
Abstract
Discovered as a contaminant of adenovirus stocks in the 1960s, adeno-associated virus (AAV) is a mono-stranded DNA virus that depends on helper factors to replicate. Even though AAV is endemic in the human population (35–80%), it is remarkable that many issues concerning the natural infection by this virus remain unanswered. In this study, we reflect on the main basic aspects of AAV biology and provide an overview of the studies exploring the impact of AAV infection on human health, focusing on three major research areas including, (i) cervical and (ii) liver cancer, and (iii) reproductive system disorders. Conflicting results have been obtained into the association of AAV infection with the occurrence of adverse reproductive outcomes, such as placental complications, spontaneous abortion, and fertility disorders, or with a protective role in HPV-related cervical carcinogenesis. Noteworthy, recent reports have identified AAV insertional mutagenesis as a novel risk factor for the development of hepatocellular carcinoma. This latest finding raises concern regarding the widespread usage of AAV vectors in liver-targeted gene therapy.
Similar content being viewed by others

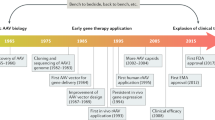

Introduction
Adeno-associated virus
Adeno-associated virus (AAV) was discovered as a contaminant in a simian adenovirus type 15 preparation in 1965 [1, 2]. Since then, AAV has been successfully developed into therapeutic vector, with Glybera (alipogene tiparvovec) becoming the first gene therapy medication to be authorized [3]. AAV infection is asymptomatic and can last a lifetime. The lack of identifiable associated disease and the unique capacity of recombinant AAV (rAAV) vectors to transduce dividing and non-dividing cells with high efficiency, long-term transgene expression, low immunogenicity, and selective tissue tropism make AAV appealing for gene therapy [4].
Within the Parvoviridae family, AAV belongs to the Dependoparvovirus genus. There are at least 13 naturally occurring serotypes, each with a different tissue tropism [5]. AAV infects various animal species, including humans, and is found worldwide, with seroprevalence varying from roughly 35–80% in the human population, depending on the AAV serotype and cohort analyzed [6, 7].
AAV can only replicate in the presence of helper factors, which are provided by helper virus coinfections. Adenovirus type 5 (AdV5) and herpes simplex virus type 1 (HSV-1) are well-studied AAV helper viruses. Although less well studied, many other members of the herpesvirus family have been shown to support productive AAV replication, such as human cytomegalovirus (HCMV), herpes simplex virus type 2 (HSV-2), varicella zoster virus (VZV), and human herpesvirus 6 (HHV-6) (reviewed in [8]). Recently, human bocavirus 1 was demonstrated as a helper virus during AAV replication [9]. Interestingly, AAV replication can be triggered by treating AAV-infected cells with physical or chemical carcinogens, indicating that it is not intrinsically dependent on viral coinfections but rather on a significant alteration in the cellular environment [10, 11]. Without helper factors, AAV transfers its genome into the host cell, where most copies are eliminated within a short time, but other AAV genomes stay indefinitely. Long-term persistence is thought to occur mainly in an episomal, circular form. Latent AAV reactivates after coinfection with a helper virus, resulting in the emergence of progeny virus [8].
AAV biology
The AAV virion is a non-enveloped icosahedral particle with a single-stranded DNA genome. The most thoroughly researched serotype of AAV is type 2 (AAV2), which serves as the AAV family’s prototype. Because most vector expertise has been gained with AAV2, we will utilize information about this serotype to explain generic AAV properties. The AAV2 virion has a diameter of around 20 nm and is made up of 60 copies of the three capsid proteins VP1, VP2, and VP3 in a 1:1:10 ratio. The VP1 and VP2 proteins share the VP3 sequence and contain extra residues at their N-termini. A conserved phospholipase A2 region found at the N-terminus of VP1 has been linked to viral escape from endosomes and is required for infectivity [12]. The VP2 protein is not required for infection or assembly [13]. The core of VP3 protein is formed by a conserved β-barrel motif consisting of antiparallel β-sheets. Other parvoviruses have this pattern, but the interstrand loops are variable, and they determine receptor usage and serology [14]. Understanding the molecular interactions of virus particles has relied heavily on structural and genetic data. X-ray crystallography and cryo-electron microscopy have been used to identify the structural images of numerous AAV capsids [14,15,16,17]. At the threefold axis, where proteins join together to create three clusters of peaks on the virion’s surface structure, there are several interactions between capsid subunits [14].
The AAV genome is a 4.7 kb molecule of single-stranded DNA. The positive and negative strands are packaged in separate premade particles with equal efficiency. Inverted terminal repeats (ITRs) create T-shaped, base-paired hairpin structures at each genome end and include cis-elements necessary for replication and packaging. Four nonstructural proteins necessary for replication (Rep78, Rep68, Rep52, and Rep40) and three structural proteins that make up the capsid (VP1, VP2, and VP3) are encoded by two genes (rep and cap). The p5, p19, and p40 viral promoters are recognized by their relative map location within the viral genome. Although different AAV serotypes have varied transcription patterns, all AAV2 transcripts have a single intron [18]. Rep78 and Rep52 are encoded by unspliced RNAs, whereas Rep68 and Rep40 are expressed by spliced RNAs (Fig. 1).

Transcriptional map of the AAV genome for major viral structural and nonstructural proteins. Spliced and unspliced forms of all three major transcripts are shown (for details, see text).
