominant [18]. The R2 fragment of the gB of PCMV/PRV was produced in E. coli BL21 cells using the pET16b expression vector encoding PCMV-R2 as described in detail [16, 18]. Cell were induced with 1 mM isopropyl-β-D-thiogalactopyranosid (Roth, Karlsruhe, Germany), harvested, and dissolved in 10 mL 8 M urea, 0.5 M NaCl, 15 mM imidazole, 20 mM Tris pH 7.5. The supernatant after centrifugation was applied to a HisTrap HP column installed on a Äkta Prime Plus system (both GE Healthcare, Chicago, Illinois, USA), and eluted after washing using 6 M urea, 0.5 M NaCl, 500 mM imidazole, 20 mM Tris pH 7.5. The purified R2 protein was characterized by a sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) using 12% gels and to obtain sharper bands 17% gels. The protein was dissolved in sample buffer (375 mM Tris–HCl, 60% glycerol, 12% SDS, 0,6 M DTT, 0.06% bromophenol blue) and denatured for 5 min at 95 °C prior to electrophoresis. The SDS PAGE was run in a Mini-Protean Tetra Vertical Electrophoresis Cell (Bio-Rad Laboratories, Incs., Hercules, CA, USA) using a 17% polyacrylamide gel and the PageRuler prestained protein ladder (Thermo Fisher Scientific, Waltham, USA). The protein was transferred for 100 min to a polyvinylidene fluoride membrane (ROTI PVDF, 8989.1, Roth, Karlsruhe, Germany) by electroblotting (100 mA) using the electroblotting device of peqlab Biotechnologie GmbH. After electroblotting the membrane was blocked for 1 h at 4 °C in 5% non-fat dry milk (Roth, Karlsruhe, Germany) in PBS with 0.05% Tween 20 (Roth, Karlsruhe, Germany) (PBS-T) (blocking buffer). The membrane was cut into strips and incubated over night at 4 °C with sera diluted 1:150 in blocking buffer. Afterwards, washing was performed with 0.05% PBS-T three times for 10 min each. Polyclonal goat anti-pig immunoglobulin G (IgG) Fc Secondary Antibody HRP (Invitrogen by Thermo Fisher Scientific, Waltham, USA) was diluted 1:15.000 in blocking buffer and strips were incubated for 1 h at room temperature, followed by three washing steps for 10 min each. Detection of the signal following incubation with the ECL Western Blotting Substrate (Cytiva, Amersham) was done with the FUSION-SL 3500 WL imaging device (peqlab Biotechnologie GmbH).
Results
Detection of PCMV/PRV in different tissues by RT-PCR
Ovarian tissues, follicular fluid, and ocytes with or without cumulus cells were collected from up to 20 slaughterhouse animals. In addition, parthenotes were obtained as described in Methods. As negative control, a commercial maturation medium was analyzed, which is used for the cultivation of oocytes instead of a medium containing follicular fluid. DNA was isolated and a duplex real-time PCR was performed using PCMV/PRV-specific primers and a probe. It is important to note that the target sequence of the real-time PCR is a sequence in the polymerase gene which is highly conserved among all PCMV/PRV. Primers and a probe specific for porcine GAPDH were used to control the presence of porcine DNA. Three samples out of 20 samples from 20 animals were PCMV/PRV-positive when ovarian tissues were analyzed (Table 2). The results of the GAPDH testing showed that sufficient DNA was present in all samples and that the DNA input was identical in all samples (Table 2, Additional file 1: Figure S1). Two out of 20 samples of follicular fluid were positive. 40 oocytes with cumulus cells from eight samples, 40 denuded oocytes from 8 samples and 30 parthenotes from 6 samples were negative in this assay. Parthenotes are oocytes that can be activated, among others, by high calcium concentrations or electric stimuli. As no sperm is required for oocyte activation, this excludes the risk of sperm-mediated PCMV/PRV transmission. Due to the higher cell number of parthenotes compared to oocytes, the DNA amount that can be obtained is much higher for subsequent PCR analysis.
Most importantly, when higher amounts of oocyte DNA (50 ng DNA per PCMV/PRV real-time PCR in triplicates) from 100 pooled oocytes was used, a positive reaction was found with the oocytes, as well as in 50 ng DNA per PCMV/PRV real-time PCR from 150 pooled oocytes, indicating that PCMV/PRV is present in or on oocytes despite the several washing steps (Table 3) (Additional file 1: Figure S1). As expected, the control maturation medium was negative (Table 2).