After binding to a primary receptor, AAV enters the cell via endocytosis [19]. It was discovered that various serotypes attach to distinct cell receptors. AAV2, AAV3, and AAV6 bind to heparan sulfate proteoglycan (HSPG), AAV1 to sialic acids, and AAV9 to N-linked galactose [19]. Coreceptors were proposed to have a role in AAV attachment and uptake, but further investigations failed to substantiate their importance [20,21,22,23]. Remarkably, AAV2 isolated directly from human tissues did not attach to HSPG, suggesting that lab strains have adapted to cell culture and that other AAV2 receptors exist in vivo [24]. A transmembrane protein termed AAV receptor (AAVR) was a necessary component for AAV transduction for many serotypes in a previous genetic screen [21]. In addition, GPR108 was another critical component for AAV entrance, which was suggested to function downstream in the same pathway as AAVR [25]. AAVR is found on the cell surface and is carried to the trans-Golgi network (TGN) in a retrograde endosomal way. Several endocytic routes have been proposed to play a role during the entrance. However, the clathrin-independent carrier (CLIC)/GPI-anchored protein-enriched early endosomal compartments (GEEC) pathway has been proven to be the most important endocytic route of infection [26, 27].
The precise method of AAV tracking from early endosomes to the cytoplasm is unknown. According to one scenario, AAV is carried from early endosomes to the TGN/Golgi apparatus, where it escapes and reaches the nucleus [28]. The transfer of AAV2 to the Golgi apparatus was revealed to be crucial for transduction, giving support to the hypothesis of retrograde endosomal transport. Retrograde transport via the endosomal system is a highly controlled and selective mechanism that allows the cell to recover and recycle proteins and lipids from the plasma membrane, allowing them to be localized to the Golgi (TGN and Golgi apparatus) and the endoplasmic reticulum (ER). Low endosomal pH and the action of proteases cause a conformational shift in the AAV capsid, exposing the N-terminal region of the major capsid protein, VP1 [29]. This so-called VP1 unique region (VP1u) comprises a phospholipase A2 domain (PLA2) and also a nuclear localization signal, enabling escape into the cytoplasm as well as nuclear import [12, 30,31,32]. Once within the nucleus, AAV2 was shown to accumulate in the nucleoli in an infectious form [33]. The process of AAV uncoating is poorly understood and appears to be a limiting step in AAV transduction [34, 35]. AAV may integrate into the host genome in the presence of Rep78, primarily at a locus on chromosome 19 designated AAVS1 [36, 37]. Although AAV genome integration is possible, multiple studies have demonstrated that integration inside AAVS1 and outside is rare, with just 0.1–0.5% of added infectious particles integrating [38, 39]. In cell culture and in vivo, the genomes of AAV-derived viral vectors were discovered to circularize over time, confirming the idea of a primarily circular episomal state during latency [40]. The binding of Rep78 and Rep68 to a particular region inside the p5 promoter, known as the Rep binding element (RBE), inhibits transcription during AAV latency, whereas the binding to the RBE sequence within the ITR stimulates transcription [41]. AAV enters its lytic stage after co-infection with a helper virus, which results in genome amplification and packaging.
During the construction of AAV-based vectors for gene therapy, a significant portion of the native AAV genome is deleted and the rep and cap genes are provided in trans. Briefly, rAAV vectors are commonly produced by triple plasmid transfection providing the AAV ITRs flanking a transgene cassette with a therapeutic gene of interest, the AAV rep and cap genes, and helper virus genes. The rAAV genomes are replicated and packaged into AAV capsids, which can be purified by different methods [42].
AAV infection in humans
Even though AAV has been studied for over 50 years, little is known about the virus’s natural infection. This fact may be even more surprising given that anti-AAV antibodies are found in up to 80% of the human population [6, 7]. AAV has been found in human blood cells, cervix uteri, penis, semen, liver, epithelial cell brushings, endometrium, amniotic fluid, and abortion material [43,44,45,46]. AAV may be transmitted through direct contact with an infected individual or indirect contact with the contaminated environment. Transmission routes include respiratory, gastrointestinal, and possibly sexual transmission. A concern for vertical transmission from mother to fetus also exists [46,47,48].
Very few studies addressing the impact of AAV infection on human health are available, and the results are conflicting. In this review, investigations on this subject were grouped into three major research topics (AAV infection and cervical cancer; AAV infection and reproductive system disorders; AAV infection and liver cancer), and were considered in the subsections below.
AAV infection and cervical cancer
With a reported 604,127 new cases and 341,831 deaths in 2020, cervical cancer is predicted to be the fourth leading cause of cancer and the third highest cause of cancer-associated mortality in women worldwide [49].
It has been proposed that AAV can be sexually transmitted, possibly in combination with herpes or papilloma viruses [54, 57]. High-risk human papillomavirus (HPV) infection plays a critical role in the natural history of cervical cancer [50]. During HPV infection, two viral oncogenic proteins, E6 and E7, play a crucial role in the development of cervical cancer by interacting with p53 and retinoblastoma protein (pRb) to render these cellular regulatory proteins inactive [51, 52]. AAV has been demonstrated to suppress HPV-induced cell transformation in vitro, which is mediated by the AAV Rep 78 protein [53,54,55]. Furthermore, the expression of H-ras and H-fos genes along with HPV oncogenes is inhibited by Rep 78 [56]. These collective studies suggest that AAV could be directly related to the suppression of HPV infection and may be thus associated with a reduced risk for HPV-related cervical neoplasia.