Detection of antibodies against PCMV/PRV by Western blot
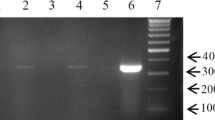
In order to analyze whether antibodies against the PCMV/PRV can be found in the follicular fluid, a Western blot assay was performed. As antigen the C-terminal R2 fragment of the gB protein of PCMV/PRV was used [18]. The R2 fragment was found to be immunodominant: In comparison with the N-terminal R1 fragment more sera were detected positive [18]. 19 of 20 samples of follicular fluid were positive in this assay (Fig. 1), indicating that these animals were infected by PCMV/PRV as shown by the positive antibody reaction. The antibodies found in the follicular fluid were either present in the follicular fluid or coming from contaminating blood when collecting the follicular fluid. The latter is unlikely because the follicular fluid was carefully collected avoiding blood contaminations.

Western blot analysis of 18 follicular fluids (1–18). A a 12% gel was used for SDS-PAGE, B, C a 17% gel was used. The molecular weight marker (M), a serum from a positive control animal (P) and the location of the recombinant R2 protein of the gB protein of PCMV/PRV are indicated. The serum dilution was 1:150
Discussion
For the generation of genetically modified pigs, usually SCNT is the method of choice, as prescreening of modified cells is possible, thereby strongly reducing the number of incorrectly modified pigs. We showed here that in PCMV/PRV positive pigs from slaughterhouses, the ovarian tissue and oocytes are infected with the virus and that PCMV/PRV is also present in the follicular fluid. Although RT-PCR analysis only could detect PCMV/PRV in a limited number of samples, almost all samples turned out to be positive for antibodies against PCMV/PRV detected by Western blot analysis. As oocytes have initially to be isolated from the follicles together with follicular fluid and all isolations are regularly pooled, even only one PCMV/PRV positive pig could potentially infect all other oocytes during or after isolation.
This data indicates that PCMV/PRV is able to infect piglets created by SCNT and this may explain how animals got infected even in previously virus-negative facilities. Usually infection with PCMV happens early in live of the piglets by contact with the infected mother [19, 20]. PCMV is found in most tissues but mainly in the nose of newly infected newborn piglets where it causes rhinitis, in addition it also causes conjunctivitis in the eyes. Most infections are sub-clinical, clinical disease is rare. Clinical signs are only seen if PCMV infects a sow for the first time when she is late in pregnancy. In this case, fetal deaths, mummified fetuses, stillbirths, and weak piglets are observed. The virus is shed from the nose and eyes, by urine and farrowing fluids, and semen of the young pig. In experimental settings, but not under natural conditions, the virus was shown to cross the placenta [21]. Therefore, the presence of PCMV/PRV in ovarian tissue and in the follicular fluid is not surprising. The fact that oocytes are virus-positive is of importance. The oocytes are usually surrounded by the zona pellucida (ZP), an extracellular matrix of glycoproteins building a three-dimensional structure which should prevent virus infection [22]. It is widely recognized that a virus infection is only possible after damaging the ZP. After infection of ZP-damaged pig oocyte with porcine circovirus 2 (PCV2) infected embryos were detected [23]. However, transmission of viruses was also observed in the presence of an intact ZP (for review see [12]). The fact that PCMV/PRV was still found in oocytes despite several washing steps indicates that these steps are not sufficient to remove the virus, either because it is already inside the oocyte or firmly attached to the glycoproteins of the ZP. Additional decontamination techniques, for example treatment with trypsin, were also not successful in the case of some viruses such as the porcine reproductive and respiratory syndrome virus (PRRSV), the porcine parvovirus (PPV), the porcine circovirus 2 (PCV2) and the encephalomyocarditis (EMCV) [24]. Treatment with hyaluronidase was more successful [24]. Since antiviral drugs effective against human cytomegalovirus such as ganciclovir are not so effective against PCMV/PRV [25], because it is a roseolovirus [1], treatment with these toxic substances is not recommended.
Although meanwhile synthetic maturation mediums are used, previously, when follicular fluid was added to the medium, the presence of PCMV/PRV in the follicular fluid as shown here could lead to an infection of the oocytes, especially after SCNT when the ZP is disrupted.