In line with the idea of a protective effect, Mayor et al. (1976) demonstrated a less likelihood of cervical cancer development in HPV-infected individuals in the presence of AAV since 85% of healthy women showed to be seropositive for AAV, while in cervical cancer patients AAV antibodies could only be detected in 14% of the cases [57]. Consistent with this observation, Georg-Fries et al. (1984) showed that sera from patients with cervical carcinoma revealed average titers of AAV antibodies well below those of age-matched control groups [58]. It was shown by Coker et al. (2001) that AAV positivity was significantly associated with a decreased risk of high-grade squamous intraepithelial lesion, suggesting that AAV may play a protective or inhibitory role in late-stage cervical carcinogenesis [59]. Furthermore, Agorastos et al. (2008) found that the prevalence of AAV was significantly lower in HPV-positive than HPV-negative patients (P = 0.0009, P = 0.00001, and P = 0.0225, for women with low-grade cervical lesions, high-grade cervical lesions, and cervical cancer, respectively). In contrast, no difference in the frequency of AAV DNA between HPV-positive and HPV-negative unaffected (control) women was observed [60]. More recently, Freitas et al. (2012) investigated the prevalence of AAV and HPV DNAs in cervical samples of HIV-seropositive and -seronegative women in Brazil. AAV-HPV co-infected women showed a lower rate of cervical intraepithelial neoplasia development compared with those infected only with HPV. HIV infection does not appear to influence AAV prevalence or AAV-HPV co-infection [61].
On the other hand, several studies did not support the protective role of AAV in cervical tumorigenesis. Strickler et al. (1999) found no relationship between AAV antibodies and the presence or grade of neoplasia in either Jamaican study subjects or women enrolled in a U.S. cervical cancer case [62]. In Odunsi et al. (2000), AAV DNA was not frequently present in either standard control cervical samples or cervical intraepithelial neoplasia, thus not supporting the hypothesis that AAV may be protective against cervical cancer [63]. Likewise, Ahn et al. (2003) found that AAV was not associated with any stages of cervical pre-cancer and cancer lesions by in situ hybridization and immunohistochemistry [64]. A case control study in pregnant and non-pregnant women found that AAV infection have any impact on cervical intraepithelial neoplasia development [65]. Moreover, a retrospective case control study by Zheng et al. (2006) found a low proportion of cervical cancer biopsies containing AAV genomes and no evidence that the presence of AAV in cervical smears of healthy women would be associated with a reduced risk of cervical cancer [66]. Finally, an Iranian study from 2017 detected a low proportion of cervical biopsies containing AAV genome (14.8% cervical cancer cases and 14% healthy controls) and no significant difference in correlation between HPV and cervical cancer in the presence or absence of AAV infection was found [67]. A description of the studies assessing the influence of AAV infection in cervical tumorigenesis is shown in Table 1.
AAV infection and reproductive system disorders
Several infections have been linked to miscarriage and other adverse reproductive outcomes. Specifically, 15% of early miscarriages and 66% of late miscarriages have been attributed to infections [68]. In particular, the common occurrence of AAV DNA (and virions) in male and female genital tissues has given rise to the hypothesis that genital AAV infection may be linked to unfavorable impacts on reproduction, including placental complications, spontaneous abortion, and fertility disorders [46, 69,70,71,72]. AAV was found fatal for mouse embryos at the early stages of gestation, and transplacental infection was established [73, 74]. In humans, AAV DNA was detected by Tobiasch et al. (1994) in 40% of abortion material during the first trimester of pregnancy but not in the material of abortion from the second or third trimester. They suggested that AAV infection in the uterine mucosa and trophoblast cells may disturb placenta development and promote early miscarriage [46]. In agreement with previous results, Malhomme et al. (1997) showed the presence of AAV DNA in 69% of materials from early abortions [75]. Koi et al. (2001) hypothesized that viral infection of extravillous trophoblast cells might hinder placental penetration of the uterine wall, resulting in situations such as spontaneous miscarriage, pre-eclampsia, and premature birth [76]. In a case-control study by Arechavaleta-Velasco et al. (2006), AAV DNA was found more frequently in trophoblast cells from cases of severe pre-eclampsia (55%) than from normal term deliveries (19%, P = 0.002) [70]. Two years later, they demonstrated that primary or reactivated AAV infection (maternal IgM seropositivity) early in pregnancy was associated with adverse reproductive outcomes linked to placental dysfunction, including pre-eclampsia, stillbirth, and spontaneous preterm delivery [69]. Kiehl et al. (2002) demonstrated AAV infection in embryo-derived tissue from Brazilian patients and further suggested a role of AAV in miscarriage and trophoblastic disease [71]. Also, in Brazil, Pereira et al. (2010) detected AAV DNA in 28.6% and 2.4% (P < 0.05) of the spontaneous and intentional abortions, respectively, suggesting an association between AAV and spontaneous miscarriage [72]. In contrast to these findings, Friedman-Einat et al. (1997) and Matovina et al. (2004) did not detect AAV DNA in any case of spontaneous abortion [77, 78]. Consistent with these studies, Sayyadi-Dehno et al. (2019), by analyzing the presence of AAV DNA in 81 therapeutic and 83 spontaneous abortions, found no statistically significant difference between the two groups [45].
Concerning to fertility factors, Rohde et al. demonstrated for the first time in 1999 the occurrence of AAV infection in human semen and suggested that sperm motility may be affected by the presence of AAV [79]. In 2001, Erles et al. showed an increased incidence of AAV infection with abnormal semen analysis, suggesting a role for AAV infection in male infertility, possibly interfering with spermatozoa development [80]. Furthermore, Mehrle et al. (2004) demonstrated that AAV DNA is integrated into testis tissue samples [81]. In contrast, Schlehofer et al. (2012) investigated AAV DNA in semen samples and endocervical material of 280 individuals of subfertile couples (146 males and 134 females), and no associations between AAV and other infectious pathogens, semen quality or subsequent fertility issues were indicated [82]. The studies investigating the association between AAV infection and adverse reproductive outcomes in humans are described in Table 2.
AAV infection and hepatocellular carcinoma
Liver cancer is expected to be the sixth most significant cause of cancer and the third leading cause of cancer-related mortality globally in 2020, with a reported 905,677 new cases and 830,180 deaths, posing a severe global health burden [83]. Hepatocellular carcinoma (HCC) represents 90% of all primary liver tumors [84]. HCC most commonly occurs due to chronic liver inflammation leading to liver cirrhosis, mainly caused by viral hepatitis, alcohol misuse, and nonalcoholic fatty liver disease [85]. The most common risk factor for developing HCC, accounting for around 50% of cases, is chronic infection with the hepatitis B virus (HBV) [86]. The evolution of a liver disease in HBV-infected patients (as well as for hepatitis C) is explained by the constant and unsuccessful attempts of the immune system to clear the virus, resulting in chronic liver damage [87]. However, apart from this common indirect mechanism, the development of 10 to 30% of HBV-associated HCC in a liver without cirrhosis suggests that HBV may also have direct oncogenic properties [88]. One of the genomic features of HBV-related HCC is the presence of frequent viral integrations in genes involved in carcinogenesis such as TERT, MLL4, and CCNE1 [88].
It has been demonstrated that the liver is the main site of infection of AAV [47]. Interestingly, in small sets of patients with HCC, AAV showed insertional oncogenic mutagenesis similar to HBV, with a common hot spot of viral insertion within TERT, CCNE1, and CCNA2 cancer driver genes, leading to their overexpression [44, 89, 90]. Two different mechanisms explained the oncogenic effects of AAV clonal integrations. First, the integrated AAV sequence typically comprises transcription factor binding sites as well as viral enhancers [91], which causes an oncogene to be strongly overexpressed nearby (CCNE1 or TERT) [44, 92]. Second, due to the usage of an alternate transcription start site (TSS) or the viral poly-A site, integrated viral sequences may cause the synthesis of truncated transcripts (for integrations in CCNA2 and TNFSF10, respectively) [93]. Remarkably, a region common to all inserted AAV sequences has recently been identified as a liver-specific enhancer-promoter element [91]. Although this region is missing in the majority of rAAV vectors currently used for gene therapy, animal studies have shown that integrated rAAV vectors can cause clonal growth and play a role in carcinogenesis [94,95,96]. On the other hand, no evidence of malignancy was observed from hundreds of normal mice treated with rAAV vectors [97, 98]. Likewise, big animals treated with relatively high doses of rAAV vectors, such as dogs and nonhuman primates, showed no malignancy [99, 100].
In La Bella et al. (2019), the analysis of tumor and non-tumor liver tissues of 1,461 patients revealed the presence of AAV DNA in 21% of patients, mainly in young women without cirrhosis [89]. AAV was found in only 8% of the examined tumors; the proportion was the same in malignant and benign tumors. However, the malignant tumors had more copies per cell due to clonal AAV integrations. In 2% of the HCC cohort, 19 AAV clonal integrations were found, and hardly any episomal virus was detected. All of these HCCs originated on non-cirrhotic livers with no known other etiologies. This study demonstrated that, although rare, AAV integration in specific regions of the human genome can promote hepatocarcinogenesis in a non-cirrhotic liver, confirming that AAV is associated with HCC occurrence without other etiologies via an insertional mutagenesis mechanism [89]. Additionally, Park et al. (2016) found AAV DNA in only 0.7% (n = 2/289) of Korean patients with HCC. Although the two patients showed no signs of liver cirrhosis, both presented other HCC risk factors, such as chronic HBV infection. The possibility of an interaction between HBV and AAV on hepatocarcinogenesis was suggested [101]. Tatsuno et al. (2019) performed virome capture sequencing using HCC and liver samples obtained from patients with prior HBV or chronic HBV infection to investigate the integration of HBV and AAV into the human genome as a possible oncogenic event. In this study, CCNE1 and CCNA2 were transcriptionally activated by AAV in prior HBV infection, suggesting that despite the seroclearance of HBV surface antigen, such patients are at risk of developing HCC [90]. Table 3 summarizes the studies demonstrating the association between AAV infection and the development of HCC in humans.
Conclusion
AAV-based gene therapy has significantly benefited from the fundamental knowledge developed in AAV research over the past 30 years. This progress has led to the development of several rAAV vectors, that are either undergoing clinical trials or have already received FDA approval, including Glybera, Luxturna, and Zolgensma. However, given that 35–80% of the world’s population is seropositive for neutralizing antibodies against one or more forms of AAV, it is remarkable that many issues concerning the AAV life cycle in vivo remain unanswered. Due to the intricate nature of the cause-and-effect link, it is challenging to make any firm inferences about the impact of AAV infection on human health. Although AAV is considered non-pathogenic, several reports describe AAV infection in association with adverse reproductive outcomes. The AAV link with tumor development is controversial. Some studies report an oncogenic effect of AAV infection (as in hepatocellular carcinoma), and others suggest a tumor suppressive role (as in HPV-related cervical cancer). Of note, natural infections with wild-type AAV have no demonstrable connection with the administration of current rAAV vectors since a significant portion of the native AAV genome is deleted during the construction of AAV-based vectors for gene therapy. In addition, no adverse event, including cancer of any kind, has ever been documented in clinical studies performed thus far using rAAV vectors [105,106]. Nevertheless, considering the extensive usage of rAAV vectors in liver-targeted gene therapy and its potential for insertion into the human genome, patients treated with rAAV vectors should be followed longitudinally to monitor long-term consequences and determine the risk of HCC development.
Data availability
Not applicable.
References
Atchison RW, Casto BC, Hammon WM. Adenovirus-Associated Defective Virus Particles. Science. 1965;149:754–6.