To use pigs from slaughterhouses as source of oocytes has many disadvantages. In addition to the high prevalence of PCMV/PRV [18], numerous other viruses were found in slaughterhouse pigs, among them hepatitis E virus (HEV) [26,27,28], parvoviruses [29], Torque teno sus virus [30], porcine lymphotropic herpes viruses [31] and circoviruses [32]. Whether these viruses (with exeption of HEV, which is a well-known zoonotic virus (for review see [33])) pose a risk for xenotransplantation and whether they can be transmitted via SCNT is unclear. However, currently the use of slaughterhouse pigs is the only feasible solution to obtain the high number of oocytes which are regularly required to perform SCNT.
PCMV/PRV was transmitted during the first pig heart transplantation to a patient in Baltimore [11]. Since the supplier of the 10-GE pig used for this transplantation, Revivicor, supplies cloned animals, it cannot be excluded that the origin of the virus was in oocytes used for cloning. PCMV/PRV is a latent herpesvirus [34] and for the detection of latent viruses special detection strategies are needed [16]. When these detection strategies will be applied and techniques of elimination of the virus from infected herds such as early weaning will be used [35], xenotransplantation could even be safer [36, 37].
Limitations of the study
To detect PCMV in our study, we used a modified real-time PCR originally developed by Mueller et al. [13], which was modified by us into a duplex real-time PCR. This PCR detects a conserved region in the polymerase gene of PCMV and was used by many laboratories, including ours [4, 13, 14, 16, 38, 39]. To improve the diagnostic, an additional PCR would be useful, either detecting another sequence in the polymerase genes as described by us [17, 40], or detecting a sequence in another gene, e.g., in the gene encoding the gB protein. This gene also contains regions which are conserved among all PCMV [41] and therefore can be used for the detection of the virus. The gB protein of PCMV/PRV contains even sequences which are related to HHV-6 to an extend that a cross-reactivity of antibodies was observed [42].
Another limitation of the study is that we did not investigate the presence of viral mRNA. In a productive infection there should be more mRNA than viral DNA and therefore mRNA could have been detected if the virus is expressed.
For the antibody testing we used a linear recombinant protein. Although recombinant proteins and even peptides are generally used for screening for virus infections, for example commercial tests for the detection of a HIV-1 infection [43, 44], the use of a properly folded protein may be more meaningful. When we used two recombinant fragments of the gB protein of PCMV/PRV, the N-terminal R1 and the C-terminal R2, we found that sera from most infected pigs recognized R2 [18]. Therefore, in later investigations, including this study, only R2 was used for testing [10, 16, 38].
Furthermore, it is unclear whether PCMV/PRV is replicating in the oocytes or whether it established latency. At present it remains unclear in which cells, tissues and organs PCMV/PRV establishes latency in pigs. When studying infected animals, the virus was found in different organs and the organ with the highest virus load was different in different animals [45]. The presence of PCMV/PRV sequences in different organs may be due to the presence of infected lymphocytes where the virus is replicating and also may establish latency.
Conclusion
The presence of PCMV/PRV in ovarian tissue, oocytes and follicular fluid of PCMV/PRV-positive pigs indicates that using oocytes and follicular fluid from these animals for SCNT may lead to PCMV/PRV-positive piglets. Therefore, it is advised to use PCMCV/PRV-negative animals as oocyte donors or to use effective decontamination techniques.
Availability of data and materials
All data are part of this manuscript.
Abbreviations
- B4GALNT2:
-
β-1,4-N-acetyl-galactosaminyl transferase 2
- CC:
-
Consumptive coagulopathy
- CMAH:
-
Cytidine monophosphate-N-acetylneuraminic acid hydroxylase
- CTAB:
-
Cetyl trimethyl ammonium bromide
- GHR:
-
Growth hormone receptor
- GGTA1:
-
α1,3-Galactosyltransferase
- hDAF:
-
Human decay-accelerating factor
- hEPCR:
-
Human endothelial protein C receptor
- hCD46:
-
Human membrane cofactor protein
- hTM:
-
Human thrombomodulin
- hEPCR:
-
Human endothelial protein C receptor
- hCD47:
-
Human signal regulatory protein alpha
- hHO1:
-
Human heme oxygenase 1
- IL-6:
-
Interleukin 6
- PCMV/PRV:
-
Porcine cytomegalovirus/porcine roseolovirus
- pGAPDH:
-
Porcine glyceraldehyde-3-phosphat3-dehydrogenase
- SCNT:
-
Somatic cell nuclear transfer
- TNF-alpha:
-
Tumor necrosis factor alpha
- tPA‑PAI‑1:
-
Tissue plasminogen activator-plasminogen activator inhibitor type 1 WM working medium
References
Denner J, Bigley TM, Phan TL, Zimmermann C, Zhou X, Kaufer BB. Comparative analysis of roseoloviruses in humans, pigs, mice, and other species. Viruses. 2019;11(12):1108.