Hoggan MD, Blacklow NR, Rowe WP. Studies of small DNA viruses found in various adenovirus preparations: physical, biological, and immunological characteristics. Proc Natl Acad Sci U S A. 1966;55:1467–74.
Yla-Herttuala S. Endgame: glybera finally recommended for approval as the first gene therapy drug in the European union. Mol Ther. 2012;20:1831–2.
Chandler RJ, Sands MS, Venditti CP. Recombinant Adeno-Associated Viral Integration and Genotoxicity: Insights from Animal Models. Hum Gene Ther. 2017;28:314–22.
Srivastava A. In vivo tissue-tropism of adeno-associated viral vectors. Curr Opin Virol. 2016;21:75–80.
Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis. 2009;199:381–90.
Erles K, Sebokova P, Schlehofer JR. Update on the prevalence of serum antibodies (IgG and IgM) to adeno-associated virus (AAV). J Med Virol. 1999;59:406–11.
Meier AF, Fraefel C, Seyffert M. The Interplay between Adeno-Associated Virus and its Helper Viruses. Viruses 2020, 12.
Wang Z, Deng X, Zou W, Engelhardt JF, Yan Z, Qiu J. Human Bocavirus 1 Is a Novel Helper for Adeno-associated Virus Replication. J Virol 2017, 91.
Yakobson B, Koch T, Winocour E. Replication of adeno-associated virus in synchronized cells without the addition of a helper virus. J Virol. 1987;61:972–81.
Yalkinoglu AO, Heilbronn R, Burkle A, Schlehofer JR, zur Hausen H. DNA amplification of adeno-associated virus as a response to cellular genotoxic stress. Cancer Res. 1988;48:3123–9.
Girod A, Wobus CE, Zadori Z, Ried M, Leike K, Tijssen P, Kleinschmidt JA, Hallek M. The VP1 capsid protein of adeno-associated virus type 2 is carrying a phospholipase A2 domain required for virus infectivity. J Gen Virol. 2002;83:973–8.
Warrington KH Jr, Gorbatyuk OS, Harrison JK, Opie SR, Zolotukhin S, Muzyczka N. Adeno-associated virus type 2 VP2 capsid protein is nonessential and can tolerate large peptide insertions at its N terminus. J Virol. 2004;78:6595–609.
Xie Q, Bu W, Bhatia S, Hare J, Somasundaram T, Azzi A, Chapman MS. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc Natl Acad Sci U S A. 2002;99:10405–10.
Nam HJ, Lane MD, Padron E, Gurda B, McKenna R, Kohlbrenner E, Aslanidi G, Byrne B, Muzyczka N, Zolotukhin S, Agbandje-McKenna M. Structure of adeno-associated virus serotype 8, a gene therapy vector. J Virol. 2007;81:12260–71.
Padron E, Bowman V, Kaludov N, Govindasamy L, Levy H, Nick P, McKenna R, Muzyczka N, Chiorini JA, Baker TS, Agbandje-McKenna M. Structure of adeno-associated virus type 4. J Virol. 2005;79:5047–58.
Walters RW, Agbandje-McKenna M, Bowman VD, Moninger TO, Olson NH, Seiler M, Chiorini JA, Baker TS, Zabner J. Structure of adeno-associated virus serotype 5. J Virol. 2004;78:3361–71.
Qiu J, Pintel D. Processing of adeno-associated virus RNA. Front Biosci. 2008;13:3101–15.
Nonnenmacher M, Weber T. Intracellular transport of recombinant adeno-associated virus vectors. Gene Ther. 2012;19:649–58.
Akache B, Grimm D, Shen X, Fuess S, Yant SR, Glazer DS, Park J, Kay MA. A two-hybrid screen identifies cathepsins B and L as uncoating factors for adeno-associated virus 2 and 8. Mol Ther. 2007;15:330–9.
Pillay S, Meyer NL, Puschnik AS, Davulcu O, Diep J, Ishikawa Y, Jae LT, Wosen JE, Nagamine CM, Chapman MS, Carette JE. An essential receptor for adeno-associated virus infection. Nature. 2016;530:108–12.
Qiu J, Brown KE. Integrin alphaVbeta5 is not involved in adeno-associated virus type 2 (AAV2) infection. Virology. 1999;264:436–40.
Summerford C, Bartlett JS, Samulski RJ. AlphaVbeta5 integrin: a co-receptor for adeno-associated virus type 2 infection. Nat Med. 1999;5:78–82.
Chen CL, Jensen RL, Schnepp BC, Connell MJ, Shell R, Sferra TJ, Bartlett JS, Clark KR, Johnson PR. Molecular characterization of adeno-associated viruses infecting children. J Virol. 2005;79:14781–92.
Dudek AM, Zabaleta N, Zinn E, Pillay S, Zengel J, Porter C, Franceschini JS, Estelien R, Carette JE, Zhou GL, Vandenberghe LH. GPR108 Is a Highly Conserved AAV Entry Factor. Mol Ther. 2020;28:367–81.
Bartlett JS, Wilcher R, Samulski RJ. Infectious entry pathway of adeno-associated virus and adeno-associated virus vectors. J Virol. 2000;74:2777–85.
Nonnenmacher M, Weber T. Adeno-associated virus 2 infection requires endocytosis through the CLIC/GEEC pathway. Cell Host Microbe. 2011;10:563–76.
Nonnenmacher ME, Cintrat JC, Gillet D, Weber T. Syntaxin 5-dependent retrograde transport to the trans-Golgi network is required for adeno-associated virus transduction. J Virol. 2015;89:1673–87.
Sonntag F, Bleker S, Leuchs B, Fischer R, Kleinschmidt JA. Adeno-associated virus type 2 capsids with externalized VP1/VP2 trafficking domains are generated prior to passage through the cytoplasm and are maintained until uncoating occurs in the nucleus. J Virol. 2006;80:11040–54.
Grieger JC, Snowdy S, Samulski RJ. Separate basic region motifs within the adeno-associated virus capsid proteins are essential for infectivity and assembly. J Virol. 2006;80:5199–210.
Stahnke S, Lux K, Uhrig S, Kreppel F, Hosel M, Coutelle O, Ogris M, Hallek M, Buning H. Intrinsic phospholipase A2 activity of adeno-associated virus is involved in endosomal escape of incoming particles. Virology. 2011;409:77–83.
Zadori Z, Szelei J, Lacoste MC, Li Y, Gariepy S, Raymond P, Allaire M, Nabi IR, Tijssen P. A viral phospholipase A2 is required for parvovirus infectivity. Dev Cell. 2001;1:291–302.
Johnson JS, Samulski RJ. Enhancement of adeno-associated virus infection by mobilizing capsids into and out of the nucleolus. J Virol. 2009;83:2632–44.
Rossi A, Dupaty L, Aillot L, Zhang L, Gallien C, Hallek M, Odenthal M, Adriouch S, Salvetti A, Buning H. Vector uncoating limits adeno-associated viral vector-mediated transduction of human dendritic cells and vector immunogenicity. Sci Rep. 2019;9:3631.
Thomas CE, Storm TA, Huang Z, Kay MA. Rapid uncoating of vector genomes is the key to efficient liver transduction with pseudotyped adeno-associated virus vectors. J Virol. 2004;78:3110–22.
Cheung AK, Hoggan MD, Hauswirth WW, Berns KI. Integration of the adeno-associated virus genome into cellular DNA in latently infected human Detroit 6 cells. J Virol. 1980;33:739–48.
Dyall J, Szabo P, Berns KI. Adeno-associated virus (AAV) site-specific integration: formation of AAV-AAVS1 junctions in an in vitro system. Proc Natl Acad Sci U S A. 1999;96:12849–54.
Huser D, Weger S, Heilbronn R. Kinetics and frequency of adeno-associated virus site-specific integration into human chromosome 19 monitored by quantitative real-time PCR. J Virol. 2002;76:7554–9.
McCarty DM, Young SM Jr, Samulski RJ. Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu Rev Genet. 2004;38:819–45.
Sun X, Lu Y, Bish LT, Calcedo R, Wilson JM, Gao G. Molecular analysis of vector genome structures after liver transduction by conventional and self-complementary adeno-associated viral serotype vectors in murine and nonhuman primate models. Hum Gene Ther. 2010;21:750–61.
Pereira DJ, McCarty DM, Muzyczka N. The adeno-associated virus (AAV) Rep protein acts as both a repressor and an activator to regulate AAV transcription during a productive infection. J Virol. 1997;71:1079–88.
Zolotukhin S. Production of recombinant adeno-associated virus vectors. Hum Gene Ther. 2005;16:551–7.
Grossman Z, Mendelson E, Brok-Simoni F, Mileguir F, Leitner Y, Rechavi G, Ramot B. Detection of adeno-associated virus type 2 in human peripheral blood cells. J Gen Virol. 1992;73(Pt 4):961–6.
Nault JC, Datta S, Imbeaud S, Franconi A, Mallet M, Couchy G, Letouze E, Pilati C, Verret B, Blanc JF, et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat Genet. 2015;47:1187–93.
Sayyadi-Dehno Z, Seyed Khorrami SM, Ghavami N, Ghotbi-Zadeh F, Khushideh M, Hosseini M, Malekshahi SS, Shafiei-Jandaghi NZ. Molecular Detection of Adeno-Associated Virus DNA in Cases of Spontaneous and Therapeutic Abortion. Fetal Pediatr Pathol. 2019;38:206–14.
Tobiasch E, Rabreau M, Geletneky K, Larue-Charlus S, Severin F, Becker N, Schlehofer JR. Detection of adeno-associated virus DNA in human genital tissue and in material from spontaneous abortion. J Med Virol. 1994;44:215–22.
Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, Wilson JM. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol. 2004;78:6381–8.
Walz CM, Anisi TR, Schlehofer JR, Gissmann L, Schneider A, Muller M. Detection of infectious adeno-associated virus particles in human cervical biopsies. Virology. 1998;247:97–105.
International Agency for Research on Cancer. Globocan 2020. 23-Cervix-uteri-fact-sheet.pdf. https://gco.iarc.fr/today/data/factsheets/cancers/23-Cervix-uteri-fact-sheet.pdf.
zur Hausen H. Papillomaviruses causing cancer: evasion from host-cell control in early events in carcinogenesis. J Natl Cancer Inst. 2000;92:690–8.
Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63:1129–1136.
Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76–9.
Hermonat PL. Adeno-associated virus inhibits human papillomavirus type 16: a viral interaction implicated in cervical cancer. Cancer Res. 1994;54:2278–81.
Hermonat PL, Plott RT, Santin AD, Parham GP, Flick JT. Adeno-associated virus Rep78 inhibits oncogenic transformation of primary human keratinocytes by a human papillomavirus type 16-ras chimeric. Gynecol Oncol. 1997;66:487–94.
Walz CM, Correa-Ochoa MM, Muller M, Schlehofer JR. Adenoassociated virus type 2-induced inhibition of the human papillomavirus type 18 promoter in transgenic mice. Virology. 2002;293:172–81.
Hermonat PL. Down-regulation of the human c-fos and c-myc proto-oncogene promoters by adeno-associated virus Rep78. Cancer Lett. 1994;81:129–36.