Cooper DKC, Hara H, Iwase H, Yamamoto T, Wang ZY, Jagdale A, Bikhet MH, Nguyen HQ, Foote JB, Paris WD, Ayares D, Kumar V, Anderson DJ, Locke JE, Eckhoff DE. Pig kidney xenotransplantation: progress toward clinical trials. Clin Transplant. 2021;35(1):e14139.
Reichart B, Längin M, Denner J, Schwinzer R, Cowan PJ, Wolf E. Pathways to clinical cardiac xenotransplantation. Transplantation. 2021;105(9):1930–43.
Denner J, Längin M, Reichart B, Krüger L, Fiebig U, Mokelke M, Radan J, Mayr T, Milusev A, Luther F, Sorvillo N, Rieben R, Brenner P, Walz C, Wolf E, Roshani B, Stahl-Hennig C, Abicht JM. Impact of porcine cytomegalovirus on long-term orthotopic cardiac xenotransplant survival. Sci Rep. 2020;10(1):17531.
Yamada K, Tasaki M, Sekijima M, Wilkinson RA, Villani V, Moran SG, Cormack TA, Hanekamp IM, Hawley RJ, Arn JS, Fishman JA, Shimizu A, Sachs DH. Porcine cytomegalovirus infection is associated with early rejection of kidney grafts in a pig to baboon xenotransplantation model. Transplantation. 2014;98(4):411–8.
Sekijima M, Waki S, Sahara H, Tasaki M, Wilkinson RA, Villani V, Shimatsu Y, Nakano K, Matsunari H, Nagashima H, Fishman JA, Shimizu A, Yamada K. Results of life-supporting galactosyltransferase knockout kidneys in cynomolgus monkeys using two different sources of galactosyltransferase knockout Swine. Transplantation. 2014;98(4):419–26.
Denner J. Xenotransplantation and porcine cytomegalovirus. Xenotransplantation. 2015;22(5):329–35.
Denner J. Reduction of the survival time of pig xenotransplants by porcine cytomegalovirus. Virol J. 2018;15(1):171.
Gollackner B, Mueller NJ, Houser S, Qawi I, Soizic D, Knosalla C, Buhler L, Dor FJ, Awwad M, Sachs DH, Cooper DK, Robson SC, Fishman JA. Porcine cytomegalovirus and coagulopathy in pig-to-primate xenotransplantation. Transplantation. 2003;75(11):1841–7.
Mueller NJ, Kuwaki K, Dor FJ, Knosalla C, Gollackner B, Wilkinson RA, Sachs DH, Cooper DK, Fishman JA. Reduction of consumptive coagulopathy using porcine cytomegalovirus-free cardiac porcine grafts in pig-to-primate xenotransplantation. Transplantation. 2004;78(10):1449–53.
Griffith BP, Goerlich CE, Singh AK, Rothblatt M, Lau CL, Shah A, Lorber M, Grazioli A, Saharia KK, Hong SN, Joseph SM, Ayares D, Mohiuddin MM. Genetically modified porcine-to-human cardiac xenotransplantation. N Engl J Med. 2022. https://doi.org/10.1056/NEJMoa2201422.
Denner J. Risk of pathogenic virus transmission by somatic cell nuclear transfer (SCNT): implications for xenotransplantation. Biol Reprod. 2022;107(3):717–22. https://doi.org/10.1093/biolre/ioac120.
Mueller NJ, Barth RN, Yamamoto S, Kitamura H, Patience C, Yamada K, Cooper DK, Sachs DH, Kaur A, Fishman JA. Activation of cytomegalovirus in pig-to-primate organ xenotransplantation. J Virol. 2002;76(10):4734–40.
Halecker S, Metzger J, Strube C, Krabben L, Kaufer B, Denner J. Virological and parasitological characterization of Mini-LEWE minipigs using improved screening methods and an overview of data on various minipig breeds. Microorganisms. 2021;9(12):2617.
Hansen S, Franzo G, Menandro ML, Krabben L, Marino SF, Kaufer B, Denner J. Prevalence of the porcine cytomegalovirus virus (PCMV), a porcine roseolovirus, in wild boars in Italy and Germany. Research Square, https://doi.org/10.21203/rs.3.rs-1898102/v1
Halecker S, Hansen S, Krabben L, Ebner F, Kaufer B, Denner J. How, where and when to screen for porcine cytomegalovirus (PCMV) in donor pigs for xenotransplantation. Sci Rep. 2022;12(1):21545.
Morozov VA, Morozov AV, Denner J. New PCR diagnostic systems for the detection and quantification of porcine cytomegalovirus (PCMV). Arch Virol. 2016;161(5):1159–68.
Plotzki E, Keller M, Ivanusic D, Denner J. A new Western blot assay for the detection of porcine cytomegalovirus (PCMV). J Immunol Methods. 2016;437:37–42.
Edington N. Porcine cytomegalovirus. Dis Swine. 1986;138:330–6.
Edington N, Broad S, Wrathall AE, Done JT. Superinfection with porcine cytomegalovirus initiate infection. Vet Microbiol. 1988;16:189–93.
Edington N, Watt RG, Plowright W. Experimental transplacental transmission of porcine cytomegalovirus. J Hyg (Lond). 1977;78:243–51.
Karimian K, Seydewitz R, Töpfer D, Böl M. Poro-viscoelastic behaviour of the zona pellucida: impact of three-dimensional modelling on material characterization. J Mech Behav Biomed Mater. 2022;131:105211.
Zhao H, Ji Q, Zhao G, Song Z, Du B, Nie Y, Chen Y, Cong P. Damage of zona pellucida reduces the developmental potential and quality of porcine circovirus type 2-infected oocytes after parthenogenetic activation. Theriogenology. 2014;82(6):790–9.
Bureau M, Dea S, Sirard MA. Evaluation of virus decontamination techniques for porcine embryos produced in vitro. Theriogenology. 2005;63(9):2343–55.
Mueller NJ, Sulling K, Gollackner B, Yamamoto S, Knosalla C, Wilkinson RA, Kaur A, Sachs DH, Yamada K, Cooper DK, Patience C, Fishman JA. Reduced efficacy of ganciclovir against porcine and baboon cytomegalovirus in pig-to-baboon xenotransplantation. Am J Transplant. 2003;3(9):1057–64.
Chelli E, Suffredini E, De Santis P, De Medici D, Di Bella S, D’Amato S, Gucciardi F, Guercio A, Ostanello F, Perrone V, Purpari G, Scavia GS, Schembri P, Varcasia BM, Di Bartolo I. Hepatitis E virus occurrence in pigs slaughtered in Italy. Animals (Basel). 2021;11(2):277.
Sooryanarain H, Heffron CL, Hill DE, Fredericks J, Rosenthal BM, Werre SR, Opriessnig T, Meng XJ. Hepatitis E virus in pigs from slaughterhouses, United States, 2017–2019. Emerg Infect Dis. 2020;26:354–7.
Boxman ILA, Verhoef L, Dop PY, Vennema H, Dirks RAM, Opsteegh M. High prevalence of acute hepatitis E virus infection in pigs in Dutch slaughterhouses. Int J Food Microbiol. 2022;379:109830.
Thuy NTD, Trung NT, Dung TQ, Khoa DVA, Thuy DTN, Opriessnig T. First investigation of the prevalence of parvoviruses in slaughterhouse pigs and genomic characterization of ungulate copiparvovirus 2 in Vietnam. Arch Virol. 2021;166(3):779–88.
Brassard J, Gagné MJ, Leblanc D. Real-time PCR study of the infection dynamics of Torque teno sus viruses in naturally infected pigs from nursery to slaughterhouse. Vet J. 2013;197(2):506–8.
Plotzki E, Keller M, Ehlers B, Denner J. Immunological methods for the detection of porcine lymphotropic herpesviruses (PLHV). J Virol Methods. 2016;233:72–7.
Yue W, Li Y, Zhang X, He J, Ma H. Prevalence of Porcine circoviruses in slaughterhouses in central Shanxi Province. China Front Vet Sci. 2022;9:820914.