Mayor HD, Drake S, Stahmann J, Mumford DM. Antibodies to adeno-associated satellite virus and herpes simplex in sera from cancer patients and normal adults. Am J Obstet Gynecol. 1976;126:100–4.
Georg-Fries B, Biederlack S, Wolf J, zur Hausen H. Analysis of proteins, helper dependence, and seroepidemiology of a new human parvovirus. Virology. 1984;134:64–71.
Coker AL, Russell RB, Bond SM, Pirisi L, Liu Y, Mane M, Kokorina N, Gerasimova T, Hermonat PL. Adeno-associated virus is associated with a lower risk of high-grade cervical neoplasia. Exp Mol Pathol. 2001;70:83–9.
Agorastos T, Chrisafi S, Lambropoulos AF, Mikos T, Constandinides TC, Schlehofer JR, Schlehofer B, Kotsis A, Bontis JN. Adeno-associated virus infection and cervical neoplasia: is there a protective role against human papillomavirus-related carcinogenesis? Eur J Cancer Prev. 2008;17:364–8.
Freitas LB, Tonani de Mattos A, Lima BM, Miranda AE, Spano LC. Adeno-associated virus may play a protective role against human papillomavirus-induced cervical lesions independent of HIV serostatus. Int J STD AIDS. 2012;23:258–61.
Strickler HD, Viscidi R, Escoffery C, Rattray C, Kotloff KL, Goldberg J, Manns A, Rabkin C, Daniel R, Hanchard B, et al. Adeno-associated virus and development of cervical neoplasia. J Med Virol. 1999;59:60–5.
Odunsi KO, van Ee CC, Ganesan TS, Shelling AN. Evaluation of the possible protective role of adeno-associated virus type 2 infection in HPV-associated premalignant disease of the cervix. Gynecol Oncol. 2000;78:342–5.
Ahn WS, Bae SM, Chung JE, Lee HK, Kim BK, Lee JM, Namkoong SE, Kim CK, Sin J. Evaluation of adenoassociated virus 2 and human papilloma virus 16 and 18 infection in cervical cancer biopsies. Gynecol Oncol. 2003;89:105–11.
Grce M, Husnjak K, Matovina M, Milutin N, Magdic L, Husnjak O, Pavelic K. Human papillomavirus, cytomegalovirus, and adeno-associated virus infections in pregnant and nonpregnant women with cervical intraepithelial neoplasia. J Clin Microbiol. 2004;42:1341–4.
Zheng BY, Li XD, Wiklund F, Chowdhry S, Angstrom T, Hallmans G, Dillner J, Wallin KL. Detection of adeno-associated virus type 2 genome in cervical carcinoma. Br J Cancer. 2006;94:1913–7.
Shafiei-Jandaghi NZ, Yavarian J, Faghihloo E, Ghavami N, Ghalejoogh ZY, Kiani SJ, Malekshahi SS, Shahsiah R, Jahanzad E, Hosseini M, Azad TM. Prevalence of adeno-associated virus and human papillomavirus DNA in Iranian women with and without cervical cancer. Pathol Res Pract. 2017;213:457–60.
Giakoumelou S, Wheelhouse N, Cuschieri K, Entrican G, Howie SE, Horne AW. The role of infection in miscarriage. Hum Reprod Update. 2016;22:116–33.
Arechavaleta-Velasco F, Gomez L, Ma Y, Zhao J, McGrath CM, Sammel MD, Nelson DB, Parry S. Adverse reproductive outcomes in urban women with adeno-associated virus-2 infections in early pregnancy. Hum Reprod. 2008;23:29–36.
Arechavaleta-Velasco F, Ma Y, Zhang J, McGrath CM, Parry S. Adeno-associated virus-2 (AAV-2) causes trophoblast dysfunction, and placental AAV-2 infection is associated with preeclampsia. Am J Pathol. 2006;168:1951–9.
Kiehl K, Schlehofer JR, Schultz R, Zugaib M, Armbruster-Moraes E. Adeno-associated virus DNA in human gestational trophoblastic disease. Placenta. 2002;23:410–5.
Pereira CC, de Freitas LB, de Vargas PR, de Azevedo ML, do Nascimento JP, Spano LC. Molecular detection of adeno-associated virus in cases of spontaneous and intentional human abortion. J Med Virol. 2010;82:1689–93.
Botquin V, Cid-Arregui A, Schlehofer JR. Adeno-associated virus type 2 interferes with early development of mouse embryos. J Gen Virol. 1994;75(Pt 10):2655–62.
Lipps BV, Mayor HD. Transplacental infection with adeno-associated virus type 1 in mice. Intervirology. 1980;14:118–23.
Malhomme O, Dutheil N, Rabreau M, Armbruster-Moraes E, Schlehofer JR, Dupressoir T. Human genital tissues containing DNA of adeno-associated virus lack DNA sequences of the helper viruses adenovirus, herpes simplex virus or cytomegalovirus but frequently contain human papillomavirus DNA. J Gen Virol. 1997;78(Pt 8):1957–62.
Koi H, Zhang J, Parry S. The mechanisms of placental viral infection. Ann N Y Acad Sci. 2001;943:148–56.
Friedman-Einat M, Grossman Z, Mileguir F, Smetana Z, Ashkenazi M, Barkai G, Varsano N, Glick E, Mendelson E. Detection of adeno-associated virus type 2 sequences in the human genital tract. J Clin Microbiol. 1997;35:71–8.
Matovina M, Husnjak K, Milutin N, Ciglar S, Grce M. Possible role of bacterial and viral infections in miscarriages. Fertil Steril. 2004;81:662–9.