Denner J. Xenotransplantation and hepatitis E virus. Xenotransplantation. 2015;22(3):167–73.
Cohen JI. Herpesvirus latency. J Clin Invest. 2020;130(7):3361–9.
Egerer S, Fiebig U, Kessler B, Zakhartchenko V, Kurome M, Reichart B, Kupatt C, Klymiuk N, Wolf E, Denner J, Bähr A. Early weaning completely eliminates porcine cytomegalovirus from a newly established pig donor facility for xenotransplantation. Xenotransplantation. 2018;25(4):e12449.
Denner J. The porcine cytomegalovirus (PCMV) will not stop xenotransplantation. Xenotransplantation. 2022;29(3):e12763.
Mueller NJ, Denner J. Porcine cytomegalovirus/porcine roseolovirus (PCMV/PRV): a threat for xenotransplantation? Xenotransplantation. 2022;8:e12775.
Halecker S, Papatsiros V, Psalla D, Krabben L, Kaufer B, Denner J. Virological characterization of pigs with erythema multiforme. Microorganisms. 2022;10(3):652.
Krüger L, Kristiansen Y, Reuber E, Möller L, Laue M, Reimer C, Denner J. A comprehensive strategy for screening for xenotransplantation-relevant viruses in a second isolated population of Göttingen minipigs. Viruses. 2019;12(1):38.
Morozov VA, Plotzki E, Rotem A, Barkai U, Denner J. Extended microbiological characterization of Göttingen minipigs: porcine cytomegalovirus and other viruses. Xenotransplantation. 2016;23:490–6.
Widen F, Goltz M, Wittenbrink N, Ehlers B, Banks M, Belak S. Identification and sequence analysis of the glycoprotein B gene of porcine cytomegalovirus. Virus Genes. 2001;23(3):339–46.
Fiebig U, Holzer A, Ivanusic D, Plotzki E, Hengel H, Neipel F, Denner J. Antibody cross-reactivity between porcine cytomegalovirus (PCMV) and human herpesvirus-6 (HHV-6). Viruses. 2017;9(11):317.
Khurana S, Norris PJ, Busch MP, Haynes BF, Park S, Sasono P, Mlisana K, Salim AK, Hecht FM, Mulenga J, Chomba E, Hunter E, Allen S, Nemo G, Rodriguez-Chavez IR, Women's Interagency HIV Study Collaborative Study Group, Margolick JB, Multicenter AIDS Cohort Study (MACS), Golding H. HIV-Selectest enzyme immunoassay and rapid test: ability to detect seroconversion following HIV-1 infection. J Clin Microbiol. 2010;48(1):281–5.
Thorn RM, Beltz GA, Hung CH, Fallis BF, Winkle S, Cheng KL, Marciani DJ. Enzyme immunoassay using a novel recombinant polypeptide to detect human immunodeficiency virus env antibody. J Clin Microbiol. 1987;25(7):1207–12.
Fiebig U, Abicht JM, Mayr T, Längin M, Bähr A, Guethoff S, Falkenau A, Wolf E, Reichart B, Shibahara T, Denner J. Distribution of porcine cytomegalovirus in infected donor pigs and in baboon recipients of pig heart transplantation. Viruses. 2018;10(2):66.
Duvigneau JC, Hartl RT, Groiss S, Gemeiner M. Quantitative simultaneous multiplex real-time PCR for the detection of porcine cytokines. J Immunol Methods. 2005;306:16–27.
Acknowledgements
We thank the embryology team of the School of Life Sciences Weihenstephan of the Technical University Munich for their technical support.
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was supported by the Deutsche Forschungsgemeinschaft (DFG), TRR127.
Author information
Authors and Affiliations
Contributions
Conceptualization, JD; methodology, SH and KF; writing—original draft preparation, SH, KF and JD; writing—review and editing, SH, KF, ARK, BeKl, LK, BK, AS and JD; funding acquisition, JD, BK, KF, AS. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Demonstration of real-time PCR testing of pig oocytes for PCMV/PRV. As positive control a gene block was used.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hansen, S., Fischer, K., Krabben, L. et al. Detection of porcine cytomegalovirus, a roseolovirus, in pig ovaries and follicular fluid: implications for somatic cells nuclear transfer, cloning and xenotransplantation. Virol J 20, 15 (2023). https://doi.org/10.1186/s12985-023-01975-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-023-01975-7