Rohde V, Erles K, Sattler HP, Derouet H, Wullich B, Schlehofer JR. Detection of adeno-associated virus in human semen: does viral infection play a role in the pathogenesis of male infertility? Fertil Steril. 1999;72:814–6.
Erles K, Rohde V, Thaele M, Roth S, Edler L, Schlehofer JR. DNA of adeno-associated virus (AAV) in testicular tissue and in abnormal semen samples. Hum Reprod. 2001;16:2333–7.
Mehrle S, Rohde V, Schlehofer JR. Evidence of chromosomal integration of AAV DNA in human testis tissue. Virus Genes. 2004;28:61–9.
Schlehofer JR, Boeke C, Reuland M, Eggert-Kruse W. Presence of DNA of adeno-associated virus in subfertile couples, but no association with fertility factors. Hum Reprod. 2012;27:770–8.
International Agency for Research on Cancer. Globocan 2020. 11-Liver-fact-sheet.pdf. https://gco.iarc.fr/today/data/factsheets/cancers/11-Liver-fact-sheet.pdf.
Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Zucman-Rossi J, Finn RS. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7:6.
Fujiwara N, Friedman SL, Goossens N, Hoshida Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J Hepatol. 2018;68:526–49.
Global Burden of Disease Liver. Cancer C, Akinyemiju T, Abera S, Ahmed M, Alam N, Alemayohu MA, Allen C, Al-Raddadi R, Alvis-Guzman N, Amoako Y, et al: The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level: Results From the Global Burden of Disease Study 2015. JAMA Oncol 2017, 3:1683–91.
Shirvani-Dastgerdi E, Schwartz RE, Ploss A. Hepatocarcinogenesis associated with hepatitis B, delta and C viruses. Curr Opin Virol. 2016;20:1–10.
Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol. 2016;64:84–101.
La Bella T, Imbeaud S, Peneau C, Mami I, Datta S, Bayard Q, Caruso S, Hirsch TZ, Calderaro J, Morcrette G, et al. Adeno-associated virus in the liver: natural history and consequences in tumour development. Gut. 2020;69:737–47.
Tatsuno K, Midorikawa Y, Takayama T, Yamamoto S, Nagae G, Moriyama M, Nakagawa H, Koike K, Moriya K, Aburatani H. Impact of AAV2 and Hepatitis B Virus Integration Into Genome on Development of Hepatocellular Carcinoma in Patients with Prior Hepatitis B Virus Infection. Clin Cancer Res. 2019;25:6217–27.
Logan GJ, Dane AP, Hallwirth CV, Smyth CM, Wilkie EE, Amaya AK, Zhu E, Khandekar N, Ginn SL, Liao SHY, et al. Identification of liver-specific enhancer-promoter activity in the 3’ untranslated region of the wild-type AAV2 genome. Nat Genet. 2017;49:1267–73.
Bayard Q, Meunier L, Peneau C, Renault V, Shinde J, Nault JC, Mami I, Couchy G, Amaddeo G, Tubacher E, et al. Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat Commun. 2018;9:5235.
Peneau C, Zucman-Rossi J, Nault JC. Genomics of Viral Hepatitis-Associated Liver Tumors. J Clin Med 2021, 10.
Chandler RJ, LaFave MC, Varshney GK, Trivedi NS, Carrillo-Carrasco N, Senac JS, Wu W, Hoffmann V, Elkahloun AG, Burgess SM, Venditti CP. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J Clin Invest. 2015;125:870–80.
Donsante A, Miller DG, Li Y, Vogler C, Brunt EM, Russell DW, Sands MS. AAV vector integration sites in mouse hepatocellular carcinoma. Science. 2007;317:477.
Nguyen GN, Everett JK, Kafle S, Roche AM, Raymond HE, Leiby J, Wood C, Assenmacher CA, Merricks EP, Long CT, et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat Biotechnol. 2021;39:47–55.
Bell P, Moscioni AD, McCarter RJ, Wu D, Gao G, Hoang A, Sanmiguel JC, Sun X, Wivel NA, Raper SE, et al. Analysis of tumors arising in male B6C3F1 mice with and without AAV vector delivery to liver. Mol Ther. 2006;14:34–44.
Li H, Malani N, Hamilton SR, Schlachterman A, Bussadori G, Edmonson SE, Shah R, Arruda VR, Mingozzi F, Wright JF, et al. Assessing the potential for AAV vector genotoxicity in a murine model. Blood. 2011;117:3311–9.
Li S, Ling C, Zhong L, Li M, Su Q, He R, Tang Q, Greiner DL, Shultz LD, Brehm MA, et al. Efficient and Targeted Transduction of Nonhuman Primate Liver With Systemically Delivered Optimized AAV3B Vectors. Mol Ther. 2015;23:1867–76.
Nichols TC, Whitford MH, Arruda VR, Stedman HH, Kay MA, High KA. Translational data from adeno-associated virus-mediated gene therapy of hemophilia B in dogs. Hum Gene Ther Clin Dev. 2015;26:5–14.
Park KJ, Lee J, Park JH, Joh JW, Kwon CH, Kim JW. Adeno-Associated Virus 2-Mediated Hepatocellular Carcinoma is Very Rare in Korean Patients. Ann Lab Med. 2016;36:469–74.
Acknowledgements
Not applicable.
Funding
This research was funded by Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), grant number E-26/210.450/2019.
Author information
Authors and Affiliations
Contributions
TBS and NMA designed the study, searched and collected the literature, wrote, revised, and finalized the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sant’Anna, T.B., Araujo, N.M. Adeno-associated virus infection and its impact in human health: an overview. Virol J 19, 173 (2022). https://doi.org/10.1186/s12985-022-01900-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-022-01900-4